

Abstract
Objective
Recent studies have shown that microRNAs (miRNAs/miRs) play key roles in adipogenesis. This study aimed to investigate the role and underlying mechanism of miR‐182 in adipogenesis.
Methods
This study used the 3T3‐L1 cell line and human visceral adipose tissue (VAT)‐derived adipocytes to determine the role of miR‐182 in adipogenesis. Adipose tissues from mice with high‐fat diet–induced obesity, ob/ob mice, or human individuals with obesity were used to determine the association of miR‐182 levels with obesity. A luciferase reporter assay was used to determine the target of miR‐182.
Results
The expression level of miR‐182 was greatly downregulated during white adipogenesis and markedly lower in the VAT of mice and humans with obesity. Ectopic expression of miR‐182 in 3T3‐L1 cells and human adipocytes suppressed the formation of lipid droplets and the expression of adipogenic genes. The luciferase reporter assay showed that miR‐182 targeted the 3′‐untranslated sequence of CCAAT/enhancer‐binding protein α (C/EBPα) directly. In addition, glucocorticoids negatively regulated miR‐182 expression, which, in turn, suppressed the glucocorticoid‐induced expression of C/EBPα.
Conclusions
Taken together, our studies identified miR‐182 as a novel negative regulator of adipogenesis and a potential therapeutic target for obesity.
Study Importance.
What is already known?
-
►
Obesity is associated with the dysregulation of adipogenesis.
-
►
MicroRNAs (miRNAs/miRs) play important roles in adipogenesis and the functional maintenance of adipocytes.
What does this study add?
-
►
miR‐182 levels are markedly lower in adipose tissues of mice and humans with obesity.
-
►
miR‐182 negatively regulates adipogenesis by directly targeting the key regulator of adipogenic genes, CCAAT/enhancer‐binding protein α (C/EBPα).
-
►
miR‐182 mediates glucocorticoid‐induced C/EBPα expression.
How might these results change the direction of research?
-
►
As miR‐182 is a novel negative regulator of adipogenesis, it might be a potential therapeutic target for obesity.
Introduction
Obesity, a risk factor for many diseases, such as insulin resistance, type 2 diabetes (T2D), and cardiovascular diseases, has become a major health problem worldwide (1).Obesity is associated with adipocyte hypertrophy (increases in cell size) and/or hyperplasia (expanding the numbers of adipocytes), 2 processes largely dependent on the dysregulation of adipocyte differentiation (2). As adipose tissue is crucial not only as an energy reservoir but also as an endocrine organ that secretes numerous biologically active factors, including adiponectin, leptin, resistin, and many other cytokines (3), phenotypic and functional changes of this tissue may lead to metabolic dysfunction (4). Thus, the elucidation of molecular mechanisms underlying adipogenesis is important, as it may lead to the identification of novel therapeutic targets for the treatment of obesity and related diseases.
Adipogenesis in vitro is a highly ordered and well‐characterized temporal process. Transcription factors, such as peroxisome proliferator–activated receptor γ (PPARγ) and CCAAT/enhancer‐binding protein (C/EBP) family members, including C/EBPα, C/EBP β, and C/EBPδ, have been shown to play critical roles in determining adipocyte differentiation and maturation (for reviews, see (5, 6, 7)). During the early stage of adipogenesis, the expression of C/EBPβ is induced rapidly, and this transcription factor initiates clonal expansion and the expression of C/EBPα and PPARγ. The expression of C/EBPα and PPARγ functions as a master regulator of various adipogenic genes, allowing cells to acquire characteristics of mature adipocytes through terminal differentiation (7).
Glucocorticoids (GCs), important anti‐inflammatory and immunosuppressive agents, are potent stimuli for adipogenesis (8). GCs promote visceral obesity in humans by increasing the number of newly formed adipocytes, and prolonged hypercortisolemia has been shown to lead to truncal obesity in patients with Cushing syndrome (9, 10). Dexamethasone (DEX), a synthetic GC, accelerates adipogenesis by promoting the expression of C/EBPδ, C/EBPβ, C/EBPα, and PPARγ in the early phase of differentiation (11). However, the molecular mechanisms underlying GC‐induced adipogenesis have yet to be elucidated.
MicroRNAs (miRNAs/miRs) play key roles in the differentiation, growth, and apoptosis of various cells. Recently, an accumulating number of miRNAs have been identified as playing a role in the regulation of adipogenesis, and some were shown to be involved in human adipocyte differentiation (12, 13). Some other miRNAs are important for lipid homeostasis (14) and the regulation of insulin sensitivity. The dysregulation of miRNAs is associated with metabolic disorders, including obesity and T2D (15, 16).
miR‐182 belongs to the miR‐183 cluster, which comprises miR‐96, miR‐182, and miR‐183. Its sequence is highly conserved among mice, humans, and zebra fish (17). In patients with T2D and in T2D animal models, the levels of miR‐182 are markedly suppressed in sera and insulin‐response tissues, such as the liver, adipose tissue, and skeletal muscle, compared with respective controls (18). The lipid‐synthesis regulator sterol regulatory element–binding protein (SREBP)2 activates miR‐182 transcription, which, in turn, causes the negative regulation of F‐box and tryptophan–aspartic acid repeat domain–containing 7 expression, a known negative factor involved in nuclear SREBP accumulation (19). To conclude, this miRNA plays an important role in the regulation of nuclear SREBP levels and endogenous lipid synthesis. Zhang et al. (20) reported that muscle‐enriched miR‐182 regulates glucose use in muscles by targeting forkhead box O1 (FoxO1) and pyruvate dehydrogenase kinase 4. Sun and his colleagues (21) found that miR‐182 is required for brown adipocyte development and the maintenance of brown fat features. However, the role of miR‐182 in adipogenesis has not been addressed.
In this study, we investigated the role of miR‐182 in adipogenesis in 3T3‐L1 cells and human visceral adipocytes. Our results have shown that, in the adipose tissues of mice and human patients with obesity, expression levels of miR‐182 were reduced greatly. Overexpression of miR‐182 inhibited adipocyte differentiation by directly targeting C/EBPα, one of the master genes that regulate lipid formation and adipocyte maturation. We also found that GCs suppressed the expression of miR‐182, which caused negative expression of GC‐induced C/EBPα and adipogenesis in vitro. Our results identified miR‐182 as a novel negative regulator of adipogenesis.
Methods
Cell culture and Oil Red O staining
3T3‐L1 preadipocytes were maintained in DMEM (Gibco, Life Technologies, Grand Island, New York) culture medium supplemented with 10% fetal bovine serum (FBS) (Gibco) and 1% penicillin and streptomycin (Gibco). After the cells reached 100% confluence (day 0), the culture medium was converted to a differentiation cocktail containing isobutylmethylxanthine, DEX, and insulin (Sigma‐Aldrich, St. Louis, Missouri). On day 4 after induction, the medium was replaced by fresh DMEM containing 10% FBS and insulin only. After 10 days of induction, 3T3‐L1 preadipocytes reached complete maturation, with large amounts of lipid formation.
Human stromal vascular fraction (SVF) cells were isolated from visceral adipose tissue (VAT) by collagenase digestion, according to our previous report (22). Isolated preadipocytes were plated and cultured in DMEM containing 10% FBS and a differentiation cocktail. The cells were induced to differentiation according to the aforementioned procedure. The adipose tissues were obtained from Chinese patients who underwent abdominal minimally invasive surgery at the Minimally Invasive Surgery Center at The Second Xiangya Hospital of Central South University. All recruited human participants underwent BMI measurement (calculated as body weight in kilograms divided by height in meters squared) and were given forms for written informed consent, and the protocol was approved by the ethics committee of The Second Xiangya Hospital of Central South University, Changsha, China. Written informed consent was received from participants prior to inclusion in the study.
For Oil Red O staining, mature adipocytes were washed twice with phosphate‐buffered saline and fixed in 4% paraformaldehyde for 1 hour, which was followed by Oil Red O staining for 1 hour. The images were visualized by light microscopy (Olympus, Tokyo, Japan) and photographed.
Transfection and treatment
For transfection, the duplex oligonucleotide (mimic) or single‐stain antisense oligonucleotide (inhibitor) designated for miR‐182 or nonspecific control (NC) (GenePharma, Shanghai, China) was added to the cultured cells with 70% to 80% confluence at a final concentration of 50 nM using FuGENE HD Transfection Reagent (Promega, Madison, Wisconsin). For hormonal treatment, the preadipocytes cultured in DMEM were treated with DEX, a synthetic GC; mifepristone (RU486; Sigma‐Aldrich), a GC receptor (GR) antagonist; or DMEM control for 6 hours for gene expression or for 6 days for lipid‐droplet formation.
Animals
Six‐week‐old male C57BL/6J mice were purchased from Shanghai Laboratory Animal Center Laboratory Animal Co., Ltd. (Shanghai, China) and housed in a temperature‐controlled environment with a 12:12‐hour light/dark cycle. The mice had access to food and water ad libitum. After an adaptive period of 1 week, the mice were randomly distributed into weight‐matched groups and fed either a normal chow diet or a high‐fat diet (HFD) (60% of kilocalories from fat; Research Diets Inc., New Brunswick, New Jersey). After 16 weeks of feeding, animals were sacrificed, and perigonadal VAT was removed rapidly, immediately frozen in liquid nitrogen, and stored at −80 °C for further analyses. The 8‐week‐old male ob/ob mice were purchased from the National Resource Center for Mutant Mice (Nanjing, China), and fat tissues were collected as described earlier. All procedures involving animals were conducted in accordance with the guidelines of the Central South University Committee on the Care and Use of Animals.
RNA isolation and real‐time quantitative reverse transcription–polymerase chain reaction
Total RNA was extracted by using TRIzol Reagent (Invitrogen, Life Technologies) and following the manufacturer’s instructions, and 1 μg of RNA was reverse‐transcribed by using the Takara One Step PrimeScript miRNA complementary DNA (cDNA) synthesis kit (Takara Bio Inc., Shiga, Japan) to obtain cDNA for miRNAs and by using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Beijing, China) to obtain cDNA for mRNA, respectively. A SYBR green (Takara) polymerase chain reaction (PCR) method with specific pairs of primers was used to determine gene expression levels on a 7900HT Fast Real‐Time PCR System (Applied Biosystems, Foster City, California). Relative expression of mRNA or miRNA was calculated using the comparative cycle‐threshold method (23). The mRNA levels of designated genes were normalized to β‐actin, and the miRNA levels were normalized to small nuclear RNA U6.
The gene‐specific primer pairs used in the experiment were: actin, forward primer (FP) 5′‐CAACGAGCG GTTCCGATG‐3′ and reverse primer (RP) 5′‐GCCACAGGATTCCA TACC CA‐3′; acetyl–coenzyme A carboxylase (Acc) α, FP 5′‐GCT ACCCCAGCAGTATTTGAAC‐3′ and RP 5′‐GAC ATCAGCCACCATCTCTGTA‐3′; Accβ, FP 5′‐TTTCTGACTTGCCTGGTTTCT‐3′ and RP 5′‐GAGATTGGCTTCCAC CTTACTG‐3′; adiponectin, FP 5′‐CCCAAGGGA ACTTGTGCAGGT TGGATG‐3′ and RP 5′‐GTTGGTATCATGGTAGAGAAGAA AGCC‐3′; C/EBPα, FP 5′‐GAAGTC GGTGGACAAGAACAG‐3′ and RP 5′‐GTTTGGCTTTATCTCGGCTCT‐3′; C/EBPβ, FP 5′‐GACT TCCTCTCCGACCTCTTC‐3′ and RP 5′‐GAGGCTCACGTAACCGTA GTC‐3′; fatty acid–binding protein 4 (Fabp4), FP 5′‐GATT TCCTTCAAACTGGGC G‐3′ and RP 5′‐CACATTCCACCACCAG CTTG‐3′; fatty acid synthase, FP 5′‐ATTCGTGATGGAGTCGTG AAG‐3′ and RP 5′‐GGT CTTGGAGATGGCAGAA AT‐3′; forkhead box protein O1 (Foxo1), FP 5′‐GGACAGCA AATCAAGTTACGG‐3′ and RP 5′‐ATTGTGGGG AGGAGAGTCAGA‐3′; and Pparγ, FP 5′‐GCCTGCGGAAGCCCTTTGGT‐3′ and RP 5′‐AAGCCTGGGCGGTCTCCACT‐3′. The mature miRNA sequence for mouse miR‐182‐5p (UUUGGCAAUGGUAGAACUCACACCG; miRbase accession number MIMAT0000211) and the quantitative PCR primers against mature miRNAs (catalog number MmiRQP0895) were purchased from GeneCopoeia (Rockville, Maryland).
Western blotting
To examine protein expression levels, cells were lysed in radioimmunoprecipitation assay lysis buffer (Beyotime, Shanghai, China) and centrifuged at 12,000g for 10 minutes at 4 °C. Protein concentrations were determined using a standard bicinchoninic acid assay. The same number of protein samples was separated on 10% sodium dodecyl sulfate–polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Bio‐Rad, Hercules, California). The primary antibody for C/EBPα was obtained from Santa Cruz Biotechnology (Santa Cruz, California), and the primary antibody for α‐tubulin was obtained from Sigma‐Aldrich. Signals were detected and analyzed using ChemiDoc XRS+ with the Image Lab system (Bio‐Rad).
Luciferase constructs and reporter assay
Mouse 3′‐untranslated sequences (UTRs) for CEBP/α were PCR‐amplified with specific primers: wild type, FP 5′‐CCGCTCGAGGGCGCGCGGCTGCGGGAC‐3′ and RP 5′‐GAATGC GGCCGCGAGTTTGA TATGTTTATATTATAGATATAATACATACGACATCTAG CATG‐3′; mutant, FP 5′‐TGATTC GACCGAGACTGAGACTCTTCACTAAC‐3′ and RP 5′‐AGTCTCAGTCTCGGTCGAAT CAGAGCAAAACC‐3′. The wild type or mutant (MUT) of 3′‐UTR was cloned into the pmiR‐RB‐Report vector (Ribobio, Guangzhou, China) between the Xhol and Notl sites downstream of the Renilla luciferase gene. To generate the MUT reporter, 5 nucleotides in the CEBP/α (1,020‐1,030) of the miR‐182 seed region were point‐mutated using the Site‐directed Gene Mutagenesis Kit (Beyotime, Suzou, China), and the mutations were verified by sequencing.
HEK 293T cells were seeded in 24‐well plates and cotransfected with 3′‐UTR reporter or MUT construct and miR‐182 mimic or NC using Lipofectamine 2000 (Invitrogen, Carlsbad, California), according to the manufacturer’s protocol. Luciferase activities in the cell lysate were measured using the Dual‐Glo luciferase assay system, in accordance with the manufacturer's instructions (Promega). The firefly luciferase activity was normalized with Renilla luciferase activity for each well.
Construction of the C/EBPα expression vector
cDNA for the mouse C/EBPα gene was obtained from total RNA, which was prepared from mouse tissues using a RevertAid First Strand cDNA Synthesis Kit–supported reverse transcription (RT)–PCR technique (Thermo Fisher Scientific, Shanghai, China). The FP sequence was 5′‐CTTGGATC CGCCATGGAGTCGGCCGACTTC TACG‐3′, and the RP sequence was 5′‐CATGAGCTCAGTGCGCG TCAACGGGTAC‐3′. The PCR products were digested with BamH I and Xho I (Takara), purified from agarose gels, and subcloned into plasmid‐cloning DNA 3.1+ (Invitrogen). The cells were transfected with expression plasmids or plasmid‐cloning DNA 3.1 control plasmids using Lipofectamine 2000.
Statistical analyses
All results were presented as means ± SEMs. Comparisons between 2 groups were assessed using the 2‐tailed Student t test for independent samples, and comparisons among multiple groups were analyzed by using 1‐way ANOVA. The linear correlation between 2 variables was analyzed by using the Pearson correlation coefficient. Statistical analyses were carried out using SPSS Statistics Software (version 19.0; IBM Corp., Armonk, New York). P < 0.05 was considered statistically significant.
Results
Expression levels of miR‐182 in adipogenesis and adipose tissues
To investigate the role of miR‐182 in adipocyte differentiation, we examined miR‐182 expression during 3T3‐L1 adipocyte differentiation. The expression levels of miR‐182 decreased significantly after induction during days 2, 4, 6, and 8 compared with the initiation of differentiation (day 0) (Figure 1A). Similar expression patterns were observed in the human visceral fat SVF–derived primary adipocytes (Figure 1B). However, a moderate increase occurred at day 10 in both 3T3‐L1 and human adipocytes (40% ± 0.396% and 32% ± 0.205%, respectively, vs. day 0), which was not significantly higher than that of day 0 (P = 0.067 and P = 0.0825, respectively, vs. day 0).
Figure 1.
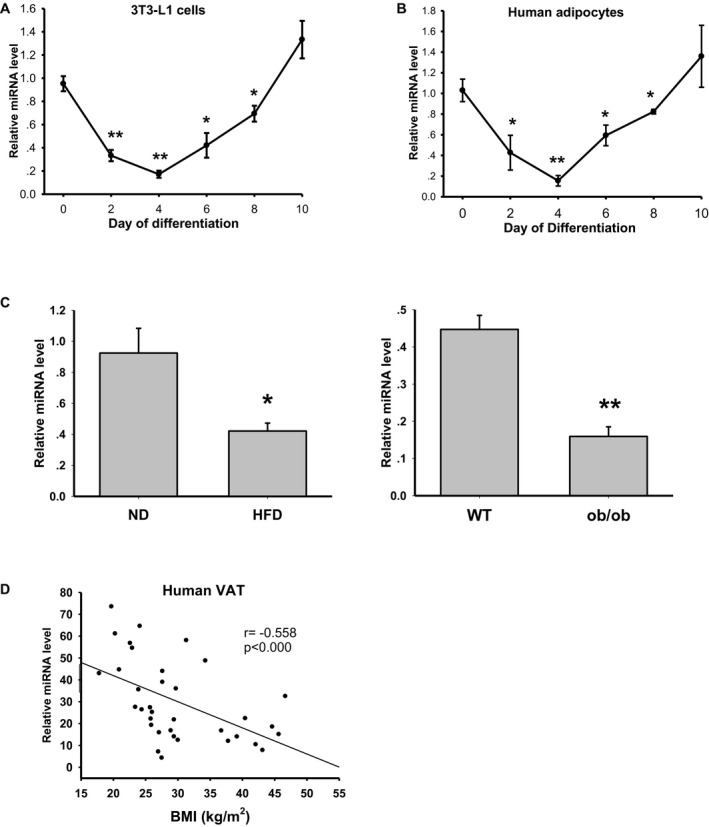
Expression levels of miR‐182 in adipogenesis and adipose tissues. (A) Expression levels of miR‐182 during the differentiation of 3T3‐L1 cells (n = 4 independent experiments; *P < 0.05, **P < 0.01 vs. day 0). (B) Expression levels of miR‐182 during the differentiation of human VAT stromal vascular fraction–derived adipocytes (n = 4; *P < 0.05, **P < 0.01 vs. day 0). (C) Expression levels of miR‐182 in the perigonadal visceral white adipose tissues of C57BL/6 mice. Left graph: HFD‐ and ND‐fed mice (n = 6; *P < 0.05, **P < 0.01 vs. ND). Right graph: ob/ob and littermate WT mice (n = 4) (D) Expression levels of miR‐182 in the human intra‐abdominal VAT of Chinese people (n = 35, Pearson correlation analysis). HFD, high‐fat diet; miR, microRNA; ND, normal chow diet; VAT, visceral adipose tissue; WT, wild type.
To determine the expression levels of miR‐182 in the fat tissues of mice with obesity, we collected perigonadal VAT from C57BJ/6 mice fed with an HFD or from ob/ob mice. We found that the mice fed an HFD for 16 weeks showed a significant increase in body weight (45.52 ± 0.68 g vs. 26.62 ± 0.88 g in controls). In the meantime, when compared with those of the control mice, it was observed that miR‐182 levels decreased dramatically in the fat pads (Figure 1C, left graph). Similarly, miR‐182 levels were also decreased in the fat tissues of ob/ob mice (Figure 1C, right graph). We also found that miR‐182 levels in human intra‐abdominal VAT decreased in overweight (BMI > 25 kg/m2) and obesity (BMI > 30 kg/m2), and there was a negative correlation between the miR‐182 level and BMI (Figure 1D), suggesting that downregulation of miR‐182 is associated with increased adiposity in obesity.
miR‐182 inhibits adipogenesis and decreases adipogenically related gene expression
To determine the role of miR‐182 in adipocyte differentiation, we transfected 3T3‐L1 or human fat SVF–derived preadipocytes with miR‐182 mimic or an NC oligo (Figure 2A) and induced cells to go through differentiation. Overexpression of miR‐182 significantly decreased triglyceride accumulation as analyzed by Oil Red O staining in both rodent and human adipocytes (Figure 2B). Compared with those of control cells, the expression levels of adipocyte‐related genes, such as early‐response gene C/EBPβ; late‐response genes PPARγ and C/EBPα; lipogenic genes Srebp1, fatty acid synthase, and Accα; and mature‐adipocyte genes Fabp4 and adiponectin, decreased dramatically in miR‐182–transfected 3T3‐L1 and human adipocytes (Figure 2C). These data suggest that miR‐182 has a negative effect on adipogenesis.
Figure 2.
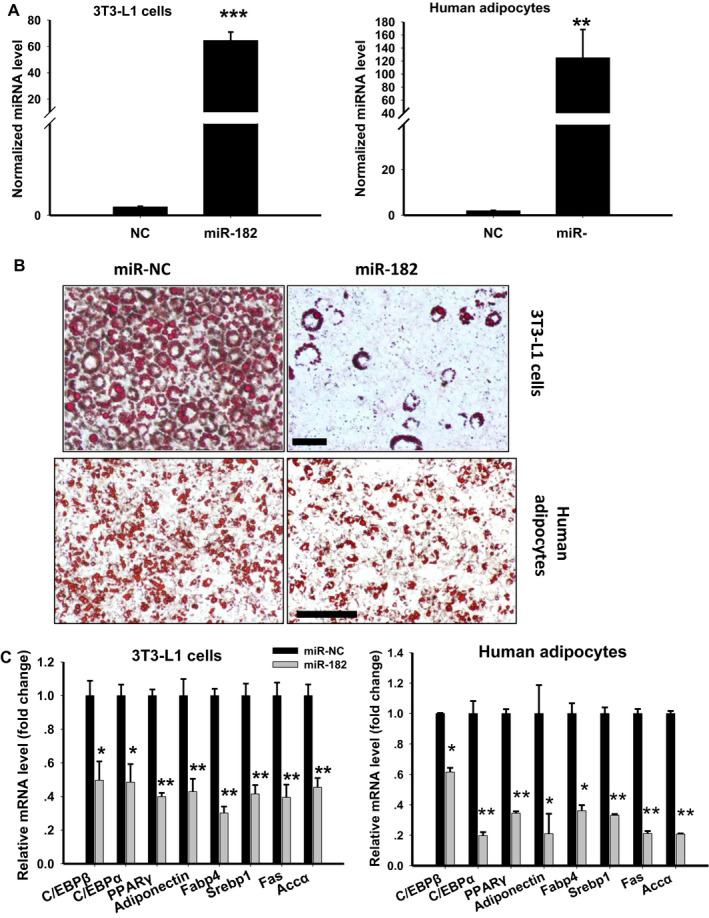
Effects of miR‐182 on adipogenesis. (A) Expression levels of miR‐182 in adipocytes transfected with miR‐182 mimic or an NC oligo (n = 4; **P < 0.01, ***P < 0.001 vs. NC). (B) Representative images of Oil Red O staining of mature 3T3‐L1 cells (upper) or adipocytes derived from human white adipose tissues (lower). The 3T3‐L1 or human adipose stromal vascular fraction–derived preadipocytes transfected with miR‐182 mimic or NC were cultured and induced to differentiation. Mature adipocytes were fixed and stained with Oil Red O (scale bar = 50 µm). (C) mRNA levels of the key adipogenic genes in 3T3‐L1 cells (left) and in human adipocytes (right) transfected with miR‐182 mimic or NC (n = 4; *P < 0.05, **P < 0.01 vs. control). Accα, acetyl–coenzyme A carboxylase; C/EBP, CCAAT/enhancer‐binding protein; Fabp4, fatty acid–binding protein 4; NC, nonspecific control; PPARγ, peroxisome proliferator–activated receptor γ; Srebp1, sterol regulatory element–binding protein 1. [Color figure can be viewed at wileyonlinelibrary.com]
miR‐182 directly targets C/EBPα
We used the TargetScan version 6.2 computational program (http://www.targetscan.org/) to analyze potential targets of miR‐182. The in silico analysis identified C/EBPα, a key regulator of adipocyte differentiation, as a potential target of miR‐182. miR‐182 recognizes the 3′‐UTR sequence of the C/EBPα gene that is highly conserved in many mammalian species, including humans, mice, rats, chimpanzees, and dogs (Figure 3A). To investigate whether miR‐182 targets C/EBPα directly, we performed luciferase reporter assays using the firefly luciferase reporter gene, which was fused with C/EBPα 3′‐UTR or 3′‐UTR binding‐site MUTs. Compared with the blank control or C/EBPα 3′‐UTR MUT, cotransfection of the miR‐182 mimic significantly reduced the luciferase activity of the firefly luciferase gene containing the C/EBPα 3′‐UTR (Figure 3B), indicating that miR‐182 could directly target the 3′‐UTR sequence of C/EBPα.
Figure 3.
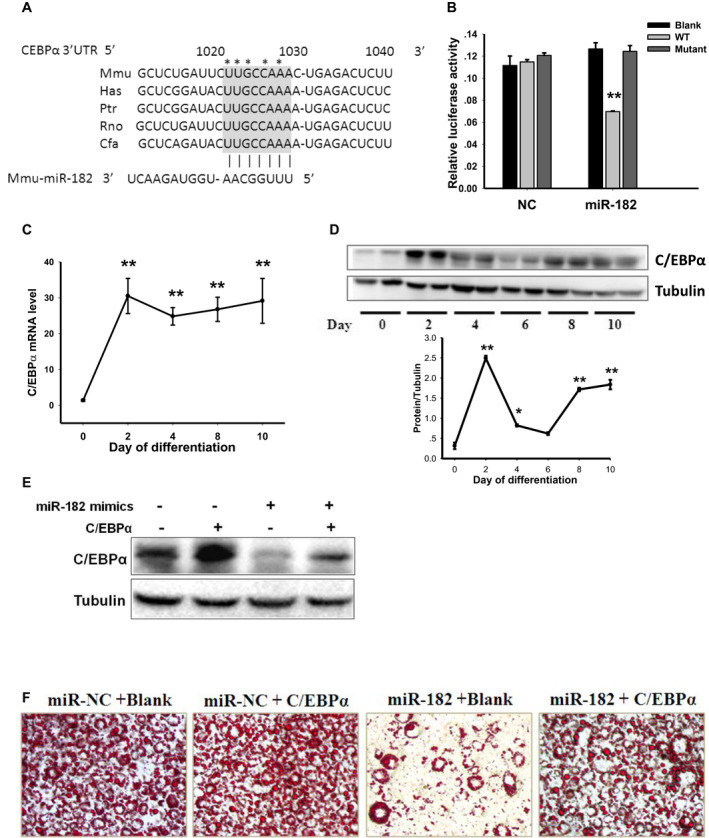
miR‐182 directly targets C/EBPα. (A) Conserved sequences of the miR‐182 binding site in the C/EBPα 3′‐UTR of mammalians. The miR‐182 seed‐match region in indicated by the gray box. Asterisks indicate the mutation sites of the seed sequence in the luciferase reporter construct. (B) Luciferase activity of the C/EBPα 3′‐UTR in the presence of miR‐182 mimic or NC (n = 5; **P < 0.01 vs. blank or mutant vector control). The (C) mRNA (n = 3; **P < 0.01 vs. day 0) and (D) protein levels of C/EBPα during the differentiation of 3T3‐L1 cells (*P < 0.05, **P < 0.01 vs. day 0). (E) Protein levels of C/EBPα in the 3T3‐L1 cells transfected with C/EBPα expression vector in the presence of miR‐182 mimic or NC. (F) Representative Oil Red O–stained microscopic images of mature adipocytes transfected with C/EBPα expression or blank vector in the presence of miR‐182 mimic or NC. C/EBPα, CCAAT/enhancer‐binding protein α; Cfa, Canis familiaris; Has, Homo sapiens; Mmu, Mus musculus; NC, nonspecific control; Ptr, chimpanzee; Rno, Rattus norvegicus; UTR, untranslated region; WT, wild type. [Color figure can be viewed at wileyonlinelibrary.com]
We observed that both mRNA and protein levels of C/EBPα were upregulated during 3T3‐L1 adipocyte differentiation started from induction day 2 (Figure 3C‐3D), which were, in part, negatively correlated with the expression pattern of miR‐182 (Figure 1A). Consistent with these findings, overexpression of miR‐182 caused C/EBPα mRNA (Figure 2B) and protein levels (Figure 3E) to decrease in the adipocytes.
Next, we examined whether C/EBPα mediates the negative effects of miR‐182 on adipogenesis in 3T3‐L1 cells. We transfected preadipocytes with C/EBPα expression vector with or without miR‐182 mimic (Figure 3E) and induced cells to differentiation. Although miR‐182 downregulated C/EBPα expression, overexpression of C/EBPα rescued the reduced protein levels (Figure 3E) in adipocytes. In line with this, the results of Oil Red O staining showed that overexpression of C/EBPα recovered the abolished lipid‐droplet formation caused by miR‐182 (Figure 3F), suggesting that C/EBPα mediates the effect of miR‐182 on adipogenesis.
miR‐182 represses GC‐induced C/EBPα expression and adipogenesis
GCs are powerful regulators of white adipocyte differentiation, but the underlying mechanisms remain elusive. To determine whether miR‐182 plays a role in GC‐induced adipogenesis, we treated 3T3‐L1 cells with the synthetic GC DEX at different concentrations and found that DEX treatment caused miR‐182 levels (Figure 4A) to reduce dramatically. However, when cells were cotreated with GR antagonist RU486, the suppressive effect of DEX on miR‐182 was reversed (Figure 4B), suggesting that GC‐GR signaling could regulate miR‐182 expression in a negative manner. We also found that insulin treatment had no such effects on miR‐182 expression (data not shown). Because DEX can induce the expression of C/EBPα, to determine whether miR‐182 plays a role in DEX‐regulated C/EBPα expression, we treated 3T3‐L1 cells with DEX with or without miR‐182 and found that DEX treatment significantly induced C/EBPα expression (Figure 4C). However, this effect had diminished with the cotreatment of miR‐182 mimic (Figure 4D), suggesting that miR‐182 mediates the effects of DEX on C/EBPα expression. Finally, to determine whether miR‐182 is involved in DEX‐induced adipogenesis, we transfected 3T3‐L1 preadipocytes with or without miR‐182 before DEX treatment and induced cells to differentiation. The analysis of Oil Red O staining showed that overexpression of miR‐182 caused a reduction in the DEX‐induced formation of lipid droplets in 3T3‐L1 cells (Figure 4E). In line with this, the DEX‐induced expression of adipogenic genes was suppressed when cotreated with miR‐182 mimic (Figure 4F). These results suggest that miR‐182 plays a negative role in DEX‐induced C/EBPα expression and adipogenesis.
Figure 4.
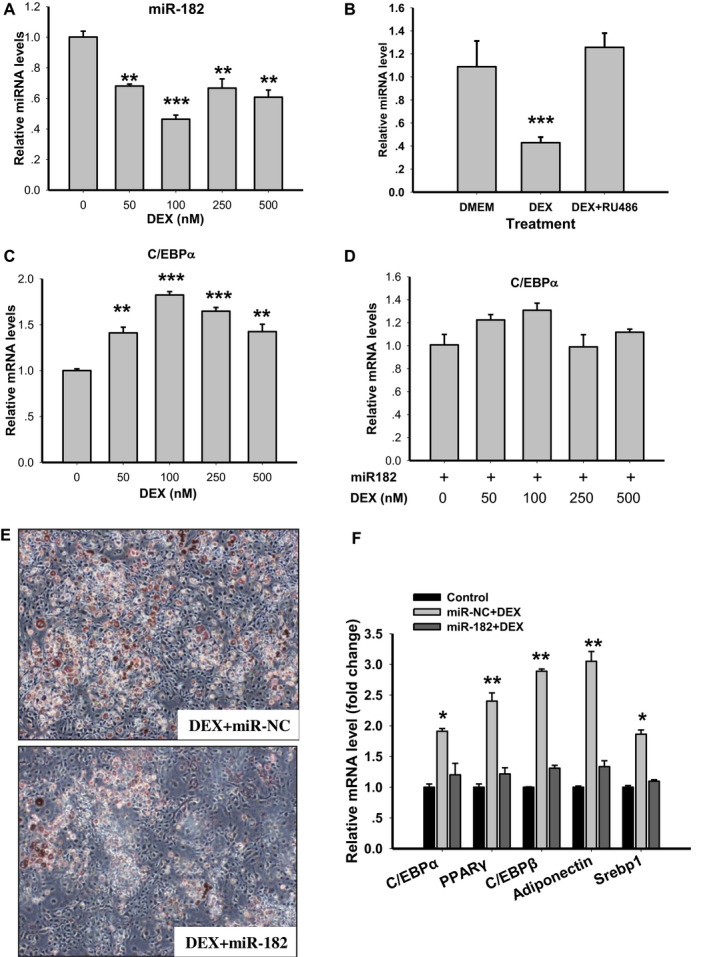
miR‐182 is negatively regulated by glucocorticoids and suppresses glucocorticoid‐induced C/EBPα expression and adipose differentiation in 3T3‐L1 cells. (A) Expression levels of miR‐182 in 3T3‐L1 cells treated with the synthetic glucocorticoid DEX for 6 hours with the designated concentration (n = 4; **P < 0.01, ***P < 0.001 vs. DMEM control). (B) Expression levels of miR‐182 in 3T3‐L1 cells treated with DEX or the combination of DEX and RU486 (n = 4; ***P < 0.001 vs. DMEM control). (C) Expression levels of C/EBPα in 3T3‐L1 cells treated with DEX for 6 hours with the designated concentration (n = 4; **P < 0.01, ***P < 0.001 vs. DMEM control). (D) Expression levels of C/EBPα treated with DEX in the presence of miR‐182 mimic (n = 4). (E) Representative Oil Red O–stained microscopic images of adipocytes transfected with miR‐182 mimic or NC and treated with DEX for 6 days. (F) mRNA levels of the adipogenic genes in 3T3‐L1 cells transfected with miR‐182 mimic or NC and treated with DEX for 6 days (n = 4; *P < 0.05, **P < 0.01 vs. control). C/EBPα, CCAAT/enhancer‐binding protein α; DEX, dexamethasone; NC, nonspecific control; PPARγ, peroxisome proliferator–activated receptor γ; Srebp1, sterol regulatory element–binding protein 1. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
miRNAs play an important role in the regulation of adipogenesis (12, 13). In the present study, we demonstrated that miR‐182 is downregulated during adipocyte differentiation and is markedly lower in the VAT of mice and humans with obesity. In addition, overexpression of miR‐182 caused a significant decrease of triglyceride accumulation and inhibited the expression of genes involved in adipocyte differentiation and lipogenesis by targeting C/EBPα. Our results suggest that miR‐182 is a negative regulator of adipogenesis.
Adipogenesis is a highly ordered and well‐characterized temporal process under the delicate control of several key transcription factors, including the early‐response genes C/EBPβ and C/EBPδ, followed by later‐response genes PPARγ and C/EBPα, which activate de novo or enhance expression of most genes that characterize the mature‐adipocyte phenotype (5, 7). It has been well established that C/EBPα plays an essential role in adipogenesis. Overexpression of C/EBPα in 3T3‐L1 preadipocytes induces cell differentiation (24), and knockdown of this gene by antisense RNA blocks this process (24). By working together with PPARγ, C/EBPα functions as a pleiotropic transcriptional activator of a large group of genes associated with adipocyte differentiation and maturation (5, 7). In this study, we found that miR‐182 decreased mRNA and protein levels of C/EBPα and also decreased levels of many adipocyte‐associated genes, including adiponectin, Fabp4, Srebp1, Fas, and Accα, suggesting that miR‐182–induced downregulation of C/EBPα might cause transcriptional repression of genes related to adipocyte phenotypes. We also identified that miR‐182 could directly target the 3′‐UTR of C/EBPα and that overexpression of this gene could reverse the anti‐adipogenic effect of miR‐182. Taken together, our data suggest that targeting C/EBPα might be the underlying mechanism of miR‐182–repressed adipogenesis. However, it should be pointed out that, except when downregulated by miR‐182, expression of C/EBPα is also regulated by some transcriptional regulators, including C/EBPβ and C/EBPδ, which are the early responsive transcription factors at the initiation of adipocyte differentiation (5, 7). It is possible that the C/EBPα level increases quickly following the induction of C/EBPβ and C/EBPδ, whose effects might exceed the action of miRNA during adipogenesis. It should also be pointed out that C/EBPα is unlikely to be the only target of miR‐182 during adipocyte differentiation. For example, FoxO1, a known target of miR‐182 in osteoblast and helper T cells (25, 26, 27), was also found to be significantly decreased at both the mRNA level and the protein level in miR‐182–overexpressed 3T3‐L1 preadipocytes (data not shown). miR‐182 could regulate FoxO3 in melanoma cells (28), BRCA1 in breast cancer cells, and fibroblast growth factor 9 and neurotrimin in Schwann cells (29). Whether these proteins are involved in regulating adipogenesis remains to be elucidated.
Studies have shown that enhancing the transcription of C/EBPα is an important mechanism of GC‐stimulated adipogenesis (30). DEX treatment activates the transcriptional activity of C/EBPβ at the C/EBPα promoter and increases adipogenesis (30). It has also been reported that DEX could shift the differentiation of bone‐marrow stromal cells to favor adipocytes over osteoblasts by upregulating the expression level of C/EBPα in vitro and in vivo (31). In this study, we discovered that DEX suppressed the expression of miR‐182, whereas it upregulated C/EBPα in adipocytes. In addition, overexpression of miR‐182 repressed DEX‐induced C/EBPα and adipogenesis, suggesting that DEX may promote adipogenesis by suppressing miR‐182 and leading to the upregulation of C/EBPα. Currently, it is not understood how the GR‐related signaling pathway suppresses miR‐182 expression in adipocytes. Interestingly, transcriptional regulator PPARγ may target binding sites of several miRNA loci, including the miR‐183 cluster, and upregulate expression of these miRNAs (32). As it is well established that there is a transcriptional network in adipogenesis involving the sequential activation of C/EBPs and PPARγ in response to hormone stimuli (5, 6, 7), it is possible that the expression of miR‐182 is under combinatorial regulation of several transcription factors or pathways that together lead to the expression pattern in adipocytes.
Previous findings have shown that miR‐182 expression levels are decreased in the liver, adipose tissue, and skeletal muscle in rats and patients with T2D (18) and that miR‐182 is involved in insulin signaling in muscle (20). We found that miR‐182 levels are markedly reduced in the adipose tissues of mice with obesity and humans with overweight or obesity. This is the first evidence showing that adipose miR‐182 is involved in adiposity. However, whether and how dysregulation of miR‐182 may contribute to adipose dysfunction in obesity is still unknown. In addition, the physiological function of miR‐182 in vivo has not been studied yet and deserves further investigation.
In conclusion, our findings demonstrate that miR‐182 is a novel negative regulator of adipogenesis and that this molecule inhibits C/EBP‐dependent adipocyte differentiation via suppression of GC‐induced C/EBPα expression. When taken together with the finding that the expression levels of miR‐182 were dramatically decreased in the adipose tissues of mice and humans with obesity, our results suggest that targeting miR‐182 may be a strategy for the treatment of obesity and obesity‐associated disorders.
Funding agencies
This work was supported by grants from the National Natural Science Foundation of China (91957113 and 31871180), the International Science & Technology Cooperation Program of China (2014DFG32490), and the Natural Science Foundation of Hunan Province (2019JJ40410 to FH).
Disclosure
The authors declared no conflict of interest.
Author contributions
MD, YY, TX, ZC, and WL performed research and analyzed data; MD wrote the draft manuscript; and FH designed the research and edited the paper.
References
- 1. Afshin A, Forouzanfar MH, Reitsma MB, et al.; GBD Obesity Collaborators . Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med 2017;377:13‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol 2006;7:885‐896. [DOI] [PubMed] [Google Scholar]
- 3. Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 2004;89:2548‐2556. [DOI] [PubMed] [Google Scholar]
- 4. Stolar MW. Insulin resistance, diabetes, and the adipocyte. Am J Health Syst Pharm 2002;59(suppl 9):S3‐S8. [DOI] [PubMed] [Google Scholar]
- 5. Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes Dev 2000;14:1293‐1307. [PubMed] [Google Scholar]
- 6. Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol 2011;12:722‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tang QQ, Lane MD. Adipogenesis: from stem cell to adipocyte. Annu Rev Biochem 2012;81:715‐736. [DOI] [PubMed] [Google Scholar]
- 8. Pantoja C, Huff JT, Yamamoto KR. Glucocorticoid signaling defines a novel commitment state during adipogenesis in vitro . Mol Biol Cell 2008;19:4032‐4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boscaro M, Barzon L, Fallo F, Sonio N. Cushing’s syndrome. Lancet 2001;357:783‐791. [DOI] [PubMed] [Google Scholar]
- 10. Tomlinson JJ, Boudreau A, Wu D, Atlas E, Haché RJG. Modulation of early human preadipocyte differentiation by glucocorticoids. Endocrinology 2006;147:5284‐5293. [DOI] [PubMed] [Google Scholar]
- 11. Park YK, Ge K. Glucocorticoid receptor accelerates, but is dispensable for, adipogenesis. Mol Cell Biol 2017;37:e00260‐16. doi:10.1128/MCB.00260‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shi C, Huang F, Gu X, et al. Adipogenic miRNA and meta‐signature miRNAs involved in human adipocyte differentiation and obesity. Oncotarget 2016;7:40830‐40845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lorente‐Cebrian S, Gonzalez‐Muniesa P, Milagro FI, Martínez JA. MicroRNAs and other non‐coding RNAs in adipose tissue and obesity: emerging roles as biomarkers and therapeutic targets. Clin Sci (Lond) 2019;133:23‐40. [DOI] [PubMed] [Google Scholar]
- 14. Zhang X, Price NL, Fernández‐Hernando C. Non‐coding RNAs in lipid metabolism. Vascul Pharmacol 2019;114:93‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trajkovski M, Hausser J, Soutschek J, et al. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 2011;474:649‐653. [DOI] [PubMed] [Google Scholar]
- 16. Rottiers V, Naar AM. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol 2012;13:239‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu S, Witmer PD, Lumayag S, Kovacs B, Valle D. MicroRNA (miRNA) transcriptome of mouse retina and identification of a sensory organ‐specific miRNA cluster. J Biol Chem 2007;282:25053‐25066. [DOI] [PubMed] [Google Scholar]
- 18. Karolina DS, Armugam A, Tavintharan S, et al. MicroRNA 144 impairs insulin signaling by inhibiting the expression of insulin receptor substrate 1 in type 2 diabetes mellitus. PLoS One 2011;6:e22839. doi:10.1371/journal.pone.0022839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jeon TI, Esquejo RM, Roqueta‐Rivera M, et al. An SREBP‐responsive microRNA operon contributes to a regulatory loop for intracellular lipid homeostasis. Cell Metab 2013;18:51‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang D, Li Y, Yao X, et al. miR‐182 regulates metabolic homeostasis by modulating glucose utilization in muscle. Cell Rep 2016;16:757‐768. [DOI] [PubMed] [Google Scholar]
- 21. Kim HJ, Cho H, Alexander R, et al. MicroRNAs are required for the feature maintenance and differentiation of brown adipocytes. Diabetes 2014;63:4045‐4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kong X, Yu J, Bi J, et al. Glucocorticoids transcriptionally regulate miR‐27b expression promoting body fat accumulation via suppressing the browning of white adipose tissue. Diabetes 2015;64:393‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pfaffl MW. A new mathematical model for relative quantification in real‐time RT‐PCR. Nucleic Acids Res 2001;29:e45. doi:10.1093/nar/29.9.e45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin FT, Lane MD. CCAAT/enhancer binding protein alpha is sufficient to initiate the 3T3‐L1 adipocyte differentiation program. Proc Natl Acad Sci U S A 1994;91:8757‐8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O'Neill LA. Outfoxing Foxo1 with miR‐182. Nat Immunol 2010;11:983‐984. [DOI] [PubMed] [Google Scholar]
- 26. Stittrich AB, Haftmann C, Sgouroudis E, et al. The microRNA miR‐182 is induced by IL‐2 and promotes clonal expansion of activated helper T lymphocytes. Nat Immunol 2010;11:1057‐1062. [DOI] [PubMed] [Google Scholar]
- 27. Kim KM, Park SJ, Jung SH, et al. miR‐182 is a negative regulator of osteoblast proliferation, differentiation, and skeletogenesis through targeting FoxO1. J Bone Miner Res 2012;27:1669‐1679. [DOI] [PubMed] [Google Scholar]
- 28. Segura MF, Hanniford D, Menendez S, et al. Aberrant miR‐182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia‐associated transcription factor. Proc Natl Acad Sci U S A 2009;106:1814‐1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yu B, Qian T, Wang Y, et al. miR‐182 inhibits Schwann cell proliferation and migration by targeting FGF9 and NTM, respectively at an early stage following sciatic nerve injury. Nucleic Acids Res 2012;40:10356‐10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuzmochka C, Abdou HS, Haché RJG, Atlas E. Inactivation of histone deacetylase 1 (HDAC1) but not HDAC2 is required for the glucocorticoid‐dependent CCAAT/enhancer‐binding protein alpha (C/EBPalpha) expression and preadipocyte differentiation. Endocrinology 2014;155:4762‐4773. [DOI] [PubMed] [Google Scholar]
- 31. Li J, Zhang N, Huang X, et al. Dexamethasone shifts bone marrow stromal cells from osteoblasts to adipocytes by C/EBPalpha promoter methylation. Cell Death Dis 2013;4:e832. doi:10.1038/cddis.2013.348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. John E, Wienecke‐Baldacchino A, Liivrand M, Heinäniemi M, Carlberg C, Sinkkonen L. Dataset integration identifies transcriptional regulation of microRNA genes by PPARgamma in differentiating mouse 3T3‐L1 adipocytes. Nucleic Acids Res 2012;40:4446‐4460. [DOI] [PMC free article] [PubMed] [Google Scholar]