

Abstract
We report the ligand‐enabled C−H activation/olefination of free carboxylic acids in the γ‐position. Through an intramolecular Michael addition, δ‐lactones are obtained as products. Two distinct ligand classes are identified that enable the challenging palladium‐catalyzed activation of free carboxylic acids in the γ‐position. The developed protocol features a wide range of acid substrates and olefin reaction partners and is shown to be applicable on a preparatively useful scale. Insights into the underlying reaction mechanism obtained through kinetic studies are reported.
Keywords: C−H activation, carboxylic acids, ligand-enabled catalysis, palladium, δ-lactones
Straight shot: A direct γ‐C−H olefination of carboxylic acids and subsequent intramolecular cyclization generates δ‐lactones. Two suitable ligand classes are identified to enable this Pd‐catalyzed transformation that features a broad scope of both reaction partners. The reaction does not require an exogenous directing group and can be performed on a synthetically useful scale. Kinetic studies provide first mechanistic insights.

The synthesis of complex carboxylic acid derivatives from simple and readily available carboxylic acids is highly attractive, due to the prevalence of the carboxylic acid moiety in compounds such as pharmaceuticals, fragrances, flavors, etc.1 However, despite some recent progress, the direct C−H activation and functionalization of free carboxylic acids remains highly challenging, due to the weak directing ability of the carboxylate group and competing coordination modes amongst other reasons, and thus requires the identification of suitable ligands and careful fine‐tuning of the associated reaction conditions.2 These challenges can be circumvented through the introduction of more strongly directing exogenous directing groups, a strategy that has enabled a variety of highly useful transformations.3 One highly attractive synthetic target in the field has been the C−H olefination of carboxylic acid derivatives. Yu and co‐workers have developed conditions for the β‐olefination of aliphatic amides bearing a perfluorinated arene substituent on the nitrogen, which delivered γ‐lactams through a C−H olefination followed by an intramolecular Michael addition (Scheme 1 A).4 Later, the same group developed ligands that allowed them to expand this reactivity to the C−H olefination of the substantially more challenging γ‐position, giving access to δ‐lactams (Scheme 1 B).5 In parallel to the development of methods relying on exogenous directing groups, substantial efforts by ourselves and others have recently been directed towards the use of free carboxylic acids in C−H activation processes and the identification of suitable ligands enabled several highly useful transformations.6 Amongst these, Yu and co‐workers have reported a direct synthesis of γ‐lactones through the β‐C(sp3)−H olefination of free carboxylic acids, followed by an intramolecular cyclization (Scheme 1 C).6l While constituting a synthetically highly attractive approach towards the valuable δ‐lactone motif,7 the analogous γ‐olefination/cyclization has remained elusive to date. It should be noted that research towards the direct γ‐C(sp3)−H activation of free carboxylic acids is still in its infancy and to the best of our knowledge only two synthetic methods relying on this type of process have been reported to date by the groups of Maiti and Shi, in both cases enabling the γ‐arylation of free carboxylic acids through PdII/PdIV‐catalytic cycles, albeit with complementary acid scopes.8 We thus became interested in developing a method for the synthesis of δ‐lactones through the direct γ‐C(sp3)−H olefination of free carboxylic acids. Herein we report the realization of this goal enabled through the identification of two suitable ligand classes: N‐acetyl anthranilic acid derivatives and N‐acetyl amino acids.
Scheme 1.

Key advances in the C(sp3)−H olefination of carboxylic acid derivatives.
Based on our experience in the development of challenging β‐C(sp3)−H functionalization processes for free carboxylic acids, we expected that the identification of suitable ligands would be key in order to establish the desired protocol. We thus initiated our studies using 3,3‐dimethylbutyric acid (1 a) and ethyl acrylate (2 a) as model substrates. After the initial identification of L10 as a particularly promising ligand, we optimized the reaction conditions using this ligand (for details see the Supporting Information). After identifying the otherwise best reaction conditions, we re‐evaluated representatives of common ligand classes in the C(sp3)−H activation (Scheme 2). We found that the anthranilic acid derivative L10 continues to deliver superior results compared to pyridone L1,9 pyridine L2,4b, 6g, 10 and the bidentate ligands L3–L7.6g, 6j, 6k, 6l, 11 Structural variants of the anthranilic acid motif L8 and L9 gave no further improvements. Finally, a re‐investigation of amino acid derived ligands L11–L14 6h, 6i, 8 showed that N‐Ac‐β‐alanine L14 gave equally good results as L10. Notably, this ligand performed substantially worse than L10 in an initial comparison under nonoptimized conditions.
Scheme 2.

Identification of suitable ligands for the γ‐C(sp3)−H olefination of free carboxylic acids. Reactions were conducted on a 0.2 mmol scale. Yields were determined by 1H NMR analysis of the crude reaction mixture with 1,3,5‐trimethoxy benzene as an internal standard.
Having identified two suitable ligands for the γ‐olefination of free carboxylic acids, we chose to investigate the scope of this transformation using L14, simply based on the broader availability of this ligand (Scheme 3). It should be noted however, that the discovery of anthranilic acid ligand L10, which has not previously been used in C−H activation to the best of our knowledge, may prove helpful in future related studies. We began by studying the substrate scope with respect to the acid substrate (Scheme 3). As expected based on our optimization studies, the model product 3 a could be obtained in good yield (64 %). This example was also used to probe the scalability of our protocol. Importantly, a virtually identical yield of 62 % was obtained on a 5 mmol scale. For structurally more complex acid substrates we found that an increased reaction time and acrylate loading were required to obtain optimal yields and used these conditions for the remainder of the acid scope studies. The alkyl‐substituted products 3 b–e were obtained in good yields and with moderate diastereoselectivities in favor of the cis‐configured isomer. The spirocyclic products 3 f and 3 g, as well as 3 h, bearing two non‐methyl substituents, were all obtained in synthetically useful yields. Finally, products 3 i–m containing aromatic substituents were again obtained in good yields and moderate to good diastereoselectivities. Several limitations were also encountered during the evaluation of the acid scope.12 When we examined the reactivity of substrates without a quaternary center at the β‐position, no conversion of the starting material was observed, presumably due to the absence of an accelerating Thorpe–Ingold effect. Furthermore, it should be noted that in the case of competition between β‐ and γ‐methyl groups, the β‐position is expected to react preferentially.6h, 6l
Scheme 3.

Acid scope of the ligand‐enabled γ‐C(sp3)−H olefination of free carboxylic acids. Reactions were conducted on a 0.2 mmol scale. a. 2 a (2.5 equiv) and Ag2CO3 (1.75 equiv) were used with 40 h reaction time. b. 2 a (7 equiv) and Ag2CO3 (2.5 equiv) were used with 72 h reaction time. c. The yield in parentheses was obtained on a 5 mmol scale.
We proceeded to study the scope of this transformation with respect to the olefinic reaction partner (Scheme 4). Various acrylates were found to react smoothly, giving products 4 a–d in good yields. Olefins bearing other electron‐withdrawing groups, such as methyl vinyl ketone (4 e), acrylonitrile (4 f), phenyl vinyl sulfone (4 g), ethenesulfonyl fluoride (4 h), and diethyl vinylphosphonate (4 i) could all be used as reaction partners. Finally, olefins containing structurally more complex subunits were also successfully employed in the reaction, giving product 4 j–l in good yields.
Scheme 4.
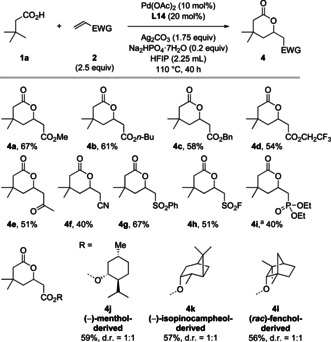
Olefin scope of the ligand‐enabled γ‐C(sp3)−H olefination of free carboxylic acids. Reactions were conducted on a 0.2 mmol scale. a. Diethyl vinylphosphate (7 equiv) was used with 72 h reaction time.
Having studied the scope of this transformation, we became interested in obtaining basic insights into the underlying reaction mechanism. We began by evaluating whether the C−H activation contributes to the overall rate of the transformation (Scheme 5 A). The clear primary kinetic isotope effect observed both in a competition experiment and in parallel experiments indicates that the C−H activation is indeed rate‐determining.13
Scheme 5.

Preliminary mechanistic studies. Experiments in (A) and (B) were conducted on a 0.2 mmol scale. The reversibility experiments were conducted on a 0.1 mmol scale.
To obtain further knowledge about this step, we proceeded to determine the kinetic orders in both reaction partners and the catalyst (Scheme 5 B). We began by determining the initial rates of the reaction by varying one of the starting concentrations. The results were analyzed by determining the slope of the double natural logarithmic plot of the initial rate vs. the starting concentration.14 The analysis revealed an order of 1 in catalyst. Importantly, monomer–dimer equilibria are known to exist for palladium catalysts with amino acid derived ligands and both monomers and dimers have been shown to be the active catalyst depending on the transformation and reaction conditions.15 The order of 1 indicates that only monomers or dimers can be present in non‐negligible amounts under the reaction conditions, such that a change in concentration is not correlated with a shift in equilibrium.16 Based on the closest literature reports,15b, 15c, 15d, 15e, 15f we propose the reaction to occur though monomeric catalyst species.
When we attempted to determine the order with respect to the acid component, we found that both an increase and a decrease in acid concentration were detrimental to the reaction outcome. In light of the known importance of speciation in the transformations discussed above, this result can easily be rationalized: The rate of product formation is influenced by the amount of substrate–palladium pre‐reactive complex formed, which in turn depends more on the acid–base balance of the reaction mixture than on the actual substrate concentration. Since we optimized the acid and base amounts during our optimization studies, deviations in both directions are detrimental. Finally, we found a small, but non‐zero (0.2) order in the olefin reaction partner. This result was unexpected, due to the previous identification of the C−H activation as the rate‐determining step, which implies a zero order in the olefin that enters the catalytic cycle after this step. We hypothesized that this result can be explained by the reversibility of the C−H activation step, together with a lower, but comparable barrier for a subsequent step involving the olefin. In such a scenario, the C−H activation step would determine the overall rate of product formation, but a small fraction of the palladacycle formed could statistically revert to the starting material, when the subsequent reaction with the olefin does not occur fast enough. In order to probe this hypothesis, we conducted two reversibility experiments (Scheme 5 C), one during the product‐forming reaction with 1 a‐d9 as substrate and one in the absence of the olefin reaction partner. In both cases the deuteration of the remaining starting material was analyzed. When no olefin is available, a strong de‐deuteration was observed, showing that the C−H activation is in principle reversible under the reaction conditions. However, the result in the presence of olefin clearly demonstrates that when product formation is possible, it mostly outcompetes the retro‐C−H activation, while a small but measurable back reaction occurs. These results are in good agreement with the observed 0.2 order in olefin. Overall, we propose the mechanism shown in Scheme 6 A, which takes into account the observations discussed above.
Scheme 6.

Proposed catalytic cycle and mechanism for side product formation.
Accordingly, the reaction would proceed through a (mostly) rate‐determining C−H activation by a mononuclear PdII catalyst. Next, a sequence of ligand exchange, carbopalladation, β‐H elimination, and decoordination would result in the formation of the product in its noncyclized form 4‐open, which could subsequently cyclize to product 4 though an intramolecular Michael addition. Concomitantly, the product decoordination would deliver a PdII–hydride species that could undergo a reductive elimination giving a Pd0 species, which would then be re‐oxidized by the silver salt employed as the terminal oxidant. A final mechanistic feature of our protocol concerns a side product observed in small quantities throughout these studies (5, Scheme 6 B). We could isolate and characterize this side product in two cases, 5 e and 5 m. Since the formation of these compounds requires a second oxidation event, we hypothesize that they are formed through a carboxylate‐directed C−H activation/oxidation starting from the open form of the primary product.
In summary, we have developed a protocol for the palladium‐catalyzed γ‐C(sp3)−H olefination of free carboxylic acids. Through an in situ Michael addition, δ‐lactones are obtained without the need to install/remove exogenous directing groups. Our protocol features a broad scope of both reaction partners. Mechanistic experiments support a PdII/Pd0 catalytic cycle, which renders this study the first report on the direct γ‐C(sp3)−H activation/ functionalization of free carboxylic acids through this redox manifold. We expect that these results will serve as a proof of principle and inspire research towards further transformations of this kind.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We gratefully acknowledge financial support from the Max Planck Society (Otto Hahn Award to M.v.G.), FCI (Liebig Fellowship to M.v.G.), DFG (SFB 858), Studienstiftung des deutschen Volkes (Felloswhip to F.G.), and WWU Münster. We thank the members of our NMR and MS departments for their excellent service. Furthermore, we are indebted to Prof. F. Glorius for his generous support.
K. K. Ghosh, A. Uttry, A. Mondal, F. Ghiringhelli, P. Wedi, M. van Gemmeren, Angew. Chem. Int. Ed. 2020, 59, 12848.
In memory of Prof. Rolf Huisgen
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv.11857857.v1).
References
- 1. He J., Wasa M., Chan K. S. L., Shao Q., Yu J.-Q., Chem. Rev. 2017, 117, 8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Uttry A., van Gemmeren M., Synlett 2018, 29, 1937; [Google Scholar]
- 2b. Uttry A., van Gemmeren M., Synthesis 2020, 52, 479; [Google Scholar]
- 2c. Pichette Drapeau M., Gooßen L. J., Chem. Eur. J. 2016, 22, 18654. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Engle K. M., Mei T.-S., Wasa M., Yu J.-Q., Acc. Chem. Res. 2012, 45, 788; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Rouquet G., Chatani N., Angew. Chem. Int. Ed. 2013, 52, 11726; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11942; [Google Scholar]
- 3c. Chen Z., Wang B., Zhang J., Yu W., Liu Z., Zhang Y., Org. Chem. Front. 2015, 2, 1107; [Google Scholar]
- 3d. Zhu R.-Y., Farmer M. E., Chen Y.-Q., Yu J.-Q., Angew. Chem. Int. Ed. 2016, 55, 10578; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10734; [Google Scholar]
- 3e. Sambiagio C., Schönbauer D., Blieck R., Dao-Huy T., Pototschnig G., Schaaf P., Wiesinger T., Zia M. F., Wencel-Delord J., Besset T., Maes B. U. W., Schnürch M., Chem. Soc. Rev. 2018, 47, 6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Wasa M., Engle K. M., Yu J.-Q., J. Am. Chem. Soc. 2010, 132, 3680; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. He J., Li S., Deng Y., Fu H., Laforteza B. N., Spangler J. E., Homs A., Yu J.-Q., Science 2014, 343, 1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Li S., Chen G., Feng C.-G., Gong W., Yu J.-Q., J. Am. Chem. Soc. 2014, 136, 5267; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Thrimurtulu N., Khan S., Maity S., Volla C. M. R., Maiti D., Chem. Commun. 2017, 53, 12457. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Kao L.-C., Sen A., J. Chem. Soc. Chem. Commun. 1991, 1242; [Google Scholar]
- 6b. Dangel B. D., Johnson J. A., Sames D., J. Am. Chem. Soc. 2001, 123, 8149; [DOI] [PubMed] [Google Scholar]
- 6c. Fraunhoffer K. J., Prabagaran N., Sirois L. E., White M. C., J. Am. Chem. Soc. 2006, 128, 9032; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Lee J. M., Chang S., Tetrahedron Lett. 2006, 47, 1375; [Google Scholar]
- 6e. Giri R., Maugel N., Li J.-J., Wang D.-H., Breazzano S. P., Saunders L. B., Yu J.-Q., J. Am. Chem. Soc. 2007, 129, 3510; [DOI] [PubMed] [Google Scholar]
- 6f. Novák P., Correa A., Gallardo-Donaire J., Martin R., Angew. Chem. Int. Ed. 2011, 50, 12236; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12444; [Google Scholar]
- 6g. Chen G., Zhuang Z., Li G.-C., Saint-Denis T. G., Hsiao Y., Joe C. L., Yu J.-Q., Angew. Chem. Int. Ed. 2017, 56, 1506; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1528; [Google Scholar]
- 6h. Ghosh K. K., van Gemmeren M., Chem. Eur. J. 2017, 23, 17697; [DOI] [PubMed] [Google Scholar]
- 6i. Zhu Y., Chen X., Yuan C., Li G., Zhang J., Zhao Y., Nat. Commun. 2017, 8, 14904; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6j. Hu L., Shen P.-X., Shao Q., Hong K., Qiao J. X., Yu J.-Q., Angew. Chem. Int. Ed. 2019, 58, 2134; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 2156; [Google Scholar]
- 6k. Shen P.-X., Hu L., Shao Q., Hong K., Yu J.-Q., J. Am. Chem. Soc. 2018, 140, 6545; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6l. Zhuang Z., Yu C.-B., Chen G., Wu Q.-F., Hsiao Y., Joe C. L., Qiao J. X., Poss M. A., Yu J.-Q., J. Am. Chem. Soc. 2018, 140, 10363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Gerth D. B., Giese B., J. Org. Chem. 1986, 51, 3726; [Google Scholar]
- 7b. Seiichi T., Youichi S., Minoru M., Kunio O., Chem. Lett. 1990, 19, 1177; [Google Scholar]
- 7c. Takano S., Shimazaki Y., Ogasawara K., Tetrahedron Lett. 1990, 31, 3325; [Google Scholar]
- 7d. Kishimoto N., Sugihara S., Mochida K. Y. O., Fujita T., Biocontrol Sci. 2005, 10, 31; [Google Scholar]
- 7e. Teiber J. F., Xiao J., Kramer G. L., Ogawa S., Ebner C., Wolleb H., Carreira E. M., Shih D. M., Haley R. W., Biochem. Biophys. Res. Commun. 2018, 505, 87. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Dolui P., Das J., Chandrashekar H. B., Anjana S. S., Maiti D., Angew. Chem. Int. Ed. 2019, 58, 13773; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13911; [Google Scholar]
- 8b. Liu L., Liu Y.-H., Shi B.-F., Chem. Sci. 2020, 11, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Wang P., Verma P., Xia G., Shi J., Qiao J. X., Tao S., Cheng P. T. W., Poss M. A., Farmer M. E., Yeung K.-S., Yu J.-Q., Nature 2017, 551, 489; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Chen X.-Y., Wu Y., Zhou J., Wang P., Yu J.-Q., Org. Lett. 2019, 21, 1426; [DOI] [PubMed] [Google Scholar]
- 9c. Liu L., Yeung K.-S., Yu J.-Q., Chem. Eur. J. 2019, 25, 2199; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Zhu R.-Y., Li Z.-Q., Park H. S., Senanayake C. H., Yu J.-Q., J. Am. Chem. Soc. 2018, 140, 3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.
- 10a. Li S., Zhu R.-Y., Xiao K.-J., Yu J.-Q., Angew. Chem. Int. Ed. 2016, 55, 4317; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4389; [Google Scholar]
- 10b. He J., Jiang H., Takise R., Zhu R.-Y., Chen G., Dai H.-X., Dhar T. G. M., Shi J., Zhang H., Cheng P. T. W., Yu J.-Q., Angew. Chem. Int. Ed. 2016, 55, 785; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 795. [Google Scholar]
- 11.
- 11a. Jerhaoui S., Djukic J.-P., Wencel-Delord J., Colobert F., ACS Catal. 2019, 9, 2532; [Google Scholar]
- 11b. Yang Y.-F., Chen G., Hong X., Yu J.-Q., Houk K. N., J. Am. Chem. Soc. 2017, 139, 8514; [DOI] [PubMed] [Google Scholar]
- 11c. Wu Q.-F., Shen P.-X., He J., Wang X.-B., Zhang F., Shao Q., Zhu R.-Y., Mapelli C., Qiao J. X., Poss M. A., Yu J.-Q., Science 2017, 355, 499; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11d. He J., Shao Q., Wu Q., Yu J.-Q., J. Am. Chem. Soc. 2017, 139, 3344; [DOI] [PubMed] [Google Scholar]
- 11e. Chen G., Gong W., Zhuang Z., Andrä M. S., Chen Y.-Q., Hong X., Yang Y.-F., Liu T., Houk K. N., Yu J.-Q., Science 2016, 353, 1023; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11f. Naksomboon K., Valderas C., Gómez-Martínez M., Álvarez-Casao Y., Fernández-Ibáñez M. Á., ACS Catal. 2017, 7, 6342; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11g. Naksomboon K., Poater J., Bickelhaupt F. M., Fernández-Ibáñez M. Á., J. Am. Chem. Soc. 2019, 141, 6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For details, see the Supporting Information.
- 13. Simmons E. M., Hartwig J. F., Angew. Chem. Int. Ed. 2012, 51, 3066; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3120. [Google Scholar]
- 14.
- 14a.“The rates of chemical reactions”: Atkins P., de Paula J., in Atkins’ Physical Chemistry. 8th ed., Oxford University Press, Oxford, New York, 2006, pp. 791–823. For representative examples, see: [Google Scholar]
- 14b. Liu B., Roisnel T., Carpentier J.-F., Sarazin Y., Angew. Chem. Int. Ed. 2012, 51, 4943–4946; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5027–5030; [Google Scholar]
- 14c. Chatelet B., Joucla L., Dutasa J.-P., Martinez A., Szeto K. C., Dafaud V., J. Am. Chem. Soc. 2013, 135, 5348–5351. An alternative exponential fitting of the non-logarithmic plot delivered analogous results. For an example, see: [DOI] [PubMed] [Google Scholar]; Cook A. K., Sanford M. S., J. Am. Chem. Soc. 2015, 137, 3109–3118. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Shi B.-F., Maugel N., Zhang Y.-H., Yu J.-Q., Angew. Chem. Int. Ed. 2008, 47, 4882; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4960; [Google Scholar]
- 15b. Engle K. M., Wang D.-H., Yu J.-Q., J. Am. Chem. Soc. 2010, 132, 14137; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Shi B.-F., Zhang Y.-H., Lam J. K., Wang D.-H., Yu J.-Q., J. Am. Chem. Soc. 2010, 132, 460; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15d. Engle K. M., Thuy-Boun P. S., Dang M., Yu J.-Q., J. Am. Chem. Soc. 2011, 133, 18183; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15e. Cheng G.-J., Yang Y.-F., Liu P., Chen P., Sun T.-Y., Li G., Zhang X., Houk K. N., Yu J.-Q., Wu Y.-D., J. Am. Chem. Soc. 2014, 136, 894; [DOI] [PubMed] [Google Scholar]
- 15f. Cheng G.-J., Chen P., Sun T.-Y., Zhang X., Yu J.-Q., Wu Y.-D., Chem. Eur. J. 2015, 21, 11180; [DOI] [PubMed] [Google Scholar]
- 15g. Haines B. E., Musaev D. G., ACS Catal. 2015, 5, 830; [Google Scholar]
- 15h. Gair J. J., Haines B. E., Filatov A. S., Musaev D. G., Lewis J. C., Chem. Sci. 2017, 8, 5746; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15i. Hill D. E., Bay K. L., Yang Y.-F., Plata R. E., Takise R., Houk K. N., Yu J.-Q., Blackmond D. G., J. Am. Chem. Soc. 2017, 139, 18500; [DOI] [PubMed] [Google Scholar]
- 15j. Plata R. E., Hill D. E., Haines B. E., Musaev D. G., Chu L., Hickey D. P., Sigman M. S., Yu J.-Q., Blackmond D. G., J. Am. Chem. Soc. 2017, 139, 9238; [DOI] [PubMed] [Google Scholar]
- 15k. Gair J. J., Haines B. E., Filatov A. S., Musaev D. G., Lewis J. C., ACS Catal. 2019, 9, 11386; [Google Scholar]
- 15l.C. A. Salazar, J. J. Gair, K. N. Flesch, I. A. Guzei, J. C. Lewis, S. S. Stahl, Angew. Chem. Int. Ed 2020, DOI: 10.1002/anie.202002484; Angew. Chem 2020, DOI: . [DOI]
- 16. Hill D. E., Pei Q.-l., Zhang E.-x., Gage J. R., Yu J.-Q., Blackmond D. G., ACS Catal. 2018, 8, 1528. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary