

Abstract
Derivatives of 4‐aminomethyl‐l‐phenylalanine with aromatic oligoamide foldamers as sidechain appendages were successfully charged on tRNA by means of flexizymes. Their subsequent incorporation both at the C‐terminus of, and within, peptide sequences by the ribosome, was demonstrated. These results expand the registry of chemical structures tolerated by the ribosome to sidechains significantly larger and more structurally defined than previously demonstrated.
Keywords: foldamers, flexizyme, in vitro translation, peptides, peptidomimetics
The ribosome tolerates aromatic oligoamide foldamer–peptide hybrids not only when the foldamer belongs to the initiation unit, but also when it is appended as a sidechain within the peptide or at the C‐terminus of the peptide. This expands the range of substrates known to be accepted by the ribosome to include significantly larger and more structurally defined substrates.
Genetic code reprogramming has enabled the incorporation of non‐proteinogenic amino acids into peptides and proteins, greatly expanding available structures, functions, and chemistries. Despite eons of evolution, the ribosome displays startling tolerance towards residues significantly deviating from the twenty canonical α‐amino acids: d‐,1, 2 β‐3, 4 and N‐methyl‐5 amino acids, and α‐amino acids equipped with bulky and exotic sidechains6 have been charged on tRNA by means of flexizymes and, when combined with a reconstituted flexible in vitro translation (FIT) system,7 have been ribosomally incorporated within α‐peptides. Capitalizing on the even greater tolerance of the ribosome during translation initiation, exotic peptides,8 terpenes9 and, recently, non‐amino acid substrates10, 11 and aromatic oligoamide foldamers,12, 13 have been incorporated as the first unit of ribosomal peptide synthesis to produce various types of hybrid molecules.14 Because of their strong propensity to adopt helical conformations,15 aromatic foldamers may transmit folding information to the peptide and control its conformation.16, 17 With masses up to 1 kDa, they constitute the bulkiest and most rigid objects ever incorporated by the translation machinery, including delivery to the ribosome and passage through its exit tunnel. Having highlighted the tolerance of the ribosome in translation initiation, we decided to explore whether incorporation of abiotic foldamers was feasible in positions other than the N‐terminus of the peptide.
Herein, we demonstrate how aromatic foldamers appended to the sidechain of a 4‐(aminomethyl)‐l‐α‐phenylalanine residue (Amf) can be recognized by flexizymes and ribosomally incorporated to produce peptide–foldamer hybrids with unique architectures (Figure 1 a). We thus identified the most rigid and structurally defined sidechains accepted by the ribosome machinery to date.18, 19 Overall, these findings not only extend by a great degree what was previously thought possible, but also call into question the limitations of nature's translational apparatus.
Figure 1.
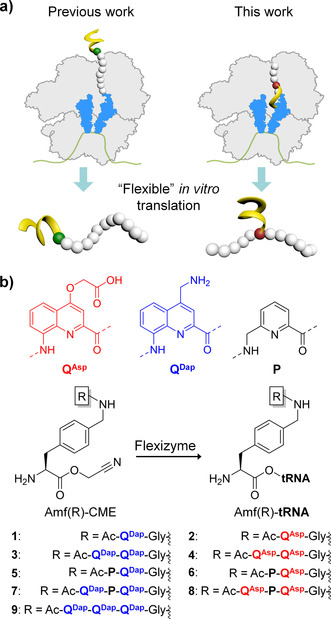
a) Schematic illustration of the ribosomal incorporation of amino acids bearing helical aromatic foldamers (left) at the N‐terminus and (right) within a peptide sequence. b) Aromatic foldamer substrates attached to the sidechain of Amf used in this study. CME: cyanomethyl ester.
Oligoamides consisting of quinoline (Q) and pyridine (P) monomers have been shown to fold into stable helices in a broad range of solvents including water (Figure 1 b).20, 21 Monomer P, which is more flexible and smaller than Q, was previously shown to ameliorate translation efficiency,12 potentially by alleviating the conformational rigidity of the resulting oligomer.22 A series of short foldamers was designed and synthesized to pursue the translation of foldamer‐type sidechains in a stepwise manner (Figure 1 b). To facilitate substrate recognition by flexizymes and owing to the distinct nature of aromatic foldamers from the arsenal of ribosomally incorporated, exotic amino acids, the foldamer segments were appended at a remote position from the peptide main chain, on the sidechain of Amf. A Gly spacer was introduced as for earlier foldamer‐based peptide initiators.12 Furthermore, having previously unveiled foldamer sidechain‐dependent behavior on both aminoacylation and translation,13 we set out to explore foldamers having either positively or negatively charged sidechains. Amf(‐Gly‐foldamer) amino acids were activated as cyanomethyl esters and the ability of flexizyme to load these new substrates onto tRNA was assessed using an established protocol involving microhelix RNA (Supporting Information, Figures S1–S3).7 The results of the aminoacylation reaction (Table 1) indicated that for Amf(‐Gly‐Q‐Ac) (1,2) and Amf(‐Gly‐Q2‐Ac) (3,4), the reaction proceeded adequately both with negatively charged QAsp and positively charged QDap. In contrast, for sequences including P, that is Amf(‐Gly‐QP‐Ac) and Amf(‐Gly‐QPQ‐Ac), the reaction only worked with QDap (5,7). No loading on tRNA was observed with QAsp (6,8). QDap was then used to also test Amf(‐Gly‐Q3‐Ac) (9), but this more rigid sequence was found to be incompatible with the flexizyme. Aminoacylation of tRNA by Amf(‐Gly‐foldamer) thus appears to depend on monomer charge, sequence, rigidity or bulkiness, and perhaps solubility.
Table 1.
The sequences of amino acid elongators used in this study and their acylation yields and translation efficiencies. (N.D. indicates the product was detected but the yield was not determined)
|
Entry |
Foldamer sequence |
Acylation yield [%] |
Translation yield [%][a] |
|
|---|---|---|---|---|
|
|
|
|
T1 |
T2 |
|
1 |
Amf(‐Gly‐QDap‐Ac) |
20 |
92 |
68 |
|
2 |
Amf(‐Gly‐QAsp‐Ac) |
55 |
82 |
62 |
|
3 |
Amf(‐Gly‐(QDap)2‐Ac) |
24 |
48 |
11 |
|
4 |
Amf(‐Gly‐(QAsp)2‐Ac) |
29 |
– |
N.D.[b] |
|
5 |
Amf(‐Gly‐QDapP‐Ac) |
24 |
35 |
10 |
|
6 |
Amf(‐Gly‐QAspP‐Ac) |
0 |
– |
– |
|
7 |
Amf(‐Gly‐QDapPQDap‐Ac) |
10 |
5 |
N.D.[b] |
|
8 |
Amf(‐Gly‐QAspPQAsp‐Ac) |
0 |
– |
– |
|
9 |
Amf(‐Gly‐(QDap)3‐Ac) |
0 |
– |
– |
[a] The values provided are based on the observed incorporation efficiency when compared with the wild type expression (absence of reprogramming). [b] Low yields and band overlap hampered their identification and subsequent quantification (Supporting Information, Figures S4 and S5).
This dependency seems stronger than for the substrates previously used for translation initiation. It may perhaps be reduced by a longer spacer than Gly but this was not tested in the context of this investigation and peptide elongation was investigated with 1–5 and 7. Aiming at introducing aromatic oligoamide‐containing Amf residues both at the C‐terminus and within the sequence, we designed mRNA templates T1 and T2 encoding a peptide 14‐mer and 15‐mer, respectively (Figure 2 a). The ACC codon, originally coding for Thr, was arbitrarily chosen and reprogrammed to code for the Amf(‐Gly‐foldamer).
Figure 2.
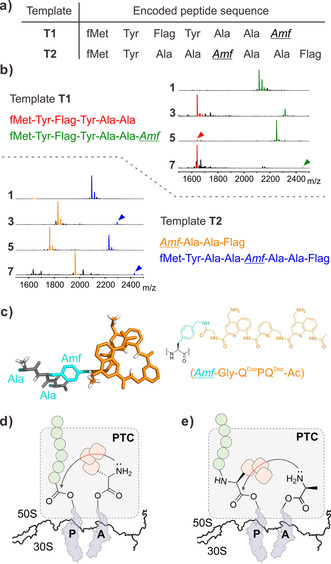
a) Template‐encoded peptides containing Amf(‐Gly‐foldamer) at the C‐terminus (from T1) and within the peptide (from T2). “Flag” stands for DYKDDDDK. b) Characteristic MALDI‐TOF‐MS spectra of resulting peptides containing 1, 3, 5 or 7 as a positively charged foldamer sidechain. Desired products are indicated by green and blue peaks. Side‐products corresponding to unsuccessful peptidyl transferase reactions are indicated by red and orange peaks (magnification of the peaks corresponding to the desired products, along with the [M+Na]+ and [M+K]+ are available in the Supporting Information). c) Molecular model of Ala‐Amf(‐Gly‐QDapPQDap‐Ac)‐Ala illustrating the size of the foldamer with respect to the peptide chain (left) and its extended chemical structure (right). Nonpolar hydrogen atoms have been omitted for clarity. d) Scheme of the nucleophilic addition of the foldamer‐containing Amf residue on the peptidyl tRNA ester at the P‐site. e) Nucleophilic addition of the Ala amine following the Amf residue. In (d) and (e) green circles and rounded squares represent elongating amino acids and aromatic residues on Amf, respectively. The red dashed part of the reaction arrow represents potential obstacles caused by the foldamer‐containing residue. PTC: peptidyl transferase center.
The Flag peptide sequence was encoded in both templates, either before or after ACC, to enable radiolabeling of the peptide using [14C]‐Asp, as well as purification. The oligoamide–tRNA conjugates were supplemented into a FIT system, from which Thr had been omitted, and the translation reactions were subjected to Flag purification prior to MALDI‐TOF‐MS analysis (Figure 2 b). Amf bearing a single Q monomer (1 and 2) could be incorporated at both internal and C‐terminal positions. The size and rigidity of these sidechains is comparable to that of other large previously reported residues.23 Amf bearing two foldamer units (3–5) was incorporated within the peptide sequence, but only the positively charged, 3 and 5, were incorporated at the C‐terminus of the peptide. Overall, these results exceeded our expectations given their size and aromatic nature.
Remarkably, we also observed the formation of the desired peptide when Amf bearing an entire trimer (7) was incorporated either at the C‐terminus or within the sequence. Though the expression level was modest (see below), compound 7 is by far the most structurally complex sidechain incorporated by the ribosome to date18, 19 (Figure 2 c). However, it is likely that the foldamer helix is unfolded during the actual process of translation, as we have previously observed the trend foldamers with lower helical propensities are accepted by the ribosome with higher efficiency.12, 13 As well as having the ability to unfold, it could be important that, when unfolded, 7 has a width comparable to that of the largest canonical aromatic amino acids.
Incorporation of Amf derivatives 1–5 and 7 was then quantified by means of radioisotopes (RI, Table 1, and Supporting Information, Figures S4 and S5). It became immediately apparent that, for both templates, the increase in the size of the appended foldamer moiety resulted in a decrease in the translation efficiency. Derivatives 1 and 2 were incorporated the best, followed by Amf bearing QQ and PQ dimers (3 and 5, respectively) and finally Amf bearing the QPQ trimer (7), which can span over a full helix turn. The incorporation of negatively charged Amf(‐Gly‐(QAsp)2‐Ac) 4 did not yield the desired product in a sufficient amount, hampering its quantification. A comparison of the incorporation efficiency for the Amf‐bearing monomers (1 and 2) and homodimers (3 and 4) clearly indicates that oligoamides composed of QDap are incorporated better than their QAsp analogues, which is consistent with our recent studies on translation initiation.13
The translated products were systematically accompanied by a truncated peptide, which depended on the template, shedding light on different aspects of elongation. The truncated peptides obtained with template T1 (Figure 2 b) revealed some difficulty in forming the Ala–Amf peptide bond: the peptide is truncated before Amf. This could come from poor delivery of Amf–tRNA to the ribosome. It may also be that the α‐amine of Amf, once accommodated at the A‐site, cannot easily attack the peptidyl tRNA ester bond in the peptidyl transferase center. Even though the foldamer unit is remote from the α‐amino group of Amf, its bulk could result in unproductive presentation of the α‐amine (Figure 2 d). The truncated peptides obtained with template T2, further highlight the difficulty to form the Ala–Amf bond (Figure 2 d): α‐amino acid residues that precede Amf are missing in the truncated sequence. Intriguingly, however, this does not prevent the sequence from being further elongated. Amf–tRNA must somehow move to the P‐site and elongation starts again from there. Formation of similar truncated peptides has been observed during the incorporation of certain d‐amino acids.1 For all substrates tested, excluding substrate 4, the incorporation efficiency is higher with T1 than with T2 (Table 1). C‐terminal incorporation of Amf‐foldamers thus appears to be more efficient than elongation but this hypothesis has not been investigated in detail.
Encouraged by these results, we set out to incorporate more than one foldamer‐containing Amf residue, either consecutively or spaced by one or two Ala residues. We thus designed templates T3–T8 (Figure 3), coding for peptides with two Amf residues at different positions. Remarkably, translation was successful in all cases with substrates 1 and 2, which contain one quinoline unit. With substrate 5, which contains a PQ segment, translation was successful only when Amf residues were not consecutive. Quantification experiments with substrates 1 and 2 also showed that yields increase upon appropriate spacing between Amf residues (Supporting Information, Figures S5 and S6, and Table S1). The double incorporation of larger substrate 3 failed in all cases, in contrast with its single incorporation. Even larger substrates were not tested.
Figure 3.
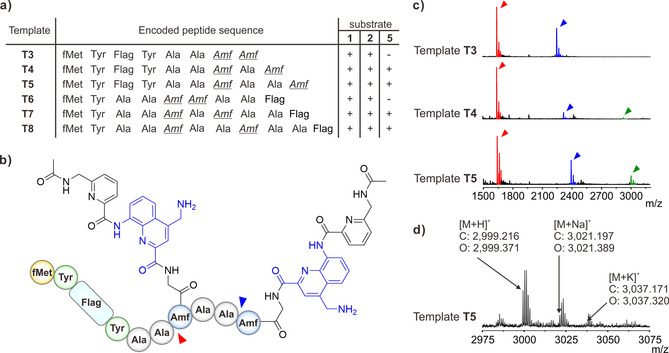
a) Peptide sequences used for the investigation of the dual incorporation of foldamer‐containing α‐amino acids. Codon assignment is the same as for the single incorporation. b) Desired product upon incorporation of 5 into template T5. Red and blue arrowheads indicate the truncation sites. c) MALDI‐TOF‐MS spectra, showing the dual incorporation of Amf(‐Gly‐QDapP‐Ac) (5) on templates T3–T5. The green arrowheads indicate peaks of the fully translated product. Red arrowheads indicate mass peaks of the peptide lacking both foldamer‐containing α‐amino acids, while blue arrowheads indicate peaks of the peptide only lacking the C‐terminal Amf residue. d) Magnified MALDI‐TOF‐MS spectrum showing the dual incorporation of Amf(‐Gly‐QDapP‐Ac) (5) on template T5. The calculated mass (C) and observed mass (O) for [M+H]+, [M+Na]+, and [M+K]+ of the product are shown.
Finally, we challenged the ribosome to combine a quinoline unit as the initiator, a second one appended within the peptide sequence, and a final one with a different sidechain appended at the C‐terminus of the peptide, in a cyclic thioether peptide. ClAc‐QDap‐Gly‐Phe‐CME was charged on tRNAfMet CAU and supplemented along with Amf(‐Gly‐QDap‐Ac)‐tRNAPro1E2 GGU and Amf(‐Gly‐QAsp‐Ac)‐tRNAPro1E2 CAU to a Met‐free and Thr‐free FIT system for the translation of T9 (Figure 4 a). This template also encodes for a Cys meant to react with the N‐terminal chloroacetamide to form the aromatic oligoamide–peptide hybrid macrocycle.24 The correct topology of the hybrid macrocycle was further confirmed by using a similar template but lacking the Cys residue (T2). The absence of cyclization (Supporting Information, Figure S9) validated the selective thiol‐chloroacetyl reaction on template T9. Despite the presence of a small number of truncated products, the complex macrocycle shown in Figure 4 b was indeed the major product, highlighting the potential of the ribosome to assemble polymers from amino acids significantly deviating from its original substrates.
Figure 4.
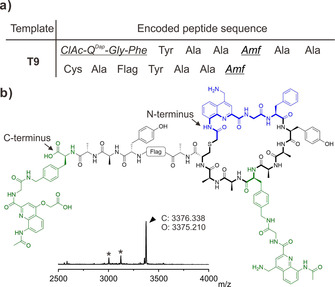
a) Sequence encoded by template T9. b) MALDI‐TOF‐MS spectrum and chemical structure of the expressed macrocycle showing the formation of the desired product. The initiation unit and Amf elongation units are depicted in blue and green, respectively. The calculated mass (C) and observed mass (O) for [M+H]+ of the fully translated products are shown. Asterisks (*) indicate translation by‐products (shown in the Supporting Information).
In summary, we have shown that the ribosome tolerates short foldamers not only as an initiation unit but also as a sidechain appendage within or at the C‐terminus of a peptide. Foldamer‐containing initiation and elongation units may be even combined to generate novel foldamer–peptide hybrids. The sidechain of Amf(‐Gly‐QPQ‐Ac), which spans a full foldamer helix turn, is by far the largest and most structurally defined ever incorporated within a peptide sequence by the ribosome. Multiple directions exist to further improve the breadth of these reactions including, but not limited to, the optimization of tRNA units.2, 25, 26 Interest in increasing the diversity of mRNA‐encoded chemical entities is driven by the possibility to implement selection from very large libraries. Non‐peptidic features may enhance resistance to protease degradation, cell penetration or, in the case of foldamers, conformational control. The benefits of foldamers in this respect are currently being investigated.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by Grant‐in‐Aid for JSPS Postdoctoral Fellowship for Research in Japan (Standard) (P18727) to C.T.; a Joint ANR‐JST grant (ANR‐14‐JITC‐2014‐003 and JST‐SICORP) to I.H and H.S., JST CREST (JPMJCR12L2) to H.S., and by Basic Science Research Programs through the National Research Foundation grant (NRF‐2018R1C1B5086457) to S.K. Open access funding enabled and organized by Projekt DEAL.
C. Tsiamantas, S. Kwon, J. M. Rogers, C. Douat, I. Huc, H. Suga, Angew. Chem. Int. Ed. 2020, 59, 4860.
Contributor Information
Prof. Ivan Huc, Email: ivan.huc@cup.lmu.de.
Prof. Hiroaki Suga, Email: hsuga@chem.s.u-tokyo.ac.jp.
References
- 1. Fujino T., Goto Y., Suga H., Murakami H., J. Am. Chem. Soc. 2013, 135, 1830–1837. [DOI] [PubMed] [Google Scholar]
- 2. Katoh T., Tajima K., Suga H., Cell Chem. Biol. 2017, 24, 46–54. [DOI] [PubMed] [Google Scholar]
- 3. Katoh T., Suga H., J. Am. Chem. Soc. 2018, 140, 12159–12167. [DOI] [PubMed] [Google Scholar]
- 4. Fujino T., Goto Y., Suga H., Murakami H., J. Am. Chem. Soc. 2016, 138, 1962–1969. [DOI] [PubMed] [Google Scholar]
- 5. Yamagishi Y., Shoji I., Miyagawa S., Kawakami T., Katoh T., Goto Y., Suga H., Chem. Biol. 2011, 18, 1562–1570. [DOI] [PubMed] [Google Scholar]
- 6. Rogers J. M., Suga H., Org. Biomol. Chem. 2015, 13, 9353–9363. [DOI] [PubMed] [Google Scholar]
- 7. Murakami H., Ohta A., Ashigai H., Suga H., Nat. Methods 2006, 3, 357–359. [DOI] [PubMed] [Google Scholar]
- 8. Goto Y., Suga H., J. Am. Chem. Soc. 2009, 131, 5040–5041. [DOI] [PubMed] [Google Scholar]
- 9. Torikai K., Suga H., J. Am. Chem. Soc. 2014, 136, 17359–17361. [DOI] [PubMed] [Google Scholar]
- 10. Ad O., Hoffman K. S., Cairns A. G., Featherston A. L., Miller S. J., Söll D., Schepartz A., ACS Cent. Sci. 2019, 5, 1289–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee J., Schwieter K. E., Watkins A. M., Kim D. S., Yu H., Schwarz K. J., Lim J., Coronado J., Byrom M., Anslyn E. V., Ellington A. D., Moore J. S., Jewett M. C., Nat. Commun. 2019, 10, 5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rogers J. M., Kwon S., Dawson S. J., Mandal P. K., Suga H., Huc I., Nat. Chem. 2018, 10, 405–412. [DOI] [PubMed] [Google Scholar]
- 13. Tsiamantas C., Kwon S., Douat C., Huc I., Suga H., Chem. Commun. 2019, 55, 7366–7369. [DOI] [PubMed] [Google Scholar]
- 14. Ezure T., Nanatani K., Sato Y., Suzuki S., Aizawa K., Souma S., Ito M., Hohsaka T., von Heijine G., Utsumi T., Abe K., Ando E., Uozumi N., PLoS One 2014, 9, e112874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiang H., Léger J.-M., Huc I., J. Am. Chem. Soc. 2003, 125, 3448–3449. [DOI] [PubMed] [Google Scholar]
- 16. Kudo M., Maurizot V., Kauffmann B., Tanatani A., Huc I., J. Am. Chem. Soc. 2013, 135, 9628–9631. [DOI] [PubMed] [Google Scholar]
- 17. Hu X., Dawson S. J., Nagaoka Y., Tanatani A., Huc I., J. Org. Chem. 2016, 81, 1137–1150. [DOI] [PubMed] [Google Scholar]
- 18. Hohsaka T., Kajihara D., Ashizuka Y., Murakami H., Sisido M., J. Am. Chem. Soc. 1999, 121, 34–40. [Google Scholar]
- 19. Kajihara D., Abe R., Iijima I., Komiyama C., Sisido M., Hohsaka T., Nat. Methods 2006, 3, 923–929. [DOI] [PubMed] [Google Scholar]
- 20. Sánchez-García D., Kauffmann B., Kawanami T., Ihara H., Takafuji M., Delville M.-H., Huc I., J. Am. Chem. Soc. 2009, 131, 8642–8648. [DOI] [PubMed] [Google Scholar]
- 21. Baptiste B., Douat-Casassus C., Laxmi-Reddy K., Godde F., Huc I., J. Org. Chem. 2010, 75, 7175–7185. [DOI] [PubMed] [Google Scholar]
- 22. Vallade M., Sai Reddy P., Fischer L., Huc I., Eur. J. Org. Chem. 2018, 5489–5498. [Google Scholar]
- 23. Murakami H., Kourouklis D., Suga H., Chem. Biol. 2003, 10, 1077–1084. [DOI] [PubMed] [Google Scholar]
- 24. Goto Y., Ohta A., Yamagishi Y., Murakami H., Suga H., J. Am. Chem. Soc. 2008, 130, 120–129. [DOI] [PubMed] [Google Scholar]
- 25. Katoh T., Iwane Y., Suga H., Nucleic Acids Res. 2017, 45, 12601–12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Katoh T., Wohlgemuth I., Nagano M., Rodnina M. V., Suga H., Nat. Commun. 2016, 7, 11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary