

Abstract
Objective
To evaluate fenebrutinib, an oral and highly selective noncovalent inhibitor of Bruton's tyrosine kinase (BTK), in patients with active rheumatoid arthritis (RA).
Methods
Patients with RA and an inadequate response to methotrexate (MTX) (cohort 1; n = 480) were randomized to receive fenebrutinib (50 mg once daily, 150 mg once daily, or 200 mg twice daily), adalimumab (40 mg every other week), or placebo. Patients with RA and an inadequate response to tumor necrosis factor inhibitors (cohort 2; n = 98) received fenebrutinib (200 mg twice daily) or placebo. Both cohorts continued MTX therapy.
Results
In cohort 1, the percentages of patients in whom American College of Rheumatology 50% improvement criteria (ACR50) was achieved at week 12 were similar in the fenebrutinib 50 mg once daily and placebo groups, and were higher in the fenebrutinib 150 mg once daily group (28%) and 200 mg twice daily group (35%) than in the placebo group (15%) (P = 0.016 and P = 0.0003, respectively). Fenebrutinib 200 mg twice daily and adalimumab (36%) were comparable (P = 0.81). In cohort 2, ACR50 was achieved in more patients receiving fenebrutinib 200 mg twice daily (25%) than placebo (12%) (P = 0.072). The most common adverse events in the fenebrutinib groups included nausea, headache, anemia, and upper respiratory tract infections. Fenebrutinib had significant effects on myeloid and B cell biomarkers (CCL4 and rheumatoid factor). Fenebrutinib and adalimumab caused overlapping as well as distinct changes in B cell and myeloid biomarkers.
Conclusion
Fenebrutinib demonstrates efficacy comparable to adalimumab in patients with an inadequate response to MTX, and safety consistent with existing immunomodulatory therapies for RA. These data support targeting both B and myeloid cells via this novel mechanism for potential efficacy in the treatment of RA.
Introduction
Rheumatoid arthritis (RA) is an autoimmune disorder characterized by persistent synovitis and systemic inflammation that can lead to severe disability and premature mortality 1, 2, 3. Great progress has been made over the last 20 years with the introduction of biologics, and subsequently, targeted synthetic disease‐modifying antirheumatic drugs (DMARDs) such as the JAK inhibitors, that have dramatically improved quality of life 4. However, sustained disease remission or low disease activity are seldom achieved, leading to patients cycling through therapies 5, 6. Hence, a need exists for new therapies with novel mechanisms of action. One potential target in RA is Bruton's tyrosine kinase (BTK) 7, 8, 9, a member of the TEC family of nonreceptor tyrosine kinases 10. BTK is highly expressed in hematopoietic cells and plays a critical role in B cell receptor signaling to control B cell function, survival, and proliferation 11. In addition, BTK is a component of the Fc receptor signaling pathway in myeloid cells such as monocytes, macrophages, and mast cells. BTK inhibitors have demonstrated clinical utility for the treatment of B cell malignancies and multiple sclerosis 12, whereas to date there exists limited clinical evidence of efficacy for RA patients 13, 14.
Fenebrutinib is a highly selective, orally administered, and reversible BTK inhibitor that has demonstrated dose‐dependent activity in an in vivo rat model of inflammatory arthritis 15. Fenebrutinib appears unique due to its high selectivity (for BTK versus nontarget kinases) relative to other BTK inhibitors as well as its noncovalent binding mode 15. Phase I clinical studies have shown fenebrutinib to be well‐tolerated with no safety signals precluding further clinical development 16, 17.
The ANDES trial was a global, randomized, double‐blind, placebo‐ and active‐controlled study in patients with moderate‐to‐severe RA. This is the first large phase II dose‐ranging study to evaluate a highly selective BTK inhibitor therapy in 2 patient populations with RA. We report the efficacy and safety of fenebrutinib compared to placebo and the tumor necrosis factor (TNF) inhibitor adalimumab in patients continuing to receive background methotrexate (MTX) therapy but with an inadequate response, as well as in patients with disease that had failed to respond to prior TNF inhibitor therapy.
Patients and methods
Patients
Patients ages 18–75 years with RA according to the 2010 American College of Rheumatology (ACR)/European League Against Rheumatism classification criteria 18 were eligible for inclusion in cohort 1 (patients with an inadequate response to MTX) or cohort 2 (patients with an inadequate response to TNF inhibitors). Other criteria included active disease (≥6 tender joints in a 68‐joint count and ≥6 swollen joints in a 66‐joint count at screening and randomization), C‐reactive protein (CRP) level ≥0.400 mg/dl (cohort 1) or ≥0.650 mg/dl (cohort 2) at screening, and rheumatoid factor (RF) and/or anti–citrullinated protein antibody (ACPA) positivity at screening or documented evidence of RF and/or ACPA positivity after diagnosis. Eligible patients had received MTX for ≥12 weeks prior to trial entry, with a stable dose of 7.5–25 mg/week during the last 8 weeks that was continued during the study, and had discontinued all other DMARDs at least 4 weeks prior to randomization. Prednisone (≤10 mg/day or equivalent), nonsteroidal antiinflammatory drugs, and folic acid ≥5 mg/week were permitted at stable doses throughout the study. Patients in cohort 1 were naive for biologic DMARDs while those in cohort 2 had experienced an inadequate response or intolerance to previous treatment with 1 or 2 TNF inhibitors, and may also have been exposed to 1 non‐TNF biologic inhibitor. Exclusion criteria included a history of or current inflammatory or autoimmune disorder other than RA, and previous treatment with any BTK inhibitor, B cell–depleting therapy, or JAK inhibitor.
Study design
This multicenter, randomized, double‐blind, double‐dummy, active‐ and placebo‐controlled phase II study was conducted at 103 centers in 10 countries in Europe, Latin America, and North America (ClinicalTrials.gov identifier: NCT02833350). All investigators are listed in Appendix A. The study was conducted in accordance with the ethical principles of the Declaration of Helsinki and Good Clinical Practice guidelines, and was approved by the appropriate institutional review boards. All patients provided written informed consent. Adalimumab was purchased through commercial sources (AbbVie).
Randomization, masking, and dose rationale
Patients in cohort 1 were randomized in a 1:1:1:1:1 ratio to oral fenebrutinib (at 50 mg once daily, 150 mg once daily, or 200 mg twice daily), 40 mg adalimumab by subcutaneous injection every other week, or placebo (Supplementary Figure 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41275/abstract). Once 40 patients had been allocated to each of the 5 treatment groups, randomization to the fenebrutinib 50 mg once daily group was closed as it was considered unlikely that this would be an effective dose; randomization then became 1:1:1:1 to the remaining treatment groups. Patients in the fenebrutinib and placebo groups received placebo injections (mimicking adalimumab). Patients randomized to receive adalimumab, placebo, fenebrutinib 150 mg once daily, or fenebrutinib 50 mg once daily received placebo tablets (mimicking fenebrutinib) so that all patients took 8 tablets per day (to match the fenebrutinib 200 mg twice daily dosing).
Patients in cohort 2 were randomized in a 1:1 ratio to receive placebo or fenebrutinib 200 mg twice daily, the highest dose evaluated in cohort 1, since patients in cohort 2 were expected to have more refractory disease than those in cohort 1. The study was 12 weeks in duration and patients who completed the study were eligible to enter a long‐term open‐label extension study in which all patients received fenebrutinib 200 mg twice daily.
The dosing regimens of fenebrutinib were selected based on pharmacokinetic/pharmacodynamic modeling of BTK inhibition based on results from ascending dose studies in healthy subjects 16 with the goal of achieving plasma concentrations reaching at least IC70, as this extent of inhibition was correlated with efficacy in a rat collagen‐induced arthritis model 15.
Efficacy assessments
The primary efficacy analysis evaluated response at week 12 according to the proportion of patients in cohort 1 in whom 50% improvement in ACR criteria (ACR50) was achieved 19, and compared fenebrutinib (50 mg once daily, 150 mg once daily, and 200 mg twice daily) to placebo (Supplementary Table 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41275/abstract). Key secondary efficacy analyses assessed the proportions of patients in whom an ACR50 response was achieved at week 12 for the comparison of each fenebrutinib regimen (150 mg once daily and 200 mg twice daily) versus adalimumab in cohort 1, and fenebrutinib 200 mg twice daily versus placebo in cohort 2. Other secondary end points (Supplementary Table 1) were assessed over time (weeks 1, 2, 4, 8, and 12).
Safety assessments
The incidence, severity, and timing of adverse events and laboratory abnormalities were assessed in the safety population (all randomized patients who received at least 1 dose of fenebrutinib, adalimumab, or placebo). Laboratory tests, assessment of vital signs, and other safety assessments were performed at scheduled visits. The National Institutes of Health Common Terminology Criteria for Adverse Events version 4.0 was used to describe the severity (grade) of adverse events and laboratory abnormalities. Unblinded safety data were evaluated on a bimonthly basis by an Internal Monitoring Committee that included employees of the sponsor who were not involved in study oversight and did not interact with sites.
Biomarker assessments
Biomarkers were evaluated in serum or plasma samples at baseline and weeks 1, 4, and 12 in patients with available samples (n = 438 in cohort 1; n = 86 in cohort 2). BTK‐dependent myeloid biomarkers were identified using peripheral blood CD14+ monocytes stimulated with immune complexes in the presence or absence of a BTK inhibitor; the biomarkers were CCL4, interleukin‐6 (IL‐6), and TNF‐like molecule 1A (TL1A). Total IgM and IgG (Siemens; Covance), IgM‐RF autoantibody (Siemens), CCL4 (Singulex; EMD Millipore), TL1A (R&D Systems; Genentech), IL‐6 (Simoa assay; Myriad RBM), and anti–cyclic citrullinated peptide autoantibody (IgG ACPA) (Euro‐Diagnostica; Genentech) levels were analyzed using immunoassays. CD19+ B cells and CD3+ T cells were measured by flow cytometric analysis (Covance). The proportion of patients in whom an ACR50 response was achieved at week 12 and the change in Disease Activity Score in 28 joints (DAS28) by week 12 were assessed relative to baseline levels of RF in both cohorts.
Statistical analysis
The purpose of both cohorts was to estimate the treatment effect of fenebrutinib as assessed by ACR response, and evaluate the underlying mechanistic effects of BTK inhibition. Further details are provided in the Supplementary Methods, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41275/abstract.
Results
Patient characteristics
Between June 2016 and February 2018, 1,248 patients were screened and 578 patients were randomized into the study (Figure 1). The main reasons for screen failure included CRP level; evidence of tuberculosis, hepatitis B, or hepatitis C; and RF and ACPA negativity at screening. Patients were stratified by geographic region (both cohorts) and prior exposure to a non‐TNF biologic inhibitor (cohort 2). The geographic distribution of cohorts 1 and 2 were similar, and baseline demographic and clinical characteristics were similar across groups (Table 1 and Supplementary Table 2, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41275/abstract). The median disease duration was shorter for cohort 1 (5.3 years) than cohort 2 (8.5 years), and mean baseline MTX doses were balanced across groups (16.1 mg/week for cohort 1 and 16.2 mg/week for cohort 2). In cohort 2, 83 patients had received 1 prior TNF inhibitor and 10 patients had received 2, while 14 had received an additional non‐TNF biologic inhibitor.
Figure 1.

Disposition of the study patients. A total of 1,248 patients with rheumatoid arthritis were screened, and 578 patients were randomized into cohort 1 (patients with an inadequate response to methotrexate [MTX‐IR]) and cohort 2 (patients with an inadequate response to tumor necrosis factor inhibitors [TNF‐IR]). In cohort 2, the intent‐to‐treat population included 50 patients in the placebo arm and 48 patients in the fenebrutinib arm. One patient in the placebo arm received fenebrutinib in error; thus, the safety population included 49 patients in each arm.
Table 1.
Demographic and baseline clinical characteristics of the patientsa
| Cohort 1 (MTX‐IR) | Cohort 2 (TNF‐IR) | ||||||
|---|---|---|---|---|---|---|---|
| Placebo (n = 110) | Fenebrutinib 50 mg once daily (n = 40) | Fenebrutinib 150 mg once daily (n = 109) | Fenebrutinib 200 mg twice daily (n = 110) | Adalimumab 40 mg every other week (n = 111) | Placebo (n = 50) | Fenebrutinib 200 mg twice daily (n = 48) | |
| Age, years | 50 ± 12 | 52 ± 12 | 50 ± 11 | 50 ± 12 | 50 ± 12 | 55 ± 12b | 51 ± 13b |
| Sex, no. (%) female | 90 (82) | 35 (88) | 92 (84) | 85 (77) | 87 (78) | 37 (76)b | 37 (76)b |
| Race, no. (%) white | 95 (86) | 36 (90) | 96 (88) | 96 (87) | 99 (89) | 42 (86)b | 42 (86)b |
| BMI, kg/m2 | 28 ± 6 | 27 ± 6 | 27 ± 5 | 28 ± 6 | 27 ± 5 | 27 ± 5b | 26 ± 5b |
| RA duration, median (range) years | 5 (0.3–25) | 8 (0.3–32) | 5 (0.3–31) | 5 (0.3–38) | 5 (0.3–30) | 9 (1.2–29)b | 7 (2.0–36)b |
| Antibody positive, no. (%) | |||||||
| ACPA | 102 (93) | 39 (98) | 100 (91) | 105 (95) | 103 (92) | 42 (84) | 46 (96) |
| RF | 98 (89) | 38 (95) | 108 (99) | 103 (94) | 104 (93) | 49 (98) | 46 (96) |
| No. of swollen joints (66‐joint count) | 16 ± 10 | 13 ± 8 | 14 ± 8 | 14 ± 8 | 14 ± 8 | 15 ± 8 | 15 ± 8 |
| No. of tender joints (68‐joint count) | 24 ± 13 | 24 ± 13 | 24 ± 13 | 22 ± 12 | 24 ± 13 | 27 ± 16 | 24 ± 12 |
| HAQ DI | 1.6 ± 0.6 | 1.8 ± 0.5 | 1.7 ± 0.5 | 1.7 ± 0.6 | 1.8 ± 0.7 | 1.8 ± 0.5 | 1.5 ± 0.6 |
| DAS28‐CRP | 5.9 ± 0.8 | 5.7 ± 0.9 | 5.9 ± 0.8 | 5.8 ± 1.0 | 5.8 ± 0.9 | 6.0 ± 1.0 | 5.9 ± 0.9 |
| CRP, mg/dl | 2.0 ± 1.9 | 1.6 ± 1.4 | 2.0 ± 2.0 | 2.5 ± 3.1 | 2.1 ± 2.6 | 3.1 ± 4.1 | 2.5 ± 2.8 |
| ESR, mm/hour | 45 ± 21 | 45 ± 20 | 43 ± 21 | 45 ± 23 | 41 ± 22 | 61 ± 32 | 52 ± 29 |
| IgM, gm/liter | 1.58 ± 0.81 | 1.43 ± 0.52 | 1.64 ± 1.02 | 1.38 ± 0.60 | 1.45 ± 0.60 | 1.72 ± 0.84 | 1.56 ± 0.72 |
| IgG, gm/liter | 12.91 ± 3.00 | 12.50 ± 2.99 | 12.45 ± 3.68 | 13.17 ± 4.71 | 12.71 ± 3.55 | 13.23 ± 3.81 | 13.33 ± 4.95 |
Except where indicated otherwise, values are the mean ± SD. RA = rheumatoid arthritis; MTX‐IR = inadequate response to methotrexate; TNF‐IR = inadequate response to tumor necrosis factor inhibitors; BMI = body mass index; ACPA = anti–citrullinated protein antibody; RF = rheumatoid factor; HAQ DI = Health Assessment Questionnaire disability index; DAS28‐CRP = Disease Activity Score in 28 joints using the C‐reactive protein level; ESR = erythrocyte sedimentation rate.
n = 49.
Primary and key secondary outcomes
In cohort 1 at week 12, ACR50 response rates were achieved in 18% of the patients receiving fenebrutinib 50 mg once daily, 28% of the patients receiving fenebrutinib 150 mg once daily, and 35% of the patients receiving fenebrutinib 200 mg twice daily, compared to 15% of the patients receiving placebo (primary outcome) (P = 0.25, P = 0.016, and P = 0.0003, respectively). The ACR50 response rate of 35% achieved in the fenebrutinib 200 mg twice daily group at week 12 was numerically similar to the 36% response rate achieved in the adalimumab group, while the ACR50 response rate for the fenebrutinib 150 mg once daily group was 28% (key secondary outcome) (P = 0.81 and P = 0.17, respectively) (Table 2). In cohort 2 at week 12, ACR50 response was achieved in 25% of the patients in the fenebrutinib 200 mg twice daily group versus 12% of the patients in the placebo group (key secondary outcome) (P = 0.072).
Table 2.
Primary, key secondary, and major secondary efficacy end points at week 12a
| Cohort 1 (MTX‐IR) | Cohort 2 (TNF‐IR) | ||||||
|---|---|---|---|---|---|---|---|
| Placebo (n = 110) | Fenebrutinib 50 mg once daily (n = 40) | Fenebrutinib 150 mg once daily (n = 109) | Fenebrutinib 200 mg twice daily (n = 110) | Adalimumab 40 mg every other week(n = 111) | Placebo (n = 50) | Fenebrutinib 200 mg twice daily (n = 48) | |
| ACR50b | |||||||
| Responders, no. (%) | 16 (15) | 7 (18) | 30 (28) | 38 (35) | 40 (36) | 6 (12) | 12 (25) |
| 95% CI, % | (8–21) | (6–29) | (19–36) | (26–43) | (27–45) | (3–21) | (13–37) |
| P c | – | 0.2503 | 0.0164 | 0.0003 | 0.0001 | – | 0.0717 |
| ACR20b | |||||||
| Responders, no. (%) | 40 (36) | 24 (60) | 61 (56) | 65 (59) | 80 (72) | 12 (24) | 28 (58) |
| 95% CI, % | (27–45) | (45–75) | (47–65) | (50–68) | (64–80) | (12–36) | (44–72) |
| P c | – | 0.0003 | 0.0020 | 0.0003 | <0.0001 | – | 0.0001 |
| ACR70b | |||||||
| Responders, no. (%) | 8 (7) | 2 (5) | 10 (9) | 14 (13) | 20 (18) | 2 (4) | 7 (15) |
| 95% CI, % | (2–12) | (0–12) | (4–15) | (7–19) | (11–25) | (0–9) | (5–25) |
| P c | – | 1.0000 | 0.6200 | 0.1901 | 0.0155 | – | 0.0983 |
| DAS28‐CRPd | |||||||
| No. of patients | 99 | 36 | 95 | 95 | 104 | 43 | 47 |
| Mean change from baseline | −1.34 | −1.74 | −1.96 | −1.96 | −2.11 | −1.43 | −2.26 |
| Difference from placebo | – | −0.41 | −0.63 | −0.62 | −0.78 | – | −0.83 |
| P c | – | 0.1696 | 0.0002 | 0.0003 | <0.0001 | – | 0.0001 |
| CRP (mg/dl)d | |||||||
| No. of patients | 102 | 37 | 97 | 97 | 106 | 44 | 47 |
| Adjusted mean | 1.63 | 1.28 | 1.28 | 1.20 | 1.12 | 1.62 | 0.59 |
| Difference from placebo | – | −0.35 | −0.35 | −0.43 | −0.51 | – | −1.02 |
| P c | – | 0.4860 | 0.2205 | 0.0850 | 0.0254 | – | 0.0003 |
| ESR (mm/hour)d | |||||||
| No. of patients | 102 | 37 | 98 | 97 | 108 | 45 | 47 |
| Adjusted mean | 36.50 | 31.83 | 33.84 | 31.00 | 31.24 | 45.48 | 32.55 |
| Difference from placebo | – | −4.67 | −2.66 | −5.50 | −5.26 | – | −12.9 |
| P c | – | 0.3739 | 0.6006 | 0.0528 | 0.0600 | – | 0.0018 |
| HAQ DI scored | |||||||
| No. of patients | 101 | 36 | 102 | 103 | 108 | 45 | 47 |
| Adjusted mean | 1.30 | 1.19 | 1.13 | 1.03 | 1.08 | 1.22 | 0.88 |
| Difference from placebo | – | −0.11 | −0.16 | −0.26 | −0.22 | – | −0.34 |
| P c | – | 0.7339 | 0.1245 | 0.0030 | 0.0198 | – | 0.0084 |
RA = rheumatoid arthritis; MTX‐IR = inadequate response to methotrexate; TNF‐IR = inadequate response to tumor necrosis factor inhibitors; ACR50 = American College of Rheumatology 50% improvement criteria; 95% CI = 95% confidence interval; DAS28‐CRP = Disease Activity Score in 28 joints using the C‐reactive protein level; ESR = erythrocyte sedimentation rate; HAQ DI = Health Assessment Questionnaire disability index.
Adjusted for region in cohort 1; adjusted for region and non‐TNF biologic status in cohort 2.
Versus placebo.
Adjusted for region, treatment, and baseline value in cohort 1; adjusted for region, treatment, baseline value, and non‐TNF biologic status in cohort 2.
Secondary outcomes
ACR20 and ACR70 responses were achieved in higher proportions of patients in the majority of the fenebrutinib groups compared to the placebo group at week 12 in both cohorts (Table 2). Longitudinal evaluation indicated greater improvements in efficacy at earlier time points with adalimumab compared to fenebrutinib 200 mg twice daily, as indicated by ACR50 response and change from baseline in DAS28 using the CRP level and Health Assessment Questionnaire disability index, but the responses were comparable by week 12 (Figure 2). Clinically meaningful improvements with fenebrutinib versus placebo were seen for other secondary outcomes in both cohorts, including DAS28 using the ESR (DAS28‐ESR), Clinical Disease Activity Index, Simplified Disease Activity Index, and low disease activity as defined by the DAS at week 12 (Supplementary Table 3, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41275/abstract).
Figure 2.
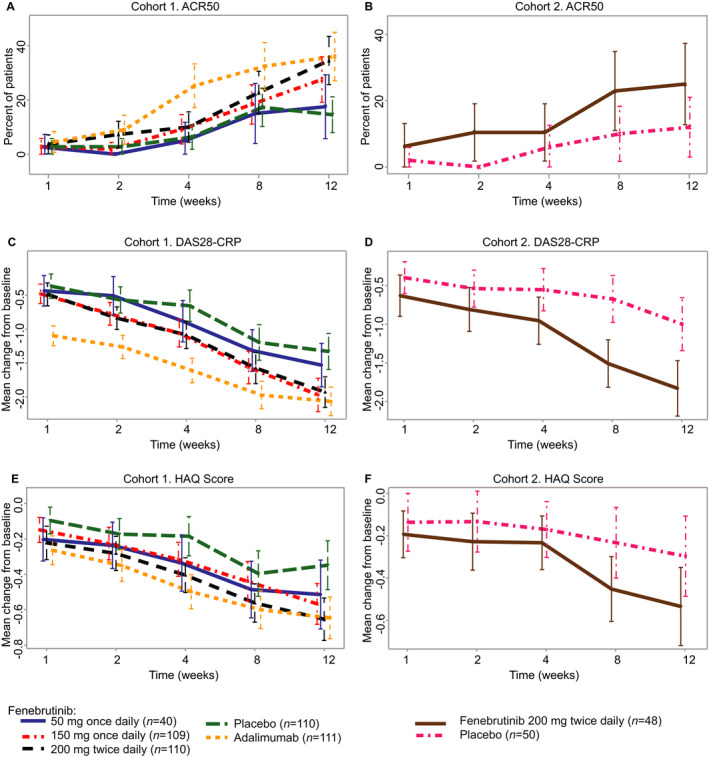
Secondary end point efficacy data over time up to week 12. A and B, American College of Rheumatology 50% improvement criteria (ACR50) response at the indicated time points in each treatment group in the cohort of rheumatoid arthritis (RA) patients with an inadequate response to methotrexate (cohort 1) (A) and the cohort of RA patients with an inadequate response to tumor necrosis factor inhibitors (cohort 2) (B). C and D, Change from baseline in the 4‐variable Disease Activity Score using the C‐reactive protein level (DAS28‐CRP) at the indicated time points in each treatment group in cohort 1 (C) and cohort 2 (D). E and F, Change from baseline in the Health Assessment Questionnaire (HAQ) disability index score at the indicated time points in each treatment group in cohort 1 (E) and cohort 2 (F). In A and B, values are the proportion of ACR50 responders and 95% confidence interval (95% CI); in C–F, values are the mean change from baseline and 95% CI. In C–F, change from baseline was not adjusted for the randomization stratification factor, “geographic region.”
Systemic markers of inflammation were reduced in the fenebrutinib 200 mg twice daily group compared to the placebo group at week 12, with a difference in CRP level of −0.43 mg/dl and a difference in ESR of −5.50 mm/hour. The mean reduction in markers of inflammation in the fenebrutinib groups was less than that observed in the adalimumab group at earlier postbaseline visits but was similar at week 12. Results of the comparison of fenebrutinib with placebo and adalimumab, for other secondary and exploratory outcomes, showed comparable benefit for fenebrutinib 200 mg twice daily and adalimumab by week 12 (Supplementary Table 3 and Supplementary Figures 2–4, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41275/abstract); these outcomes included ACR20, ACR70, DAS28‐ESR, Boolean remission, quality of life assessments, and individual components of the ACR response rate.
Safety
A majority of patients completed the 12‐week study, with a completion rate of ≥90% per group (Figure 1). One death due to myocardial infarction was reported in the fenebrutinib 200 mg twice daily group in cohort 1. Rates of adverse events for all 3 fenebrutinib doses were comparable to those for adalimumab (Table 3). The most common adverse events in the fenebrutinib groups included nausea, headache, anemia, and upper respiratory tract infections. Serious adverse events were infrequent and occurred in 1%, 3%, and 2% of patients in the fenebrutinib 150 mg once daily, fenebrutinib 200 mg twice daily, and adalimumab groups in cohort 1, respectively (Supplementary Table 4, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41275/abstract). There were no serious adverse events reported in cohort 2. Four patients experienced a serious infection: 1 patient with pneumonia in the fenebrutinib 150 mg once daily group, 1 patient each with cellulitis and pyelonephritis in the fenebrutinib 200 mg twice daily group, and 1 patient with pneumonia in the placebo group. One patient in cohort 1 (in the fenebrutinib 200 mg twice daily group) had herpes zoster, and 2 patients in cohort 2 (1 each in the placebo and fenebrutinib 200 mg twice daily groups) had herpes zoster. No tuberculosis cases were reported (Table 3).
Table 3.
Adverse events and laboratory parameters from baseline to week 12a
| Cohort 1 (MTX‐IR) | Cohort 2 (TNF‐IR) | ||||||
|---|---|---|---|---|---|---|---|
| Placebo (n = 110) | Fenebrutinib 50 mg once daily (n = 40) | Fenebrutinib 150 mg once daily (n = 109) | Fenebrutinib 200 mg twice daily (n = 110) | Adalimumab 40 mg every other week (n = 111) | Placebo (n = 49)b | Fenebrutinib 200 mg twice daily (n = 49)b | |
| Adverse eventsc | 50 (46) | 15 (38) | 46 (42) | 56 (51) | 50 (45) | 22 (45) | 11 (22) |
| Serious adverse eventsc | 1 (1) | 0 (0) | 1 (1) | 3 (3) | 2 (2) | 0 (0) | 0 (0) |
| Adverse events grade 3 or higherc | 2 (2) | 0 (0) | 2 (2) | 9 (8) | 4 (4) | 0 (0) | 0 (0) |
| ALT adverse events grade 3 or higherc | 0 (0) | 0 (0) | 1 (1) | 3 (3) | 1 (1) | 0 (0) | 0 (0) |
| AST adverse events grade 3 or higherc | 1 (1) | 0 (0) | 0 (0) | 2 (2) | 2 (2) | 0 (0) | 0 (0) |
| Infection adverse eventsc | 16 (15) | 3 (8) | 16 (15) | 12 (11)d | 25 (23) | 7 (14)d | 3 (6)d |
| Serious infectionsc | 1 (1) | 0 (0) | 1 (1) | 2 (2) | 0 (0) | 0 (0) | 0 (0) |
| Deaths | 0 (0) | 0 (0) | 0 (0) | 1 (1)e | 0 (0) | 0 (0) | 0 (0) |
| Events occurring in ≥5% of patients | |||||||
| Increased ALT | 1 (1) | 2 (5) | 5 (5) | 4 (4) | 1 (1) | 1 (2) | 0 (0) |
| Increased AST | 1 (1) | 2 (5) | 4 (4) | 3 (3) | 1 (1) | 0 (0) | 0 (0) |
| Headache | 5 (5) | 2 (5) | 4 (4) | 5 (5) | 0 (0) | 0 (0) | 1 (2) |
| Nausea | 5 (5) | 2 (5) | 5 (5) | 3 (3) | 1 (1) | 1 (2) | 3 (6) |
| Upper respiratory tract infection | 2 (2) | 1 (3) | 3 (3) | 3 (3) | 6 (5) | 0 (0) | 0 (0) |
| Urinary tract infection | 2 (2) | 2 (5) | 2 (2) | 0 (0) | 8 (7) | 3 (6) | 0 (0) |
Values are the number (%). RA = rheumatoid arthritis; MTX‐IR = inadequate response to methotrexate; TNF‐IR = inadequate response to tumor necrosis factor inhibitors; ALT = alanine transaminase; AST = aspartate aminotransferase.
In cohort 2, the intent‐to‐treat population included 50 patients in the placebo arm and 48 patients in the fenebrutinib arm. One patient in the placebo group received fenebrutinib in error; thus, the safety population included 49 patients in each arm.
Patients with ≥1 event.
Includes 1 herpes zoster event.
Due to myocardial infarction.
Infrequent and reversible grade 3 transaminase elevations were observed in the fenebrutinib 150 mg once daily group (1%), fenebrutinib 200 mg twice daily group (3%), and adalimumab group (1%); these elevations were asymptomatic and did not coincide with elevations in bilirubin levels. A review of concomitant medications also indicated no obvious association between MTX dose and the cases of transaminase elevations 20. There was a small dose‐dependent increase from baseline to week 12 in serum creatinine levels in the fenebrutinib groups (mean ± SD −0.4 ± 9.5, 3.9 ± 9.8, and 8.6 ± 38.7 μmoles/liter for the 3 ascending doses, respectively), which was not considered clinically meaningful (Supplementary Tables 5 and 6, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41275/abstract). No patients in any of the fenebrutinib arms developed adverse events of acute kidney injury. No clinically relevant changes were observed in hematology parameters (hemoglobin/hematocrit, white blood cells, platelets, neutrophils, lymphocytes, or monocytes) (Supplementary Table 6).
Biomarkers
Total IgM, IgG, and IgM‐RF levels were reduced in the fenebrutinib 150 mg once daily and 200 mg twice daily groups relative to placebo by week 12 in cohort 1 (Figures 3A–C). Total IgM levels were also decreased in the patients treated with fenebrutinib 50 mg once daily compared to those treated with placebo in cohort 1 (Figure 3A). The largest reductions in total IgM, IgG, and IgM‐RF levels were observed in the fenebrutinib 200 mg twice daily groups in cohorts 1 and 2. In cohort 1, treatment with fenebrutinib 200 mg twice daily resulted in a median reduction in IgM‐RF of −29% relative to placebo by week 12. Median absolute reductions in immunoglobulin levels were −22 mg/dl for total IgM and −115 mg/dl for total IgG in the fenebrutinib 200 mg twice daily group compared to placebo by week 12 in cohort 1. By week 12, patients treated with fenebrutinib 200 mg twice daily in cohort 2 had similar median reductions in total IgM and IgG levels (−17 mg/dl and −101 mg/dl, respectively, relative to placebo).
Figure 3.
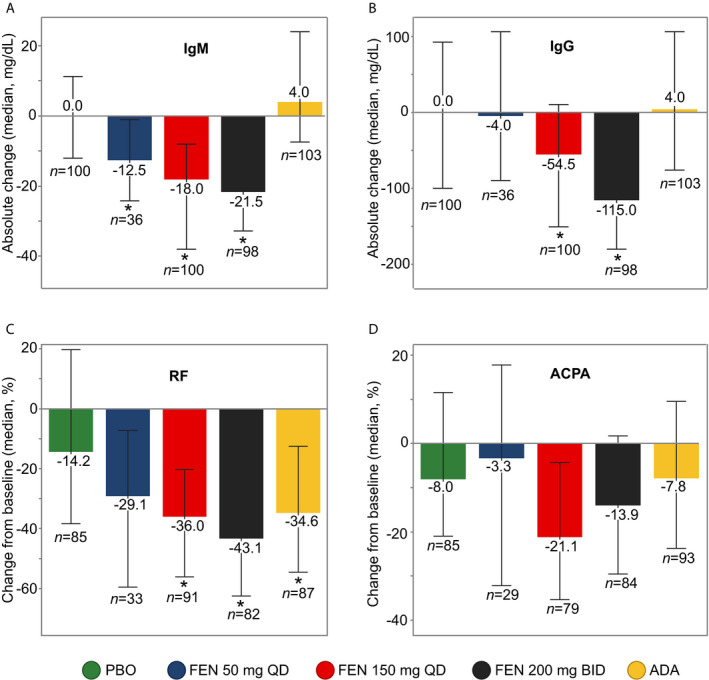
Changes in immunoglobulin and autoantibody levels in patients with rheumatoid arthritis with an inadequate response to methotrexate treated with placebo (PBO), fenebrutinib (FEN) 50 mg once daily (QD), fenebrutinib 150 mg once daily, fenebrutinib 200 mg twice daily (BID), or adalimumab (ADA) (cohort 1). A and B, Absolute change from baseline to week 12 in levels of total IgM (A) and IgG (B). C and D, Percent change from baseline to week 12 in levels of rheumatoid factor (RF) (C) and anti–citrullinated protein antibody (ACPA) (D). RF and ACPA analyses included patients who were positive at screening. Values are the median and interquartile range. * = P < 0.05 versus placebo.
Two patients with normal baseline levels of IgG had reductions to below the lower limit of normal (LLN) following randomization: 1 patient in the fenebrutinib 50 mg once daily group in cohort 1 and 1 patient in the fenebrutinib 200 mg twice daily group in cohort 2. One patient in the adalimumab arm of cohort 1 had a baseline IgG level below the LLN and a decline from baseline following treatment. Eight patients (4 in the fenebrutinib 150 mg once daily arm, 2 in the fenebrutinib 200 mg arm, and 1 in the placebo arm of cohort 1; and 1 in the fenebrutinib 200 mg arm of cohort 2) had reductions in IgM from normal levels to below the LLN. Three additional patients (1 each in the fenebrutinib 150 mg once daily, fenebrutinib 200 mg twice daily, and placebo arms in cohort 1) had baseline levels of IgM below the LLN and a further reduction in IgM levels during the study. Adalimumab treatment (cohort 1) reduced median IgM‐RF levels −20% relative to placebo by week 12 (Figure 3C), but, in contrast to fenebrutinib, did not reduce total IgM or IgG levels (Figures 3A and B). IgG ACPA levels trended downward with fenebrutinib treatment, but were not significantly changed relative to placebo in cohort 1 (Figure 3D). In contrast, by week 12, median IgG ACPA levels were reduced −24% in the fenebrutinib 200 mg twice daily group compared to +15% in the placebo group in cohort 2 (Supplementary Figure 5, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41275/abstract).
Elevated levels of CCL4, IL‐6, and TL1A can reflect Fcγ receptor (FcγR) activation in myeloid cells 21, 22, 23. We found immune complex–stimulated expression of these biomarkers to be BTK‐dependent in monocytes assayed in vitro (Supplementary Figure 6, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41275/abstract).
All doses of fenebrutinib reduced CCL4 levels compared to placebo by week 1 in cohort 1 (Supplementary Figure 7, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41275/abstract) and cohort 2 (Supplementary Figure 8, Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41275/abstract); the fenebrutinib 200 mg twice daily group in cohort 1 had the largest median reduction, of −28% relative to placebo. Similar to fenebrutinib, adalimumab treatment reduced CCL4 levels in cohort 1 (median −36% relative to placebo). In addition, median reductions in CCL4 were greater in the adalimumab group than in the fenebrutinib treatment groups at weeks 4 and 12. Although IL‐6 levels trended downward over 12 weeks, none of the 3 dose levels of fenebrutinib affected IL‐6 levels relative to placebo in cohort 1 (Supplementary Figure 7). However, fenebrutinib 200 mg twice daily reduced IL‐6 levels relative to placebo (median –50%) by week 12 in cohort 2 (Supplementary Figure 8). Adalimumab treatment resulted in a reduction in IL‐6 levels relative to placebo (median −28%) at week 12 in cohort 1 (Supplementary Figure 7). TL1A was not detectable in the patient samples tested (n = 33).
By week 1, a transient increase in peripheral CD19+ B cells occurred in the fenebrutinib‐treated groups (median 45–79% relative to placebo) in cohort 1 (Supplementary Figure 7) and cohort 2 (Supplementary Figure 8), with levels returning toward pretreatment ranges by week 12 (median −4% to 13% relative to placebo) (Supplementary Figure 7). Although CD3+ T cell numbers were higher at week 1 following fenebrutinib treatment (median +10–15% relative to placebo) in cohort 1 (Supplementary Figure 7) and cohort 2 (Supplementary Figure 8), these elevations were not significant. Adalimumab treatment was also accompanied by an increase in CD19+ B cells (median +44%) and CD3+ T cells (median +22%) relative to placebo by week 1 in cohort 1 (Supplementary Figure 7); levels also appeared elevated relative to placebo after 12 weeks of treatment in cohort 1 (median +42% for B cells and +13% for T cells).
Since this study primarily enrolled autoantibody‐positive patients, the question of whether fenebrutinib has differential efficacy in autoantibody‐positive versus antibody‐negative patients could not be robustly assessed. However, higher baseline levels of RF were associated with a small increase in the proportion of ACR50 responders in the fenebrutinib 200 mg twice daily group in both cohorts (Supplementary Figure 9, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41275/abstract).
Discussion
Patients with RA who develop an inadequate response to their current therapy may benefit from the addition of or replacement with another therapy. Clinical studies have shown that switching to a therapy with a different mechanism of action is more effective than cycling through therapies within the same mechanism‐of‐action group. BTK is a cytoplasmic tyrosine kinase largely restricted to the hematopoietic system that may be exploited as a potential drug target due to its expression and role in B cells and myeloid cells, which are implicated in the pathogenesis of RA. Fenebrutinib is a highly selective and reversible BTK inhibitor previously shown to suppress B cell– and myeloid cell–mediated components of the disease and with dose‐dependent activity in an in vivo rat model of inflammatory arthritis.
This phase II placebo‐ and active‐controlled study of fenebrutinib was designed as a proof‐of‐concept and signal‐generating trial to inform the utility of BTK inhibition with fenebrutinib in patients with active RA. Comparing fenebrutinib to a standard therapy, such as adalimumab, provided important efficacy data to inform future potential studies of fenebrutinib. This phase II trial represents the first study to demonstrate the treatment benefits of inhibiting BTK, a novel therapeutic target for RA, including efficacy in patients with refractory disease that has failed to respond to TNF therapy. Meaningful improvements in clinical responses were observed in patients with MTX‐refractory RA who received fenebrutinib compared to those who received placebo, as reflected by ACR50 scores at week 12. Fenebrutinib was also more efficacious than placebo in the patients with more treatment‐refractory disease that had previously failed to respond to prior TNF inhibitor therapies. The dosing regimens evaluated in this study were selected based on pharmacokinetic/pharmacodynamic results from phase I studies, and the highest dose of fenebrutinib 200 mg twice daily is expected to achieve maximal target inhibition in the periphery. Dose‐ and exposure‐response analyses of ACR50 at week 12 indicated that fenebrutinib 200 mg twice daily was associated with maximal efficacy 24. Thus, the fenebrutinib doses assessed in this study support the hypothesis that BTK inhibition contributes to efficacy in the treatment of RA.
Adalimumab was used as an active comparator to further contextualize the efficacy and safety of fenebrutinib; results showed fenebrutinib to have efficacy comparable to that of adalimumab at week 12. However, a relatively slower onset of response was observed for fenebrutinib at time points earlier than 12 weeks across several end points. This may be due to a delayed effect of BTK inhibition on systemic inflammation. Consistent with this hypothesis, fenebrutinib reduced the ESR, CRP level, and IL‐6 level relative to placebo, but this effect was only apparent after 8–12 weeks of treatment, whereas adalimumab reduced all 3 markers of inflammation by week 1.
The safety profiles of adalimumab and fenebrutinib were similar in this study. Four serious infections were observed, including 3 in the fenebrutinib groups and 1 in the placebo group. While events in the fenebrutinib groups may reflect the immunomodulatory effect of BTK inhibition, the number of infections was small and the numbers of patients with infections were similar across treatment groups. More grade 3 transaminase elevations were observed in the fenebrutinib 200 mg twice daily group in cohort 1, suggesting a dose‐response relationship. However, there was a low rate of alanine aminotransferase elevations overall, and the analysis is limited by the small number of events.
Fenebrutinib elicited changes in B cell and myeloid cell biomarkers, consistent with the proposed dual mechanism of action of a BTK inhibitor capable of inhibiting B cell and FcγR signaling in B cells and myeloid cells, respectively 25. The inhibition of FcγR signaling–regulated myeloid biomarkers also indicates that activation of myeloid cells via crosslinking of these receptors may be a feature of RA pathophysiology. It will be informative to assess changes in these biomarkers in other disease indications where BTK inhibitors are utilized. Both fenebrutinib and adalimumab affected myeloid cell biomarkers (CCL4 and IL‐6) and B cell biomarkers (RF). However, the effects of fenebrutinib on myeloid markers were similar to or weaker than those of adalimumab. In contrast, fenebrutinib treatment elicited effects on IgM and IgG that were not observed with adalimumab. These data highlight mechanistic changes that, while different between fenebrutinib and adalimumab, may contribute to their clinical activity.
Despite reductions in IgM and IgG levels, levels in most patients treated with fenebrutinib remained above the LLN. There were no notable differences in the safety and efficacy profiles of patients found to have IgG levels below the LLN. More patients in the fenebrutinib 150 mg once daily and 200 mg twice daily arms developed treatment‐related reductions in IgM to levels below the LLN. However, none of these patients developed serious or opportunistic infections. It is unknown whether longer exposure to fenebrutinib would lead to a greater reduction in immunoglobulin levels or a higher incidence of hypogammaglobulinemia, and whether this would increase the risk of infections. The reductions in autoantibody levels indicate that fenebrutinib can affect autoreactive B cells, similar to rituximab 26. However, in contrast to rituximab, the effects of fenebrutinib on autoreactive B cells were not accompanied by reductions in B cell numbers. The initial effect of fenebrutinib on peripheral B cells results in a transient increase in peripheral B cell numbers, followed by an inhibitory effect on B cells as supported by subsequent decreases in the level of autoantibodies. Interestingly, the early transient increase in B cell numbers in fenebrutinib‐treated patients appears similar to the increase in peripheral B cell numbers observed in patients with mantle cell lymphoma (MCL) treated with the covalent BTK inhibitor ibrutinib 27, 28. In MCL patients, the increase in peripheral B cell numbers is thought to reflect a redistribution of B cells from tissue to the periphery due to the role of BTK in B cell homing/retention 29.
Previous studies have shown a correlation between autoantibody production and BTK protein expression in patients with systemic autoimmune disease 30. The efficacy results may reflect the direct effect of BTK inhibition on the development and proliferation of autoreactive B cells, a reduction in the underlying inflammatory pathology in RA mediated through the effect of BTK inhibition on myeloid cells, or a combination of both mechanisms 31.
Limitations of the study include the requirement that patients have evidence of seropositivity upon study entry, which could have excluded patients with otherwise active RA. Given the trial was 3 months in duration, the efficacy of fenebrutinib beyond this time point is not known, nor has the safety profile with longer‐term treatment been established. Specifically, longer trials will be required to determine whether the reduction in immunoglobulin levels may become more severe with prolonged BTK inhibition and whether that may be accompanied by a risk for infections. Patients were not evaluated radiographically; thus, the utility of fenebrutinib for inhibition of joint damage progression is unknown. There is also the potential for regional bias in results since a large proportion of patients were recruited from Eastern Europe 32; regional subgroup analysis was not feasible due to limited sample size in some regions.
In this randomized, phase II trial in patients with MTX‐refractory RA, greater efficacy was observed with fenebrutinib 150 mg once daily or 200 mg twice daily compared to placebo, and response rates with fenebrutinib were numerically similar to those observed with adalimumab. Moreover, fenebrutinib showed activity in patients who had disease that was refractory to therapies beyond MTX. The results of this proof‐of‐concept study provide clinical evidence supporting the rationale of targeting of BTK by fenebrutinib in 2 different patient populations with active RA. Additional evaluation of fenebrutinib in patients with biologic DMARD–refractory RA, who represent a population with significant unmet need, will be useful to further characterize the safety and efficacy profile of fenebrutinib.
Author contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Cohen had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Katsumoto, Zhao, Chinn, Townsend, Morimoto.
Acquisition of data
Cohen, Zhao, Berman, Damjanov, Fedkov, Jeka, Genovese.
Analysis and interpretation of data
Tuckwell, Zhao, Galanter, Lee, Rae, Toth, Ramamoorthi, Hackney, Townsend, Morimoto.
ROLE OF THE STUDY SPONSOR
This study was funded by Genentech, Inc., the sponsor. The sponsor and the authors were involved in the study design and/or conduct. The investigators and their respective research teams collected all the data. Statistical analyses were conducted by biostatisticians at Genentech, Inc., in accordance with a prespecified statistical analysis plan. All authors participated in the analysis and interpretation of the data, reviewed and provided feedback on the draft manuscript, and made the decision to submit the manuscript for publication. All authors vouch for the completeness and accuracy of the data and analyses. Writing and editing assistance were provided by A. Daisy Goodrich, PhD (Genentech, Inc.) according to Good Publication Practice guidelines. Publication of this article was contingent upon approval by Genentech, Inc.
Supporting information
Acknowledgments
We thank the patients and their families. We thank Alexandra Ward, Joel Mathews, and Rich Erickson for biomarker technical support. We also thank David Musselman and Edmond Teng for medical monitoring support.
APPENDIX A. THE ANDES TRIAL INVESTIGATORS
The Andes Trial investigators are as follows: in Argentina, Alberto Berman, Alejandro Porto, Amelia Granel, Cecilia Asnal, Eduardo Fabian Mysler, Gladys Alicia Testa, Jose Luis Velasco Zamora, Jose Luis Cristian Moreno, Juan Pablo Gulin, Julio Hofman, Maria Rosa Ulla, Mirtha Sabelli, Pablo Alejandro Mannucci, and Pablo Jorge Maid; in Brazil, Ana Cláudia Cauceglia Melazzi, Antônio Scafuto Scotton, Antônio Carlos Ximenes, Elisete Funes, Emerson Alves Gimenez, Flora Maria D'Andrea Marcolino, João Francisco Marques Neto, Mauro Waldemar Keiserman, Sebastião Cézar Radominski, Sônia Maria Alvarenga Anti Loduca Lima, Thaís Rohde Pavan, and Valderílio Feijó Azevedo; in Bulgaria, Aneliya Koleva, Antoaneta Toncheva, Daniela Bichovska, Delina Ivanova, Dimitar Penev, Emil Dimitrov, Mariyana Mihaylova, Nadezhda Kapandjieva, Natalia Marinova, Tanya Aleksieva, Tanya Tsvetanova, Tsvetanka Petranova, Valentina Popova, and Yuliy Spasov; in Colombia, Carlos Enrique Toro, Carlos Ernesto Arteaga Unigarro, Edwin Jauregui, Javier Dario Marquez Hernandez, Juan Jose Jaller Raad, and Patricia Julieta Velez Sanchez; in the Republic of Korea, Chang Keun Lee, Chang‐Hee Suh, Eun Young Lee, Sang‐Heon Lee, Seong Wook Kang, Shin‐Seok Lee, and Yun Jong Lee; in Mexico, Beatriz Elena Zazueta Montiel, Blanca Irma Pinzon de la O, Daniel Xibille Friedmann, Francisco Rosas Lopez, Isaura Rodriguez Torres, Luis Jara Quezada, Marco Maradiaga Ceceña, Miguel Cortes Hernandez, and Miguel Saavedra Salinas; in Poland, Agnieszka Rapa, Agnieszka Pawtel, Agnieszka Zielinska, Anna Dudek, Anna Rychlewska‐Hanczewska, Anna Strzelecka, Artur Racewicz, Barbara Stasiuk, Katarzyna Gruszecka, Krystyna Dworak, Slawomir Jeka, and Tomasz Lowenhoff; in the Russian Federation, Alexey Maslyanskiy, Andrey Rebrov, Diana Krechikova, Elena Zhugrova, Evgeniya Shmidt, Galina Matsievskaya, Irina Vinogradova, Irina Ler, Larisa Eliseeva, Ludmila Savina, Marina Stanislav, Mikhail Sandin, Natalia Zyablova, Nikolay Korshunov, Nino Mosesova, Oksana Polovnikova, Olga Nesmeyanova, Ruzana Samigullina, Sergey Moiseev, Sergey Noskov, Tatiana Raskina, Tatiana Popovaf, and Valeriy Marchenko; in Serbia, Aleksandar Jovanovski, Bojana Stamenkovic, Gorica Ristic, Milijanka Lazarevic, Mirjana Veselinovic, Nada Vujasinovic‐Stupar, Nemanja Damjanov, and Predrag Ostojic; in Ukraine, Andriy Yagensky, Andriy Gnylorybov, Dmytro Rekalov, Dmytro Reshotko, Dmytro Fedkov, Georgiy Dzyak, Iurii Gasanov, Ludmila Khimion, Mykola Stanislavchuk, Natalya Prykhodko, Oleg Nadashkevych, Oleg Bortkevych, Orest Abrahamovych, Roman Yatsyshyn, Samvel Turyanytsya, Svitlana Smiyan, Vadym Vizir, Victoria Kachur, Vira Tseluyko, Vladyslav Povoroznyuk, Volodymyr Koshlia, Vyacheslav Zhdan, Yurii Lymar, and Yuriy Mostovoy; and in the US, Angela Hawkes, Arthur Mabaquiao, Cong‐Qiu Chu, Craig Scoville, David Wyatt, Debra Weinstein, Harris McIlwain, Jacqueline Vo, Jeffrey Poiley, Joseph Forstot, Kathryn Dao, Mark Turner, Mark Genovese, Michael Borofsky, Paul Caldron, Philip Waller, Robert Levin, Samy Metyas, Scott Stein, Sharukh Shroff, Shirley Pang, Stanley Cohen, Tauseef Syed, and Vishala Chindalore.
ClinicalTrials.gov identifier: NCT02833350.
Supported by Genentech, Inc.
Dr. Cohen has received consulting fees, speaking fees, and/or honoraria from Amgen, AbbVie, Gilead, Pfizer, Eli Lilly, and Genentech (less than $10,000 each) and research support from Genentech. Dr. Tuckwell owns stock or stock options in Genentech/Roche. Dr. Katsumoto has received consulting fees from AbbVie and Genentech (less than $10,000 each) and from Principia (more than $10,000) and owns stock or stock options and holds a pending patent with Genentech/Roche. Drs. Zhao, Galanter, and Townsend own stock or stock options in Genentech/Roche. Dr. Lee owns stock or stock options in Genentech/Roche and Eli Lilly. Ms Rae, Mr. Toth, and Drs. Ramamoorthi and Hackney own stock or stock options in Genentech/Roche. Dr. Damjanov has received consulting fees, speaking fees, and/or honoraria from AbbVie, Boehringer Ingelheim, MSD, and Pfizer (less than $10,000 each). Dr. Fedkov has received consulting fees, speaking fees, and/or honoraria from Laboratoires Expanscience and Fidia Pharma Group (less than $10,000 each) and from MSD, Pfizer, AbbVie, ProPharma, and Janssen (more than $10,000 each). Dr. Chinn owns stock or stock options and has a patent pending with Genentech/Roche. Dr. Morimoto owns stock or stock options and has a patent pending with Genentech/Roche. Dr. Genovese has received consulting fees from EMD Serono, Principia, and AbbVie (less than $10,000 each) and from Genentech/Roche, Eli Lilly, and Novartis (more than $10,000 each) and research support from Genentech/Roche, EMD Serono, Eli Lilly, and AbbVie. No other disclosures relevant to this article were reported.
Qualified researchers may request access to individual patient–level data through the Vivli Center for Global Clinical Research Data platform https://vivli.org/ http://www.clinicalstudydatarequest.com. Further details on Roche's criteria for eligible studies are available online at https://clinicalstudydatarequest.com/Study‐Sponsors/Study‐Sponsors‐Roche.aspx. For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.
References
- 1. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet 2010;376:1094–108. [DOI] [PubMed] [Google Scholar]
- 2. Renaudineau Y, Jamin C, Saraux A, Youinou P. Rheumatoid factor on a daily basis. Autoimmunity 2005;38:11–6. [DOI] [PubMed] [Google Scholar]
- 3. Mahendra A, Yang X, Abnouf S, Adolacion JR, Park D, Soomro S, et al. Beyond autoantibodies: biologic roles of human autoreactive B cells in rheumatoid arthritis revealed by RNA‐sequencing. Arthritis Rheumatol 2019;71:529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jegatheeswaran J, Turk M, Pope JE. Comparison of Janus kinase inhibitors in the treatment of rheumatoid arthritis: a systemic literature review. Immunotherapy 2019;11:737–54. [DOI] [PubMed] [Google Scholar]
- 5. Chastek B, Becker LK, Chen CI, Mahajan P, Curtis JR. Outcomes of tumor necrosis factor inhibitor cycling versus switching to a disease‐modifying anti‐rheumatic drug with a new mechanism of action among patients with rheumatoid arthritis. J Med Econ 2017;20:464–73. [DOI] [PubMed] [Google Scholar]
- 6. Favalli EG, Raimondo MG, Becciolini A, Crotti C, Biggioggero M, Caporali R. The management of first‐line biologic therapy failures in rheumatoid arthritis: current practice and future perspectives. Autoimmun Rev 2017;16:1185–95. [DOI] [PubMed] [Google Scholar]
- 7. Goess C, Harris CM, Murdock S, McCarthy RW, Sampson E, Twomey R, et al. ABBV‐105, a selective and irreversible inhibitor of Bruton's tyrosine kinase, is efficacious in multiple preclinical models of inflammation. Mod Rheumatol 2019;29:510–22. [DOI] [PubMed] [Google Scholar]
- 8. Gillooly KM, Pulicicchio C, Pattoli MA, Cheng L, Skala S, Heimrich EM, et al. Bruton's tyrosine kinase inhibitor BMS‐986142 in experimental models of rheumatoid arthritis enhances efficacy of agents representing clinical standard‐of-care. PLoS One 2017;12:e0181782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Norman P. Investigational Bruton's tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Expert Opin Investig Drugs 2016;25:891–9. [DOI] [PubMed] [Google Scholar]
- 10. Smith CI, Islam TC, Mattsson PT, Mohamed AJ, Nore BF, Vihinen M. The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. Bioessays 2001;23:436–46. [DOI] [PubMed] [Google Scholar]
- 11. Corneth OB, Klein Wolterink RG, Hendriks RW. BTK signaling in B cell differentiation and autoimmunity. Curr Top Microbiol Immunol 2016;393:67–105. [DOI] [PubMed] [Google Scholar]
- 12. Montalban X, Arnold DL, Weber MS, Staikov I, Piasecka‐Stryczynska K, Willmer J, et al. Placebo‐controlled trial of an oral BTK inhibitor in multiple sclerosis. N Engl J Med 2019;380:2406–17. [DOI] [PubMed] [Google Scholar]
- 13. Genovese MC, Spindler A, Sagawa A, Park W, Dudek A, Kivitz A, et al. SAT0089 safety and efficacy of LY3337641, a Bruton's tyrosine kinase inhibitor in patients with rheumatoid arthritis: a randomized, double‐blind, placebo‐controlled, 2‐part phase 2 study. Ann Rheum Dis 2019;78:1109–10. [DOI] [PubMed] [Google Scholar]
- 14. Kivitz AJ, Gupta R, Valenzuela G, Smith E, Rehman Q, El Kadi H, et al. A phase 2a, 4‐week double‐blind, proof‐of-concept efficacy and safety study of CC‐292 versus placebo as co‐therapy with methotrexate in active rheumatoid arthritis (RA) [abstract]. Arthritis Rheumatol 2016;68 Suppl 10 URL: https://acrabstracts.org/abstract/a-phase-2a-4-week-double-blind-proof-of-concept-efficacy-and-safety-study-of-cc-292-versus-placebo-as-co-therapy-with-methotrexate-in-active-rheumatoid-arthritis-ra/. [Google Scholar]
- 15. Crawford JJ, Johnson AR, Misner DL, Belmont LD, Castanedo G, Choy R, et al. Discovery of GDC‐0853: a potent, selective, and noncovalent Bruton's tyrosine kinase inhibitor in early clinical development. J Med Chem 2018;61:2227–45. [DOI] [PubMed] [Google Scholar]
- 16. Herman AE, Chinn LW, Kotwal SG, Murray ER, Zhao R, Florero M, et al. Safety, pharmacokinetics, and pharmacodynamics in healthy volunteers treated with GDC‐0853, a selective reversible Bruton's tyrosine kinase inhibitor. Clin Pharmacol Ther 2018;103:1020–8. [DOI] [PubMed] [Google Scholar]
- 17. Byrd JC, Smith SD, Wagner‐Johnston N, Sharman J, Chen AI, Advani RH, et al. First‐in-human phase 1 study of the BTK inhibitor GDC‐0853 in relapsed or refractory B‐cell NHL and CLL. Oncotarget 2018;9:13023–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO III, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. [DOI] [PubMed] [Google Scholar]
- 19. Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, et al. American College of Rheumatology preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum 1995;38:727–35. [DOI] [PubMed] [Google Scholar]
- 20. Jones NS, Winter H, Katsumoto TR, Florero M, Murray E, Walker H, et al. Absence of pharmacokinetic interactions between the Bruton's tyrosine kinase inhibitor fenebrutinib and methotrexate. J Pharmacol Exp Ther 2019;371:202–7. [DOI] [PubMed] [Google Scholar]
- 21. Krutmann J, Kirnbauer R, Kock A, Schwarz T, Schopf E, May LT, et al. Cross‐linking Fc receptors on monocytes triggers IL‐6 production: role in anti‐CD3-induced T cell activation. J Immunol 1990;145:1337–42. [PubMed] [Google Scholar]
- 22. Fernandez N, Renedo M, Garcia‐Rodriguez C, Sanchez Crespo M. Activation of monocytic cells through Fcγ receptors induces the expression of macrophage‐inflammatory protein (MIP)‐1 α, MIP‐1 β, and RANTES. J Immunol 2002;169:3321–8. [DOI] [PubMed] [Google Scholar]
- 23. Prehn JL, Thomas LS, Landers CJ, Yu QT, Michelsen KS, Targan SR. The T cell costimulator TL1A is induced by FcγR signaling in human monocytes and dendritic cells. J Immunol 2007;178:4033–8. [DOI] [PubMed] [Google Scholar]
- 24. Chan P, Yu J, Chinn L, Prohn M, Huisman J, Matzuka B, et al. Population pharmacokinetics, efficacy exposure‐response analysis, and model‐based meta‐analysis of fenebrutinib in subjects with rheumatoid arthritis. Pharm Res 2020;37:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Whang JA, Chang BY. Bruton's tyrosine kinase inhibitors for the treatment of rheumatoid arthritis [review]. Drug Discov Today 2014;19:1200–4. [DOI] [PubMed] [Google Scholar]
- 26. Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, et al. Rituximab for rheumatoid arthritis refractory to anti‐tumor necrosis factor therapy: results of a multicenter, randomized, double‐blind, placebo‐controlled, phase III trial evaluating primary efficacy and safety at twenty‐four weeks. Arthritis Rheum 2006;54:2793–806. [DOI] [PubMed] [Google Scholar]
- 27. Chang BY, Francesco M, de Rooij MF, Magadala P, Steggerda SM, Huang MM, et al. Egress of CD19+CD5+ cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood 2013;122:2412–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, et al. The Bruton tyrosine kinase inhibitor PCI‐32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood 2012;119:1182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Spaargaren M, Beuling EA, Rurup ML, Meijer HP, Klok MD, Middendorp S, et al. The B cell antigen receptor controls integrin activity through Btk and PLCγ2. J Exp Med 2003;198:1539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rip J, van Der Ploeg EK, Hendriks RW, Corneth OB. The role of Bruton's tyrosine kinase in immune cell signaling and systemic autoimmunity [review]. Crit Rev Immunol 2018;38:17–62. [DOI] [PubMed] [Google Scholar]
- 31. Nigrovic PA, Lee DM. Mast cells in inflammatory arthritis. Arthritis Res Ther 2005;7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Keebler D, Teng E, Chia J, Galanter J, Peake J, Tuckwell K. Regional variations in adverse event reporting rates and ACR responses in placebo/standard‐of-care arms of rheumatoid arthritis trials. Rheumatology 2020. E‐pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials