

Summary
Use of haematopoietic cell transplantation (HCT) in the treatment of haematologic and neoplastic diseases may lead to life‐threatening complications that cause substantial morbidity and mortality if untreated. In addition to patient‐ and disease‐related factors, toxicity associated with HCT puts patients at risk for complications that share a similar pathophysiology involving endothelial cells (ECs). Normally, the endothelium plays a role in maintaining homeostasis, including regulation of coagulation, vascular tone, permeability and inflammatory processes. When activated, ECs acquire cellular features that may lead to phenotypic changes that induce procoagulant, pro‐inflammatory and pro‐apoptotic mediators leading to EC dysfunction and damage. Elevated levels of coagulation factors, cytokines and adhesion molecules are indicative of endothelial dysfunction, and endothelial damage may lead to clinical signs and symptoms of pathological post‐HCT conditions, including veno‐occlusive disease/sinusoidal obstruction syndrome, graft‐versus‐host disease, transplant‐associated thrombotic microangiopathy and idiopathic pneumonia syndrome/diffuse alveolar haemorrhage. The endothelium represents a rational target for preventing and treating HCT complications arising from EC dysfunction and damage. Additionally, markers of endothelial damage may be useful in improving diagnosis of HCT‐related complications and monitoring treatment effect. Continued research to effectively manage EC activation, injury and dysfunction may be important in improving patient outcomes after HCT.
Keywords: endothelial cell dysfunction, endothelial‐related disorders, haematopoietic cell transplantation, chimeric antigen receptor T‐cell (CAR‐T) therapy, treatment for post‐HCT complications
Haematopoietic cell transplantation (HCT) is used to treat a variety of haematologic and neoplastic diseases; however, complications can be life‐threatening and cause substantial morbidity and mortality if untreated. 1 Early HCT‐related complications, including veno‐occlusive disease/sinusoidal obstruction syndrome (VOD/SOS), graft‐versus‐host disease (GvHD), transplant‐associated thrombotic microangiopathy (TA‐TMA) and idiopathic pneumonia syndrome (IPS)/diffuse alveolar haemorrhage (DAH), have a common origin in endothelial cell (EC) activation. Similarly, chimaeric antigen receptor T cell (CAR‐T) therapy, associated with cytokine release syndrome (CRS) and neurotoxicity, is also known to activate ECs.
Understanding these pathologies and their association with the endothelium is important in identifying treatment strategies and developing novel therapies. This review describes normal EC function, evaluates endothelial changes that occur after insults, and discusses the role of the endothelium in the pathogenesis and treatment of post‐HCT and CAR‐T–associated complications.
Role of the endothelium
The endothelium is composed of heterogeneous cells that line the inner wall of blood and lymph vasculature. 2 , 3 These cells have many functions (Fig 1), including regulating transport from the blood to underlying cells and tissues, haemostasis and coagulation, host defence and inflammation, and angiogenesis. 4 , 5 Normal EC function is crucial for homeostasis, and ECs help maintain blood fluidity and balance coagulation and fibrinolysis by synthesising tissue factor (TF) pathway inhibitors, which help regulate activation of coagulation factors VII and X and formation of thrombin. 4 , 6 Additionally, von Willebrand factor (vWF), essential for coagulation and platelet function, is primarily derived from the endothelium. ECs regulate blood flow, affect changes in vascular tone and surrounding smooth muscle and control vessel permeability through adhesion molecules and maintenance of the barrier function between the blood and underlying cells. Vasoactive mediators produced by ECs include nitric oxide, prostacyclin and endothelin. 4 , 6
Fig 1.
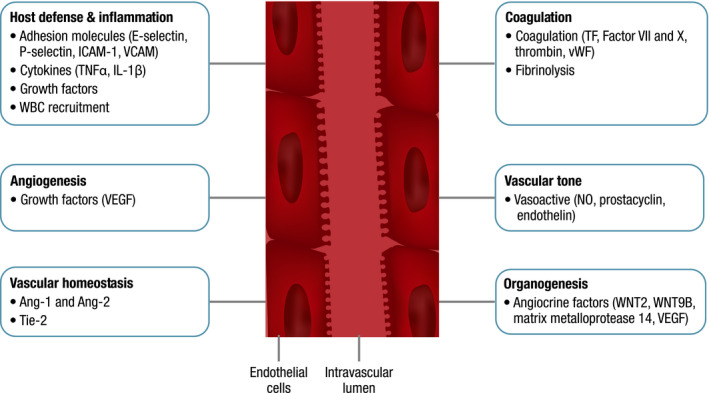
Normal function and physiology of endothelial cells. Ang‐1, angiopoetin‐1; Ang‐2, angiopoetin‐2; ICAM‐1, intercellular adhesion molecule 1; IL‐1β, interleukin 1 beta; NO, nitric oxide; TF, tissue factor; Tie‐2, receptor tyrosine kinase; TNFα, tumour necrosis factor alpha; VCAM, vascular adhesion molecule; VEGF, vascular endothelial growth factor; vWF, von Willebrand factor; WBC, white blood cell; WNT2 and WNT9B, protein coding genes.
Angiopoetin‐1 (Ang‐1) and Ang‐2, an agonist/antagonist peptide pair, are key regulators of vascular homeostasis through modulation of vascular growth and the endothelial barrier. These ligands bind the receptor tyrosine kinase, Tie‐2, found on ECs, 7 and the Ang‐1–Tie‐2 interaction promotes EC survival and vascular stability. This interaction may also suppress the inflammatory response via downregulation of surface adhesion molecules. 7
ECs produce regulators of inflammation and host defence mechanisms and play a key role in recruiting white blood cells and adhesion and chemoattractants to sites of inflammation. 8 Key adhesion molecules expressed on ECs include E‐selectin, P‐selectin, intracellular cell adhesion molecule 1 (ICAM‐1) and vascular cell adhesion molecule (VCAM). 4 , 9
In response to stress stimuli, resting ECs are activated and acquire new cellular functions. 5 , 6 , 7 , 8 There are two types of activation. Type 1 is rapid, independent of new gene expression and transient, with limited inflammation associated with histamine release. Type 2 is slower, dependent on new gene expression and provides a more sustained inflammatory stimulus through cytokines, including tumour necrosis factor alpha (TNFα) and interleukin (IL) 1β. These cytokines induce expression of the adhesion molecules ICAM‐1, VCAM‐1 and E‐selectin.
The adaptive functions of activated ECs can be beneficial. 2 Tissue‐specific ECs express unique angiocrine factors that support homeostasis and can generate different endothelial and non‐vascular cell types, including haematopoietic stem cells in vitro. 2 , 3 , 10 Angiocrine factors derived from ECs, including WNT2, WNT9B, matrix metalloprotease 14 and vascular endothelial growth factor (VEGF), have been shown to regenerate liver and lung tissue. 2 , 10 Tissue‐specific ECs contribute to organogenesis and homeostasis and, thus, may be essential for organ repair and regeneration.
EC activation may also be detrimental. 6 , 7 , 8 , 11 Loss of barrier function may lead to oedema and passage of soluble effectors (e.g. TNFα) that may damage the endothelium. Increased adhesion molecule expression (e.g. ICAM‐1, selectins, integrins) leads to increased vascular adhesion and leukocyte transmigration, promoting an inflammatory response leading to increased vascular leakage, further enhancing oedema. 7 Thrombogenic potential may also increase, leading to occlusion and vascular thrombotic events, such as ischaemia or stroke. 8 Red blood cell fragmentation, anaemia, thrombocytopenia and renal and neurological dysfunction may also occur as a result of increased shear effect. 12
Endothelial damage and dysfunction after HCT
Prolonged EC activation and damaging stimuli can lead to EC dysfunction, hypothesised to be critical in procoagulant, pro‐inflammatory and pro‐apoptotic responses that may result in early post‐HCT complications. 13 Either loss of or inappropriate EC function can lead to pathological changes. 14 Circulating markers of endothelial activation have been measured in a number of studies and highlight the widespread effects of endothelial activation and damage across functional areas of the endothelium (Table 1). The procoagulant state of damaged ECs is evident from increased concentrations of vWF and thrombomodulin. 11 Levels of TNFα and adhesion molecules are also generally elevated after HCT and in patients with HCT complications, but results may vary based on type of transplant (autologous versus allogeneic), conditioning, timing of measurement or assay methodology. 6 Another marker of activated ECs, Ang‐2, is released following stimulation by inflammatory cytokines and hypoxia. 30 Ang‐2 induces EC apoptosis and is significantly associated with transplant‐related complications and poor overall survival. 19 , 30
Table 1.
Changes in soluble markers of EC activation and damage in patients before and after HCT and with HCT‐associated complications. 6 , 13 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31

Levels of circulating ECs (CECs) also may reflect the extent of endothelial damage after HCT conditioning and transplantation. 32 , 33 , 34 Similar to markers of endothelial damage, CEC levels are higher following allogeneic transplantation, high‐intensity conditioning regimens and use of total body irradiation (TBI). Elevated CEC levels have been correlated with endothelial complications, including VOD/SOS, transplant‐associated thrombotic microangiopathy (TA‐TMA) and capillary leak syndrome. Elevated endothelial‐derived microparticles have also been identified as a marker for endothelial damage. Endothelial microparticles were found to be elevated in patients who developed graft‐versus‐host disease (GvHD) following allogeneic transplantation, but not in patients who received a transplant and did not develop GvHD. 35 In addition to microparticles, elevated vWF has been identified as a marker for activated or damaged ECs, 36 along with endocan and high mobility group box 1. 37
Factors leading to endothelial damage
In the HCT setting, EC damage occurs from sustained activation due to a number of factors. 7 , 32 Blood diseases, such as multiple myeloma, lymphoma or leukaemia, activate ECs independent of transplantation. 5 The toxicity of chemotherapy used in HCT conditioning regimens also activates ECs and stimulates inflammatory processes. 38 TBI induces EC apoptosis and upregulates ICAM‐1. 39 , 40 In evaluations of patients before and after HCT, the intensity of the conditioning regimen and type of transplant were associated with the degree of change in EC markers. 6 Alloreactivity likely plays a role, as HCT complications are more frequent following allogeneic versus autologous transplantation. 32 Chemotherapy agents that are particularly toxic include high‐dose regimens of cyclophosphamide and/or busulfan in combination with TBI; 41 , 42 however, even the non‐myeloablative immunosuppressant fludarabine has been shown to upregulate major histocompatibility complex (MHC) class 1 on ECs and enhance their lysis by cytotoxic T lymphocytes (CTLs). 6 , 11 , 27 , 38
Endothelial damage is exacerbated by cytokines that are released by injured tissues and the complex process of engraftment. 43 , 44 Additionally, bacterial endotoxins [lipopolysaccharide (LPS)] move through damaged intestinal mucosa and can increase EC activation. Other agents used during HCT, such as sirolimus and calcineurin inhibitors (CNIs; ciclosporin, tacrolimus), potentiate endothelial injury and accelerate senescence. 17 , 44 , 45 Higher peak tacrolimus concentrations after transplantation increase the risk of HCT complications related to endothelial damage. 45
Inflammation also drives alterations in capillary permeability, resulting in extravasation of fluid from the vascular space into tissues. 7 ECs are the primary barrier separating donor‐derived leukocytes and allogeneic target tissue. As such, ECs are a target for blood‐borne executors of the immune system (T cells and antibodies). 7 , 32
Endothelial damage following HCT may lead to complications that can evolve into multiorgan dysfunction (MOD) if left untreated (Table 2, Fig 2). 32 The development of a particular condition is based on several factors including the type of insult, phenotypic change in ECs (inflammatory, procoagulant or apoptotic) and the affected organ or system.
Table 2.
Summary of common EC dysfunction syndromes after HCT. 15 , 16 , 25 , 42 , 43 , 46 , 47 , 48 , 49 , 50 , 51 , 52 , 53 , 54 , 55 , 56 , 57 , 58 , 59 , 60
| Syndrome | Incidence | Risk factors | Organ complications | Mortality |
|---|---|---|---|---|
| VOD/SOS | 14% (up to 40%) |
|
|
Severe VOD/SOS with MOD: up to 50–70% |
| GvHD | 40–50% |
|
|
15–30% |
| TA‐TMA | 10–35% |
|
|
Rates range widely due to the lack of clear diagnostic guidelines |
| IPS/DAH |
IPS: 2–15% DAH: 5–12% |
|
|
60–80% |
AGvHD, acute graft‐versus‐host disease; CNS, central nervous system; EC, endothelial cell; GI, gastrointestinal; GvHD; graft‐versus‐host disease; HCT, haematopoietic cell transplantation; HLA, human leukocyte antigen, IPS/DAH, idiopathic pneumonia syndrome/diffuse alveolar haemorrhage; MOD, multiorgan dysfunction; TA‐TMA, transplant‐associated thrombotic microangiopathy; TBI, total body irradiation; VOD/SOS, veno‐occlusive disease/sinusoidal obstruction syndrome.
Fig 2.
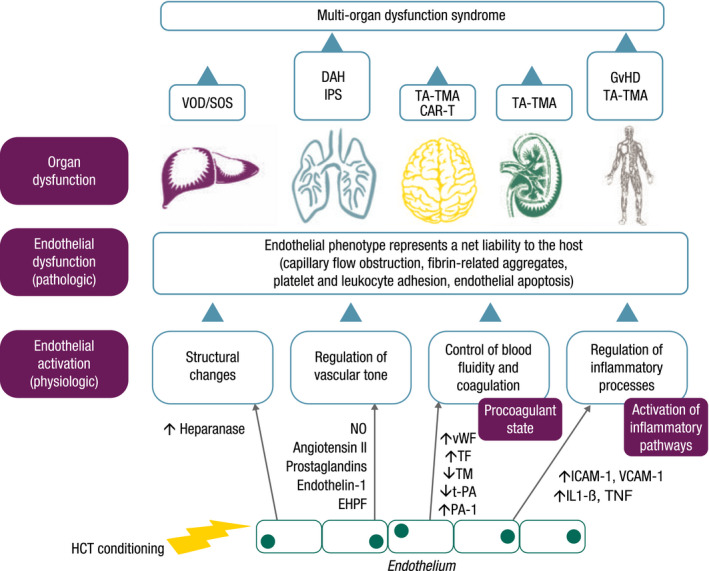
Progression of endothelial activation to endothelial dysfunction leading to different complications associated with HCT. Figure adapted and reprinted by permission from Springer Nature Customer Service Centre GmbH: Springer Nature Bone Marrow Transplantation. 2011;46(12):1495–1502. Carreras, E. & Diaz‐Ricart, M. The role of the endothelium in the short‐term complications of haematopoietic SCT. ©2011. CAR‐T, CAR‐T‐associated neurotoxicity; DAH, diffuse alveolar haemorrhage; EHPF, endothelial hyperpolarizing factor; GvHD, graft‐versus‐host disease; HCT, haematopoietic cell transplantation; ICAM‐1, intercellular adhesion molecule 1; IL1‐β, interleukin 1 beta; IPS, idiopathic pneumonia syndrome; NO, nitric oxide; PA‐1, plasminogen activator inhibitor 1; TA‐TMA, transplant‐associated thrombotic microangiopathy; TF, tissue factor; TM, thrombomodulin; TNF, tumour necrosis factor; t‐PA, tissue plasminogen activator; VCAM‐1, vascular adhesion molecule 1; VOD/SOS, veno‐occlusive disease/sinusoidal obstruction syndrome; vWF, von Willebrand factor.
Clinical conditions linked to endothelial dysfunction
Hepatic veno‐occlusive disease/sinusoidal obstruction syndrome
Hepatic veno‐occlusive disease/sinusoidal obstruction syndrome (VOD/SOS) develops as a result of primary injury to sinusoidal/central venous ECs followed by coagulative necrosis of hepatocytes. 58 The incidence of VOD/SOS following HCT varies, with some studies reporting incidences up to 40%. 47 , 61 Data from 1979 to 2007 provide a mean incidence rate of 14%, but this varies by age, primary disease, conditioning regimen, criteria used for diagnosis and type of transplant received. 47 The overall mortality rate during this time period was 84%, and death was more commonly associated with severe VOD/SOS and development of MOD. 47 In a large, prospective study conducted by the European Society for Blood and Marrow Transplantation (EBMT), autologous HCT was associated with a lower rate of VOD/SOS (3%) compared with allogeneic HCT (9%). 62
Risk factors for developing VOD/SOS include the conditioning regimen, type and number of transplants, older age, lower performance status and pre‐existing liver disease. 42 , 53 , 58 Traditionally, VOD/SOS is diagnosed using Baltimore or modified Seattle criteria, which evaluate hepatomegaly or right upper‐quadrant pain, ascites, weight gain and hyperbilirubinaemia. While both sets of criteria include the presence of hyperbilirubinaemia, only Baltimore criteria requires it for diagnosis. 53 , 63 Because these criteria monitor symptoms within 21 days of transplant, the EBMT developed separate criteria for adults and children that account for the potential late onset of VOD/SOS. 42 , 61
The pathophysiology of VOD/SOS is complex and multifactorial; however, EC injury caused by radiation or toxic metabolites of chemotherapy is typically the inciting event. 48 , 64 , 65 Common conditioning regimens include busulfan and cyclophosphamide, which are metabolised by cytochrome P450 and glutathione. Depletion of glutathione, in response to elevated TNF, has been shown to result in increased lipid hydroperoxides. 66 In turn, sinusoidal ECs and hepatocytes are damaged by these toxic agents and their metabolites. 65 , 67 Other sources of toxicity include monoclonal antibody–drug conjugates (e.g. inotuzumab ozogamicin). 55 , 68 CNIs and sirolimus may enhance endothelial injury, accelerate EC senescence and prevent healing by reducing levels of VEGF. 5 , 17 , 69 , 70
Damage to ECs leads to a hypercoagulable state, with synthesis of TF and release of vWF facilitating platelet aggregation. 64 Damaged ECs also express inflammatory mediators (e.g. ICAM‐1, TNFα), matrix metalloproteinases and VEGF, and release heparanase. 48 , 71 Heparanase triggers loss of cytoskeletal structure. 71 The sinusoidal endothelium further deteriorates as a result of inflammatory cytokines and increased metalloproteinase activity, leading to gaps in the endothelial lining. 32 , 48 , 55 , 72 These gaps allow passage of cellular and extracellular debris into the space of Disse and result in progressive detachment of the endothelial lining, producing sinusoidal narrowing and blockage by embolised sinusoidal ECs. 46 , 48 , 55 The obstruction of sinusoidal flow causes postsinusoidal hypertension, resulting in the clinical manifestations of VOD/SOS.
Graft‐versus‐host disease
GvHD occurs when immunocompetent T cells from donor tissues recognise and target tissues in the transplant recipient. 51 The incidence of acute GvHD following HCT ranges from 40% to 50% and is dependent on a number of factors, including the degree of human leukocyte antigen (HLA) mismatch, patient age, GvHD prophylaxis, gender disparity, multiparous female donors, the presence of pretransplant comorbidities, intensity of the conditioning regimen, use of TBI and source of donor graft. 54
In a recent database analysis of 729 patients, overall mortality from acute GvHD was 16%; however, mortality varies by GvHD grade, with rates as high as 92% for stage IV disease. 51 , 60 The onset of acute GvHD typically occurs before Day 100 post‐transplant but may develop later. 73 Changes in CEC levels may predict the onset of acute GvHD. 73 In a study of 90 allogeneic HCT recipients, CEC peaks were consistently observed at acute GvHD onset and returned to pretransplant values after treatment response, suggesting the potential use of CEC counts at the onset of GvHD symptoms to assist in diagnosis; however, these results need to be confirmed in larger studies.
GvHD arises from natural functions of the immune system. 43 , 51 A recipient's immune system is compromised by the HCT conditioning regimen, resulting in tissue damage that releases pro‐inflammatory cytokines and activates host antigen presenting cells (APCs). Most of the alloreactivity occurs when host APCs present host antigens to donor T cells. Moreover, innate lymphoid cells, primarily in the gut, also participate in this process. Proliferation and differentiation of T cells into Th1 and Th17 cells in turn activate cytotoxic T cells that mediate pro‐inflammatory cytokines and endothelial damage. Loss of microbial diversity in the gut may also lead to disruption of epithelial homeostasis. 43 LPS and other pathogens from damaged gastrointestinal mucosa provide another mechanism for APC activation. These activated APCs promote T‐cell proliferation and differentiation into CTLs that induce cellular and pro‐inflammatory factors including TNFα and IL‐1, IL‐2 and IL‐6 leading to target organ damage and development of GvHD clinical symptoms. 13 , 19 , 43 , 51 Although ECs are usually not recognised by T cells, the pro‐inflammatory environment following HCT and the location of ECs leads to continuous exposure to immune mediators. Under certain conditions, T cells have been shown to exhibit anti‐EC reactivity. Specifically, GvHD‐associated thymic damage resulted in T‐cell anti‐host activity in a murine model of allogeneic transplantation. 74 Endothelial damage may also contribute to steroid resistance and failure to recover from GvHD. 43 Circulating endothelial factors, such as angiopoietin before transplant and VEGF after transplant, may predict development of GvHD as well as response to therapy. 43 , 75 In the post‐HCT period, high Ang‐2/VEGF ratios indicate EC toxicity resulting in apoptosis. 19
Transplant‐associated thrombotic microangiopathy
TA‐TMA is a microangiopathic haemolytic anaemia without coagulopathy that may progress to hypertension, renal failure, central nervous system dysfunction and other organ system injury. 50 , 52 Manifestations of TA‐TMA can mimic GvHD, making recognition of TA‐TMA challenging. 43 , 50 , 52 The incidence of TA‐TMA ranges from 10% to 35% in the literature and usually presents between 20 and 100 days after transplantation. 52 , 56 , 76 HCT recipient data collected from 1990 to 2017 identified an incidence of TA‐TMA of 16%, with the syndrome presenting at a median of 86 days (range, 9–721) post‐transplant. 50 TA‐TMA is associated with increased morbidity and mortality. Reported mortality rates range widely due to the lack of clear diagnostic guidelines. Development of neurological dysfunction is associated with significantly lower overall survival in TA‐TMA patients. 50
Risk factors for TA‐TMA include patient age, donor type and degree of HLA mismatch, HCT conditioning regimen and use of TBI. 50 , 56 Withdrawal of CNIs/sirolimus may be an effective treatment or lead to reversal of disease. While sirolimus worsens the effect of CNIs, it has not been shown to damage ECs. A recent study found an association between refractory GvHD and TA‐TMA characterised by endothelial markers, protein suppressor of tumorigenicity‐2 and soluble thrombomodulin. 77 Infectious complications, particularly viral infections such as cytomegalovirus and human herpes virus 6, increase the risk of TA‐TMA by releasing cytokines that further damage the endothelium. 76 Recent studies have demonstrated increased plasma levels of complement proteins C3b, C5b‐9 and CH50, implicating the complement system in the pathogenesis of TA‐TMA. As a result, the recombinant, monoclonal humanised IgG4 antibody, eculizumab, which blocks the complement component of C5, has been identified as a potential TA‐TMA therapy. 78
In TA‐TMA, similar to other complications of HCT, vascular endothelial injury is multifactorial. 12 , 52 , 56 A procoagulant state in the endothelium results in direct endothelial damage that is pathologically intrinsic to TA‐TMA. 76 The process progresses with intimal swelling and fibrinoid necrosis of the microvascular wall that leads to intravascular platelet aggregation and narrowing of the vessel lumen. Erythrocytes are sheared by the platelet‐rich thrombi, resulting in microangiopathic haemolytic anaemia. Elevated levels of vWF have been observed along with other plasma markers of EC injury and inflammation, including thrombomodulin, plasminogen activator inhibitor‐1, ICAM‐1, VCAM‐1, E‐selectin, IL‐1, TNFα, interferon gamma (IFN‐γ) and IL‐8. 12 , 56 Unlike classical thrombocytopenic purpura, patients with TA‐TMA have activity of ADAMTS13 and have inflammatory, fibrin‐rich thrombi with evidence of complement dysfunction. 56 The generation of endothelial microparticles following EC activation and apoptosis may also contribute to TA‐TMA. 12
Idiopathic pneumonia syndrome/diffuse alveolar haemorrhage
Idiopathic pneumonia syndrome (IPS) is defined by widespread alveolar injury in the absence of an active lower respiratory tract infection or another aetiology for pulmonary dysfunction (e.g. cardiac dysfunction, acute renal failure or iatrogenic fluid overload). 15 , 25 The incidence of IPS in the first 120 days after allogeneic HCT ranges from 2% to 15%; lower rates of IPS are associated with use of reduced‐intensity conditioning regimens. 15 , 25 , 49 , 59 Historically, IPS has been reported to occur at a median of 6–7 weeks after HCT, but recent studies suggest onset can occur within three weeks of HCT. 25 , 49 Mortality rates are high, ranging from 60% to 80%. 49 , 59 IPS may complicate autologous HCT; however, in the autologous setting, the incidence of IPS is lower, onset is later and the prognosis is more favourable. 1 , 25
Full intensity conditioning, TBI, older age at transplant, presence of acute GvHD and an underlying diagnosis of acute leukaemia or myelodysplastic syndrome are all considered risk factors for developing IPS following allogeneic HCT. 25 , 57 diffuse alveolar haemorrhage (DAH) develops in a small subset of patients with IPS (5–12%) with a median time to onset of 12–19 days. Patients with mucopolysaccharidosis have a higher incidence of DAH after HCT.
IPS is associated with GvHD, suggesting an immunologic pathophysiology for IPS. Recent evidence suggests that inflammation can increase platelet adhesion to the endothelium and platelets can in turn amplify the inflammatory response. 79 Platelet transfusions have also been associated with a greater risk of IPS following myeloablative HCT. 80 Endothelial apoptosis occurs with T‐cell–mediated injury in GvHD; however, this has not been consistently observed in IPS. 25
The pathophysiology of IPS remains to be fully elucidated. TNFα appears to contribute to the development of IPS through a number of mechanisms, including increasing MHC expression, facilitating leukocyte migration and cell‐mediated cytotoxicity, as well as direct cytotoxicity via cytokines. 81 TNFα leads to pulmonary vascular EC apoptosis. Elevated levels of TNFα and neutrophils in the lungs of HCT recipients, without evidence of infection, suggest a role of host‐flora–derived LPS in IPS pathophysiology. 25 LPS levels are also elevated and stimulate cytokine release, leading to lung injury. Increased pulmonary chemokine expression results in recruitment of donor T lymphocytes and monocytes/macrophages. 82 , 83 , 84 , 85 Similar to other HCT complications, endothelial injury is clinically and experimentally observed in IPS by endothelial leak and vascular permeability, demonstrated as pulmonary oedema, enhanced total protein levels in bronchoalveolar fluid and increased wet‐to‐dry lung weight ratios. 15 Histological evidence of type II endothelial activation, with increased ICAM‐1 and/or VCAM‐1, has been demonstrated. 15 , 81 Elevated Ang‐2 levels result in pulmonary inflammation and increased permeability during acute exacerbation of IPS. 16
Endothelial damage following CAR‐T therapy
Haematopoietic cell transplantation is not the only therapy to induce EC damage and dysfunction. Genetically modified CAR‐T cells are used to target cancer‐specific antigens and are also associated with complications rooted in EC damage: CRS and CAR‐T–associated neurotoxicity. CRS is characterised by a systemic inflammatory response resulting from T‐cell activation associated with fever, fatigue, headache, rash, arthralgia and myalgia. 86 As CRS progresses, it can also lead to high fever and hypotension. CRS typically occurs within the first few weeks following CAR‐T infusion. 87
The pathophysiology of CRS is only partially understood. Binding of the CAR‐T receptor to its target leads to activation of immune cells, in particular T cells, as well as ECs. 87 This activation results in the release of many cytokines, highlighted by elevated levels of IL‐6, IL‐10 and IFN‐γ. 87 , 88 The increase in serum IL‐6 leads to many of the crucial symptoms of CRS including vascular leakage, activation of the complement pathway and intravascular coagulation. 87
Blinatumomab therapy is associated with CRS and neurotoxicity 89 which are both at least partially related to endothelial injury. Within the first 36–72 h after starting blinatumomab, serum cytokine levels of TNF, IFN‐γ, IL‐6, IL‐10 and Ang‐2 are increased. 90 , 91 While IL‐10 has mainly anti‐inflammatory effects on the endothelium, TNF, IL‐6, Ang‐2 and to some extent IFN‐γ promote inflammatory responses, such as upregulation of adhesion molecules and decrease in endothelial barrier function. These findings align with a recent report showing that neurotoxicity after blinatumomab is associated with increased levels of P‐selectin, VCAM‐1 and ICAM‐1 and increased T‐cell adhesiveness to the brain endothelium. 90
CAR‐T–associated neurotoxicity has recently been renamed ICANS and presents as toxic encephalopathy; it is often associated with severe CRS. 86 , 92 The condition is characterised by confusion, disorientation, agitation, aphasia, somnolence, tremors, seizures, motor weakness and cerebral oedema. 92 The incidence of CAR‐T–associated neurotoxicity varies based on patient population, disease type and the CAR‐T cell platform used, with incidence rates ranging from 43% to 87%. 88 , 93 , 94
The symptoms of CAR‐T–associated neurotoxicity appear within days of the infusion, 88 and the pathophysiology involves increased permeability of the blood–brain barrier, which is believed to be caused by endothelial activation and damage. 88 , 92 This results in the diffusion of cytokines into the brain. An elevated level of CAR‐T cells in the cerebrospinal fluid has also been reported in these patients, 88 , 92 as well as high levels of IL‐5 and IL‐6 in the serum of patients following CAR‐T therapy. A rapid increase of serum IL‐6 concentrations following CAR‐T infusion was found to be associated with neurotoxicity in these patients. Other cytokines elevated in CAR‐T–related neurotoxicity included TNFα, IFN‐γ, vWF and Ang‐2, all markers of endothelial damage and dysfunction. 92 Additional potential biomarkers of CAR‐T–related neurotoxicity include platelet count, disease burden and mean corpuscular haemoglobin concentration. 88
Treatment strategies targeting EC dysfunction
Due to the role of the endothelium in the pathophysiology of post‐HCT complications, ECs represent a promising target for prophylaxis of and therapeutic intervention for HCT complications. Several therapies have either been approved or are being investigated.
Defibrotide is approved to treat adult and paediatric patients with hepatic VOD/SOS with renal or pulmonary dysfunction post‐HCT in the United States, 95 and to treat patients aged >1 month with severe hepatic VOD/SOS post‐HCT in the European Union. 96 While the mechanism of action of defibrotide is not fully understood, it has been shown to reduce EC activation and restore the thrombotic/fibrinolytic balance. 97 In an historically controlled, phase 3 trial, Day 100 survival was 38% in patients with VOD/SOS post‐HCT and multiorgan failure treated with defibrotide versus 25% in historical controls. 98 An expanded access study investigating defibrotide in VOD/SOS patients with and without MOD demonstrated favourable Day 100 survival post‐HCT (59%). 99 A phase 3 study (NCT02851407) is currently evaluating defibrotide versus best supportive care for VOD/SOS prophylaxis.
Treatment of acute GvHD is based on symptoms and the disease stage. Initial therapy ranges from topical corticosteroids to systemic corticosteroid treatment; 54 however, these are simply anti‐inflammatory options, not endothelium‐specific. A phase 2 study is currently evaluating defibrotide, which is known to reduce EC activation, for the prevention of acute GvHD (NCT03339297). Alpha‐1 antitrypsin (AAT), a serine protease inhibitor, is also being considered as a potential treatment option. AAT has been shown to induce responses in GvHD target organs with minimal toxicity. 100 A randomised, double‐blind, placebo‐controlled, multicentre, phase 3 study (NCT04167514) is currently evaluating AAT combined with corticosteroids versus corticosteroids alone for the treatment of patients with high‐risk acute GvHD after allogeneic HCT. Additionally, a phase 2/3 study (NCT03805789) of AAT for the prevention of GvHD in patients receiving HCT is ongoing.
Following TA‐TMA diagnosis, patients often discontinue use of CNIs and sirolimus and begin supportive care. 101 Changing immunosuppressive therapy may increase the risk of precipitating GvHD and further complicating TA‐TMA management. Patients can also receive plasma exchange; however, this strategy does not address the pathological process, has limited efficacy and is not considered standard of care. 101 While some patients receive the CD20 antibody rituximab, more recently the complement protein C5 antibody eculizumab has been utilised in TA‐TMA. 102 In high‐risk patients with proteinuria, activated terminal complement and multiorgan impairment, eculizumab significantly improved survival at one year. 103 Several approaches for TA‐TMA prophylaxis that target endothelial health and aim to repair endothelial injury are under investigation, including vitamin D, eicosapentaenoic acid, allopurinol, statins and N‐acetyl‐L‐cysteine. 104
Corticosteroids have anti‐inflammatory effects that may mitigate endothelial damage and are commonly used to manage the pro‐inflammatory state associated with endothelial‐related HCT complications, such as IPS; however, no treatment advantage has been documented. 1 Preclinical data demonstrate that TNFα contributes to EC dysfunction both directly (e.g. through apoptosis of EC cells) and indirectly (e.g. through effects on inflammatory chemokine expression), providing a rationale for the use of anti‐TNFα agents for IPS; however, studies investigating etanercept have produced mixed results. 105 , 106 Veno‐venous haemofiltration may also be considered for IPS; however, there are no treatments for DAH outside of steroids, although the addition of aminocaproic acid, an antifibrinolytic agent, has been reported to help improve DAH mortality. 107
Similar to many post‐HCT complications, corticosteroids are the most common treatment for patients with CRS along with interruption or discontinuation of CAR‐T therapy, depending on severity. Treatment strategies for CAR‐T–associated neurotoxicity include supportive care and corticosteroid use. 88 Evidence suggests that cytokine‐mediated endothelial activation contributes to CAR‐T–associated neurotoxicity. Management with an IL‐6 receptor antagonist, tocilizumab, has been successful in many patients and may be useful in the early stages of CAR‐T–associated neurotoxicity when it is associated with CRS. 86 , 88 , 92 A phase 2 study (NCT03954106) is currently evaluating defibrotide for the prevention of CAR‐T–associated neurotoxicity.
Future directions
The endothelium plays multifunctional roles in homeostasis by regulating coagulation, blood flow, vascular tone, vessel permeability and inflammatory responses. To facilitate these functions, ECs produce a multitude of factors both at rest and in response to cellular damage. Patients undergoing HCT are exposed to a variety of stressors that can lead to EC activation and direct endothelial damage. These insults stem from conditioning regimens, the engraftment process, allogeneic reactions and infection. Sustained or intense EC activation leads to pathological changes that manifest as different complications based on the phenotypic change (pro‐inflammatory, procoagulant or pro‐apoptotic) and the affected organ. Although each HCT‐related complication has unique characteristics, there is substantial overlap in their clinical presentation as a result of shared endothelial pathophysiology.
Following HCT, injury to the endothelium induces coagulation factors (vWF and thrombomodulin), pro‐inflammatory cytokines (TNFα, interleukins, IFN‐γ), and adhesion molecules (E‐selectin, ICAM‐1, VCAM‐1); however, the relationship between concentrations of these markers and the clinical course of HCT complications is not always consistent, mostly due to different conditioning regimens, which largely influence the degree of endothelial damage. Levels of soluble markers may also differ based on the assay, time of measurement and specific HCT complication.
Monitoring CECs and endothelial‐derived microparticles may offer valuable information for treating patients receiving HCT; however, routine use of soluble biomarkers for the diagnosis and monitoring of EC activation and injury‐related disease in the clinic is limited. C5b‐9, a soluble membrane attacking complex (MAC), is currently established to identify and monitor TA‐TMA. C5b‐9 levels serve as a direct marker for response to off‐label treatment with eculizumab, a long‐acting humanized monoclonal antibody against complement 5 (C5), 108 , 109 and potentially for another C5‐directed antibody, ravulizumab. Of note, while the 50% haemolytic complement activity (CH50) can also be utilized as a biomarker for TA‐TMA in patients receiving eculizumab, this biomarker has significant limitations and is not valid for assessing complete terminal complement blockade in patients receiving ravulizumab, 110 underscoring the need for thorough understanding and careful application of biomarkers in the clinic. PAI‐1 may have the potential to be a marker for response to treatment in VOD/SOS, based on results from a phase 2 dose‐finding study showing that patients who responded to treatment (achieved a complete response [CR]) had decreased levels of serum PAI‐1 at Days 7 and 14 post‐HCT (although not statistically significant) while patients who did not achieve a CR showed no change in PAI‐1 levels. 111 However, this finding needs to be validated in a larger patient cohort and monitoring PAI‐1 is not currently used in clinical practice. Other markers, such as vWF, thrombomodulin, ICAM‐1, TNFα and Ang‐2, have been associated with GvHD; 13 , 112 however, they are not yet routinely being used in the clinic to guide preventive or therapeutic interventions (e.g. the administration of recombinant thrombomodulin).
The endothelium represents a promising target for the prophylaxis and treatment of pathologies arising from HCT complications. As treatment strategies continue to evolve, knowledge of the key mediators of HCT complications may assist with diagnosis and treatment decisions. Increased understanding and evaluation of the mechanisms of endothelial dysfunction are also likely to suggest a wider array of therapeutic targets for future therapeutic interventions for HCT‐related complications.
Author contributions
GCH and NC contributed to the writing and editing of the manuscript and gave final approval to submit.
Conflicts of interest
GCH has stocks/ownership interests in Sangamo Bioscience, Axim Biotechnologies, Juno Therapeutics, Kite Pharma, Novartis, Insys Therapeutics, Abbvie, GW Pharmaceuticals, Cardinal Health, Immunomedics, Endocyte, Clovis Oncology, Cellectis, Aetna, CVS Health, Celgene, Bluebird Bio, Bristol‐Myers Squibb/Medarex, crispr Therapeutics, IDEXX Laboratories, Johnson & Johnson, Pfizer, Procter & Gamble, Vertex, Bayer, Scotts‐Miracle, Charlottes Webb CWBHF, Almmune Therapeutics Inc AIMT, Medical PPTYS TR Inc. MPW, Caretrust Reit Inc CTRE, ANGI Homeservices Inc ANGI, and Bayer AG BAYRY; has served in advisory/consulting roles for Pfizer, Kite Pharma, Incyte, and Jazz Pharmaceuticals; has received research funding from Takeda, Astellas, Incyte, Jazz Pharmaceuticals, and Pharmacyclics; and has received travel, accommodations, and/or expense reimbursement from Kite Pharma, Incyte, Pfizer, Falk Foundation, Jazz Pharmaceuticals, and Astellas Pharma. NC has nothing to disclose.
Acknowledgements
Medical writing and editorial support were provided by Michelle McDermott, PharmD, and Michael Whitley, PhD, of SciFluent Communications, and were financially supported by Jazz Pharmaceuticals.
The copyright line for this article was changed on 29 April 2020 after original online publication
References
- 1. Pagliuca S, Michonneau D, Sicre de Fontbrune F, Sutra Del Galy A, Xhaard A, Robin M, et al. Allogeneic reactivity‐mediated endothelial cell complications after HSCT: a plea for consensual definitions. Blood Adv. 2019;3:2424–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gomez‐Salinero JM, Rafii S. Endothelial cell adaptation in regeneration. Science. 2018;362:1116–7. [DOI] [PubMed] [Google Scholar]
- 3. Nolan DJ, Ginsberg M, Israely E, Palikuqi B, Poulos MG, James D, et al. Molecular signatures of tissue‐specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell. 2013;26:204–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galley HF, Webster NR. Physiology of the endothelium. Br J Anaesth. 2004;93:105–13. [DOI] [PubMed] [Google Scholar]
- 5. Vion AC, Rautou PE, Durand F, Boulanger CM, Valla DC. Interplay of inflammation and endothelial dysfunction in bone marrow transplantation: focus on hepatic veno‐occlusive disease. Semin Thromb Hemost. 2015;41:629–43. [DOI] [PubMed] [Google Scholar]
- 6. Palomo M, Diaz‐Ricart M, Carbo C, Rovira M, Fernandez‐Aviles F, Martine C, et al. Endothelial dysfunction after hematopoietic stem cell transplantation: role of the conditioning regimen and the type of transplantation. Biol Blood Marrow Transplant. 2010;16:985–93. [DOI] [PubMed] [Google Scholar]
- 7. Cooke KR, Jannin A, Ho V. The contribution of endothelial activation and injury to end‐organ toxicity following allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2008;14:23–32. [DOI] [PubMed] [Google Scholar]
- 8. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–15. [DOI] [PubMed] [Google Scholar]
- 9. Langer HF, Chavakis T. Leukocyte‐endothelial interactions in inflammation. J Cell Mol Med. 2009;13:1211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lebaschi A, Nakagawa Y, Wada S, Cong GT, Rodeo SA. Tissue‐specific endothelial cells: a promising approach for augmentation of soft tissue repair in orthopedics. Ann N Y Acad Sci. 2017;1410:44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blix ES, Husebekk A. Raiders of the lost mark – endothelial cells and their role in transplantation for hematologic malignancies. Leuk Lymphoma. 2016;57:2752–62. [DOI] [PubMed] [Google Scholar]
- 12. Batts ED, Lazarus HM. Diagnosis and treatment of transplantation‐associated thrombotic microangiopathy: real progress or are we still waiting? Bone Marrow Transplant. 2007;40:709–19. [DOI] [PubMed] [Google Scholar]
- 13. Mir E, Palomo M, Rovira M, Pereira A, Escolar G, Penack O, et al. Endothelial damage is aggravated in acute GvHD and could predict its development. Bone Marrow Transplant. 2017;52:1317–25. [DOI] [PubMed] [Google Scholar]
- 14. Pierce RW, Giuliano JS Jr, Pober JS. Endothelial cell function and dysfunction in critically Ill children. Pediatrics. 2017;140(1):e20170355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Altmann T, Slack J, Slatter MA, O'Brien C, Cant A, Thomas M, et al. Endothelial cell damage in idiopathic pneumonia syndrome. Bone Marrow Transplant. 2018;53:515–8. [DOI] [PubMed] [Google Scholar]
- 16. Ando M, Miyazaki E, Abe T, Ehara C, Goto A, Masuda T, et al. Angiopoietin‐2 expression in patients with an acute exacerbation of idiopathic interstitial pneumonias. Respir Med. 2016;117:27–32. [DOI] [PubMed] [Google Scholar]
- 17. Cutler C, Kim HT, Ayanian S, Bradwin G, Revta C, Aldridge J, et al. Prediction of veno‐occlusive disease using biomarkers of endothelial injury. Biol Blood Marrow Transplant. 2010;16:1180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lesterhuis WJ, Rennings AJ, Leenders WP, Nooteboom A, Punt CJ, Sweep FC, et al. Vascular endothelial growth factor in systemic capillary leak syndrome. Am J Med. 2009;122:e5–7. [DOI] [PubMed] [Google Scholar]
- 19. Luft T, Dietrich S, Falk C, Conzelmann M, Hess M, Benner A, et al. Steroid‐refractory GVHD: T‐cell attack within a vulnerable endothelial system. Blood. 2011;118:1685–92. [DOI] [PubMed] [Google Scholar]
- 20. Lunn RA, Sumar N, Bansal AS, Treleaven J. Cytokine profiles in stem cell transplantation: possible use as a predictor of graft‐versus‐host disease. Hematology. 2005;10:107–14. [DOI] [PubMed] [Google Scholar]
- 21. Matsuda Y, Hara J, Osugi Y, Tokimasa S, Fujisaki H, Takai K, et al. Serum levels of soluble adhesion molecules in stem cell transplantation‐related complications. Bone Marrow Transplant. 2001;27:977–82. [DOI] [PubMed] [Google Scholar]
- 22. Moiseev IS, Lapin SV, Surkova EA, Lerner MY, Vavilov VN, Afanasyev BV. Level of vascular endothelial growth factor predicts both relapse and nonrelapse mortality after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2013;19:1677–82. [DOI] [PubMed] [Google Scholar]
- 23. Nomura S, Ishii K, Inami N, Kimura Y, Uoshima N, Ishida H, et al. Evaluation of angiopoietins and cell‐derived microparticles after stem cell transplantation. Biol Blood Marrow Transplant. 2008;14:766–74. [DOI] [PubMed] [Google Scholar]
- 24. Nurnberger W, Michelmann I, Burdach S, Gobel U. Endothelial dysfunction after bone marrow transplantation: increase of soluble thrombomodulin and PAI‐1 in patients with multiple transplant‐related complications. Ann Hematol. 1998;76:61–5. [DOI] [PubMed] [Google Scholar]
- 25. Panoskaltsis‐Mortari A, Griese M, Madtes DK, Belperio JA, Haddad IY, Folz RJ, et al. An official American Thoracic Society research statement: noninfectious lung injury after hematopoietic stem cell transplantation: idiopathic pneumonia syndrome. Am J Respir Crit Care Med. 2011;183:1262–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Richard S, Seigneur M, Blann A, Adams R, Renard M, Puntous M, et al. Vascular endothelial lesion in patients undergoing bone marrow transplantation. Bone Marrow Transplant. 1996;18:955–9. [PubMed] [Google Scholar]
- 27. Salat C, Holler E, Kolb HJ, Pihusch R, Reinhardt B, Hiller E. Endothelial cell markers in bone marrow transplant recipients with and without acute graft‐versus‐host disease. Bone Marrow Transplant. 1997;19:909–14. [DOI] [PubMed] [Google Scholar]
- 28. Salat C, Holler E, Kolb HJ, Pihusch R, Reinhardt B, Penovici M, et al. The relevance of plasminogen activator inhibitor 1 (PAI‐1) as a marker for the diagnosis of hepatic veno‐occlusive disease in patients after bone marrow transplantation. Leuk Lymphoma. 1999;33:25–32. [DOI] [PubMed] [Google Scholar]
- 29. Spitzer TR. Engraftment syndrome: double‐edged sword of hematopoietic cell transplants. Bone Marrow Transplant. 2015;50:469–75. [DOI] [PubMed] [Google Scholar]
- 30. Ueda N, Chihara D, Kohno A, Tatekawa S, Ozeki K, Watamoto K, et al. Predictive value of circulating angiopoietin‐2 for endothelial damage‐related complications in allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2014;20:1335–40. [DOI] [PubMed] [Google Scholar]
- 31. Zeigler ZR, Rosenfeld CS, Andrews DF 3rd, Nemunaitis J, Raymond JM, Shadduck RK, et al. Plasma von Willebrand Factor Antigen (vWF:AG) and thrombomodulin (TM) levels in Adult Thrombotic Thrombocytopenic Purpura/Hemolytic Uremic Syndromes (TTP/HUS) and bone marrow transplant‐associated thrombotic microangiopathy (BMT‐TM). Am J Hematol. 1996;53:213–20. [DOI] [PubMed] [Google Scholar]
- 32. Carreras E, Diaz‐Beya M, Rosinol L, Martinez C, Fernandez‐Aviles F, Rovira M. The incidence of veno‐occlusive disease following allogeneic hematopoietic stem cell transplantation has diminished and the outcome improved over the last decade. Biol Blood Marrow Transplant. 2011;17:1713–20. [DOI] [PubMed] [Google Scholar]
- 33. Ruggeri A, Paviglianiti A, Volt F, Kenzey C, Rafii H, Rocha V, et al. Endothelial and circulating progenitor cells in hematological diseases and allogeneic hematopoietic stem cell transplantation. Curr Med Chem. 2018;25:4535–4 4. [DOI] [PubMed] [Google Scholar]
- 34. Woywodt A, Scheer J, Hambach L, Buchholz S, Ganser A, Haller H, et al. Circulating endothelial cells as a marker of endothelial damage in allogeneic hematopoietic stem cell transplantation. Blood. 2004;103:3603–5. [DOI] [PubMed] [Google Scholar]
- 35. Pihusch V, Rank A, Steber R, Pihusch M, Pihusch R, Toth B, et al. Endothelial cell‐derived microparticles in allogeneic hematopoietic stem cell recipients. Transplantation. 2006;81:1405–9. [DOI] [PubMed] [Google Scholar]
- 36. Buser TA, Martinez M, Drexler B, Tschan‐Plessl A, Heim D, Passweg J, et al. Biological markers of hemostasis and endothelial activation in patients with a haematological malignancy with or without stem cell transplants. Eur J Haematol. 2019;103:472–7. [DOI] [PubMed] [Google Scholar]
- 37. Walborn A, Rondina M, Mosier M, Fareed J, Hoppensteadt D. Endothelial dysfunction is associated with mortality and severity of coagulopathy in patients with sepsis and disseminated intravascular coagulation. Clin Appl Thromb Hemost. 2019;25:1076029619852163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eissner G, Multhoff G, Gerbitz A, Kirchner S, Bauer S, Haffner S, et al. Fludarabine induces apoptosis, activation, and allogenicity in human endothelial and epithelial cells: protective effect of defibrotide. Blood. 2002;100:334–40. [DOI] [PubMed] [Google Scholar]
- 39. Eissner G, Lindner H, Behrends U, Kolch W, Hieke A, Klauke I, et al. Influence of bacterial endotoxin on radiation‐induced activation of human endothelial cells in vitro and in vivo: protective role of IL‐10. Transplantation. 1996;62:819–27. [DOI] [PubMed] [Google Scholar]
- 40. Langley RE, Bump EA, Quartuccio SG, Medeiros D, Braunhut SJ. Radiation‐induced apoptosis in microvascular endothelial cells. Br J Cancer. 1997;75:666–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gyurkocza B, Sandmaier BM. Conditioning regimens for hematopoietic cell transplantation: one size does not fit all. Blood. 2014;124:344–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mohty M, Malard F, Abecassis M, Aerts E, Alaskar AS, Aljurf M, et al. Revised diagnosis and severity criteria for sinusoidal obstruction syndrome/veno‐occlusive disease in adult patients: a new classification from the European Society for Blood and Marrow Transplantation. Bone Marrow Transplant. 2016;51:906–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ghimire S, Weber D, Mavin E, Wang XN, Dickinson AM, Holler E. Pathophysiology of GvHD and other HSCT‐related major complications. Front Immunol. 2017;8:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Palomo M, Diaz‐Ricart M, Carbo C, Rovira M, Fernandez‐Aviles F, Escolar G, et al. The release of soluble factors contributing to endothelial activation and damage after hematopoietic stem cell transplantation is not limited to the allogeneic setting and involves several pathogenic mechanisms. Biol Blood Marrow Transplant. 2009;15:537–46. [DOI] [PubMed] [Google Scholar]
- 45. Morishita T, Okabe M, Kawaguchi Y, Lee Y, Ohbiki M, Osaki M, et al. Higher peak tacrolimus concentrations after allogeneic hematopoietic stem cell transplantation increase the risk of endothelial cell damage complications. Biol Blood Marrow Transplant. 2018;24:2509–16. [DOI] [PubMed] [Google Scholar]
- 46. Carreras E, Diaz‐Ricart M. The role of the endothelium in the short‐term complications of hematopoietic SCT. Bone Marrow Transplant. 2011;46:1495–502. [DOI] [PubMed] [Google Scholar]
- 47. Coppell JA, Richardson PG, Soiffer R, Martin PL, Kernan NA, Chen A, et al. Hepatic veno‐occlusive disease following stem cell transplantation: incidence, clinical course, and outcome. Biol Blood Marrow Transplant. 2010;16:157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fan CQ, Crawford JM. Sinusoidal obstruction syndrome (hepatic veno‐occlusive disease). J Clin Exp Hepatol. 2014;4:332–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fukuda T, Hackman RC, Guthrie KA, Sandmaier BM, Boeckh M, Maris MB, et al. Risks and outcomes of idiopathic pneumonia syndrome after nonmyeloablative and conventional conditioning regimens for allogeneic hematopoietic stem cell transplantation. Blood. 2003;102:2777–85. [DOI] [PubMed] [Google Scholar]
- 50. Gavriilaki E, Sakellari I, Batsis I, Mallouri D, Bousiou Z, Vardi A, et al. Transplant‐associated thrombotic microangiopathy: incidence, prognostic factors, morbidity, and mortality in allogeneic hematopoietic cell transplantation. Clin Transplant. 2018;32:e13371. [DOI] [PubMed] [Google Scholar]
- 51. Jamil MO, Mineishi S. State‐of‐the‐art acute and chronic GVHD treatment. Int J Hematol. 2015;101:452–66. [DOI] [PubMed] [Google Scholar]
- 52. Jodele S, Laskin BL, Dandoy CE, Myers KC, El‐Bietar J, Davies SM, et al. A new paradigm: diagnosis and management of HSCT‐associated thrombotic microangiopathy as multi‐system endothelial injury. Blood Rev. 2015;29:191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McDonald GB, Hinds MS, Fisher LD, Schoch HG, Wolford JL, Banaji M, et al. Veno‐occlusive disease of the liver and multiorgan failure after bone marrow transplantation: a cohort study of 355 patients. Ann Intern Med. 1993;118:255–67. [DOI] [PubMed] [Google Scholar]
- 54. Nassereddine S, Rafei H, Elbahesh E, Tabbara I. Acute graft versus host disease: a comprehensive review. Anticancer Res. 2017;37:1547–55. [DOI] [PubMed] [Google Scholar]
- 55. Richardson PG, Carreras E, Iacobelli M, Nejadnik B. The use of defibrotide in blood and marrow transplantation. Blood Adv. 2018;2:1495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Seaby EG, Gilbert RD. Thrombotic microangiopathy following haematopoietic stem cell transplant. Pediatr Nephrol. 2018;33:1489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shankar G, Scott Bryson J, Darrell Jennings C, Kaplan AM, Cohen DA. Idiopathic pneumonia syndrome after allogeneic bone marrow transplantation in mice. Role of pretransplant radiation conditioning. American Journal of Respiratory Cell and Molecular Biology. 1999;20:1116–24. [DOI] [PubMed] [Google Scholar]
- 58. Valla DC, Cazals‐Hatem D. Sinusoidal obstruction syndrome. Clin Res Hepatol Gastroenterol. 2016;40:378–85. [DOI] [PubMed] [Google Scholar]
- 59. Yadav H, Nolan ME, Bohman JK, Cartin‐Ceba R, Peters SG, Hogan WJ, et al. Epidemiology of acute respiratory distress syndrome following hematopoietic stem cell transplantation. Crit Care Med. 2016;44:1082–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yu J, Parasuraman S, Shah A, Weisdorf D. Mortality, length of stay and costs associated with acute graft‐versus‐host disease during hospitalization for allogeneic hematopoietic stem cell transplantation. Curr Med Res Opin. 2019;35:983–8. [DOI] [PubMed] [Google Scholar]
- 61. Corbacioglu S, Carreras E, Ansari M, Balduzzi A, Cesaro S, Dalle JH, et al. Diagnosis and severity criteria for sinusoidal obstruction syndrome/veno‐occlusive disease in pediatric patients: a new classification from the European society for blood and marrow transplantation. Bone Marrow Transplant. 2018;53:138–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Carreras E, Bertz H, Arcese W, Vernant JP, Tomas JF, Hagglund H, et al. Incidence and outcome of hepatic veno‐occlusive disease after blood or marrow transplantation: a prospective cohort study of the European Group for Blood and Marrow Transplantation. European Group for Blood and Marrow Transplantation Chronic Leukemia Working Party. Blood. 1998;92:3599–604. [PubMed] [Google Scholar]
- 63. Jones RJ, Lee KS, Beschorner WE, Vogel VG, Grochow LB, Braine HG, et al. Venoocclusive disease of the liver following bone marrow transplantation. Transplantation. 1987;44:778–83. [DOI] [PubMed] [Google Scholar]
- 64. Bearman SI. The syndrome of hepatic veno‐occlusive disease after marrow transplantation. Blood. 1995;85:3005–20. [PubMed] [Google Scholar]
- 65. Dalle JH, Giralt SA. Hepatic veno‐occlusive disease after hematopoietic stem cell transplantation: risk factors and stratification, prophylaxis, and treatment. Biol Blood Marrow Transplant. 2016;22:400–9. [DOI] [PubMed] [Google Scholar]
- 66. Toborek M, Barger SW, Mattson MP, McClain CJ, Hennig B. Role of glutathione redox cycle in TNF‐alpha‐mediated endothelial cell dysfunction. Atherosclerosis. 1995;117:179–88. [DOI] [PubMed] [Google Scholar]
- 67. Srivastava A, Poonkuzhali B, Shaji RV, George B, Mathews V, Chandy M, et al. Glutathione S‐transferase M1 polymorphism: a risk factor for hepatic venoocclusive disease in bone marrow transplantation. Blood. 2004;104:1574–7. [DOI] [PubMed] [Google Scholar]
- 68. Akil A, Zhang Q, Mumaw CL, Raiker N, Yu J, Velez de Mendizabal N, et al. Biomarkers for diagnosis and prognosis of sinusoidal obstruction syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2015;21:1739–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cutler C, Stevenson K, Kim HT, Richardson P, Ho VT, Linden E, et al. Sirolimus is associated with veno‐occlusive disease of the liver after myeloablative allogeneic stem cell transplantation. Blood. 2008;112:4425–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Roeker LE, Kim HT, Glotzbecker B, Nageshwar P, Nikiforow S, Koreth J, et al. Early clinical predictors of hepatic veno‐occlusive disease/sinusoidal obstruction syndrome after myeloablative stem cell transplantation. Biol Blood Marrow Transplant. 2019;25:137–44. [DOI] [PubMed] [Google Scholar]
- 71. Richardson PG, Corbacioglu S, Ho VT, Kernan NA, Lehmann L, Maguire C, et al. Drug safety evaluation of defibrotide. Expert Opin Drug Saf. 2013;12:123–36. [DOI] [PubMed] [Google Scholar]
- 72. Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247–50. [DOI] [PubMed] [Google Scholar]
- 73. Almici C, Skert C, Bruno B, Bianchetti A, Verardi R, Di Palma A, et al. Circulating endothelial cell count: a reliable marker of endothelial damage in patients undergoing hematopoietic stem cell transplantation. Bone Marrow Transplant. 2017;52:1637–42. [DOI] [PubMed] [Google Scholar]
- 74. van den Brink MR, Moore E, Ferrara JL, Burakoff SJ. Graft‐versus‐host‐disease‐associated thymic damage results in the appearance of T cell clones with anti‐host reactivity. Transplantation. 2000;69:446–9. [DOI] [PubMed] [Google Scholar]
- 75. Holtan SG, Verneris MR, Schultz KR, Newell LF, Meyers G, He F, et al. Circulating angiogenic factors associated with response and survival in patients with acute graft‐versus‐host disease: results from Blood and Marrow Transplant Clinical Trials Network 0302 and 0802. Biol Blood Marrow Transplant. 2015;21:1029–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Khosla J, Yeh AC, Spitzer TR, Dey BR. Hematopoietic stem cell transplant‐associated thrombotic microangiopathy: current paradigm and novel therapies. Bone Marrow Transplant. 2018;53:129–37. [DOI] [PubMed] [Google Scholar]
- 77. Zeisbrich M, Becker N, Benner A, Radujkovic A, Schmitt K, Beimler J, et al. Transplant‐associated thrombotic microangiopathy is an endothelial complication associated with refractoriness of acute GvHD. Bone Marrow Transplant. 2017;52:1399–405. [DOI] [PubMed] [Google Scholar]
- 78. Qi J, Wang J, Chen J, Su J, Tang Y, Wu X, et al. Plasma levels of complement activation fragments C3b and sC5b‐9 significantly increased in patients with thrombotic microangiopathy after allogeneic stem cell transplantation. Ann Hematol. 2017;96:1849–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Middleton EA, Weyrich AS, Zimmerman GA. Platelets in pulmonary immune responses and inflammatory lung diseases. Physiol Rev. 2016;96:1211–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Vande Vusse LK, Madtes DK, Guthrie KA, Gernsheimer TB, Curtis JR, Watkins TR. The association between red blood cell and platelet transfusion and subsequently developing idiopathic pneumonia syndrome after hematopoietic stem cell transplantation. Transfusion. 2014;54:1071–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gerbitz A, Nickoloff BJ, Olkiewicz K, Willmarth NE, Hildebrandt G, Liu C, et al. A role for tumor necrosis factor‐alpha‐mediated endothelial apoptosis in the development of experimental idiopathic pneumonia syndrome. Transplantation. 2004;78:494–502. [DOI] [PubMed] [Google Scholar]
- 82. Hildebrandt GC, Corrion LA, Olkiewicz KM, Lu B, Lowler K, Duffner UA, et al. Blockade of CXCR3 receptor:ligand interactions reduces leukocyte recruitment to the lung and the severity of experimental idiopathic pneumonia syndrome. J Immunol. 2004;173:2050–9. [DOI] [PubMed] [Google Scholar]
- 83. Hildebrandt GC, Duffner UA, Olkiewicz KM, Corrion LA, Willmarth NE, Williams DL, et al. A critical role for CCR2/MCP‐1 interactions in the development of idiopathic pneumonia syndrome after allogeneic bone marrow transplantation. Blood. 2004;103:2417–26. [DOI] [PubMed] [Google Scholar]
- 84. Hildebrandt GC, Olkiewicz KM, Choi S, Corrion LA, Clouthier SG, Liu C, et al. Donor T‐cell production of RANTES significantly contributes to the development of idiopathic pneumonia syndrome after allogeneic stem cell transplantation. Blood. 2005;105:2249–57. [DOI] [PubMed] [Google Scholar]
- 85. Hildebrandt GC, Olkiewicz KM, Corrion LA, Chang Y, Clouthier SG, Liu C, et al. Donor‐derived TNF‐α regulates pulmonary chemokine expression and the development of idiopathic pneumonia syndrome after allogeneic bone marrow transplantation. Blood. 2004;104:586–93. [DOI] [PubMed] [Google Scholar]
- 86. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric antigen receptor T‐cell therapy – assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15:47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Shimabukuro‐Vornhagen A, Godel P, Subklewe M, Stemmler HJ, Schlosser HA, Schlaak M, et al. Cytokine release syndrome. J ImmunoTher Cancer. 2018;6:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Jain T, Litzow MR. No free rides: management of toxicities of novel immunotherapies in ALL, including financial. Blood Adv. 2018;2:3393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Frey NV, Porter DL. Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematology Am Soc Hematol Educ Program. 2016;2016:567–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Klinger M, Zugmaier G, Nagele V, Goebeler ME, Brandl C, Stelljes M, et al. Adhesion of T cells to endothelial cells facilitates blinatumomab‐associated neurologic adverse events. Cancer Res. 2020;80:91–101. [DOI] [PubMed] [Google Scholar]
- 91. Näegele V, Kratzer A, Zugmaier G, Holland C, Hijazi Y, Topp MS, et al. Changes in clinical laboratory parameters and pharmacoynamic markers in response to blinatumomab treatment of patients with relapsed/refractory ALL. Exp Hematol Oncol. 2017;6:14 10.1186/s40164-017-0074-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez‐Cuyar LF, et al. Endothelial activation and blood‐brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR‐T Cells. Cancer Discov. 2017;7:1404–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. KYMRIAH™ . KYMRIAH™ (tisagenlecleucel) suspension for intravenous infusion [package insert]. Basel: Novartis AG; 2017. [Google Scholar]
- 94. YESCARTA® . YESCARTA® (axicabtagene ciloleucel) suspension for intravenous infusion [package insert]. Los Angeles, CA: Kite Pharma, Inc.; 2017. [Google Scholar]
- 95. Defitelio . Defitelio (defibrotide sodium) injection [packet insert]. Dublin: Jazz Pharmaceuticals, Inc.; 2016. [Google Scholar]
- 96. Defitelio . Summary of product characteristics. Dublin: Jazz Pharmaceuticals, Inc.; 2019. [cited 2019 April 2]. [Google Scholar]
- 97. Falanga A, Vignoli A, Marchetti M, Barbui T. Defibrotide reduces procoagulant activity and increases fibrinolytic properties of endothelial cells. Leukemia. 2003;17:1636–42. [DOI] [PubMed] [Google Scholar]
- 98. Richardson PG, Riches ML, Kernan NA, Brochstein JA, Mineishi S, Termuhlen AM, et al. Phase 3 trial of defibrotide for the treatment of severe veno‐occlusive disease and multi‐organ failure. Blood. 2016;127:1656–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kernan NA, Grupp S, Smith AR, Arai S, Triplett B, Antin JH, et al. Final results from a defibrotide treatment‐IND study for patients with hepatic veno‐occlusive disease/sinusoidal obstruction syndrome. Br J Haematol. 2018;181:816–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Magenau JM, Goldstein SC, Peltier D, Soiffer RJ, Braun T, Pawarode A, et al. alpha1‐Antitrypsin infusion for treatment of steroid‐resistant acute graft‐versus‐host disease. Blood. 2018;131:1372–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Rosenthal J. Hematopoietic cell transplantation‐associated thrombotic icroangiopathy: a review of pathophysiology, diagnosis, and treatment. J Blood Med. 2016;7:181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Elsallabi O, Bhatt VR, Dhakal P, Foster KW, Tendulkar KK. Hematopoietic stem cell transplant‐associated thrombotic microangiopathy. Clin Appl Thromb Hemost. 2016;22:12–20. [DOI] [PubMed] [Google Scholar]
- 103. Jodele S, Dandoy CE, Myers KC, El‐Bietar J, Nelson A, Wallace G, et al. New approaches in the diagnosis, pathophysiology, and treatment of pediatric hematopoietic stem cell transplantation‐associated thrombotic microangiopathy. Transfus Apher Sci. 2016;54:181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Dvorak CC, Higham C, Shimano KA. Transplant‐associated thrombotic microangiopathy in pediatric hematopoietic cell transplant recipients: a practical approach to diagnosis and management. Front Pediatr. 2019;7:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Thompson J, Yin Z, D'Souza A, Fenske T, Hamadani M, Hari P, et al. Etanercept and corticosteroid therapy for the treatment of late‐onset idiopathic pneumonia syndrome. Biol Blood Marrow Transplant. 2017;23:1955–60. [DOI] [PubMed] [Google Scholar]
- 106. Yanik GA, Horowitz MM, Weisdorf DJ, Logan BR, Ho VT, Soiffer RJ, et al. Randomized, double‐blind, placebo‐controlled trial of soluble tumor necrosis factor receptor: enbrel (etanercept) for the treatment of idiopathic pneumonia syndrome after allogeneic stem cell transplantation: blood and marrow transplant clinical trials network protocol. Biol Blood Marrow Transplant. 2014;20:858–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wanko SO, Broadwater G, Folz RJ, Chao NJ. Diffuse alveolar hemorrhage: retrospective review of clinical outcome in allogeneic transplant recipients treated with aminocaproic acid. Biol Blood Marrow Transplant. 2006;12:949–53. [DOI] [PubMed] [Google Scholar]
- 108. Jodele S, Dandoy CE, Lane A, Laskin BL, Teusink‐Cross A, Myers KC, et al. Complement blockade for TA‐TMA: lessons learned from large pediatric cohort treated with eculizumab. Blood. 2020;135:1049–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Rudoni J, Jan A, Hosing C, Aung F, Yeh J. Eculizumab for transplant‐associated thrombotic microangiopathy in adult allogeneic stem cell transplant recipients. Eur J Haematol. 2018;101:389–98. [DOI] [PubMed] [Google Scholar]
- 110. Cataland S, Ariceta G, Chen P, Dixon B, Garlo K, Greenbaum L, et al. Discordance between free c5 and ch50 complement assays in measuring complement c5 inhibition in patients with aHUS treated with ravulizumab. Blood. 2019;134:1099–1099. [Google Scholar]
- 111. Richardson PG, Soiffer RJ, Antin JH, Uno H, Jin Z, Kurtzberg J, et al. Defibrotide for the treatment of severe hepatic veno‐occlusive disease and multiorgan failure after stem cell transplantation: a multicenter, randomized, dose‐finding trial. Biol Blood Marrow Transplant. 2010;16:1005–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Nomura S, Ishii K, Fujita S, Nakaya A, Satake A, Ito T. Associations between acute GVHD‐related biomarkers and endothelial cell activation after allogeneic hematopoietic stem cell transplantation. Transpl Immunol. 2017;43–44:27–32. [DOI] [PubMed] [Google Scholar]