

Abstract
Stiff‐stilbene, a sterically restricted fused ring analogue of stilbene, has been regularly used as a model compound in theoretical studies of stilbene photoisomerization. Lately, owing to its excellent photoswitching properties, it is increasingly being applied to reversibly control the properties and function of chemical as well as biological systems. Stiff‐stilbene photoswitches possess a number of advantageous properties including a high quantum yield for photoisomerization and a high thermal stability. Furthermore, they undergo a large geometrical change upon isomerization and their synthesis is straightforward. Herein, we provide an overview of the basic properties of stiff‐stilbene and of recent applications in supramolecular chemistry, catalysis, and biological systems.
Keywords: catalysis, molecular switches, photochromism, self-assembly, stiff-stilbene
Stiff‐stilbene is increasingly applied to reversibly control the properties and function of chemical as well as biological systems. In this Minireview an overview is provided of the basic properties of this promising photoswitch, which is followed by a survey of recent applications.

1. Introduction
Along with the growing interest in dynamic functional molecular systems,1 the development and application of photochromic molecular switches (molecular photoswitches) has become one of the most active fields of research in the chemical sciences.2 Incorporation of molecular photoswitches into materials,3, 4, 5, 6, 7 and biological systems8, 9, 10 has led to unprecedented control of properties and function. In this regard, the use of light as a stimulus seems to be ideal as it can be delivered with high spatiotemporal precision, light of a specific wavelength can be selected, and no chemical waste is produced in the system. Among the nowadays most frequently studied and used photoswitches are azobenzene,11 stilbene,12 diarylethene,13 spiropyran,14 hemithioindigo,15 and hydrazone.16
Photoisomerization of stilbene has been thoroughly investigated as a model system for condensed‐phase reaction dynamics.12 Photoexcitation of E stilbene using UV light leads to the formation of Z stilbene (Scheme 1 a). However, instead of the reverse Z→E photoisomerization, Z stilbene can undergo 6π‐electrocyclization to give dihydrophenanthrene (>10 % yield), which is easily oxidized to give phenanthrene. This electrocyclization, in addition to other possible photo‐oxidation and photocyclization pathways, have significantly hampered application of stilbene as a molecular photoswitch.
Scheme 1.
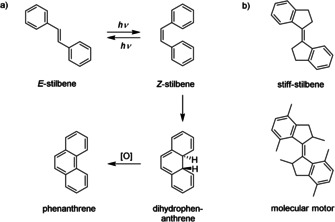
a) Stilbene isomerization, cyclization and oxidation and b) chemical structures of stiff‐stilbene and molecular motor.
The 5‐membered fused ring analogue of stilbene (named stiff‐stilbene, Scheme 1 b) was found to be much more stable, that is, clean photoisomerization occurs when exposed to UV light.17 This fused ring analogue has been used mostly as a prototype for investigating the photoisomerization path in stilbene as this process is simplified in this case due to steric restrictions. Furthermore, stiff‐stilbene is best recognized as a structural motif in Feringa‐type molecular motors, of which the properties and applications have been comprehensively reviewed elsewhere.18, 19, 20 However, it should be noted that the isomerization behavior differs significantly and that, in contrast to molecular motors, stiff‐stilbene is characterized by a very high quantum yield for photoisomerization and straightforward synthesis. Also, unlike other photoswitches that are able to undergo double bond isomerization (such as azobenzene and hemithioindigo), the higher energy Z isomer of stiff‐stilbene possesses an excellent thermal stability. This thermal stability facilitates the synthesis and isolation of the Z isomer as well as the investigation of its behavior. Despite these advantageous properties and the fact that stiff‐stilbene is already known for a long time, it is only since recently that applications of stiff‐stilbene, other than as molecular motor, have appeared in the literature. In this Minireview, our aim is to give an overview of the properties of stiff‐stilbene and to highlight recent applications in host–guest chemistry, supramolecular self‐assembly, catalysis, and biological systems.
2. Synthetic Procedures
The first description of the synthesis of a 5‐membered fused ring analogue of stilbene that we encountered was in a 1980 paper by Lenoir and Lemmen.21 They applied McMurry's method22 to achieve homocoupling of either 1‐indanone or 2,2′‐dimethyl‐1‐indanone using TiCl4 and Zn (Scheme 2 a). This homocoupling reaction afforded the E and Z isomers of products 1 and 2 in a ratio (E/Z) of 92:8 and 88:12, respectively.23
Scheme 2.

Different approaches to the synthesis of stiff‐stilbene.
Where the E isomer is usually the major product in the McMurry reaction,22, 23 the group of Boulatov came up with an elegant intramolecular approach to exclusively synthesize the Z isomer of stiff‐stilbene.24 In a typical example, they first carried out O‐alkylation of 1‐indanone‐6‐carboxylic acid with 1,5‐dibromopentane to yield bis‐indanone 3 (Scheme 2 b). Then, McMurry coupling under dilute conditions afforded cyclic stiff‐stilbene Z‐4 and subsequent reduction afforded dibenzylic alcohol Z‐5. The same strategy was applied to synthesize a diol‐functionalized stiff‐stilbene as well as various desymmetrized stiff‐stilbenes.4, 25, 26
A different route toward desymmetrized stiff‐stilbenes is through a mixed McMurry reaction. In 1992, Lapouyade and co‐workers reported such a mixed coupling between 5‐dimethylamino‐1‐indanone and either 1‐indanone or 5‐bromo‐1‐indanone (Scheme 2 c).27, 28 This afforded stiff‐stilbenes 6 and 7 of which the latter was cyanated using CuCN. However, full synthetic details and characterization have not been reported for these mixed McMurry reactions. Our group recently described the mixed McMurry coupling of 6‐bromoindanone with TBS‐protected 6‐hydroxyindanone.29 When the reaction was carried out in a 1:1 stoichiometry, the homocoupled product of 6‐bromoindanone, homocoupled TBS‐protected 6‐hydroxyindanone, and the desired heterocoupled product were obtained in a 10:34:27 ratio. Other successful mixed McMurry couplings were recently reported by the groups of Boulatov30 and Wu.31
In an attempt to synthesize dibromo‐functionalized tetramethyl‐stiff‐stilbene 12, the group of Shinmyozu used McMurry conditions, but the reaction failed.32 The reason for this has not been specified but, nevertheless, the desired compound could be synthesized using Barton–Kellog methodology (Scheme 2 d). First, 5‐bromo‐2,2‐dimethyl‐1‐indanone was reacted with Lawesson's reagent to give the corresponding thioketone 8. Simultaneously, the same indanone was converted into hydrazone 9 by using hydrazone monohydrate and, upon the addition of Ag2O, diazo compound 10 was generated. Thereafter, the diazo compound and the thioketone were combined giving episulfide 11. Subsequent desulfurization in the presence of Cu powder afforded the desired tetramethyl‐stiff‐stilbene 12 as a mixture of E and Z isomers in a 5:1 ratio.
3. Basic Properties
3.1. Structural Features
Early force‐field calculations by the group of Lenoir suggested that E ‐1 and E‐2 are nearly planar, while for the corresponding Z isomers there is an out‐of‐plane deformation.21 The latter Z isomers therefore possess helical chirality and, where the enantiomers of Z ‐1 undergo fast racemization, the interconversion between the enantiomers of Z ‐2 is slow on the NMR timescale.23 Later on, Tomoda and co‐workers provided deep insight into the structural features of sterically and non‐sterically congested stiff‐stilbenes (Figure 1) by solid state studies and force‐field calculations.33, 34 They demonstrated by X‐ray crystallography that the non‐sterically congested tolyl‐derivatives E‐13, E‐14, and E‐15 have a planar geometry as is reflected by the C−Ph torsion angles (ϕ) being close to zero.33 For the sterically congested E‐2 and E‐16, having methyl substituents close to the ethylene bond, the C−Ph bonds are twisted and the benzene rings do not lie in the same plane. Furthermore, the ethylene bond is twisted and the C atoms are slightly pyramidalized. The same group also reported crystal structures of tolyl and xylyl derivatives Z‐15 and Z‐17, which were found to be non‐planar with a considerably twisted ethylene bond (θ=5.7° for Z‐15 and 17.2° for Z‐17).34 The larger steric interaction between the methyl groups in Z‐17, than between the hydrogen atoms in Z‐15, is reflected in a larger torsion angle. Solid state analysis and calculations were also reported by Oelgemöller et al. and are in agreement with the work described above.17, 35
Figure 1.
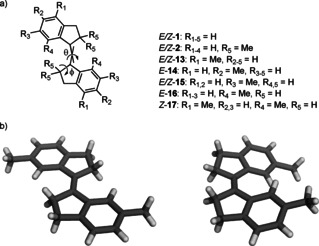
a) Line drawings of stiff‐stilbenes 1, 2 and 13–17 and b) X‐ray crystal structures of E‐15 (left) and Z‐15 (right).
It may be concluded that the E isomer is the thermodynamically most stable form since it is less sterically congested than the Z isomer. Density functional theory (DFT) calculations at the B3LYP/6‐31G(d,p) level of theory, which were recently performed by our group, revealed that E‐1 is 1.5 kcal mol−1 lower in energy than Z‐1.36 Interestingly, the group of Boulatov described that the half‐life of Z‐1 is very long (i.e. 109 years at 300 K). The corresponding energy barrier for thermal Z→E isomerization is 43 kcal mol−1 making this process negligible.37
3.2. Photophysical Properties
Stiff‐stilbene has been used in a number of studies as a sterically restricted analogue of stilbene in order to gain insight into the mechanism of stilbene photoisomerization. To the best of our knowledge, the first absorption study of stiff‐stilbene (i.e. E‐1) was reported by Saltiel and D'Agostino in 1972.38 The absorption spectrum turned out to be slightly red‐shifted with respect to that of parent E‐stilbene. It has been suggested that after excitation of the E isomer, a twisted (phantom) geometry is formed, before evolvement to the Z‐isomer. Interestingly, Saltiel and D'Agostino observed that the fluorescence quantum yield, which can be coupled to the rate constant of twisting, is affected by a change in solvent viscosity. More than ten years later, fluorescence quantum yield measurements by Rothenberger et al. further confirmed that the isomerization rate of stiff‐stilbene is viscosity dependent.39 Overall, the fluorescence quantum yield increases in more viscous solvents indicating that the quantum yield for photoisomerization is reduced. This means that the barrier for twisting in the first singlet excited state (S1) is elevated as a result of increased friction exerted by the solvent on the twisting molecule.
A time‐resolved spectroscopic study of the isomerization process by Doany et al. showed a transient absorption at 351 nm, which was ascribed to the twisted (phantom) intermediate.40 In a related study, the group of Fuss showed that the lifetime of the twisted intermediate of stiff‐stilbene (800 fs) is longer than that of parent stilbene (135–160 fs).41 The reason for this difference is that for stilbene a hula twist mechanism is the lower‐energy path, whereas for stiff‐stilbene, the hula‐twist mechanism can be discarded because of the fixation of the single bonds in the five membered ring. It is envisaged that for stiff‐stilbene the conical intersection is reached via pyramidalization of one ethylenic C atom involving a beginning H migration.
Time‐dependent density functional theory (TD‐DFT) calculations by Santoro and Improta pointed to a smaller energy barrier to isomerization for stiff‐stilbene than for stilbene,42, 43 in line with experimental studies.41 The groups of Ernsting and Kovalenko further investigated the photoisomerization dynamics by combining experimental and theoretical studies on the S1 state.44 It was concluded that evolvement to the Z isomer occurs in the excited state, where most of the molecules that reach the intermediate state do not relax to the ground state but continue the torsion process. What is interesting to note is that for their study, Ernsting and Kovalenko prepared Z‐1 by 313 nm irradiation of a solution of E‐1 in degassed hexane, which gave a 37:63 E/Z ratio at the photostationary state (PSS). The corresponding spectral changes are shown in Figure 2. Furthermore, the quantum yields for E→Z and Z→E isomerizations (λ irr=339 nm) were calculated as 0.36 and 0.31 in hexane and acetonitrile, respectively.
Figure 2.
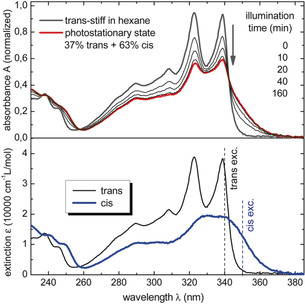
UV/Vis spectral changes of E‐1 upon 313 nm irradiation (top) and UV/Vis absorption spectra of E‐1 and Z‐1 (bottom). Adapted with permission from Ref. 44. Copyright 2014 American Chemical Society.
The same photoisomerization process was monitored earlier by Oelgemoller et al.17 They found that irradiation with 254 nm light of a solution of E‐1 in degassed hexane led to a PSS mixture containing the E and Z isomer in a 77:23 ratio. Alternatively, irradiation with 300 nm light resulted in the formation of more Z isomer, that is, a PSS ratio 37:63 (E/Z), identical to the PSS313 ratio reported by Ernsting and Kovalenko.44
4. Emergent Applications
4.1. Switchable Host–Guest Systems
Among the strategies to modify the affinity of a host toward a guest molecule, the use of light to generate structural changes in molecular receptors has proven to be very promising.4, 45, 46, 47 In this regard, stiff‐stilbene is highly suitable for incorporation into synthetic hosts owing to its rigidity and large geometrical change upon photoisomerization.
In an early example, the group of Shinmyozu decorated stiff‐stilbene with two binaphthol moieties which served as hydrogen bonding motifs for anions.48 Irradiation of the E isomer with 365 nm light in degassed MeCN‐d3 led to a PSS ratio of 25:75 (E/Z). The reverse isomerization process was achieved by irradiation with 410 nm light, which resulted in a PSS ratio of 91:9 (E/Z). Calculations indicated that in the Z isomer a binding pocket is formed because of the binaphthol moieties pointing in the same direction. Binding studies were conducted in CDCl3 with dihydrogen phosphate, fluoride and chloride. Surprisingly, the E isomer showed an 8‐fold stronger affinity for chloride (K a=4.6×102 m −1) than the Z isomer (K a=5.9×10 m −1).
Recently, our group reported stiff‐stilbene based bis‐urea receptor 18 (Scheme 3 a).49 The Z isomer of this receptor was found to strongly bind dihydrogen phosphate (K a=1.40×103 m −1) and acetate (K a=2.02×103 m −1) in [D6]DMSO/0.5 %H2O. In this Z isomer, the urea groups are in close proximity and therefore able to form four hydrogen bonds with the anion. In the E isomer, on the other hand, the relative distance between the urea units is too large to bind anions in a cooperative manner. As a result, the binding affinity is much lower (i.e. K a =77 m −1 for dihydrogen phosphate and 1.04×102 m −1 for acetate). Furthermore, irradiation of a solution of E‐18 in [D6]DMSO with 365 nm light led to the formation of a PSS with a 49:51 E/Z ratio and irradiation of a solution of Z‐18 with 385 nm light led to nearly quantitative conversion to E‐18 (97 %).
Scheme 3.
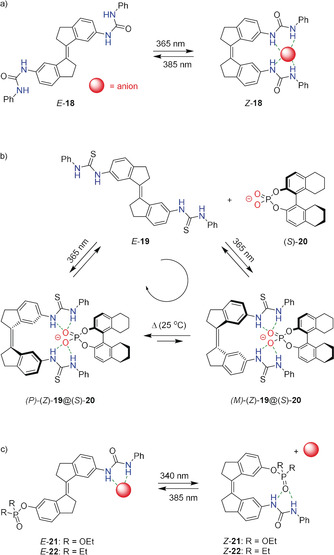
a) Switchable anion binding to bis‐urea stiff‐stilbene 18, b) supramolecularly induced rotational motion in bis‐thiourea stiff‐stilbene 19 and c) phototriggered blockage and exposure of a urea binding site in 21 and 22.
In a subsequent study, we employed the binding of a chiral phosphate anion to thiourea variant 19 for inducing a unidirectional rotational motion upon photoisomerization (Scheme 3 b).36 A helical conformation is adopted by the Z isomer due to steric congestion, as was earlier described by Lemmen and Lenoir.21 The P and M helical isomers of Z‐19 rapidly interconvert and hence, form a racemate. However, addition of chiral phosphate (S)‐20 to racemic Z‐19 promoted the preferential formation of the P helical isomer and vice versa. Where the E isomer is planar (achiral), irradiation with 365 nm light in CD2Cl2 leads to the formation of P and M helical Z isomers with equal probability. In the presence of the chiral phosphate, the backwards Z→E isomerization process takes place predominantly from the P helical isomer resulting in a net rotary motion around the central double bond. Interestingly, the PSS365 ratio in the absence of the chiral phosphate anion was determined as 42:58 (E/Z), while in the presence of the chiral phosphate anion it was elevated to 24:76 (E/Z).
Chirality induction in the Z isomer of stiff‐stilbene was also achieved by the group of Gogoll using bis‐porphyrin substituents. One of the helical isomers was preferentially formed upon the binding of chiral ditopic and monotopic amines.50, 51 However, the switching properties of these bis‐porphyrin receptors have not yet been exploited.
In an attempt to fully block the binding site of a stiff‐stilbene based receptor in one of its photoaddressable states, we attached a urea hydrogen bond donating group and either a phosphate or phosphinate hydrogen bond accepting group (receptor 21 and 22 in Scheme 3 c).29 Irradiation of E‐21 and E‐22 with 340 nm light in [D6]DMSO led to PSS mixtures containing the E and Z isomer in a 47:53 and a 42:58 ratio, respectively. The reverse process was achieved by using 385 nm light, resulting in a PSS ratio (E/Z) of 94:6 for 21 and 93:7 for 22. It was envisioned that upon E→Z isomerization, the binding affinity would decrease significantly, as in the Z isomer the urea binding site would be blocked by intramolecular hydrogen bonding. Binding studies were performed with dihydrogen phosphate, acetate and chloride in [D6]DMSO/0.5 %H2O. In general, binding to the E isomer was found to be stronger, however, the differences in binding affinity between the E and Z isomers were not as large as expected. For example, binding of acetate to Z‐22 (K a=8.4×102 m −1) is only slightly weaker than to E‐22 (K a=1.4×103 m −1).
Yang and co‐workers reported a crown ether based photoresponsive cryptand 23 that was obtained via intramolecular McMurry coupling (Scheme 4).52 Irradiation of the Z isomer with 382 nm light in MeCN‐d3 yielded the E isomer in 98 %. In its Z form, the macrocycle strongly binds a 2,7‐diazapyrenium guest (K a=3.26×103 m −1 in MeCN‐d3), while for the E isomer the affinity is about 14 times lower (K a=227 m −1). This large difference in affinity is explained by the fact that the cavity provided by the E isomer of the macrocycle is too small to encapsulate the large 2,7‐diazapyrenium guest.
Scheme 4.
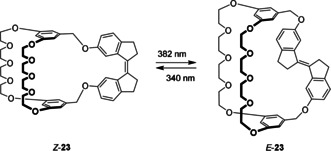
Crown ether based photoresponsive cryptand 23.
In a different study, Yang and co‐workers connected a pillar[5]arene macrocycle and an axle, able to thread the macrocycle, to Z stiff‐stilbene giving rotaxane‐like structure 24 (Scheme 5).53 The role of stiff‐stilbene was to trigger translation of the pillar[5]arene while, at the same time, it acted as a stopper to prevent dethreading. With stiff‐stilbene in its Z‐form, the pillar[5]arene was preferentially located at region A of the thread. Irradiation with 387 nm light in CDCl3 produced 90 % of the E isomer and promoted translocation of the pillar[5]arene across the axle to region B. Subsequent irradiation with 360 nm light to promote E→Z isomerization resulted in the reverse translocation event.
Scheme 5.
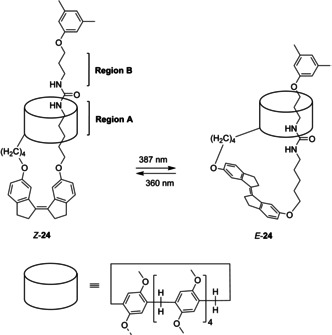
Pillar[5]arene translocation in rotaxane‐like structure 24.
The group of Zhang also explored the use of stiff‐stilbene in the construction of a rotaxane system.54 In their approach, stiff‐stilbene was incorporated into an oligoether axle, which furthermore contained either a naphthalene or biphenyl unit. In the E isomer, a tetracationic macrocycle, threaded by the oligoether axle, shuttles between stiff‐stilbene and the second recognition unit. Upon 340 nm irradiation in MeOD, PSS ratios of about 55:45 (E/Z) were obtained. The E→Z isomerization process triggers movement of the macrocycle from the stiff‐stilbene to the anthracene or biphenyl unit, which is associated with a color change due to different charge‐transfer interactions. Backwards photochemical isomerization could be achieved by irradiation with 375 nm light resulting in a PSS ratio of 92:8 (E/Z).
4.2. Dynamic Supramolecular Assembly
Supramolecular self‐assembly has attracted major interest for the utilization of photoswitches to create materials of which the properties can be modified by a light source.3, 55 The group of Yang has developed various supramolecular polymers based on stiff‐stilbene monomers (Figure 3). In a seminal paper in 2013, they functionalized stiff‐stilbene with ureidopyrimidone (UPy) giving monomer 25.56 At low concentration (50 mm) in CDCl3, the Z isomer was found to dimerize through hydrogen bonding as indicated by 1H and DOSY NMR studies as well as HR‐MS. Above a concentration of 0.1 mm, however, also polymers could be formed (Scheme 6). On the contrary, even at low concentrations (50 mm), E‐25 forms a supramolecular polymer. After irradiation of a solution of Z ‐25 in CDCl3 with 387 nm light, the E isomer was obtained in 99 %. The reverse isomerization process was triggered by irradiation with 360 nm light. By alternating these irradiation wavelengths, the transformation from cyclic dimers to supramolecular polymers and vice versa took place (Scheme 6). Interestingly, a concentrated solution of E‐25 in CHCl3 formed a gel and irradiation of this gel with 360 nm light resulted in gel dissolution as a result of E→Z isomerization.
Figure 3.
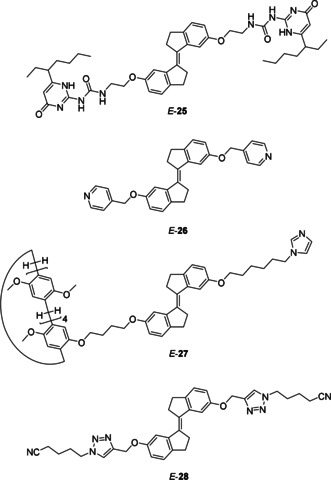
Different stiff‐stilbene monomers able to self‐assemble into a polymer.
Scheme 6.
Supramolecular assembly processes of compound 25. Reproduced with permission from Ref. 56. Copyright 2013 Wiley‐VCH.
In a subsequent study by the groups of Huang, Yang and Stang pyridyl appended stiff‐stilbene monomer 26 was used (Figure 3). The E and Z isomers of this monomer self‐assembled with di‐PtII acceptors in CH2Cl2 into two distinct supramolecular coordination complexes (Scheme 7).57 The use of Z‐26 favored the formation of metallacycles, whereas irradiation of these metallacycles with 387 nm light led to almost quantitative conversion to E‐26 and concomitant transformation into a metallosupramolecular polymer. These changes in self‐assembly behavior were confirmed by NMR and UV/Vis spectroscopy, in addition to HR‐MS. When the polymer was irradiated with 360 nm light, a mixture of Z‐26 and E‐26 in a 53:47 ratio was obtained. Due to the fact that there is still E isomer present, the original metallacycle did not reform but instead, a larger cyclic oligomer was obtained. Furthermore, at very high concentration, the metallacycles were found to aggregate into spherical nanoparticles, which could be transformed into a nanofiber network upon Z→E isomerization.
Scheme 7.
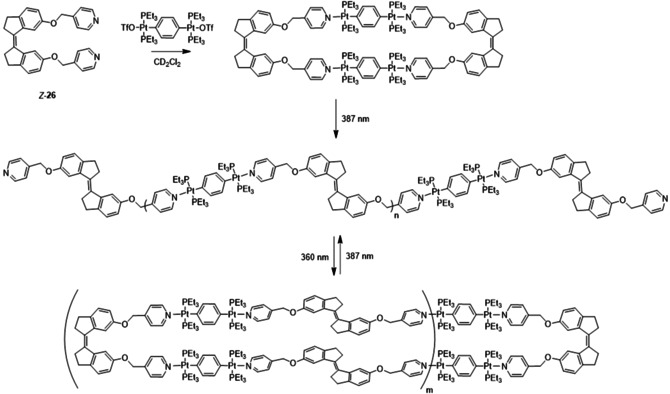
Di‐PtII‐induced formation of metallacycles and metallosupramolecular polymers.
In addition, the group of Yang developed a desymmetrized system in which stiff‐stilbene is functionalized on one side with pillar[5]arene and on the other side with a triazole unit (compound 27, Figure 3).58 The alkyl chains bearing the triazole unit can be threaded into the pillar[5]arene. For the Z isomer, this threading leads to self‐assembly into a mixture of daisy chains and pseudo rotaxanes (in CDCl3). Irradiation with 387 nm light of a solution containing Z‐27 led to 97 % conversion to E‐27, which at a concentration of 150 mm assembled into a supramolecular polymer. Similarly, the same group combined the stiff‐stilbene‐bridged guest 28 (Figure 3), containing two triazole chains, with a disulfide‐bridged bis‐pillar[5]arene host.59 The E and Z isomers of 28 were found to promote very different assembly events. The bent shape of Z‐28 caused the formation of low molecular weight structures and irradiation of Z‐28 with 387 nm light, which gave a PSS ratio of 97:3 (E/Z), led to the formation of supramolecular polymers.
In a different approach towards self‐assembled materials, Shi and co‐workers prepared a photoresponsive supramolecular amphiphile from stiff‐stilbene 29 and a water soluble pillar[5]arene (Scheme 8).60 Irradiation of Z‐29 in a 1:1 [D6]DMSO/D2O mixture with 387 nm light yielded E‐29 in 90 %. The reverse isomerization process could be induced by 360 nm irradiation. Encapsulation of Z ‐29 by the water soluble pillar[5]arene was confirmed with NMR spectroscopy and isothermal titration calorimetry (ITC) and led to the formation of a gemini‐type supra‐amphiphile. This supra‐amphiphile self‐assembled into nanocubes for the Z isomer and crumpled films for the E isomer, as was concluded from TEM measurements.
Scheme 8.

Stiff‐stilbene 29 and water soluble pillar[5]arene.
4.3. Control of Catalysis
When it comes to application of stiff‐stilbene in catalysis, two approaches may be distinguished. In the first approach, stiff‐stilbene isomerization is used to induce strain on a substrate in order to accelerate its chemical transformation. In the second approach, stiff‐stilbene is used as a ligand in metal‐catalyzed transformations. Both of these approaches are discussed in this paragraph.
In 2009, the group of Boulatov described the concept of a molecular force probe, that is, they studied the relationship between the restoring force in a stretched stiff‐stilbene macrocycle and the rate of C−C bond dissociation within the macrocycle.61, 62 Hence, stiff‐stilbene was bridged by trans‐3,4‐dimethylcyclobutene using various linkers giving a set of 9 differently sized macrocycles (compounds 30–38, Scheme 9). Irradiation of the strain‐free Z isomers in hexane using 365 nm or 375 nm light yielded strained E isomers. After the PSS was reached the resulting E/Z mixtures were analyzed by HPLC. The amounts of E isomer were found to be very low, except for the largest macrocycle. A similar observation was very recently reported by Gogoll et al. who studied the effect of ring size on the photoisomerization behavior of cyclic stiff‐stilbene.63 Remarkably, Boulatov and co‐workers demonstrated that light irradiation promotes an acceleration of up to 5×106‐fold of the C−C bond dissociation of cyclobutene. The same concept was applied to accelerate thiol/disulfide exchange,25 disulfide reduction,64 sulfonate hydrolysis,65 and the cleavage of a phosphorus oxygen bond.26
Scheme 9.
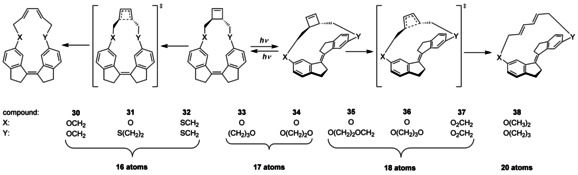
Use of a molecular force probe to accelerate C−C bond dissociation.
In order to control metal‐catalyzed transformations, the groups of Widenhoefer, Boulatov and Craig connected stiff‐stilbene to a bis‐phosphine ligand (compound 39, Scheme 10 a).66 DFT calculations showed that upon stiff‐stilbene isomerization, the dihedral angle in the bis‐phosphine ligand changes from 83° for Z‐39 to 106° for E‐39, while the biaryl geometry in a higher energy atropisomer of the E isomer (i.e. E‐39′) is virtually undistorted (98°). Irradiation of the Z isomer with 365 nm light in CH2Cl2 yielded a PSS mixture consisting of Z ‐39, E ‐39 and E ‐39′ in a 68:23:9 ratio. The isomers were separated and tested in asymmetric Heck and Trost reactions. In the Heck arylation of 2,3‐dihydrofuran (Scheme 10 b), the use of the Z isomer resulted in the formation of (S)‐40 in 96 % ee, while the use of the E isomer yielded (S)‐40 in 79 % ee. It has been well established that a change in the dihedral angle of bis‐phosphine ligands influences the outcome of metal‐catalyzed reactions67 and this feature is elegantly exploited in this design.
Scheme 10.
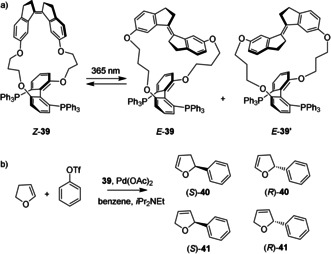
a) Isomers of bis‐phosphine ligand 39 and b) asymmetric Heck arylation reaction.
In a related study, the group of Ma synthesized macrocycles constituted of stiff‐stilbene and a binaphthyl moiety that were bridged by alkyl chains of different length.68 The corresponding macrocycles could be switched between E and Z forms by alternating with 387 nm and 360 nm light, leading to a change in the twist angle of the binaphthyl unit.
4.4. Toward Biological Application
The regulation of proton transport across lipid bilayers is of fundamental importance to many biological reactions. In an effort to control proton transport by light, Gewirth and Zimmerman developed stiff‐stilbene based proton carrier E‐42, which possesses a boronic acid head group and an alkyl tail (Scheme 11 a).69 When a solution of E‐42 in CD2Cl2 was irradiated with 360 nm light, the Z isomer was obtained in 40 % yield and subsequent irradiation with 390 nm light led to nearly quantitative conversion back to the E isomer. The Z isomer was found to be capable of proton transport from an aqueous solution across a lipid bilayer to a self‐assembled monolayer of CuBTT, which acted as an O2 reduction catalyst. The reduction rate of O2 was unaffected when the E isomer was incorporated in the bilayer membrane, showing that it is incapable of transport. Proton transport occurs via a flip‐flop diffusion mechanism of the proton carrier and the energy barrier for this process is suggested to be higher for the E isomer than for the Z isomer.
Scheme 11.
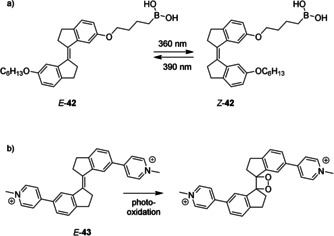
a) Stiff‐stilbene based proton carrier 42 and b) bis‐pyridinium ligand 43.
Recently, the group of Galan synthesized the E and Z isomers of pyridinium functionalized stiff‐stilbene 43 (Scheme 11 b), which were able to stabilize G‐quadruplex DNA in potassium buffer.70 In stark contrast, when sodium buffer was used, the E isomer was found to induce unfolding into a single‐stranded helix. Unfortunately, 400 nm light irradiation of E‐43 provoked decomposition instead of E→Z isomerization, which was ascribed to photo‐oxidation followed by fragmentation. Similar photo‐oxidation of a stiff‐stilbene based fluorophore was very recently used by the group of Boulatov for O2 sensing.30 Nevertheless, Galan and co‐workers noted that decomposition of E‐43 caused the G‐quadruplex DNA to re‐adopt its original topology and hence, by cycles of irradiation and E‐43 addition, the conformation could be controlled in a reversible manner. In a subsequent study, the same group demonstrated that the strong binding of E‐43 to G‐quadruplex DNA promotes toxicity towards cancerous cells.71
In another recent study, Míšek and Luptak described that the photocontrol of binding of a stiff‐stilbene amino alcohol to m‐RNA can be used to regulate protein expression.72
5. Summary and Outlook
It is clear that the number of applications of stiff‐stilbene photoswitches in various fields is rapidly increasing. This is not surprising considering the advantageous high quantum yield for photochemical isomerization, the high thermal stability of the Z isomer, the straightforward synthesis, and the large geometrical change upon isomerization. These features make stiff‐stilbene highly suitable for use in the development of smart materials and for exerting control over biological processes.
However, application of stiff‐stilbene photoswitches is still in an early stage and certain drawbacks need to be addressed in the coming years. For instance, the PSS ratio for E→Z isomerization is typically low. Better PSS ratios may be obtained by enlarging the separation of the absorption bands of the E and Z isomers. Furthermore, stiff‐stilbene photoswitches are operated with UV light, which is damaging to tissue and can cause material degradation, for example, by photo‐oxidation. Therefore, visible or even near IR light is desired for most applications in materials science and biology. Strategies to red‐shift the absorption of stiff‐stilbene should thus be explored. Another point of attention is to develop improved synthetic methods for desymmetrization and to widen the scope of functional groups that can be introduced. These issues, which may explain why stiff‐stilbene is underexplored in comparison to other molecular photoswitches, are currently being addressed in our lab. With that, we are confident that many more exciting applications are still to come.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
David Villarón obtained his Bachelor's degree in chemistry at the University of Salamanca in 2017. He completed his Master's degree at the Institute of Chemical Research of Catalonia in 2018, where he worked in the group of Prof. Pablo Ballester. After that, he moved to Leiden University to carry out his PhD research in the group of Dr. Sander Wezenberg. His work focuses on the development of photoresponsive anion receptors and transporters.

Biographical Information
Sander J. Wezenberg studied chemistry at the University of Nijmegen where he carried out his Master's research in the group of Prof. Roeland Nolte. He then moved to the Institute of Chemical Research of Catalonia for his PhD studies in the group of Prof. Arjan Kleij. During this period he spent three months as a visiting researcher in the group of Prof. Joseph Hupp at Northwestern University. After receiving his PhD in 2011, he joined the group of Prof. François Diederich at ETH Zurich as a postdoctoral fellow. He moved to the University of Groningen in 2013 to work with Prof. Ben Feringa and was later appointed Assistant Professor. In 2019, He moved to Leiden University to establish his independent research group. His main research interests are in the areas of anion binding, molecular switches, and self‐assembled materials.

Acknowledgements
Financial support from the European Research Council (Starting Grant no. 802830 to S.J.W.) and the Netherlands Organization for Scientific Research (NWO‐ENW, Vidi Grant no. VI.Vidi.192.049 to S.J.W.) is gratefully acknowledged.
D. Villarón, S. J. Wezenberg, Angew. Chem. Int. Ed. 2020, 59, 13192.
References
- 1. Lubbe A. S., van Leeuwen T., Wezenberg S. J., Feringa B. L., Tetrahedron 2017, 73, 4837–4848. [Google Scholar]
- 2. Molecular Switches, 2nd ed. (Eds.: B. L. Feringa, W. R. Browne), Wiley-VCH, Weinheim, 2011. [Google Scholar]
- 3. Russew M.-M., Hecht S., Adv. Mater. 2010, 22, 3348–3360. [DOI] [PubMed] [Google Scholar]
- 4. Qu D.-H., Wang Q.-C., Zhang Q.-W., Ma X., Tian H., Chem. Rev. 2015, 115, 7543–7588. [DOI] [PubMed] [Google Scholar]
- 5. Nie H., Self J. L., Kuenstler A. S., Hayward R. C., Read de Alaniz J., Adv. Opt. Mater. 2019, 7, 1900224. [Google Scholar]
- 6. Baroncini M., Groppi J., Corra S., Silvi S., Credi A., Adv. Opt. Mater. 2019, 7, 1900392. [Google Scholar]
- 7. Dattler D., Fuks G., Heiser J., Moulin E., Perrot A., Yao X., Giuseppone N., Chem. Rev. 2020, 120, 310–433. [DOI] [PubMed] [Google Scholar]
- 8. Szymański W., Beierle J. M., Kistemaker H. A. V., Velema W. A., Feringa B. L., Chem. Rev. 2013, 113, 6114–6178. [DOI] [PubMed] [Google Scholar]
- 9. Lerch M. M., Hansen M. J., van Dam G. M., Szymanski W., Feringa B. L., Angew. Chem. Int. Ed. 2016, 55, 10978–10999; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11140–11163. [Google Scholar]
- 10. Hüll K., Morstein J., Trauner D., Chem. Rev. 2018, 118, 10710–10747. [DOI] [PubMed] [Google Scholar]
- 11. Bandara H. M. D., Burdette S. C., Chem. Soc. Rev. 2012, 41, 1809–1825. [DOI] [PubMed] [Google Scholar]
- 12. Waldeck G. H., Chem. Rev. 1991, 91, 415–436. [Google Scholar]
- 13. Irie M., Fukaminato T., Matsuda K., Kobatake S., Chem. Rev. 2014, 114, 12174–12277. [DOI] [PubMed] [Google Scholar]
- 14. Klajn R., Chem. Soc. Rev. 2014, 43, 148–184. [DOI] [PubMed] [Google Scholar]
- 15. Wiedbrauk S., Dube H., Tetrahedron Lett. 2015, 56, 4266–4274. [Google Scholar]
- 16. Aprahamian I., Chem. Commun. 2017, 53, 6674–6684. [DOI] [PubMed] [Google Scholar]
- 17. Oelgemöller M., Frank R., Lemmen P., Lenoir D., Lex J., Inoue Y., Tetrahedron 2012, 68, 4048–4056. [Google Scholar]
- 18. van Leeuwen T., Lubbe A. S., Štacko P., Wezenberg S. J., Feringa B. L., Nat. Rev. Chem. 2017, 1, 0096. [Google Scholar]
- 19. Kassem S., van Leeuwen T., Lubbe A. S., Wilson M. R., Feringa B. L., Leigh D. A., Chem. Soc. Rev. 2017, 46, 2592–2621. [DOI] [PubMed] [Google Scholar]
- 20. Roke D., Wezenberg S. J., Feringa B. L., Proc. Natl. Acad. Sci. USA 2018, 115, 9423–9431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lenoir D., Lemmen P., Chem. Ber. 1980, 113, 3112–3119. [Google Scholar]
- 22. McMurry J. E., Chem. Rev. 1989, 89, 1513–1524. [Google Scholar]
- 23. Lenoir D., Lemmen P., Chem. Ber. 1984, 117, 2300–2313. [Google Scholar]
- 24. Akbulatov S., Tian Y., Boulatov R., J. Am. Chem. Soc. 2012, 134, 7620–7623. [DOI] [PubMed] [Google Scholar]
- 25. Kucharski T. J., Huang Z., Yang Q.-Z., Tian Y., Rubin N. C., Concepcion C. D., Boulatov R., Angew. Chem. Int. Ed. 2009, 48, 7040–7043; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7174–7177. [Google Scholar]
- 26. Akbulatov S., Tian Y., Huang Z., Kucharski T. J., Yang Q.-Z., Boulatov R., Science 2007, 357, 299–303. [DOI] [PubMed] [Google Scholar]
- 27. Lapouyade R., Czeschka K., Majenz W., Rettig W., Gilabert E., Rulliere C., J. Phys. Chem. 1992, 96, 9643–9650. [Google Scholar]
- 28. Letard J. F., Lapouyade R., Rettig W., J. Am. Chem. Soc. 1993, 115, 2441–2447. [Google Scholar]
- 29. de Jong J., Feringa B. L., Wezenberg S. J., ChemPhysChem 2019, 20, 3306–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang J.-X., Niu L.-Y., Chen P.-Z., Chen Y.-Z., Yang Q.-Z., Boulatov R., Chem. Commun. 2019, 55, 7017–7020. [DOI] [PubMed] [Google Scholar]
- 31. Wu Y.-H., Huang K., Chen S.-F., Chen Y.-Z., Tung C.-H., Wu L.-Z., Sci. China Chem. 2019, 62, 1194–1197. [Google Scholar]
- 32. Shimasaki T., Kato S., Shinmyozu T., J. Org. Chem. 2007, 72, 6251–6254. [DOI] [PubMed] [Google Scholar]
- 33. Ogawa K., Harada J., Tomoda S., Acta Crystallogr. Sect. B 1995, 51, 240–248. [Google Scholar]
- 34. Ogawa K., Harada J., Tomoda S., Acta Crystallogr. Sect. C 1995, 51, 2125–2127. [Google Scholar]
- 35. Oelgemöller M., Brem B., Frank R., Schneider S., Lenoir D., Hertkorn N., Origane Y., Lemmen P., Lex J., Inoue Y., J. Chem. Soc. Perkin Trans. 2 2002, 1760–1771. [Google Scholar]
- 36. Wezenberg S. J., Feringa B. L., Nat. Commun. 2018, 9, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kucharski T. J., Boulatov R., J. Mater. Chem. 2011, 21, 8237–8255. [Google Scholar]
- 38. Saltiel J., D‘Agostino J. T., J. Am. Chem. Soc. 1972, 94, 6445–6456. [Google Scholar]
- 39. Rothenberger G., Negus D. K., Hochstrasser R. M., J. Chem. Phys. 1983, 79, 5360–5367. [Google Scholar]
- 40. Doany F. E., Heilweil E. J., Moore R., Hochstrasser R. M., J. Chem. Phys. 1984, 80, 201–206. [Google Scholar]
- 41. Fuss W., Kosmidis C., Schmid W. E., Trushin S. A., Angew. Chem. Int. Ed. 2004, 43, 4178–4182; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 4273–4277. [Google Scholar]
- 42. Improta R., Santoro F., Dietl C., Papastathopoulos E., Gerber G., Chem. Phys. Lett. 2004, 387, 509–516. [Google Scholar]
- 43. Improta R., Santoro F., J. Phys. Chem. A 2005, 109, 10058–10067. [DOI] [PubMed] [Google Scholar]
- 44. Quick M., Berndt F., Dobryakov A. L., Ioffe I. N., Granovsky A. A., Knie C., Mahrwald R., Lenoir D., Ernsting N. P., Kovalenko S. A., J. Phys. Chem. B 2014, 118, 1389–1402. [DOI] [PubMed] [Google Scholar]
- 45. Shinkai S., Manabe O., Top. Curr. Chem. 1984, 121, 67–104. [Google Scholar]
- 46. Lee S., Flood A. H., J. Phys. Org. Chem. 2013, 26, 79–86. [Google Scholar]
- 47. Díaz-Moscoso A., Ballester P., Chem. Commun. 2017, 19, 324–327. [DOI] [PubMed] [Google Scholar]
- 48. Shimasaki T., Kato S., Ideta K., Goto K., Shinmyozu T., J. Org. Chem. 2007, 72, 1073–1087. [DOI] [PubMed] [Google Scholar]
- 49. Wezenberg S. J., Feringa B. L., Org. Lett. 2017, 19, 324–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Blom M., Norrehed S., Andersson C.-H., Huang H., Light M., Bergquist J., Grennberg H., Gogoll A., Molecules 2016, 21, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Olsson S., Schäfer C., Blom M., Gogoll A., ChemPlusChem 2018, 83, 1169–1178. [DOI] [PubMed] [Google Scholar]
- 52. Xu J. F., Chen Y.-Z., Wu L.-Z., Tung C.-H., Yang Q.-Z., Org. Lett. 2014, 16, 684–687. [DOI] [PubMed] [Google Scholar]
- 53. Wang Y., Tian Y., Chen Y.-Z., Niu L.-Y., Wu L.-Z., Tung C.-H., Yang Q.-Z., Boulatov R., Chem. Commun. 2018, 54, 7991–7994. [DOI] [PubMed] [Google Scholar]
- 54. Zhan T.-G., Yin H.-H., Zheng S.-T., Lin W.-C., Shen N.-L., Cui J., Kong L.-C., Liu L.-J., Zhang K.-D., Chem. Commun. 2018, 54, 9356–9359. [DOI] [PubMed] [Google Scholar]
- 55. Ma X., Tian H., Acc. Chem. Res. 2014, 47, 1971–1981. [DOI] [PubMed] [Google Scholar]
- 56. Xu J.-F., Chen Y.-Z., Wu D., Wu L. Z., Tung C. H., Yang Q.-Z., Angew. Chem. Int. Ed. 2013, 52, 9738–9742; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9920–9924. [Google Scholar]
- 57. Yan X., Xu J.-F., Cook T. R., Huang F., Yang Q.-Z., Tung C.-H., Stang P. J., Proc. Natl. Acad. Sci. USA 2014, 111, 8717–8722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang Y., Xu J.-F., Chen Y.-Z., Niu L.-Y., Wu L.-Z., Tung C.-H., Yang Q.-Z., Chem. Commun. 2014, 50, 7001–7003. [DOI] [PubMed] [Google Scholar]
- 59. Wang Y., Sun C.-L., Niu L.-Y., Wu L.-Z., Tung C.-H., Chen Y.-Z., Yang Q.-Z., Polym. Chem. 2017, 8, 3596–3602. [Google Scholar]
- 60. Zhu H., Shangguan L., Xia D., Mondal J. H., Shi B., Nanoscale 2017, 9, 8913–8917. [DOI] [PubMed] [Google Scholar]
- 61. Yang Q.-Z., Huang Z., Kucharski T. J., Khvostichenko D., Chen J., Boulatov R., Nat. Nanotechnol. 2009, 4, 302–306. [DOI] [PubMed] [Google Scholar]
- 62. Huang Z., Yang Q.-Z., Khvostichenko D., Kucharski T. J., Chen J., Boulatov R., J. Am. Chem. Soc. 2009, 131, 1407–1409. [DOI] [PubMed] [Google Scholar]
- 63. Olsson S. K., Benito Perez O., Blom M., Gogoll A., Beilstein J. Org. Chem. 2019, 15, 2408–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tian Y., Kucharski T. J., Yang Q.-Z., Boulatov R., Nat. Commun. 2013, 4, 2538. [DOI] [PubMed] [Google Scholar]
- 65. Kucharski T. J., Yang Q.-Z., Tian Y., Boulatov R., J. Phys. Chem. Lett. 2010, 1, 2820–2825. [Google Scholar]
- 66. Kean Z. S., Akbulatov S., Tian Y., Widenhoefer R. A., Boulatov R., Craig S. L., Angew. Chem. Int. Ed. 2014, 53, 14508–14511; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14736–14739. [Google Scholar]
- 67. van Leeuwen P. W. N. M., Kamer P. C. J., Reek J. N. H., Dierkes P., Chem. Rev. 2000, 100, 2741–2770. [DOI] [PubMed] [Google Scholar]
- 68. Zhu N., Li X., Wang Y., Ma X., Dyes Pigm. 2016, 125, 259–265. [Google Scholar]
- 69. Li Y., Tse E. C. M., Barile C. J., Gewirth A. A., Zimmerman S. C., J. Am. Chem. Soc. 2015, 137, 14059–14062. [DOI] [PubMed] [Google Scholar]
- 70. O′Hagan M. P., Haldar S., Duchi M., Oliver T. A. A., Mulholland A. J., Morales J. C., Galan M. C., Angew. Chem. Int. Ed. 2019, 58, 4334–4338; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4378–4382. [Google Scholar]
- 71. O'Hagan M. P., Peñalver P., Gibson R. S. L., Morales J. C., Galan M. C., Chem. Eur. J. 2020, 10.1002/Chem.201905753. [DOI] [Google Scholar]
- 72. Rotstan K. A., Abdelsayed M. M., Passalacqua L. F. M., Chizzolini F., Sudarshan K., Chamberlin A. R., Míšek J., Luptak A., eLife 2020, 9, e51737. [DOI] [PMC free article] [PubMed] [Google Scholar]