

Abstract
Objective
To evaluate the potential for pharmacokinetic interaction and the safety and tolerability when ubrogepant and sumatriptan are coadministered in a Phase 1 study in healthy participants, and to inform the safety and tolerability of ubrogepant alone and in combination with triptans in Phase 3 trials in participants with migraine.
Background
Calcitonin gene–related peptide is a potent vasodilatory neurotransmitter believed to play a key role in the pathophysiology of migraine. Ubrogepant (UBRELVY™) is a potent and selective antagonist of the human calcitonin gene–related peptide receptor approved for the acute treatment of migraine. Sumatriptan is a serotonin receptor agonist and the most commonly used triptan for the acute treatment of migraine. Ubrogepant could be prescribed with triptans.
Design
The Phase 1 study was a single‐center, open‐label, randomized, 3‐way crossover, single‐dose, pharmacokinetic interaction study, where participants received each of 3 oral treatments with a 7‐day washout period between treatments: single dose of ubrogepant 100 mg, single dose of sumatriptan 100 mg, and ubrogepant 100 mg plus sumatriptan 100 mg. Pharmacokinetic parameters were calculated using a model‐independent approach. The ACHIEVE I and II trials were 2 multicenter, single‐attack, randomized, Phase 3 trials in adults with a history of migraine with or without aura. Participants had the option to take a second dose of study medication or rescue medication to treat a nonresponding migraine or a migraine recurrence from 2 to 48 hours after the initial dose of study medication. Rescue medication options included acetaminophen, nonsteroidal anti‐inflammatory drugs, opioids, anti‐emetics, or triptans. Treatment‐emergent adverse events were evaluated up to 30 days after the last dose in the Phase 1 and Phase 3 studies.
Results
Ubrogepant median time to maximum plasma concentration was delayed (3 hours [range: 1‐5 hours] vs 1.5 hours [range: 1‐4 hours]), mean maximum plasma concentration was reduced by 24% (coefficient of variation: 37.4%) when ubrogepant was coadministered with sumatriptan (n = 29) compared with ubrogepant administered alone (N = 30). No significant effect was observed on the area under the plasma concentration‐time curve of ubrogepant. Sumatriptan area under the curve and maximum plasma concentration showed no significant change when sumatriptan was coadministered with ubrogepant (n = 29), but the sumatriptan time to maximum plasma concentration was delayed (1 hour [range: 0.5‐5 hours] vs 3 hours [range: 0.5‐6 hours]. No treatment‐emergent adverse events were reported with the coadministration of ubrogepant 100 mg and sumatriptan 100 mg in the Phase 1 study. The pooled safety data from ACHIEVE trials (N = 1938) showed similar rates of treatment‐related treatment‐emergent adverse events between participants who took ubrogepant alone and participants who took ubrogepant and a triptan as a rescue medication (14.9% [53/355] vs 12.8% [5/39] in the ubrogepant 100 mg treatment group, respectively).
Conclusions
Although there were slight alterations in ubrogepant pharmacokinetic parameters when coadministered with sumatriptan, such changes are expected to have minimal clinical relevance, especially because no changes were seen in sumatriptan area under the curve and maximum plasma concentration when coadministered with ubrogepant. Coadministration of ubrogepant with sumatriptan was well tolerated in healthy participants in the Phase 1 study, and coadministration of ubrogepant with triptans was well tolerated in participants with migraine in the Phase 3 trials. No new safety concerns for ubrogepant were identified across all trials.
Keywords: migraine attack, acute treatment, combination drug therapy, ubrogepant, sumatriptan, triptans
Abbreviations
- 5‐HT1B/1D
5‐hydroxytryptamine (serotonin)
- AE
adverse event
- AUC0‐∞
area under the plasma drug concentration vs time curve from time 0 to infinity
- AUC0‐t
area under the plasma drug concentration vs time curve from time 0 to time t
- BLOQ
below the limit of quantification
- CGRP
calcitonin gene–related peptide
- Cmax
maximum plasma drug concentration
- GMR
geometric mean ratio
- MedDRA
Medical Dictionary for Regulatory Activities
- PK
pharmacokinetic
- t½
mean apparent terminal elimination half‐life
- TEAE
treatment‐emergent adverse event
- tmax
time to maximum plasma drug concentration
Introduction
Migraine, a chronic, neurologic disease with recurrent episodes, is associated with often‐incapacitating symptoms, such as nausea, pain, photophobia, and phonophobia. 1 Sumatriptan, a 5‐hydroxytryptamine (5‐HT1B/1D) receptor agonist, is the most commonly used triptan for the acute treatment of migraine attacks. 2 , 3 Despite their utility, triptans can be ineffective in or poorly tolerated by some people with migraine and are avoided by others, including those with cardiovascular risk factors. 4
The need for novel and effective therapies has spurred research into the complex pathophysiology of migraine, which has led to the development of new treatment approaches. 5 , 6 In particular, calcitonin gene–related peptide (CGRP) and the CGRP receptor have been recognized as key mediators in the pathophysiology of migraine. 5 , 6 Blocking CGRP has emerged as a novel and specific strategy for treating migraine. 4 , 7
Ubrogepant is an oral CGRP receptor antagonist approved by the Food and Drug Administration for the acute treatment of migraine with or without aura in adults at doses of 50 and 100 mg. 8 Phase 3 trial results have shown ubrogepant to be statistically superior to placebo in providing pain relief, pain freedom, and a reduction in most bothersome symptom when used as an acute treatment for migraine attacks. 9 , 10
Individualizing acute treatment for migraine attacks is common and can involve combinations of agents. 2 , 11 Given the potential for people with migraine to take ubrogepant and sumatriptan concomitantly for acute treatment of migraine attacks, it is important to determine if there are any clinically meaningful pharmacokinetic (PK) interactions between these 2 treatments. The monoamine oxidase A isozyme is considered to be the primary enzyme that metabolizes sumatriptan, and sumatriptan is primarily excreted in the urine as major metabolite indole acetic acid. 12 The time to maximum plasma drug concentration (C max) (ie, t max) of sumatriptan is approximately 2.0 hours, and the mean C max following an oral dose of 100 mg is 51 ng/mL (range: 28‐100 ng/mL). 12 Cytochrome P450 3A4 is the primary enzyme that metabolizes ubrogepant. 13 Ubrogepant is rapidly absorbed, with a median t max of 1.5 hours. 13 A significant interaction between ubrogepant and sumatriptan is not expected based on their distinct metabolic pathways.
The primary objective of the Phase 1 study was to evaluate the potential for PK interactions between ubrogepant (100 mg) and sumatriptan (100 mg). The secondary objective was to assess the safety and tolerability profiles of ubrogepant and sumatriptan when administered alone and when coadministered in healthy participants in the Phase 1 study. Additionally, the safety and tolerability of ubrogepant administered with a triptan rescue medication after 2 hours were evaluated in participants with migraine in a pooled analysis of the ACHIEVE I and ACHIEVE II Phase 3 trials.
Methods
Phase 1 Study
Study Design
The Phase 1 study was a single‐center, single‐dose, open‐label, randomized, 3‐way crossover study to assess the potential of a PK interaction between ubrogepant and sumatriptan in healthy participants in the United States. A list of participant randomization codes was generated by Allergan plc and was provided to the investigator. Each 4‐digit randomization number corresponded to 1 of the 6 treatment sequences; block randomization with block size 6 was used. Randomization numbers were assigned sequentially. Eligible participants were randomized to receive treatments A (single oral 100 mg ubrogepant dose [2 × 50 mg hot‐melt extrudate‐oral compressed tablets]), B (single oral 100 mg sumatriptan dose [1 × 100 mg; Imitrex® Tablets, GlaxoSmithKline, Research Triangle Park, NC, USA]), and C (100 mg ubrogepant + 100 mg sumatriptan) in 1 of 6 treatment sequences (A‐B‐C, A‐C‐B, B‐A‐C, B‐C‐A, C‐A‐B, or C‐B‐A) under fasted conditions after a screening period of up to 14 days. Study treatments were administered orally on Days 1, 8, and 15. The duration of each participant’s participation in the study was up to 45 days (excluding the screening period), including a 30‐day safety follow‐up visit after the administration of the last dose of study treatment on Day 15. A washout period of 7 days occurred between treatments.
The study was conducted in accordance with the ethical principles of the Declaration of Helsinki and the International Council for Harmonisation’s Guideline for Good Clinical Practice. Institutional review board approval was obtained for the study protocol. All participants provided written informed consent. All authors had full access to the study data.
Study Population
Planned enrollment in the Phase 1 study was approximately 30 participants. Eligible participants were healthy males or females aged 18 to 45 years, with body mass index ≥18 and ≤30 kg/m2 and sitting pulse rate ≥50 and ≤100 bpm during the assessment of the vital signs at screening, and nonsmokers (never smoked or had not smoked within the previous 2 years). Males and females of childbearing potential agreed to use an effective method of contraception; if female had a negative result from a serum pregnancy test at screening and a negative result from a serum or urine pregnancy test on Day –1. Participants were excluded if they had a known hypersensitivity to CGRP receptor antagonists or sumatriptan; had sitting systolic blood pressure ≥140 or ≤90 mm Hg or sitting diastolic blood pressure ≥90 or ≤50 mm Hg at screening; had an abnormal electrocardiogram result thought to be potentially clinically significant according to the investigator, or a QT prolongation (QTcF ≥450 ms for male participants; QTcF ≥470 ms for female participants or uncorrected QT ≥500 ms) at screening; or had a positive urine test result for drugs, alcohol, or cotinine at screening or Day –1. Additional exclusion criteria included the consumption of herbal preparations containing St. John’s wort within 4 weeks before study treatment administration or any concomitant medications (including over‐the‐counter medications) within 14 days or hormonal drug products within 30 days of study treatment administration; any clinical condition or previous surgery that may have affected the absorption, distribution, biotransformation, or excretion of ubrogepant or sumatriptan; and consumption of caffeine or xanthine‐containing compounds within 48 hours, grapefruit‐containing products, vegetables from the mustard green family, or charbroiled meats within 14 days, or alcohol within 72 hours before the administration of study treatment. Participants were also excluded if they had a clinically significant disease, in the opinion of the examining physician, in any body system, or were breastfeeding.
Study Procedures
In the Phase 1 study, participants checked into the study center on Days –1, 7, and 14 and stayed the nights of Days –1, 1, 7, 8, 14, and 15. All study treatments were administered under fasting conditions (ie, overnight for at least 10 hours) with 240 mL of water on Days 1, 8, and 15, with additional water restricted 1 hour before and 1 hour after administration of each dose. Participants fasted for an additional 4 hours after dosing and remained seated upright and awake. Participants were released on the mornings of Days 2, 9, and 16. Meals were provided at appropriate times each day.
Pharmacokinetic Assessments
Blood samples for the determination of plasma ubrogepant and sumatriptan concentrations in the Phase 1 study were collected on Days 1, 8, and 15 at the following time points: 0 hour (before dosing) and 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, 14, and 24 hours postdose. A total of 39 PK blood samples was intended to be drawn from each participant. Collected blood for PK analysis was centrifuged at 2500 g for 10 minutes at 4°C within 30 minutes. Plasma samples were then flash‐frozen and stored at −70°C or colder. PK plasma samples were analyzed by Algorithme Pharma (Laval, Quebec, Canada) using validated LC‐MS/MS methods for ubrogepant and sumatriptan. The lower limit of quantitation (LLOQ) for ubrogepant was 1 ng/mL and for sumatriptan was 1 ng/mL.
Pharmacokinetic and Statistical Analysis
All standard noncompartmental PK parameters were calculated using WinNonlin Phoenix® Version 8.0 (Pharsight Corporation, Mountain View, CA). Concentrations below the limit of quantification (BLOQ) were set to zero for the analyses; they mainly occurred at the last time point of 24 hours postdose for ubrogepant and at 14 and 24 hours postdose for sumatriptan. For ubrogepant, BLOQ values were observed once in 1 of 30 participants in the ubrogepant alone group and in 3 instances in 2 of 30 participants in the ubrogepant with sumatriptan treatment group. For sumatriptan, there were 15 instances in 15 of 30 participants in the sumatriptan alone group and 17 instances in 16 of 30 participants in the ubrogepant with sumatriptan treatment group.
WinNonlin Phoenix® was used to calculate descriptive statistics (mean, standard deviation, coefficient of variation, maximum, median, and minimum) for plasma concentrations and for all plasma PK parameters. C max and area under the plasma drug concentration vs time curve from time 0 to time t (AUC0‐ t) and from time 0 to infinity (AUC0‐∞) for ubrogepant and sumatriptan were compared using a linear mixed‐effects model, with treatment, period, and sequence as fixed effects and participant within sequence as a random effect. Log‐transformed values for C max and AUC parameters were used for statistical analysis. The 2‐sided 90% CI was constructed for the ratio of least squares geometric means of C max, AUC0‐ t, and AUC0‐∞ of ubrogepant in combination with sumatriptan (test) vs ubrogepant alone (reference); the same analysis was performed for sumatriptan in combination with ubrogepant vs sumatriptan alone. No effect of sumatriptan on the PK of ubrogepant or vice versa was concluded if the 90% CIs for the ratios of the PK parameters for the drugs administered in combination (test) vs drug administered alone (reference) were within the limits of 80 to 125% per the Food and Drug Administration guidance. 14 If the 90% CIs were outside the 80 to 125% range, then the totality of the data, such as the efficacious dose range and safety margins, was taken into account as per regulatory guidance recommendations. 14
Safety Assessments
Safety and tolerability in the Phase 1 study were monitored by physical examination, vital signs (blood pressure, heart rate), routine laboratory tests (hematology, blood chemistry [fasted], urinalysis), and 12‐lead electrocardiogram. Participants were monitored for adverse events (AEs) throughout the study. AE assessments were conducted using an open‐ended, non‐leading question. AEs were coded by the Medical Dictionary for Regulatory Activities (MedDRA®) version 20.1.
Pooled Phase 3 ACHIEVE Trials Safety Assessment
Full methods for the ACHIEVE I (NCT02828020) and ACHIEVE II (NCT02867709) trials have been previously described. 9 , 10 Briefly, the ACHIEVE trials were 2 multicenter, single‐attack, randomized, Phase 3 trials in adults with a history of migraine with or without aura. Data from the ubrogepant 50 mg and placebo treatment groups of ACHIEVE I and ACHIEVE II were pooled for this subanalysis. Data from the 25 and 100 mg groups were not pooled and represent data from the individual studies. An optional second dose or rescue medication was allowed for the treatment of moderate or severe headache starting from 2 to 48 hours after the initial dose of study medication. Rescue medication options included acetaminophen, nonsteroidal anti‐inflammatory drugs, opioids, anti‐emetics, or triptans. In this subanalysis, we evaluated the prevalence of treatment‐emergent AEs (TEAEs) in participants who received ubrogepant without taking any rescue medication and participants who took a triptan as a rescue medication. Eligible triptans included almotriptan, eletriptan, frovatriptan, naratriptan, rizatriptan, sumatriptan, and zolmitriptan.
Results
Phase 1 Study
Participant Disposition and Demographics
Thirty healthy participants were enrolled in the Phase 1 study, and all were included in the safety analyses. There were 30 participants in the PK population for ubrogepant, 29 in the PK population for sumatriptan, and 29 for ubrogepant coadministered with sumatriptan. One participant (assigned to sequence A‐C‐B) discontinued the study during the treatment phase after receiving a single dose of ubrogepant 100 mg and did not receive the other 2 treatments. Demographics and baseline characteristics are summarized in Table 1. Participants had a mean age of 33.2 years (range, 21 to 44 years) and there were more males (66.7%; 20/30) than females (33.3%; 10/30). A majority of participants were Hispanic (63.3%; 19/30) and a higher proportion were white (63.3%; 19/30) than were black (36.7%; 11/30).
Table 1.
Overall Participant Demographics and Baseline Characteristics
| Ubrogepant 100 mg Treatment Group (Representative of All Participants) (N = 30) | |
|---|---|
| Age,† years | |
| Mean (SD) | 33.2 (7.2) |
| Sex, n (%) | |
| Male | 10 (33.3) |
| Female | 20 (66.7) |
| Race, n (%) | |
| White | 19 (63.3) |
| Black or African‐American | 11 (36.7) |
| All other races | 0 |
| Ethnicity, n (%) | |
| Hispanic or Latino | 19 (63.3) |
| Not Hispanic or Latino | 11 (36.7) |
| Weight, kg | |
| Mean (SD) | 78.65 (12.13) |
| Height, cm | |
| Mean (SD) | 171.2 (10.35) |
| Body mass index, kg/m2 | |
| Mean (SD) | 26.75 (2.79) |
N = number of participants in the safety population.
Percentages are calculated as 100 × (n/N).
Age at screening.
Ubrogepant and Sumatriptan Pharmacokinetics
The mean (SD) plasma concentrations of ubrogepant following the administration of ubrogepant alone and in combination with sumatriptan are shown in Figure 1. For ubrogepant, the median t max was delayed by 1.5 hours, and the mean apparent terminal elimination half‐life (t 1/2) was slightly shorter when coadministered with sumatriptan than when administered alone (Table 2).
Fig. 1.
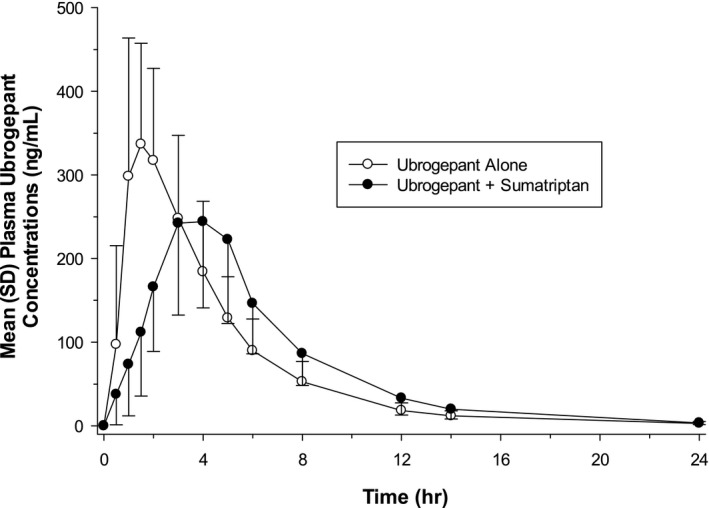
Mean (SD) plasma ubrogepant concentration‐time profile following single‐dose oral administration of 100 mg ubrogepant alone (N = 30) or in combination with single‐dose oral administration of 100 mg sumatriptan (n = 29) to fasted healthy participants (linear scale).
Table 2.
Mean (SD) Pharmacokinetic Parameters of Ubrogepant Following Single‐Dose Oral Administration of Ubrogepant Alone or in Combination With Sumatriptan in Healthy Adult Participants
| PK Parameter, Mean (SD) | Ubrogepant 100 mg (N = 30) | Ubrogepant 100 mg + Sumatriptan 100 mg (n = 29) |
|---|---|---|
| C max, ng/mL | 400.31 (149.63) | 299.71 (88.77) |
| AUC0‐ t, ng•h/mL | 1566.36 (478.03) | 1606.94 (545.96) |
| AUC0‐∞, ng•h/mL | 1590.47 (480.60) | 1628.20 (550.27) |
| t max, hours† | 1.50 (1.00‐4.00) | 3.00 (1.00‐5.00) |
| t ½, hours‡ | 4.79 (1.48) | 3.87 (0.90) |
| Vz/F, L | 478.73 (227.35) | 394.81 (210.88) |
| CL/F, L/h | 69.06 (23.57) | 69.62 (26.99) |
AUC0‐∞ = area under the plasma drug concentration vs time curve from time 0 to infinity; AUC0‐ t = area under the plasma drug concentration vs time curve from time 0 to time t; CL/F = apparent total clearance of the drug from plasma after oral administration; Cmax = maximum plasma drug concentration; t ½ = mean apparent terminal elimination half‐life; t max = time to maximum plasma drug concentration; Vz/F = apparent volume of distribution during terminal phase after non‐intravenous administration.
Median (range).
Mean apparent terminal elimination half‐life.
The mean (SD) plasma concentrations of sumatriptan following administration of sumatriptan alone and in combination with ubrogepant are shown in Figure 2. For sumatriptan, the median t max was delayed by 2 hours when coadministered with ubrogepant than when administered alone (Table 3). The mean apparent terminal elimination t 1/2 of sumatriptan was slightly longer when coadministered with ubrogepant.
Fig. 2.
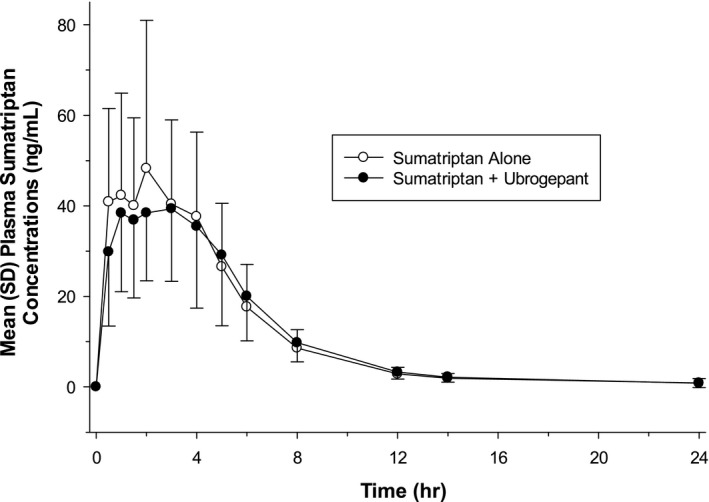
Mean (SD) plasma sumatriptan concentration‐time profile following single‐dose oral administration of 100 mg sumatriptan alone or in combination with single‐dose oral administration of 100 mg ubrogepant to fasted healthy participants (n = 29) (linear scale).
Table 3.
Mean (±SD) Pharmacokinetic Parameters of Sumatriptan Following Single‐Dose Oral Administration of Sumatriptan Alone or in Combination With Ubrogepant in Healthy Adult Participants
| PK Parameter, Mean (SD) | Sumatriptan 100 mg (n = 29) | Ubrogepant 100 mg + Sumatriptan 100 mg (n = 29) |
|---|---|---|
| C max, ng/mL | 59.13 (30.99) | 53.51 (17.73) |
| AUC0‐ t, ng•h/mL | 270.18 (105.21) | 261.16 (85.29) |
| AUC0‐∞, ng•h/mL | 279.78 (106.66) | 273.85 (87.00) |
| t max, hours† | 1.00 (0.50‐5.00) | 3.00 (0.50‐6.00) |
| t ½, hours‡ | 4.08 (2.57) | 5.09 (4.59) |
| Vz/F, L | 2377.90 (1609.15) | 2952.18 (3096.92) |
| CL/F, L/h | 416.54 (190.28) | 408.43 (154.66) |
AUC0‐∞ = area under the plasma drug concentration vs time curve from time 0 to infinity; AUC0‐ t = area under the plasma drug concentration vs time curve from time 0 to time t; CL/F = apparent total clearance of the drug from plasma after oral administration; Cmax = maximum plasma drug concentration; t ½ = mean apparent terminal elimination half‐life; t max = time to maximum plasma drug concentration; Vz/F = apparent volume of distribution during terminal phase after non‐intravenous administration.
Median (range).
Mean apparent terminal elimination half‐life.
The geometric mean ratios (GMRs) and their 90% CIs for the comparison of C max and AUC parameters of ubrogepant and sumatriptan administered in combination vs alone are shown in Table 4. The GMRs for ubrogepant were contained within the range of 0.80 and 1.25 for AUC0‐ t and AUC0‐∞; however, the C max of ubrogepant was reduced by 24% (GMR = 0.76). The lower bound of the 90% CI for this effect on C max was 0.69 and the upper bound of the 90% CI did not include 1.0, which suggests a statistically significant drug‐drug interaction. The GMRs and 90% CIs indicated no significant change in the Cmax, AUC0‐ t, or AUC0‐∞ of sumatriptan (GMRs of 0.96, 0.98, and 1.00, respectively) after coadministration with ubrogepant vs being administered alone, given that the 90% CIs were contained within the range of 0.80 and 1.25 (Table 4). Taken together, these results show that the ubrogepant C max decreased with the coadministration of sumatriptan, but sumatriptan C max and AUC did not significantly change when administered with ubrogepant.
Table 4.
Summary of Statistical Analysis Results: (1) Comparison of Plasma Ubrogepant PK Parameters When Administered Alone and in Combination With Sumatriptan; (2) Comparison of Plasma Sumatriptan PK Parameters When Administered Alone and in Combination With Ubrogepant in Healthy Adult Participants
| Statistical Analysis | PK Parameter | Geometric LSM | Geometric Mean Ratio (Test/Reference) | 90% Lower CI | 90% Upper CI | |
|---|---|---|---|---|---|---|
| Test | Reference | |||||
| Plasma ubrogepant (ubrogepant = reference; ubrogepant + sumatriptan = test) | C max, ng/mL | 284.00 | 373.13 | 0.76 | 0.69 | 0.85 |
| AUC0‐ t, ng•h/mL | 1516.11 | 1496.54 | 1.01 | 0.94 | 1.09 | |
| AUC0‐∞, ng•h/mL | 1538.91 | 1520.52 | 1.01 | 0.94 | 1.09 | |
| Plasma sumatriptan (sumatriptan = reference; sumatriptan + ubrogepant = test) | C max, ng/mL | 50.72 | 53.06 | 0.96 | 0.85 | 1.08 |
| AUC0‐ t, ng•h/mL | 246.14 | 250.00 | 0.98 | 0.92 | 1.05 | |
| AUC0‐∞, ng•h/mL | 259.13 | 259.52 | 1.00 | 0.94 | 1.06 | |
AUC0‐∞ = area under the plasma drug concentration vs time curve from time 0 to infinity; AUC0‐ t = area under the plasma drug concentration vs time curve from time 0 to time t; C max = maximum plasma drug concentration; LSM = least squares mean; PK = pharmacokinetic.
Safety
Phase 1 Study
The incidence of TEAEs was low, and all TEAEs were mild in severity and resolved on the day of onset (Table 5). No TEAEs were reported when ubrogepant was coadministered with sumatriptan. There were no serious AEs, deaths, or premature discontinuations due to AEs during the study. For all electrocardiograms, clinical laboratory parameters, and vital signs, mean changes from baseline were small, and no clinically relevant trends or patterns were observed. No participants had laboratory values with an increase in aminotransferase levels over 3× the upper limit of normal.
Table 5.
Overall Summary of Adverse Events by Treatment in the Phase 1 Study (Safety Population†)
| n (%) | Ubrogepant 100 mg (n = 30) | Sumatriptan 100 mg (n = 29) | Ubrogepant 100 mg + Sumatriptan 100 mg (n = 29) | Total‡ (N = 30) |
|---|---|---|---|---|
| Any TEAE | 2 (6.7) | 4 (13.8) | 0 | 6 (20.0) |
| Dry mouth | 0 | 2 (6.9) | 0 | 2 (6.7) |
| Rhinitis | 1 (3.3) | 0 | 0 | 1 (3.3) |
| Headache | 0 | 2 (6.9) | 0 | 2 (6.7) |
| Somnolence | 1 (3.3) | 0 | 0 | 1 (3.3) |
TEAE = treatment‐emergent adverse event.
One participant discontinued after receiving a single dose of ubrogepant and did not receive sumatriptan alone or in combination with ubrogepant.
Participants who took any study treatment are counted only once in the total. Participants were counted only once within each category.
Phase 3 Trials
A pooled analysis of ACHIEVE trial data was conducted to evaluate safety in 202 participants randomized to ubrogepant who took a triptan as an optional rescue medication. This analysis also included 1344 participants who did not take any rescue medication after receiving ubrogepant for their migraine attack. Adverse event data for these participants are summarized by the subgroup in Table 6. Overall, the frequency of TEAEs, treatment‐related TEAEs, and treatment‐emergent serious AEs was similar among participants that took ubrogepant alone and those that took ubrogepant and a triptan rescue medication. A summary of specific TEAEs occurring in ≥2% of participants in any subgroup is included in Supporting Information Table S1. TEAEs occurring in ≥2% of participants who took ubrogepant with a triptan included nausea, upper respiratory tract infection, musculoskeletal stiffness, and nasopharyngitis.
Table 6.
Overall Summary of Treatment‐Emergent Adverse Events from ACHIEVE Trials for Triptan as Rescue Medication Within 30 Days
| n (%) | Ubrogepant Alone | Ubrogepant and Triptan† Rescue Medication | ||||
|---|---|---|---|---|---|---|
| Ubrogepant 25 mg (n = 319) | Ubrogepant 50 mg‡ (n = 670) | Ubrogepant 100 mg (n = 355) | Ubrogepant 25 mg (n = 54) | Ubrogepant 50 mg‡ (n = 109) | Ubrogepant 100 mg (n = 39) | |
| Any TEAE | 66 (20.7) | 182 (27.2) | 106 (29.9) | 14 (25.9) | 32 (29.4) | 10 (25.6) |
| Any treatment‐related TEAE | 24 (7.5) | 64 (9.6) | 53 (14.9) | 1 (1.9) | 8 (7.3) | 5 (12.8) |
| Serious AE | 1 (0.3) | 3 (0.4) | 1 (0.3) | 0 | 0 | 1 (2.6) |
AE = adverse event; TEAE = treatment‐emergent adverse event.
Triptans included almotriptan, eletriptan, frovatriptan, naratriptan, rizatriptan, sumatriptan, and zolmitriptan.
Pooled data across ACHIEVE I and ACHIEVE II trials.
Discussion
Findings from the Phase 1 study, designed to determine if there was a PK interaction between ubrogepant and sumatriptan when administered concurrently in healthy participants, suggest that there was no clinically relevant PK interaction. Ubrogepant AUC0‐ t and AUC0‐∞ were similar when coadministered with sumatriptan, even though a lower C max was observed. Ubrogepant mean C max was reduced by 24% and accompanied by a delay in t max when coadministered with sumatriptan.
The PK changes for ubrogepant when coadministered with sumatriptan are expected to have minimal clinical relevance because the overall systemic exposure (AUC) was within the pre‐defined no‐effect boundary of 80 to 125% and the reduction in C max, which was likely due to the delay in t max, was not considered significant based on the totality of the data. Specifically, ubrogepant was found to be efficacious in the range of 50‐100 mg and safety was demonstrated over a wide range of doses, including single doses as high as 400 mg. 15 The ubrogepant C max and AUC values observed in past studies were similar to those obtained for ubrogepant alone in the current study. In other studies, the typical C max after a ubrogepant 100 mg dose was approximately 250 to 300 ng/mL, with an AUC ~1250 ng•h/mL (data on file, Allergan plc). The concentrations needed to achieve effective coverage of the CGRP receptor are attained within 10 to 15 minutes after the dose of ubrogepant and thus the slightly delayed C max values are unlikely to be clinically relevant (data on file, Allergan plc). Furthermore, the reported EC90 value for ubrogepant for the inhibition of capsaicin‐induced dermal vasodilation in humans was 23 nM (13 ng/mL), which is well below the lower C max observed when coadministered with sumatripan. 16
Sumatriptan PK parameters observed in this study were similar to values reported in other studies. 12 , 17 Sumatriptan C max, AUC0‐ t, and AUC0‐∞ were unchanged when sumatriptan was coadministered with ubrogepant as compared with sumatriptan alone. In addition, sumatriptan median t max was delayed by 2 hours (3 vs 1 hour) when sumatriptan was coadministered with ubrogepant, compared with sumatriptan administered alone. Both approaches align with previously reported t max values for sumatriptan alone (2 hours in package insert 12 and 1.2 hours in Scott 17 ). The mean apparent terminal elimination t 1/2 of sumatriptan was slightly longer when sumatriptan was coadministered with ubrogepant; however, the data were more variable, which could explain the change in mean apparent terminal elimination t 1/2 observed.
The coadministration of ubrogepant and sumatriptan was well tolerated in this Phase 1 study, with no serious AEs, deaths, or premature discontinuations. Overall, the incidence of AEs was low and all TEAEs reported were mild in severity. No TEAEs were reported by the participants in the Phase 1 study when ubrogepant and sumatriptan were coadministered. In the Phase 3 ACHIEVE pooled safety analysis, participants were required to wait at least 2 hours following ubrogepant administration to take any rescue medication (including sumatriptan). Safety data from this study may better reflect real‐world use, where people with migraine often require a second acute medication for treatment only after experiencing incomplete benefit from their first acute medication. In this pooled safety analysis of Phase 3 data, similar rates of TEAEs and treatment‐related TEAEs were observed in participants who took ubrogepant alone and those who took ubrogepant and a triptan rescue medication. This similarity across subgroups was observed in all dose groups (25, 50, and 100 mg), and no specific AEs showed a consistent trend toward increased rates in the combined ubrogepant and triptan subgroup. Although some specific TEAEs were only reported in the combined ubrogepant and triptan subgroup, these events were infrequent and none occurred in more than 2 participants in any ubrogepant dose group.
A higher overall rate of TEAEs was observed in the ACHIEVE trials than the Phase 1 study. The Phase 3 ACHIEVE trials included a larger number of participants, were placebo‐controlled outpatient trials, and exclusively enrolled participants with migraine. In contrast, the Phase 1 study included only 30 participants, was conducted as an inpatient study, and enrolled healthy adult volunteers. Although overall rates of TEAEs differed between studies, the within‐study comparisons of the rates of TEAEs associated with the administration of ubrogepant alone or ubrogepant with a triptan were both similar, showing no additional TEAEs associated with coadministration. Taken together, these results suggest that ubrogepant is well tolerated and safe when administered alone or in combination with a triptan rescue medication.
The Phase 1 study has several strengths. Most of the participants received all 3 study treatments (ie, ubrogepant alone, sumatriptan alone, and ubrogepant and sumatriptan together) and thus served as their own control. This design has the potential to improve the precision of estimated treatment differences and reduces the number of participants needed for the study. The washout period of 7 days was sufficient between treatments because both ubrogepant and sumatriptan have short elimination half‐lives.
A limitation of the Phase 1 study is that although it was statistically powered to detect differences in the PK profiles, assessment of changes in the safety endpoints was based only on a small number of participants without migraine because of the coadministration of ubrogepant with sumatriptan. However, the AEs from the pooled analysis of the ACHIEVE trials suggested no additional safety concerns among those who used triptans as rescue medication. The Phase 1 study population included a relatively small sample size, more males than females, and more Hispanic participants, which may not be the representative of the migraine population in clinical practice. Moreover, the Phase 3 studies included a much larger population of people with migraine and were approximately 90% women. 9 , 10 A population PK analysis indicated no significant effect of age, sex, race, and body weight on the PK of ubrogepant, 8 thus demographic differences between the population in the Phase 3 studies compared with that in the Phase 1 study are unlikely to have had an impact on the PK of ubrogepant. In the real world, many people with migraine exhibit comorbidities that require various pharmacologic treatments. Future studies evaluating the real‐world use of ubrogepant in people with migraine and associated comorbidities will be important to further evaluate potential interaction effects and overall safety.
Conclusion
The sumatriptan AUC and C max were unchanged and the t max was delayed when sumatriptan was coadministered with ubrogepant, whereas ubrogepant mean C max was slightly reduced, and t max was delayed. These effects of coadministration are expected to have minimal clinical relevance; however, additional real‐world use studies may be needed to fully characterize the impact on safety and efficacy of the coadministration of ubrogepant and sumatriptan. When sumatriptan is taken in combination with ubrogepant for acute treatment of migraine attacks, the results of this PK interaction study and the pooled safety analysis from the ACHIEVE trials show that the coadministration of these medications is well tolerated, with no additional safety concerns identified.
Statement of Authorship
Category 1
(a) Conception and Design
Abhijeet Jakate, Ramesh Boinpally, Danielle McGeeney, Antonia Periclou
(b) Acquisition of Data
Danielle McGeeney, Kaifeng Lu
(c) Analysis and Interpretation of Data
Abhijeet Jakate, Kaifeng Lu, Matthew Butler, Ramesh Boinpally, Antonia Periclou
Category 2
(a) Drafting the Manuscript
Abhijeet Jakate, Ramesh Boinpally, Matthew Butler, Kaifeng Lu, Danielle McGeeney, Antonia Periclou
(b) Revising It for Intellectual Content
Abhijeet Jakate, Ramesh Boinpally, Matthew Butler, Kaifeng Lu, Danielle McGeeney, Antonia Periclou
Category 3
(a) Final Approval of the Completed Manuscript
Abhijeet Jakate, Ramesh Boinpally, Matthew Butler, Kaifeng Lu, Danielle McGeeney, Antonia Periclou
Supporting information
Table S1
Acknowledgments
Medical writing and editorial assistance were provided to the authors by Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ, USA, and was funded by Allergan plc.
Conflict of Interest: Abhijeet Jakate, Ramesh Boinpally, Matthew Butler, Kaifeng Lu, Danielle McGeeney, and Antonia Periclou are employees and stockholders of Allergan plc.
Funding: This study was sponsored by Allergan plc, Dublin, Ireland. All authors met the ICMJE authorship criteria. Neither honoraria nor other form of payments was made for authorship.
REFERENCES
- 1. Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Annu Rev Physiol. 2013;75:365‐391. [DOI] [PubMed] [Google Scholar]
- 2. Becker WJ. Acute migraine treatment in adults. Headache. 2015;55:778‐793. [DOI] [PubMed] [Google Scholar]
- 3. Ferrari MD, Roon KI, Lipton RB, Goadsby PJ. Oral triptans (serotonin 5‐HT1B/1D agonists) in acute migraine treatment: A meta‐analysis of 53 trials. Lancet. 2001;358:1668‐1675. [DOI] [PubMed] [Google Scholar]
- 4. Holland PR, Goadsby PJ. Targeted CGRP small molecule antagonists for acute migraine therapy. Neurotherapeutics. 2018;15:304‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Iyengar S, Ossipov MH, Johnson KW. The role of calcitonin gene‐related peptide in peripheral and central pain mechanisms including migraine. Pain. 2017;158:543‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Edvinsson L. CGRP receptor antagonists and antibodies against CGRP and its receptor in migraine treatment. Br J Clin Pharmacol. 2015;80:193‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bigal ME, Walter S, Rapoport AM. Calcitonin gene‐related peptide (CGRP) and migraine current understanding and state of development. Headache. 2013;53:1230‐1244. [DOI] [PubMed] [Google Scholar]
- 8. Ubrelvy [package insert]. Madison, NJ: Allergan USA, Inc.; 2019. [Google Scholar]
- 9. Lipton RB, Dodick DW, Ailani J, et al. Effect of ubrogepant versus placebo on pain and the most bothersome associated symptom in the acute treatment of migraine: The ACHIEVE II Randomized Clinical Trial. JAMA. 2019;322:1887‐1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dodick DW, Lipton RB, Ailani J, et al. Ubrogepant for the treatment of migraine. N Engl J Med. 2019;381:2230‐2241. [DOI] [PubMed] [Google Scholar]
- 11. American Headache Society . The American Headache Society position statement on integrating new migraine treatments into clinical practice. Headache. 2019;59:1‐18. [DOI] [PubMed] [Google Scholar]
- 12. Imitrex [package insert]. Research Triangle Park, NC: GlaxoSmithKline; 2017. [Google Scholar]
- 13. Voss T, Lipton RB, Dodick DW, et al. A phase IIb randomized, double‐blind, placebo‐controlled trial of ubrogepant for the acute treatment of migraine. Cephalalgia. 2016;36:887‐898. [DOI] [PubMed] [Google Scholar]
- 14. Clinical Drug Interaction Studies – Study Design, Data Analysis, and Clinical Implications Guidance for Industry; October 24, 2017. Available at:https://www.fda.gov/files/drugs/published/Clinical‐Drug‐Interaction‐Studies‐%E2%80%94‐Study‐Design‐‐Data‐Analysis‐‐and‐Clinical‐Implications‐Guidance‐for‐Industry.pdf. Accessed November 28, 2017. [Google Scholar]
- 15. Ankrom W, Bondiskey P, Li CC, et al. Ubrogepant is not associated with clinically meaningful elevations of alanine aminotransferase in healthy adult males. Clin Transl Sci. 2020;13:462‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moore E, Fraley ME, Bell IM, et al. Characterization of ubrogepant: A potent and selective antagonist of the human calcitonin gene‐related peptide receptor. J Pharmacol Exp Ther. 2020. Jan 28. doi: jpet.119.261065. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 17. Scott AK. Sumatriptan clinical pharmacokinetics. Clin Pharmacokinet. 1994;27:337‐344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1