

Abstract
Purpose
Evaluation of phenotype and treatment outcome of retinal haemangioblastomas (RH) in von Hippel–Lindau (VHL) disease and correlation of these features with the genotype of VHL germline mutation carriers.
Methods
Retrospective analysis of a longitudinal cohort of 21 VHL germline mutation carriers and RH. Clinical and genetic data were obtained to analyse the correlation of genotype with phenotype and treatment outcomes.
Results
All patients were categorized in two genotypic categories: missense mutations (MM) and truncating mutations (TM). Mean follow‐up duration was 16.3 years and did not differ significantly between mutation groups (p = 0.383). Missense mutations (MM) carriers (n = 6) developed more progression‐related complications compared to TM carriers (n = 15) (p = 0.046). Vitreoretinal surgery was more often applied in MM carriers (p = 0.036). Moderate (visual acuity (VA)20/80 to 20/200) to severe (VA < 20/200) visual impairment was observed in 53.3% of the eyes of MM carriers and 28.1% of the eyes of TM carriers at last recorded visit.
Conclusion
Missense mutations in VHL patients seem to have a higher prevalence of progression‐related complications. Missense mutations (MM) carriers required therefore more often vitreoretinal surgical treatment with a worse treatment outcome. Genetic analysis may play a role in determining a pro‐active treatment strategy and prognosis for RH.
Keywords: benign tumours, clinical genetics, haemangioblastoma, retina, von Hippel – Lindau, Lindau
Introduction
Retinal haemangioblastomas (RH) are benign vascular neoplasms originating in the neurosensory retina or optic disc. Retinal haemangioblastomas (RH) can occur as a solitary tumour or in the setting of von Hippel–Lindau (VHL) disease (Online Mendelian Inheritance in Man 193300), an autosomal dominant inherited, multisystem neoplastic syndrome caused by mutations in the VHL tumour suppressor gene. Manifestations of VHL disease include RH, central nervous system haemangioblastoma, renal cell carcinoma, phaeochromocytoma, pancreatic neuroendocrine tumours, an endolymphatic sac tumour, cystadenoma of the epididymis and broad ligament, and renal and pancreatic cysts (Lonser et al. 2003). The incidence of VHL is approximately 1 in 36 000 live births per year and is highly penetrant, as 90% of the carriers of VHL mutations develop disease‐related symptoms (Maher et al. 1990). Retinal haemangioblastomas (RH) appear as reddish‐orange vascular lesions in the peripheral or juxtapapillary regions of the retina (Fig. 1). The frequency of RH varies from 49% to 85% as ocular involvement in VHL disease and is the first manifestation of VHL disease in 43% of patients (Maher et al. 1990; Singh et al. 2001). Retinal haemangioblastomas (RH) are asymptomatic in the early stage of development. Vision loss is generally caused by the exudative aspect of the tumour with development of hard exudates and retinal oedema affecting the macula. Furthermore, the larger size of RH can result in vitreomacular traction and preretinal fibrotic proliferation. These complications may lead to exudative and/or tractional retinal detachment (Chew 2005). Individuals with extended ocular VHL involvement can develop retinal neovascularization, neovascular glaucoma and eventually phthisis bulbi. Treatment of peripheral RH includes ablative modalities as laser photocoagulation, cryotherapy, radiation, photodynamic therapy (PDT) and transpupillary thermotherapy or radical intervention through vitreoretinal surgery (Haddad et al. 2013). Whether RH will develop progression‐related complications and fail to react to treatment are yet not well understood.
Figure 1.
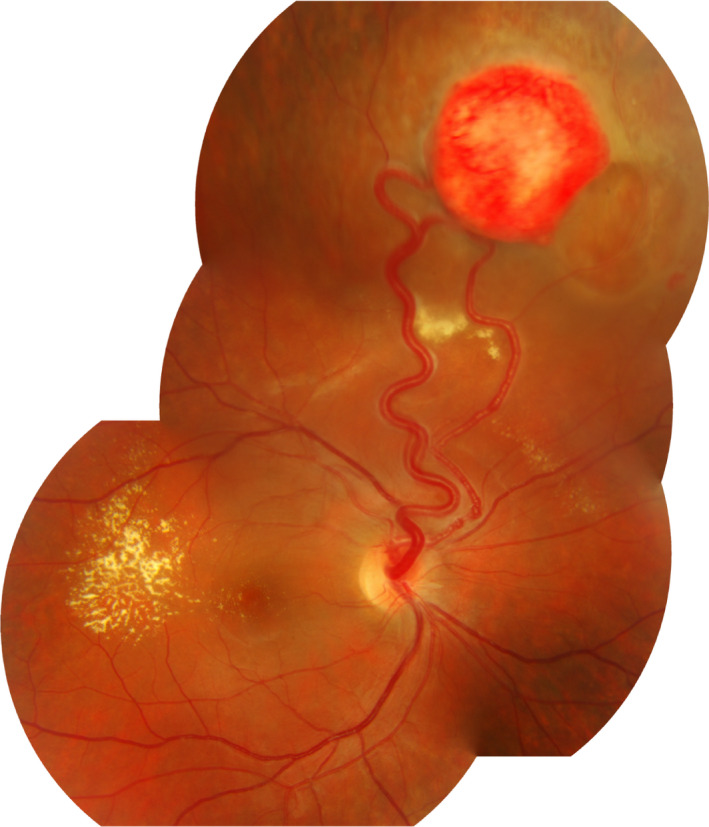
Peripheral retinal haemangioblastoma with dilated and tortuous feeding vessels, surrounded by detachment and exudation that extends into the macula.
The VHL tumour suppressor gene is located on chromosome 3p25.3.2, consists of three exons and encodes the VHL protein (pVHL) (Latif et al. 1993). von Hippel–Lindau (VHL) germline mutations are heterogeneous between VHL pedigrees and are identified at all three exons and splice site regions (Nordstrom‐O'Brien et al. 2010). Among the targets of pVHL are the hypoxia‐inducible factor 1α (HIF‐1α) and hypoxia‐inducible factor 2α (HIF‐2α), which are prolyl‐hydroxylated under normal oxygen conditions. This allows for binding to pVHL and attachment of ubiquitin peptides, resulting in proteasomal degradation of HIF‐1α and HIF‐2α (Maxwell et al. 1999; Min et al. 2002). In the absence or dysfunction of pVHL, HIF‐1α and HIF‐2α are not degraded, but form heterodimers with hypoxia‐inducible factor 1β (HIF‐1β) and upregulate proteins promoting cell proliferation and angiogenesis, including vascular endothelial growth factor (VEGF), plasminogen activator inhibitor‐1, erythropoietin (Epo), platelet‐derived growth factor and transforming growth factor α (Gossage et al. 2015). These factors have been hypothesized to contribute in vascular growth and tumour formation in VHL disease, including RH (Bohling et al. 1996; Carmeliet et al. 1998; Chan et al. 2007). Genotype–phenotype correlations have been established for further molecular understanding of the aberrant VHL protein function and its influence on formation and progression of RH. Earlier studies show that individuals with a complete deletion of the VHL gene may have a significantly decreased risk of developing RH. The individuals with a complete deletion that do develop RH have a lower extent of visual morbidity than patients with other types of VHL mutations (Chew 2005; Wong et al. 2007; Toy et al. 2012). Another study showed that the prevalence of RH in individuals with missense mutations in the functional α‐domain of the VHL gene was significantly higher than for patients with missense mutations in the functional β‐domain (Mettu et al. 2010). Severe ocular involvement, defined as enucleation of the eye with ocular VHL disease or phthisical and prephthisical changes, was likewise correlated with having a germline mutation in previous studies, but did not show a significant association with the mutation type or location (Wong et al. 2007; Mettu et al. 2010). However, whether a complicated progression of RH is related to the genotype of VHL patients has not yet been investigated. A progression accompanied by complications requires a different treatment approach and will possibly lead to different treatment outcomes. Therefore, a better understanding of the complex pathophysiologic process of RH is essential for a better treatment strategy. In the present study, we analysed the correlations between genotypic VHL categories and tumour progression, treatment approach and outcome. This study contributes to a better understanding of how VHL gene mutations affect the progression of RH based on their presumed effect on pVHL and their impact on prognosis and may therefore provide a window for a patient‐tailored treatment approach and follow‐up strategy.
Methods
Patient selection and ascertainment
We conducted a retrospective cohort study at the Department of Ophthalmology of the Erasmus Medical Centre and the Rotterdam Eye Hospital (Rotterdam, the Netherlands). The local Medical Ethical Committee approved this study. All data were extracted from medical records of patients and the research adhered to the tenets of the Declaration of Helsinki.
von Hippel–Lindau germline mutation carriers who had an ophthalmologic examination at the Erasmus Medical Centre or the Rotterdam Eye Hospital between 1984 and 2017, and were diagnosed with RH were included in this study. The diagnosis of RH was set based on the clinical appearance of a vasoproliferative retinal tumour with or without the presence of dilated and tortuous feeder vessels during fundoscopic examination with a pharmacologically dilated pupil. From the 66 confirmed VHL germline mutation carriers, 30 cases did have an ophthalmologic examination elsewhere and were therefore not included in the study. The electronic and paper medical records of 36 individuals from 16 different VHL families were retrospectively reviewed based on VHL gene mutations reported by the Department of Clinical Genetics of the Erasmus Medical Centre. Fifteen of the 36 VHL germline mutation carriers were excluded from further analysis as they did not develop any ocular manifestations. We obtained data of 21 patients from 10 families including sex, age at baseline visit, age at first manifestation of RH during follow‐up and presence of bilateral eye involvement. Furthermore, data on progression and growth of the tumours and applied treatment during follow‐up were collected.
Ocular manifestation assessment
A complete ophthalmologic examination of all patients was performed, including measurement of the best‐corrected visual acuity (BCVA), slit‐lamp examination and indirect ophthalmoscopy. The visual acuity was categorized into the classifications of visual impairment of the World Health Organization. Mild or no visual impairment is a BCVA equal to or better than 20/70. Moderate visual impairment is considered a BCVA from 20/80 to 20/200, and severe visual impairment is defined as a BCVA worse than 20/200 (Organization 2015). The rating of ocular manifestations, their complications and regression are based on fundus photographs which have been assessed by an ophthalmologist. If fundus photographs were not available, the ocular manifestations, their complications and regression were noted based on the report of the ophthalmologic examination by an ophthalmologist in the medical records. The number and location of the RH were recorded as well as the presence of complicated progression of RH. The exact number of tumours was not described in the medical records of two individuals. However, the records of these patients did contain the remaining data concerning phenotype, progression and treatment. All the data of these individuals are therefore included in the analysis with the exception of the number of tumours. The location of the RH was classified as peripheral or juxtapapillary (within one disc diameter from the disc margin). The extent of peripheral retinal involvement was described as number of quadrant involvement of the peripheral retina. The presence or absence of complications due to progression of RH, such as retinal exudation, (extra)macular oedema, retinal traction, retinal detachment, preretinal fibrosis, preretinal or vitreous haemorrhage, retinal neovascularization and neovascular glaucoma, was noted. Therapy consisted of observation, laser photocoagulation, cryotherapy, transpupillary thermotherapy, photodynamic therapy, intravitreal anti‐VEGF, radiation therapy, vitreoretinal surgery (subclassified with or without endoresection of the tumour) or a multiple treatment approach. Tumour regression was recorded and the tumour was considered regressed if at last recorded visit the size of the tumour decreased with a change of colour and pigmentation, and/or if the diameter of the feeding vessel was reduced. Treatment‐related complications were defined as exudation, retinal traction, retinal detachment, (extra)macular oedema, preretinal or vitreous haemorrhage and preretinal fibrosis within a month after the procedure according to the criteria of treatment complications defined by Kim et al. (2014). Mild transient haemorrhages and exudation were not classified as complications as these are expected responses to ablative treatments.
Genotype analysis
The VHL genotype analyses were executed as part of general clinical management to confirm the diagnosis of VHL disease, and the results were requested for research purposes. Peripheral blood samples from every member with clinically suspected VHL disease of each pedigree were collected for VHL mutation analysis. Mutational analyses were carried out at the certified diagnostic laboratory of the Department of Clinical Genetics at the Erasmus University Medical Centre (certified by the Dutch Accreditation Council; EN ISO 15189:2012,M 105) by Sanger sequencing of the exons and flanking regions. Copy number analyses were carried out by multiplex ligation‐dependent probe amplification (MLPA) and fluorescence in situ hybridization (FISH). Protocols are available upon request. Mutations were described as complete deletion, partial deletion, missense mutation, nonsense mutation, splice site mutation, duplication and insertion.
Statistical analysis
IBM SPSS Statistics software version 24.0 was used for statistical analysis. To compare baseline and clinical characteristics between genotypic categories as phenotype and the categorized visual acuity, the Fisher’s exact test and chi‐square test were used for statistical analysis of categorical data. As our sample size was less than 30, continuous data were tested for normality by the Shapiro–Wilk test. The independent samples t‐test was used to assess the relationship of the genotypic categories and age at baseline visit and age of diagnosis. The Mann–Whitney U‐test and Kruskal–Wallis test were used for the analysis of the number of applied treatments as nonparametric data. The odds ratios were calculated with their 95% confidence interval. p‐values less than 0.05 were considered to be statistically significant.
Results
VHL germline mutation data
Medical records of 66 individuals with a known VHL gene germline mutation were collected and retrospectively analysed. Thirty‐six patients (55%) had an ophthalmologic examination at the Erasmus Medical Centre or the Eye Hospital Rotterdam. The VHL gene mutation status of the individuals who underwent an ophthalmic examination is shown in Table 1. The nomenclature of the found mutations according to the Human Genome Variation Society is shown in Table S1, which shows the VHL gene mutation status of the analysed pedigrees on complementary and genomic DNA level. From the 36 individuals who underwent a complete ophthalmic examination, a total of 21 patients (58.3%) had evidence of ocular manifestations of VHL disease (Fig. S1). For further analyses, the germline mutation status of these patients was categorized in two genotypic categories, based on their presumed effect on the VHL protein structure: significant deletions, splice site mutations, frameshift mutations and insertions were categorized as the truncating mutation (TM) group, and the missense mutations were categorized as missense mutation (MM) group. The prevalence of RH as manifestation of VHL disease was highest in individuals with a TM, as 15 of the 22 (68%) individuals developed RH, in comparison with the patients with a MM where 6 of the 14 (43%) individuals had ocular involvement of VHL disease, but did not differ significantly (OR = 2.9; 95% CI: 0.7–11.4, p = 0.175, chi‐squared test).
Table 1.
Germline mutation status of the von Hippel–Lindau gene of patients with an ophthalmologic examination (n = 36).
| Pedigrees | Mutation | Expected pVHL change | Individuals with RH development | Individuals without RH development |
|---|---|---|---|---|
| 1 | Partial deletion | Deletion of exon 1 and 2 | 1 | 0 |
| 2 | Partial deletion | Deletion of exon 1 and 2 | 1 | 0 |
| 3 | Deletion | p.(Phe76del) | 1 | 0 |
| 4 | Missense | p.(Asn78Ser) | 1 | 0 |
| 5 | Indel | Deletion of exon 2 | 4 | 1 |
| 6 | Deletion | Deletion of exon 2 | 1 | 0 |
| 7 | Missense | p.(Val130Phe) | 5 | 1 |
| 8 | Splice site | Deletion exon 3 | 1 | 0 |
| 9 | Frameshift | p.(Lys159Glufs*15) | 5 | 3 |
| 10 | Complete deletion | VHL gene deletion | 1 | 2 |
| 11 | Missense | p.(Arg167Gln) | 0 | 2 |
| 12 | Missense | p.(Ile151Ser) | 0 | 1 |
| 13 | Missense | p.(Leu89Pro) | 0 | 1 |
| 14 | Missense | p.(Arg64Pro) | 0 | 2 |
| 15 | Complete deletion | VHL gene deletion | 0 | 2 |
Arg = arginine, Asn = asparagine, del = deletion, fs = frameshift, Gln = glutamine, Glu = glutamic acid, Ile = isoleucine, Leu = leucine, Lys = Lysine, Phe = phenyalanine, Pro = proline, pVHL = von Hippel–Lindau protein, RH = retinal haemangioblastoma, Ser = Serine, Val = valine, VHL = von Hippel–Lindau.
Ocular manifestations
Demographic data of the included patients are summarized in Table 2. Bilateral involvement of ocular VHL disease was described in 14 of the 21 cases. Ocular VHL disease progressed severely in three eyes of three patients, leading to phthisical changes. Two of the three eyes were enucleated as a result of the severe involvement. One TM carrier that had his eye enucleated underwent the procedure in his first decade, taking into account that some of the current treatment modalities were not yet available in that period. The remaining two individuals with phthisical changes were MM carriers and developed structural disruptions due to ocular VHL disease despite of application of current treatment modalities.
Table 2.
Demographics and ocular status of patients with ocular von Hippel–Lindau disease.
| Missense mutation carriers (n = 6) | Truncating mutation carriers (n = 15) | p | ||
|---|---|---|---|---|
| Sex | Male/female | 3/3 | 6/9 | |
| Age at baseline visit (years), mean ± SD | 22.4 ± 10.3 | 26.7 ± 16.4 | 0.570 | |
| Age of diagnosis RHs (years), mean ± SD | 26.1 ± 8.1 | 25.5 ± 15.5 | 0.931 | |
| Follow‐up duration (years), mean ± SD | 19.4 ± 7.4 | 15.1 ± 10.6 | 0.383 | |
| Ocular status, n | Unilateral involvement | 2 | 5 | |
| Bilateral involvement | 4 | 10 | ||
| Severe involvement | 2 | 1 |
The ocular phenotype of VHL patients was extracted from the medical records, and we analysed the number of RH during follow‐up in either eye. Table 3 summarizes the data of the ocular phenotype comparing the genotypic categories. The RH count was divided into three categories: two or less, between three and five RH, and more than five RH in either eye. In TM carriers, the number of RH is small, (6/13 (46%)) compared MM carriers (1/6 (17%)) with two or less RH development during follow‐up. Data related to the number of RH of two TM carriers were missing and were therefore not included in the analysis. The rate of having more than five RH was higher in the MM group (50% versus 39%). Furthermore, the extent of involvement of the peripheral retina, expressed in quadrants, was analysed. The majority of the patients (4/6, 67%) with a MM had ocular VHL involvement of all four quadrants, whereas 50.0% (7/14) of the TM group had an extent of no more than one quadrant during follow‐up. (Data on the extent of retinal involvement and location of RH in one TM carrier were missing). Location of RH was divided in the juxtapapillary and peripheral area. The two patients who developed RH in the juxtapapillary region were TM carriers. We examined ocular complications due to growth and progression of RH and compared the complications and clinical characteristics between the genotypic categories. The entire MM group (6/6, 100%) developed complications due to progression. The most common complication in this group was vitreoretinal traction (5/6, 83%), followed by preretinal fibrosis (4/6, 67%), neovascularization (4/6, 67%) and subretinal fluid (4/6, 67%). The TM group had a lower rate of complication development (7/15, 47%). In addition, each separate complication had a lower rate in the TM group compared to the MM group.
Table 3.
Comparison of ocular phenotype in von Hippel–Lindau patients between genotypic categories.
| Missense mutation carriers | Truncating mutation carriers | ||
|---|---|---|---|
| Number in either eye (%) | ≤2 RH | 1/6 (16.7) | 6/13 (46.2) |
| 3–5 RH | 2/6 (33.3) | 2/13 (15.4) | |
| >5 RH | 3/6 (50.0) | 5/13 (38.5) | |
| Quadrant involvement in either eye (%) | 1 Quadrant | 1/6 (16.7) | 7/14 (50) |
| 2 Quadrants | 1/6 (16.7) | 1/14 (7.1) | |
| 3 Quadrants | 0/6 (0.0) | 1/14 (7.1) | |
| All quadrants | 4/6 (66.7) | 5/14 (35.7) | |
| Location of RHs in either eye (%) | Juxtapapillary | 0/6 (0.0) | 1/14 (7.1) |
| Peripheral | 6/6 (100.0) | 12/14 (85.7) | |
| Both | 0/6 (0.0) | 1/14 (7.1) | |
| Progression‐related complication in either eye (%) | 6/6 (100.0) | 7/15 (46.7) | |
| Vitreoretinal traction | 5 (83.3) | 5 (35.7) | |
| Retinal detachment | 3 (50) | 3 (21.4) | |
| Haemorrhage | 3 (33.3) | 3 (21.4) | |
| Exudation | 2 (33.3) | 3 (21.4) | |
| CME | 3 (50.0) | 0 (0.0) | |
| Intraretinal oedema | 2 (33.3) | 3 (21.4) | |
| Subretinal fluid | 4 (66.7) | 2 (14.3) | |
| Retinal neovascularization | 4 (66.7) | 1 (7.1) | |
| Neovascular glaucoma | 1 (16.7) | 1 (7.1) | |
| Preretinal fibrosis | 4 (66.7) | 3 (21.4) |
CME = cystoid macular oedema, RH = retinal haemangioblastomas.
Treatment modalities and outcome
Choice for treatment depended on the location and size of RH and the existing complications due to progression of RH. The VHL patients with RH were most frequently treated with laser photocoagulation in both genotypic categories (resp. 100% (6/6) of MM group versus 93% (14/15) of TM group) (Table 4). A single treatment method was not sufficient in 62% (13/21) of the cases. Due to lack of tumour regression or relapse of tumours in the affected eye, various treatment modalities were applied in these patients. Surgical intervention was more often required in MM carriers, as 83% (5/6) underwent vitreoretinal surgery, in contrast to TM carriers of whom only 20% (3/15) needed surgical treatment. In five patients, endoresection of RH during vitreoretinal surgery was performed. In this cohort, surgical treatment and PDT were merely applied if the extent of ocular VHL disease involved more than one quadrant and the number of tumours was three or more in at least one eye.
Table 4.
Therapeutic modalities applied for retinal haemangioblastomas in patients with von Hippel–Lindau disease.
| Missense mutation carriers (n = 6, %) | Truncating mutation (n = 15, %) | ||
|---|---|---|---|
| Expectative | 1 (16.7) | 2 (13.3) | |
| Laser photocoagulation | 6 (100.0) | 14 (93.3) | |
| Cryocoagulation | 3 (50.0) | 3 (20.0) | |
| Vitreoretinal surgery | 5 (83.3) | 3 (20.0) | |
| Endoresection | 4 (66.7) | 1 (6.7) | |
| Photodynamic therapy | 2 (33.3) | 0 (0.0) | |
| Anti‐VEGF | 3 (50.0) | 3 (20.0) | |
| Radiation therapy | Ruthenium‐106 brachytherapy | 0 (0.0) | 1 (6.7) |
| Multiple treatments | 6 (100.0) | 7 (46.7) |
VEGF = Vascular Endothelial Growth factor.
Twenty of the 21 patients included in this study underwent a therapeutic intervention. One patient was clinically observed, as his ocular VHL disease remained stable without the need of treatment. The six patients with a MM underwent 63 therapeutic interventions whereas 14 TM carriers received 116 interventions. The mean ratio of treatment per patient is thus 10.5 treatments per patient in the MM group versus 8.3 treatments per patient with a TM. The number of vitreoretinal surgical interventions was significantly higher in MM carriers (p = 0.036, Mann–Whitney U‐test) with a difference in the ratio vitreoretinal surgeries per patient of 2.7 in patients with a MM versus 0.9 in TM carriers (Table 5). Phenotypic features as number of RH and the extent of retinal involvement were analysed regarding the number of interventions applied. An increase in number of RH and extent of retinal involvement seems to be related to an increase in application of therapeutic intervention (p = 0.016 and p = 0.027, respectively, Kruskal–Wallis test).
Table 5.
Number of therapeutic interventions applied for retinal haemangioblastomas in patients with von Hippel–Lindau disease.
| Interventions per patient in missense group (n = 6) | Interventions per patient in truncating group (n = 14) | p | |
|---|---|---|---|
| Therapeutic interventions, mean (range) | 10.5 (3–22) | 7.7 (1–46) | 0.080 |
| Laser | 4.7 (2–11) | 6.0 (1–29) | 0.267 |
| Cryocoagulation | 1.2 (0–3) | 0.7 (0–5) | 0.381 |
| Vitreoretinal surgery | 2.7 (0–7) | 0.9 (0–8) | 0.036 |
| Photodynamic therapy | 0.5 (0–2) | 0.0 (0–0) | 0.267 |
| Anti‐VEGF | 1.5 (0–6) | 0.6 (0–5) | 0.302 |
| Radiation therapy (Ruthenium‐106 brachytherapy) | 0 (0–0) | 1 (0–1) | 0.850 |
VEGF = Vascular Endothelial Growth factor.
The bold values are considered statistically significant.
Therapeutic complications within a month after procedure occurred in 28.6% (6/21) of the patients. In both genotypic categories, three patients developed therapeutic complications. The prevalence of therapeutic complications was highest after PDT. However, the complications occurred within a month after treatment in one of the merely three PDT sessions applied in this study. The prevalence of complications after surgical treatment is 29% (8/28 surgical interventions), followed by cryotherapy (25%, 4/16 treatments). No therapeutic complications were observed in patients with less than a quadrant retinal involvement or less than three RH in either eye.
Complete tumour regression at last recorded visit was observed in 15 of the 21 patients (71%). Regression rate decreased as the number of RH in either eye increased (Fig. 2). All patients with a lower extent of one or two quadrant retinal involvement had a complete regression of RH at last recorded visit. Patients with a complicated progression of RH had a regression rate of 54% (7/13). As regards the germline mutation status, the MM group had a regression rate of 50% (3/6) versus a rate of 80% in the TM group (12/15).
Figure 2.
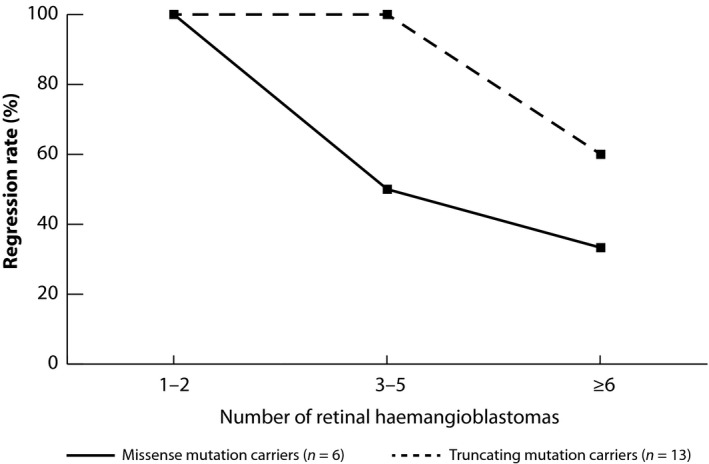
Line graph showing the regression rate of treated haemangioblastomas relative to the number of tumours arised during follow‐up. In both genotypic categories, the regression rate was 100% if merely one or two tumours arised. Missense mutation carriers showed a faster decrease of the regression rate when tumour number increased.
Best‐corrected visual acuity of the better‐ and worse‐seeing eye at baseline and last recorded visit was analysed and compared between genotypic categories (Fig. 3). All patients of both groups had a BCVA equal to or better than 20/70 in the better‐seeing eye at baseline visit. In the worse‐seeing eye, 100% (6/6) of the MM group had mild or no visual impairment versus 77% (10/13) of the TM group, respectively. The BCVA of patients in both genotypic groups deteriorated in the better‐seeing eyes, and one patient of the MM group went from good vision to severe visual impairment during follow‐up. One patient of the TM group had a deterioration of visual acuity from a good vision to moderate visual impairment. At baseline visit, there were missing data of 3 eyes from patients with a TM concerning the BCVA. At final visit, no data on the visual acuity were available of two eyes, one eye of a patient with a MM, and the other eye of a patient with a TM, and were therefore not included in the analysis.
Figure 3.
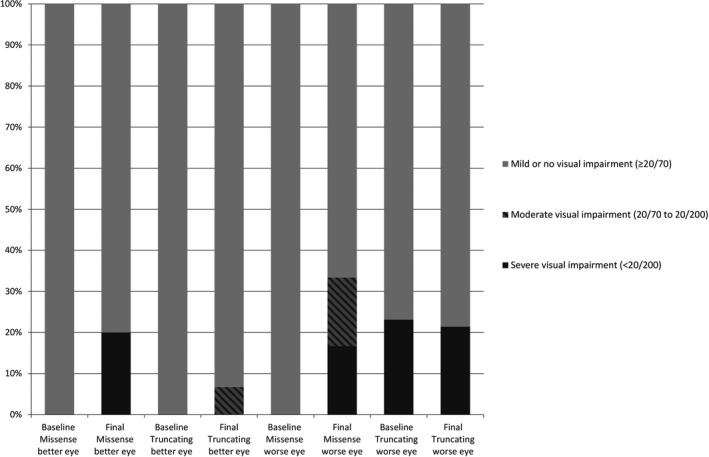
Bar graph showing the distribution of the initial and last recorded best‐corrected visual acuities between genotypic categories. Whereas missense mutation carriers started with a better visual acuity in best‐ and worse‐seeing eye, at last recorded visit a larger percentage of severe visual impairment is observed in this genotypic category.
Discussion
This study provides a clinical and genetic analysis of the progression of retinal haemangioblastomas in the context of von Hippel–Lindau disease, the applied treatments and therapeutic outcomes, measured in visual function, tumour regression and therapeutic complications. Our findings suggest that MM carriers develop a higher number of RH and a greater extent of retinal involvement of ocular VHL disease. This is in accordance with the earlier genotype–phenotype correlation study of Dollfus et al. (2002), who described a statistically significant higher number of RH in patients with a MM. Dolffus et al. give a comprehensive evaluation of the phenotype and ocular complications. Complications as extramacular exudation and tractional retinal detachment were present in 22% of the patients. However, the ocular complications are not included in the genotype analysis. Conclusions on different progression of the retinal lesions between genotypes cannot therefore be drawn. In the present study, we compared the ocular complications and the applied treatments between genotypic categories as an addition to the traditional genotype–phenotype correlation studies. This study may therefore be a first step to a personalized follow‐up and treatment approach based on the genotype. In our study, progression‐related complications were observed more often in the MM group compared to the TM group. The distribution of the participants into the genotypic categories was based on the effect of the mutation on the VHL protein (Binderup et al. 2016). We observed that the prevalence of RH is highest in VHL patients with a TM, concurrent with the results observed in a previous cohort study (Wong et al. 2007), followed by MM. Phenotypic features like complicated progression, a high number of RH and an extensive retinal involvement are known to complicate the treatment approach; therefore, standard ablative treatment will not suffice given the poor success rate and significant risks.
Several methods of vitreoretinal surgery, such as resection of the RH, can be applied for severe ocular VHL disease (Schlesinger et al. 2007; Gaudric et al. 2011). Vitreoretinal surgery is indicated when RH are accompanied by complications such as epiretinal membrane development, vitreous haemorrhage, tractional and/or exudative retinal detachment, preretinal fibrosis or proliferative vitreoretinopathy. There is not yet a standard treatment strategy for RH. Treatment methods vary depending on location, size and complications of the tumour. In addition, current treatment methods are limited by iatrogenic damage caused to normal eye structures. Treatment outcomes and complications due to treatment of RH vary, and whether RH will respond to treatment is not well understood (Kim et al. 2014; Avci et al. 2017). In our cohort (Table 4), patients with a MM required more frequently a radical treatment approach through vitreoretinal surgery and multiple treatment modalities than patients with a TM. Despite a higher rate of radical treatment approach in the MM group, regression was more often achieved in the TM group. However, visual acuity of the worse eye of patients with a germline MM remained stable. Patients with complicated ocular VHL disease might therefore benefit from radical treatment as preservation of visual function. Krzystolik et al. (2016) described the effectiveness of pars plana vitrectomy as treatment for advanced RH development and also noted a positive effect on improvement or preservation of visual function in eyes with complicated RH. The number of RH seems to be inversely related to regression rate as an increase in number is accompanied by a decrease in regression rate. Therapeutic complications were merely observed if progression‐related complications were present. We observed in most cases a mild and transient course of complications after treatment with a small chance of permanently affecting the visual function. Moderate to severe visual impairment in at least one eye was at both baseline and last recorded visit more prevalent in the MM group. This analysis shows that patients with a germline MM have worse therapeutic and visual outcome and may therefore require a more frequent follow‐up.
Two patients received surgical treatment in an early stage of disease instead of the more common less invasive treatment modalities as laser photocoagulation or cryotherapy. These patients needed a smaller number of interventions than patients who received surgical intervention in a later stage of ocular VHL disease. Complete tumour regression and a good visual outcome were achieved at last recorded visit in these patients. However, these patients had a TM. Removing the vitreous humour during vitreoretinal surgery might lead to a reduction of vitreous‐related complication as vitreoretinal traction and formation of fibrosis. These common progression‐related complications in ocular VHL disease may therefore be prevented with the expectation of a lesser need of multiple interventions and a greater chance of treatment complications. Treatment through vitreoretinal surgical intervention in an early stage might therefore be beneficial for MM carriers as they seem to have a more complicated progression of ocular VHL disease (van Overdam et al. 2017).
Truncating mutations most likely abrogate the VHL protein, or a substantial structural change of the protein is expected. Missense mutations are expected not to result in a gross pVHL disruption, influencing one or a few functions of the VHL protein. Due to the various impacts on pVHL binding sites, missense mutations may lead to diverse tumour aggressiveness and response to treatment. In renal cell carcinoma, a lower response rate to anti‐angiogenic treatment is observed in tumours with a MM, possibly because of the effect on diverse pathways caused by different mutation types. Therefore, targeted therapy for patients with a MM in the VHL gene may be more effective (Razafinjatovo et al. 2016). Our findings suggest that such difference in mutation status indeed may require a different treatment approach in VHL‐related RH.
Although this study may be limited by the relatively small sample size, which made statistical analysis challenging, the duration of follow‐up with a mean of 16.3 years is the longest longitudinal cohort of VHL patients with RH. This longitudinal analysis provides a large amount of extended clinical data of VHL patients giving a deeper insight into the progression of RH. Another limitation of this study is the retrospective aspect as treatment modalities have improved over time and the same treatment may lead to better treatment outcomes nowadays. The current follow‐up guideline has not been applied to all individuals in this study as some individuals have been followed up for 30 years. Significant progression of ocular disease can be caused by insufficient treatment or inadequate follow‐up intervals and therefore lead to dissimilar treatment strategies and outcomes between VHL patients with RH. The retrospective analysis of this study was also limited by inaccurate recordkeeping, leading to an incomplete comparison of the longitudinal data between genotypic groups.
In conclusion, MM carriers are more likely to develop more aggressive RH with progression‐related complications, which consequently leads to a more radical treatment approach. Treatment outcomes were worse as less tumour regression was achieved and a higher rate of visual impairment and therapeutic complications was observed. This study provides data of patients with a VHL mutation and the development of RH, and depicts the progression of ocular VHL disease and the effect of treatment for each genotypic category. The observations in this study imply that genotype analysis may be used as a tool for determination of treatment approach and prognosis, and indicate the importance of genetic counselling and an ophthalmologic screening protocol based on genotypic analysis. A more radical treatment approach in an early stage of the disease and shorter follow‐up intervals might lead to a better tumour control in patients with a genotype possibly leading to a more progressive phenotype.
Supporting information
Figure S1. Flow chart indicating VHL patients meeting the inclusion criteria.
Table S1. VHL gene mutation status of the analysed pedigrees with the Human Genome Variation Society nomenclature on complementary and genomic DNA level.
Part of the study was presented at the 211th Annual Meeting of the Dutch Ophthalmic Society, 31 March 2017, Maastricht, the Netherlands, and the Association for Research in Vision and Ophthalmology (ARVO) Annual Meeting, 30 April 2019, Vancouver, Canada. We acknowledge funding support from the Professor Henkes Foundation, Rotterdam, the Netherlands, and the Rotterdamse Stichting Blindenbelangen, Rotterdam, the Netherlands (grant no.: B20180038).
References
- Avci R, Yilmaz S, Inan UU, Kaderli B & Cevik SG (2017): Vitreoretinal surgery for patients with severe exudative and proliferative manifestations of retinal capillary hemangioblastoma because of Von Hippel‐Lindau disease. Retina 37: 782–788. [DOI] [PubMed] [Google Scholar]
- Binderup ML, Budtz‐Jorgensen E & Bisgaard ML (2016): Risk of new tumors in von Hippel‐Lindau patients depends on age and genotype. Genet Med 18: 89–97. [DOI] [PubMed] [Google Scholar]
- Bohling T, Hatva E, Kujala M, Claesson‐Welsh L, Alitalo K & Haltia M (1996): Expression of growth factors and growth factor receptors in capillary hemangioblastoma. J Neuropathol Exp Neurol 55: 522–527. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Dor Y, Herbert JM et al. (1998): Role of HIF‐1alpha in hypoxia‐mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 394: 485–490. [DOI] [PubMed] [Google Scholar]
- Chan CC, Collins AB & Chew EY (2007): Molecular pathology of eyes with von Hippel‐Lindau (VHL) Disease: a review. Retina 27: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew EY (2005): Ocular manifestations of von Hippel‐Lindau disease: clinical and genetic investigations. Trans Am Ophthalmol Soc 103: 495–511. [PMC free article] [PubMed] [Google Scholar]
- Dollfus H, Massin P, Taupin P et al. (2002): Retinal hemangioblastoma in von Hippel‐Lindau disease: a clinical and molecular study. Invest Ophthalmol Vis Sci 43: 3067–3074. [PubMed] [Google Scholar]
- Gaudric A, Krivosic V, Duguid G, Massin P, Giraud S & Richard S (2011): Vitreoretinal surgery for severe retinal capillary hemangiomas in von hippel‐lindau disease. Ophthalmology 118: 142–149. [DOI] [PubMed] [Google Scholar]
- Gossage L, Eisen T & Maher ER (2015): VHL, the story of a tumour suppressor gene. Nat Rev Cancer 15: 55–64. [DOI] [PubMed] [Google Scholar]
- Haddad NM, Cavallerano JD & Silva PS (2013): Von Hippel‐Lindau disease: a genetic and clinical review. Semin Ophthalmol 28: 377–386. [DOI] [PubMed] [Google Scholar]
- Kim H, Yi JH, Kwon HJ, Lee CS & Lee SC (2014): Therapeutic outcomes of retinal hemangioblastomas. Retina 34: 2479–2486. [DOI] [PubMed] [Google Scholar]
- Krzystolik K, Stopa M, Kuprjanowicz L et al. (2016): Pars plana vitrectomy in advanced cases of Von Hippel‐Lindau eye disease. Retina 36: 325–334. [DOI] [PubMed] [Google Scholar]
- Latif F, Tory K, Gnarra J et al. (1993): Identification of the von Hippel‐Lindau disease tumor suppressor gene. Science 260: 1317–1320. [DOI] [PubMed] [Google Scholar]
- Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM & Oldfield EH (2003): von Hippel‐Lindau disease. Lancet 361: 2059–2067. [DOI] [PubMed] [Google Scholar]
- Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT & Ferguson‐Smith MA (1990): Clinical features and natural history of von Hippel‐Lindau disease. Q J Med 77: 1151–1163. [DOI] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW et al. (1999): The tumour suppressor protein VHL targets hypoxia‐inducible factors for oxygen‐dependent proteolysis. Nature 399: 271–275. [DOI] [PubMed] [Google Scholar]
- Mettu P, Agron E, Samtani S, Chew EY & Wong WT (2010): Genotype‐phenotype correlation in ocular von Hippel‐Lindau (VHL) disease: the effect of missense mutation position on ocular VHL phenotype. Invest Ophthalmol Vis Sci 51: 4464–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min JH, Yang H, Ivan M, Gertler F, Kaelin WG Jr & Pavletich NP (2002): Structure of an HIF‐1alpha ‐pVHL complex: hydroxyproline recognition in signaling. Science 296: 1886–1889. [DOI] [PubMed] [Google Scholar]
- Nordstrom‐O'Brien M, van der Luijt RB, van Rooijen E et al. (2010): Genetic analysis of von Hippel‐Lindau disease. Hum Mutat 31: 521–537. [DOI] [PubMed] [Google Scholar]
- Organization WH (2015): Change the Definition of Blindness.
- van Overdam KA, Missotten T, Kilic E & Spielberg LH (2017): Early surgical treatment of retinal hemangioblastomas. Acta Ophthalmol 95: 97–102. [DOI] [PubMed] [Google Scholar]
- Razafinjatovo C, Bihr S, Mischo A, Vogl U, Schmidinger M, Moch H & Schraml P (2016): Characterization of VHL missense mutations in sporadic clear cell renal cell carcinoma: hotspots, affected binding domains, functional impact on pVHL and therapeutic relevance. BMC Cancer 16: 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlesinger T, Appukuttan B, Hwang T, Atchaneeyakasul LO, Chan CC, Zhuang Z, Stout JT & Wilson DJ (2007): Internal en bloc resection and genetic analysis of retinal capillary hemangioblastoma. Arch Ophthalmol 125: 1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AD, Shields CL & Shields JA (2001): von Hippel‐Lindau disease. Surv Ophthalmol 46: 117–142. [DOI] [PubMed] [Google Scholar]
- Toy BC, Agron E, Nigam D, Chew EY & Wong WT (2012): Longitudinal analysis of retinal hemangioblastomatosis and visual function in ocular von Hippel‐Lindau disease. Ophthalmology 119: 2622–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WT, Agron E, Coleman HR et al. (2007): Genotype‐phenotype correlation in von Hippel‐Lindau disease with retinal angiomatosis. Arch Ophthalmol 125: 239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Flow chart indicating VHL patients meeting the inclusion criteria.
Table S1. VHL gene mutation status of the analysed pedigrees with the Human Genome Variation Society nomenclature on complementary and genomic DNA level.