

Summary
Whole‐genome annotation error that omits essential protein‐coding genes hinders further research.
We developed Target Gene Family Finder (tgfam‐finder), an alternative tool for the structural annotation of protein‐coding genes containing target domain(s) of interest in plant genomes. tgfam‐finder took considerably reduced annotation run‐time and improved accuracy compared to conventional annotation tools.
Large‐scale re‐annotation of 50 plant genomes identified an average of 150, 166 and 86 additional far‐red‐impaired response 1, nucleotide‐binding and leucine‐rich‐repeat, and cytochrome P450 genes, respectively, that were missed in previous annotations. We detected significantly higher number of translated genes in the new annotations using mass spectrometry data from seven plant species compared to previous annotations.
tgfam‐finder along with the new gene models can provide an optimized platform for comprehensive functional, comparative, and evolutionary studies in plants.
Keywords: CYP450, FAR1, plant defense, plant genomics, NLR, structural gene annotation
Introduction
The structural annotation of protein‐coding gene is an essential post‐assembly process for further research (Jones, 2006). To date, hundreds of plant genomes have been sequenced and the sequences were deposited in public databases. Researchers generally consider that whole genome annotations contain all translated, transcribed and inactive genes and thus use them for subsequent functional genomics, evolutionary analyses, and biotechnology applications. However, there are continuous reports of annotation errors, including imperfect gene models and missing functional genes (Lee et al., 2007; Pertea & Salzberg, 2010; Lagarde et al., 2017; Bayer et al., 2018; Pertea et al., 2018). These reports demonstrate that the accidental omission of essential genes can ultimately generate biases in downstream research.
Cataloging translated genes into proteins is an important step in identification of proteins that control cellular processes and essential functions of organisms. To detect whole proteins in genomes, researchers generally exploit publicly available gene models, together with experimental methods such as mass spectrometry (Wilhelm et al., 2014; Gupta et al., 2018). However, recent studies reported crucial problems with imperfect annotations that had omitted numerous translated genes: previously undiscovered proteins were identified that had been absent from existing gene models (Marx et al., 2016; Frankish et al., 2018; Pertea et al., 2018). For example, Marx et al. (2016) reported the detection of hundreds of novel proteins that have not been described for Medicago truncatula before, suggesting the importance of continuous updates for improving annotation quality. Although researchers agree that improving existing gene models is essential to identify all protein‐coding genes and gene candidates, such as expressed genes or pseudogenes, the requirements of human labor, computational resources, and experimental validation of the results make this process difficult to achieve. Pre‐existing gene models are continuously being improved using manual, computational, and experimental analyses only for certain model species, such as human, mouse, and Arabidopsis (see URLs). However, the majority of published gene annotations is not updated. Hence, inaccurate or missing gene models remain in the annotations and will likely cause problems in downstream applications.
In general, the potential function of a gene is predicted based on the identification of conserved domains or motifs. Studies focusing on specific genes or gene families often begin working with annotated gene models by identifying those genes of interest that contain the appropriate target domain(s) or motifs (Lee et al., 2007; Jupe et al., 2013). Therefore, researchers have designed novel approaches to identify specific genes or gene families (Teer & Mullikin, 2010; Jupe et al., 2013; Lagarde et al., 2017; Li et al., 2018). For example, Jupe et al. (2013) developed resistance gene enrichment and sequencing (RenSeq), a high‐throughput sequencing method used for the selective capture and sequencing of nucleotide‐binding and leucine‐rich‐repeat (NLR) genes without whole‐genome sequencing. Although such methods enable the detection of candidate regions containing target genes, further annotation to determine an accurate gene structure in these regions remains a bottleneck.
Here, we present Target Gene Family Finder (tgfam‐finder), an alternative tool for the automatic structural annotation of all protein‐coding genes containing specific target domain(s) in assembled genomes. We verified that tgfam‐finder had enhanced performance via prediction for far‐red‐impaired response 1 (FAR1) transcription factor, NLR, and cytochrome P450 (CYP450) gene families in Arabidopsis, rice and maize genomes compared to maker2 (Holt & Yandell, 2011) and gemoma (Keilwagen et al., 2016), popular annotation tools, in terms of annotation accuracy, coverage, and run‐time. Then, we evaluated tgfam‐finder through a massive re‐annotation of FAR1, NLR, and CYP450 gene families in 50 plant genomes. Many predictions of tgfam‐finder do not overlap with any currently annotated gene model. Furthermore, proteomic analyses of seven plant species using publicly available mass spectrometry data revealed that significantly more protein‐coding genes of those families were abundant in the newly annotated genes compared to previous annotations. tgfam‐finder, a domain search‐based gene annotation tool, could provide an alternative solution for target‐gene family annotation in functional, comparative, and evolutionary studies.
Materials and Methods
Overview of tgfam‐finder
tgfam‐finder was developed to run in the Linux OS environment. For novices in bioinformatics‐based analyses, we constructed an installation package that allows auto‐installation of prerequisite tools needed to run tgfam‐finder (Supporting Information Fig. S1). Using the install package, prerequisite tools, including exonerate‐2.2.0 (Slater & Birney, 2005), augustus‐3.2.3 (Stanke et al., 2006), ISGAP Pipeline (Kim et al., 2015), tophat‐2.1.1 and cufflinks‐2.2.1 (Ghosh & Chan, 2016), bowtie2‐2.3.1 (Langmead & Salzberg, 2012), hmmer‐3.1b2 (Mistry et al., 2013), blast 2.6.0+ (Camacho et al., 2009), interproscan‐5.22‐61.0 (Jones et al., 2014), blat v.35 (Kent, 2002), scipio‐1.4 (Keller et al., 2008), and clustalw‐2.1(Larkin et al., 2007) are provided for further annotation. To run tgfam‐finder, users need to configure the location information of genomic resources and the prerequisite programs in ‘RESOURCE.config’ and ‘PROGRAM_PATH.config’. Basically, ‘PROGRAM_PATH.config’ is automatically generated through the auto‐installation process. Whereas, users should enter the location of the target genome, peptide sequences of target or allied species, and peptide sequences including target domains in various species as minimum resources. To classify specific proteins having target domain(s) of interest, tgfam‐finder requires the location of functional annotation information of target or allied species formatted as tsv and target domain ID(s) in ‘RESOURCE.config’. Moreover, users can input ‘EXTENSION_LENGTH’ to determine target regions for further annotation and ‘MAX_INTRON_LENGTH’ for alignment processes using proteins. For extra configuration, users can also register the location of transcriptome, genomic position of genes and coding DNA sequences of target species in ‘RESOURCE.config’. Because interproscan is not suitable for identification and classification of genes having short target domain(s) such as C2H2 zinc finger, tgfam‐finder provides an additional search option using hmmer. If users register HMM_MATRIX_NAME for the location of hmm matrix for specific domain(s), tgfam‐finder annotates target genes based on searches using hmmer as well as interproscan.
The annotation pipeline of tgfam‐finder consists of three steps: (1) determination of target regions using ‘0.SixFrameTranslation.pl’ and ‘1.Domain_Identification.pl’, (2) gene prediction in the target regions via ‘2.Auto_ProteinMapping.pl’, ‘3.Auto_ISGAP.pl’ and ‘4.Auto_Augustus.pl’, and (3) generation of the final gene model through ‘5.Generating_FinalGeneModel.pl’ (Fig. 1). To identify the positions of target domains in an assembled genome, tgfam‐finder generates six‐frame‐translated genome sequences. Then, a hidden Markov model matrix is constructed through alignments among target domain(s) in protein sequences of target or allied species using clustalw2 (Larkin et al., 2007). After identification of genomic regions containing target domain(s) using hmmer (Mistry et al., 2013), target regions including the target domain(s) and their flanking sequences are determined.
Fig. 1.
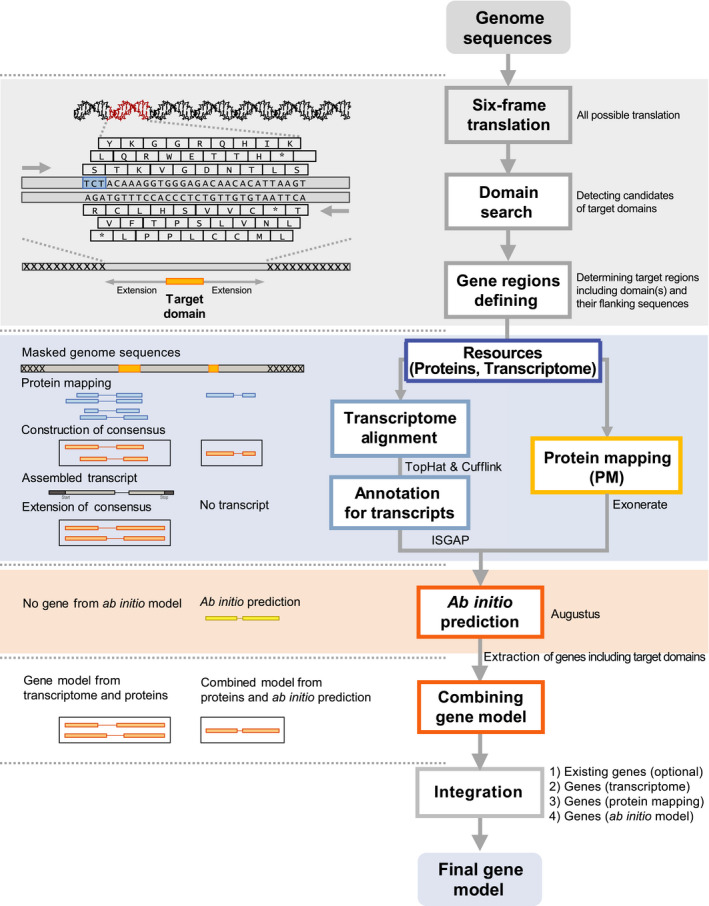
Annotation process of tgfam‐finder. An automated process for new identification of target‐gene families using tgfam‐finder is depicted. The diagram shows serial processes starting from six‐frame translation to generation of the final gene model. The gray block of the diagram shows the determination of target regions containing target domain(s) and their flanking sequences for further annotation. The blue and pink blocks indicate structural annotation using proteins and transcriptomes (blue), and the ab initio method (pink), respectively. Names of representative tools (Slater & Birney, 2005; Stanke et al., 2006; Kim et al., 2015; Ghosh & Chan, 2016) for structural annotation are given in the blue and pink blocks. Initial gene models are integrated from the structural annotation as depicted in the white block.
Structural annotation is conducted via the processes of protein mapping, transcriptome‐based annotation, and ab initio prediction to identify full‐length coding sequences having both start and stop codon or partial genes containing start or stop codon in the target regions. For efficient protein mapping, tgfam‐finder detects proteins with homology to target regions in the resource peptide sequences using Blast+, and aligns between the proteins and matched target regions using exonerate (Slater & Birney, 2005). Transcriptome‐based annotation is implemented in the order of reference‐guided transcriptome assembly using tophat and cufflinks (Ghosh & Chan, 2016), and annotation via ISGAP Pipeline (Kim et al., 2015). For ab initio gene prediction, the training set is constructed using the protein sequences of target or allied species having target domain(s), and the gene models generated from protein mapping and transcriptome‐based annotation. Then, augustus (Stanke et al., 2006) generates the gene model based on the training set. After generation of initial gene models, tgfam‐finder classified genes with target domain(s) in the initial gene models and combined partial genes from transcriptome‐based annotation or protein mapping with intact genes from augustus if those partial genes have corresponding exon/ intron structure with intact genes from augustus. Finally, the final gene model is generated in the following order: from (1) intact genes from transcriptome‐based annotation and (2) protein mapping, (3) intact genes by combining partial genes from transcriptome‐based annotation with augustus and (4) partial genes from protein‐mapping with augustus, (5) intact genes from augustus and (6) partial genes from transcriptome‐based annotation and (7) protein mapping.
Comparison of annotation accuracy for tgfam‐finder and publicly available annotation tools
To estimate annotation accuracy of tgfam‐finder with maker2 v.2.31.10 (Holt & Yandell, 2011) and gemoma v.1.6.2 (Keilwagen et al., 2016), we first annotated FAR1, NLR, and CYP450 genes in Arabidopsis, rice and maize genomes using those tools. As input resources, we exploited same proteins of those families in 50 plant genomes except for the target genomes and transcriptome of target species described in Table S1. In total, we generated five versions of predicted gene models using maker2 and gemoma with different parameters and one gene model from tgfam‐finder using default parameter for each family in each genome as described in Dataset S1. Then, we used those gene models as predictions and the previous annotations as references to evaluate annotation performance of those tools. After finishing the annotation, we counted the total number of matched and unmatched features (nucleotide, exon and gene) in references as well as predictions using gffcompare with ‐r ‐o parameters (see URLs). Then, sensitivity, specificity, positive predictive values (PPVs) and negative predictive values (NPVs) of the predictions from those tools were calculated at the nucleotide, exon and gene levels as described in Fig. S2. All annotation parameters, statistics of annotations and sensitivity, specificity, PPVs and NPVs with classification of non‐overlapping and overlapping genes are provided in Dataset S1. Fragments per kilobase of transcript per million reads mapped (FPKM) values of the annotated genes from tgfam‐finder, gemoma and maker2 were estimated using RNA‐Seq described in Table S1 by hisat2 (Kim et al., 2019) and stringtie (Pertea et al., 2015) with options ‐e ‐B ‐G ‐o. Manual inspection of the annotated genes was performed using Integrative Genomics Viewer (Robinson et al., 2011).
Structural annotation of target‐gene families
We used assembled genomes and proteins described in Table S1 as ‘TARGET_GENOME’ and ‘PROTEINS_FOR_DOMAIN_IDENTIFICATION’ in ‘RESOURCE.config’. After performing functional annotation using interproscan‐5 (Jones et al., 2014) for the proteins, generated tsv files for the proteins were used as ‘TSV_FOR_DOMAIN_IDENTIFICATION’. PF03101 (FAR1), PF00931 (NLR) and PF00067 (CYP450) were selected as ‘TARGET_DOMAIN_ID’ for classification of target‐gene families. Because lengths of full genomic DNA sequences for existing FAR1, NLR, and CYP450 genes in maize, pepper, barley, and wheat genomes as large plant genomes were < 70 kb, ‘EXTENSION_LENGTH’ and ‘MAX_INTRON_LENGTH’ were determined as 100 kb. We extracted target genes in the existing gene models of plants having the Pfam IDs in the tsv files and then merged them to use as ‘RESOURCE_PROTEIN’. Location of transcriptome, gff3, and coding DNA sequences of the plant genomes were also recorded in ‘RESOURCE.config’.
Genes in new gene models generated from tgfam‐finder are classified as three categories: (1) pre‐existing genes, (2) newly identified genes that do not share any genomic position with any existing target genes, and (3) new intact genes overlapping with existing partial target genes which have no start or stop codon. In this study, we considered (2) and (3) as newly annotated genes from tgfam‐finder. Because tgfam‐finder replaced existing partial target genes in previous annotations to new intact gene structures, the numbers of pre‐existing target genes in previous annotations and final gene models are different. For example, the average numbers of existing NLR genes in the previous annotations and new gene model from tgfam‐finder were 328 and 316, respectively, because the 12 partial NLR genes in the previous annotations were replaced by 13 new intact NLR genes overlapping with the 12 genes from tgfam‐finder in the new gene models.
Phylogenetic analyses of the new gene models
To perform a phylogenetic comparison of the new gene models for FAR1, NLR and CYP450 in plant genomes, we aligned the amino acid sequences of target domains in each genome using clustalw2 (Larkin et al., 2007), and constructed the phylogenetic trees of each gene family in the specific plant genomes with mega7 (Kumar et al., 2016) using the neighbor‐joining method with 1000 bootstraps and partial deletion options (90%).
Proteomic validation of the newly annotated genes
To validate translation of the previously and newly annotated genes, we collected raw mass spectrometry data of rice, barley, pepper, grape, bean, apple and Eucalyptus genomes from the PRIDE database (Vizcaino et al., 2014; Li et al., 2016; de Santana Costa et al., 2017; Guo et al., 2017; Mahalingam, 2017; Min et al., 2017; Gupta et al., 2018; Kambiranda et al., 2018; Meng et al., 2018). We performed proteome mapping against the previously and newly annotated genes in the seven plant genomes, respectively using maxquant v.1.6.2.3 (Cox & Mann, 2008). The list of validated protein‐coding genes in ‘proteinGroup.txt’ is provided in Dataset S2 with parameter information in Dataset S3.
Identification of genes overlapping with repeat sequences
To annotate genomic sequences containing non‐overlapping genes along with previously annotated genes, we performed repeat annotation for the genomic regions using repeatmodeler and repeatmasker (see URLs). De novo repeat libraries of each plant genome were constructed using repeatmodeler, and then repeatmasker was used to repeat masking on the repeat libraries. If specific repeat sequences covered > 50% of a non‐overlapping gene, the gene is considered as resided in the genomic region containing the specific repeat sequence.
Computational resources used to run tgfam‐finder
For the annotation of FAR1, NLR and CYP450 genes in the 50 plant genomes, we used two computer servers with the following specifications: Intel Xeon CPU E5‐2697 v2 at 2.70 GHz, 48 processors, and 264 Gb memory, and Intel Xeon CPU E5‐4650 v2 at 2.40 GHz, 80 processors, and 512 Gb memory. The re‐annotation of each gene family was completed within 1 wk using those servers. To estimate the annotation run‐time of tgfam‐finder, gemoma and maker2, we performed annotation on default parameters using the server computer (Intel Xeon CPU E5‐4650 v2 at 2.40 GHz, 80 processors, and 512 Gb memory) with four processors for FAR1, NLR and CYP450 gene families in Arabidopsis, rice, bean, maize, pepper and wheat genomes, respectively. For efficient test, we randomly extracted and used c. 10 Gb of whole transcriptome data and used the same previously annotated genes.
Data availability
The newly annotated gene sequences in plants are deposited in Dataset S4. The new gene models including peptide and coding DNA sequences with gff3 and tsv are accessible at http://tgfam‐finder.snu.ac.kr/.
Code availability
tgfam‐finder program package including the auto‐installation and annotation scripts with sample data is accessible at https://github.com/tgfam‐finder and http://tgfam‐finder.snu.ac.kr/.
URLs
GENECODE (human and mouse), https://www.gencodegenes.org/
TAIR (Arabidopsis), https://www.arabidopsis.org/
repeatmodeler, http://www.repeatmasker.org/RepeatModeler/
repeatmasker, http://www.repeatmasker.org/
gffcompare, https://ccb.jhu.edu/software/stringtie/gffcompare.shtml
Results
Conceptual overview of tgfam‐finder
tgfam‐finder is a refined annotation tool designed to identify any target‐gene family of interest in assembled genomes. tgfam‐finder was developed for ease of use from installation to completion of structural annotation, even for novice bioinformaticians. To this end, we provide additional tool packages enabling automated installation of prerequisite tools for further structural gene annotation using tgfam‐finder without any manual configuration (Fig. S1).
An automated annotation process using tgfam‐finder consists of the following three steps: (1) genome‐wide identification of target genomic regions containing specific target‐gene sequences of interest; (2) structural annotation of target regions using available proteins, transcriptomes, and ab initio prediction; and (3) construction of the final gene model (Fig. 1). One of the distinct features of tgfam‐finder is the extraction of target regions containing sequences of a particular target‐gene family. To reduce annotation time and unnecessary computation, tgfam‐finder identifies all genomic regions containing domain(s) of the target genes using hmmer (Mistry et al., 2013) from six‐frame‐translated genome sequences. The target regions are determined after masking unnecessary sequences as ‘X’, except for the identified genomic regions and their flanking sequences (Fig. 1). Then, the structural annotation of target regions is performed to generate the initial gene model through serial processes of protein mapping, transcriptome annotation, and ab initio prediction (Fig. 1). Based on the evidence gathered in the previous steps, tgfam‐finder combines the initial models and determines the final gene model of target gene families (Fig. 1).
tgfam‐finder improves upon pre‐existing gene models via the identification of missing essential genes, providing a refined model of target‐gene families. To evaluate tgfam‐finder, we collected genomic data for 50 plants, including assemblies, annotated genes, and transcriptome data from public databases (Table S1). In the plant genomes, we searched for FAR1 transcription factor family that modulates phytochrome A signaling (Hudson et al., 1999), NLR gene family that typically contains plant cytoplasmic immune receptors (Jacob et al., 2013), and CYP450 gene family that is involved in the biosynthesis of plant hormones, secondary metabolites, and defensive compounds (Schuler & Werck‐Reichhart, 2003) as target‐gene families. Because it is known that those genes are co‐localized with transposable elements in plant genomes (Feschotte, 2008; Jacob et al., 2013; Kim et al., 2017; Kim & Choi, 2018), we assumed their accidental omission during annotation and thus selected those families as target gene families for re‐annotation in this study.
Evaluation of annotation accuracy and run‐time of tgfam‐finder
Before re‐annotation of 50 plant genomes, we evaluated the annotation accuracy of tgfam‐finder compared to that of gemoma (Keilwagen et al., 2016) and maker2 (Holt & Yandell, 2011. We annotated FAR1, NLR and CYP450 gene families to compare the prediction of tgfam‐finder with gemoma (v.1.6.2) and maker2 (v.2.3.10) in three plant genomes comprising Arabidopsis thaliana, rice and maize. For gemoma and maker2, we used five different parameter sets to generate the predictions for each species and each gene family and compared them to the predictions of tgfam‐finder with default parameters (Dataset S1). In total, 99 predictions were generated for three families in three genomes from 11 different trials (one, five and five for tgfam‐finder, gemoma and maker2, respectively). To assess and compare each tool accurately, we did not use the previously annotated target genes of target genomes during the annotation process. We matched those predicted gene models from each tool to the previously annotated genes and calculated sensitivity, specificity, PPV and NPV at the nucleotide, exon, and gene levels as described in the Methods section (Dataset S1; Figs S2–S4).
We first grouped the gene models by 11 distinct trials to determine the effect of different parameters on the resulting gene models (Figs 2, S3A). As a result, higher sensitivity and NPV were observed in TGFam‐Finder_1st, GeMoMa_3rd, GeMoMa_4th, GeMoMa_5th, Maker_4th and Maker_5th (Figs 2a,b, S3A). By contrast, their specificity and PPV were lower than those of other gene models (Figs 2a,b, S3A). Because those gene models contained a higher number of newly identified genes absent in references, their lower specificity and PPV were a direct result of a higher number of newly identified genes (unmatched features in predictions) considered as false positive (Fig. 2c). Therefore, we mainly considered sensitivity (NPV) to evaluate how many features in references (predictions) are correctly matched in predictions (references) excluding the confounding effect of newly identified genes. Specifically, both individual and average sensitivity and NPV were higher in the gene model of tgfam‐finder than in other tools at nucleotide, exon, gene levels, indicating that tgfam‐finder could more accurately annotate all features in the reference than gemoma and maker2 (Figs 2a,b, S3A). Moreover, tgfam‐finder could annotate a number of newly identified genes absent in the reference with almost no missing reference genes compared to gene models from other tools (Fig. 2c,d). Considering the number of genes overlapping with references, their overlapping degree and the number of newly identified and omitted genes, our analyses demonstrate that annotation of tgfam‐finder had better coverage than those of other tools (Fig. 2c–e).
Fig. 2.

Comparison of annotation accuracy for gene models grouped by distinct trials from tgfam‐finder, gemoma and maker2. (a, b) Average sensitivity and specificity (a) with average positive predictive values (PPVs) and negative predictive values (NPVs) (b) of annotated genes from tgfam‐finder, gemoma and maker2 in Arabidopsis, rice and maize are depicted. The x‐ and y‐axes represent trial names and average of those evaluation values, respectively. (c) The number of newly identified genes (i.e. predicted genes absent in references; x‐axis) and the number of missed genes (i.e. reference genes omitted in predicted gene models; y‐axis) are depicted as dot plots. (d) The ratio of the number of newly identified genes in predictions to the number of omitted reference genes for each trial. (e) The x‐ and y‐axes indicate the number of predicted genes overlapping with references and the ratio of the number of overlapping predicted genes to the number of overlapping reference genes, respectively. The left (right) plot depicts genes sharing any (over 90%) coding sequence regions between references and gene models.
Secondly, we grouped 99 gene models by family or species for each trial to build 33 combinations of gene models for analysis. Similar to the result of the 11 combined gene models described earlier, overall sensitivity and NPV were higher in the gene models of tgfam‐finder for both families and genomes than in gene models from gemoma and maker2 (Figs 3a,b, S3B,C). This indicates that tgfam‐finder could improve gene models for not only those families in each genome but also each family in those genomes. In particular, we observed notable differences in accuracy between tgfam‐finder and others in the results of NLR and maize genome, indicating enhanced performance of tgfam‐finder especially for the annotation of NLR and the maize genome (Fig. 3a,b). Furthermore, tgfam‐finder could detect a number of omitted genes in references as newly identified genes with a few missing reference genes especially for FAR1 and maize genome (Fig. 3c,d). The number of predicted genes overlapping with references was higher in gene model of tgfam‐finder with higher overlapping proportions for each family and each genome (Fig. 3d,e). When we finally investigated 99 individual gene models, higher sensitivity and NPV of gene models from tgfam‐finder were observed compared to other tools except for Arabidopsis FAR1 and rice CYP450 genes (Fig. S4). Those values were similar in rice CYP450 genes between tgfam‐finder and other tools but lower in Arabidopsis FAR1 annotation of tgfam‐finder.
Fig. 3.
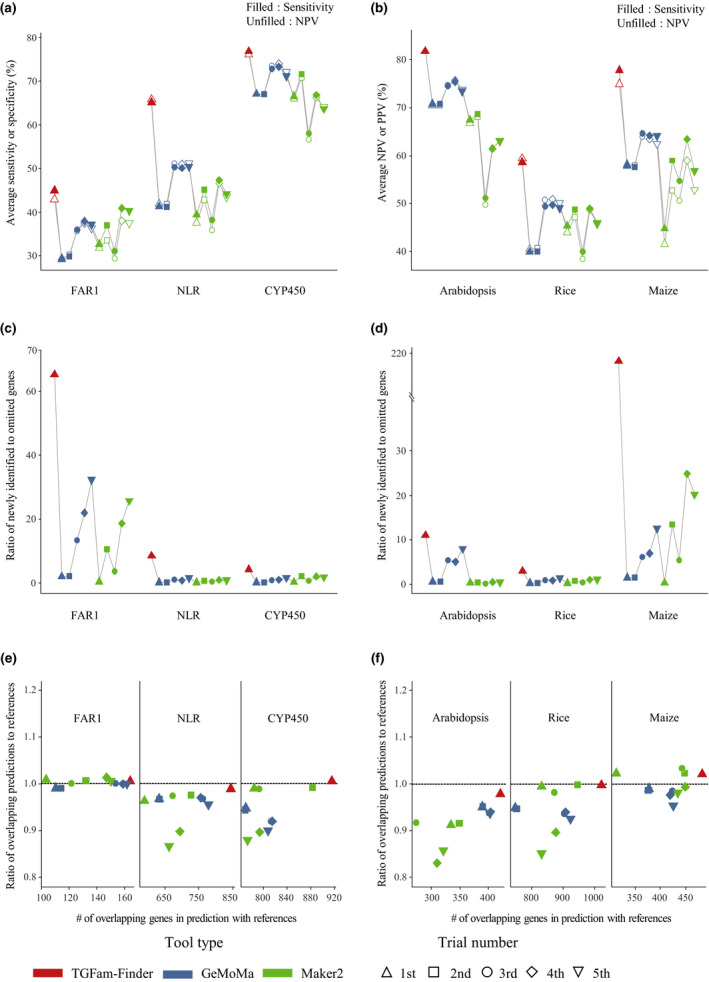
Evaluation of annotation from tgfam‐finder, gemoma and maker2 considering families and species. (a, b) Sensitivity and negative predictive values (NPVs) of 33 gene models grouped by (a) families and (b) species from tgfam‐finder, gemoma and maker2 are depicted as line graphs. (c, d) The line graphs indicate the ratio of the number of newly identified genes (i.e. predicted genes absent in references) to the number of omitted genes (i.e. reference genes omitted in predicted gene models) for gene models grouped by (c) families and (d) species. (e, f) The dot plots represent the number of predicted genes overlapping with reference genes (x‐axis) and the ratio of the number of overlapping predicted genes to the number of overlapping reference genes (y‐axis) for gene models combined by families (e) and species (f).
Manual curation of gene models from tgfam‐finder revealed that the omission of reference genes in the tgfam‐finder annotation was likely due to deficient annotation evidence, such as no or partial protein mapping, or elimination during automated final gene model generation considering the order of priority based on annotation evidence (Fig. S5). Moreover, we verified a false positive annotation case from tgfam‐finder as well as correct annotations (Fig. S6). This represents the limitation of current methods for automatic gene annotation process as well as the importance of manual annotation. We also verified expression of 296, 233 and 301 genes absent in references from tgfam‐finder, gemoma _5th and Maker2_5th with FPKM values ≥ 10, respectively, containing higher numbers of newly identified genes than other trials from each tool (Table S2). This suggests that many of the newly identified genes were truly expressed.
The full annotation of a large genome can take several weeks or longer to complete and requires an enormous amount of computational resources (Yandell & Ence, 2012). To evaluate the performance of tgfam‐finder, we estimated its actual annotation run‐time for representative plant genomes ranging from c. 100 Mb to c. 13 Gb and compared it to that of gemoma and maker2 (Fig. 4). When we used our computer server with four processors, it took an average of 9 h to 2.25 d for each gene family of Arabidopsis (c. 100 Mb) to wheat (c. 13 Gb) (Fig. 4). Specifically, we verified that the annotation of maize (2 Gb) and pepper (3 Gb) required fewer than 24 h, indicating that users can efficiently annotate their target genes in most genomes using tgfam‐finder within a day except for huge genomes such as wheat. In contrast to the run‐time of tgfam‐finder, gemoma and maker2 took from 18 h and 2 d (Arabidopsis) to 2 wk (maize) and over 6 months (wheat) to complete the annotation, respectively. In particular, annotation run‐times of tgfam‐finder for maize, pepper, and wheat were at least over 60 times faster than those of maker2 (Fig. 4). tgfam‐finder was also faster than gemoma especially for maize genome. These results represent the efficiency of tgfam‐finder for the rapid completion of annotation, especially for large genomes containing chromosome‐scale sequences. Taken together, our analyses demonstrate that tgfam‐finder enables an accurate annotation with rapid completion considering higher annotation performance in accuracy, coverage and runtime compared to the publicly available annotation tools.
Fig. 4.
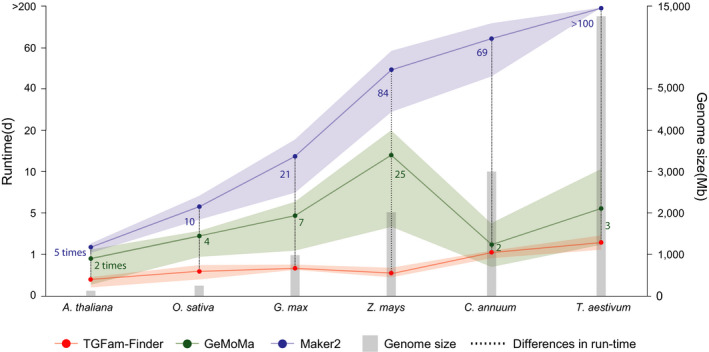
Annotation run‐times of tgfam‐finder and maker2. Line graphs indicate average annotation run‐times of FAR1, NLR, and CYP450 families in six plant genomes using tgfam‐finder (red), gemoma (green) and maker2 (navy). Red, green and navy shadings represent maximum and minimum run‐times of tgfam‐finder and maker2, respectively. The numeric values between the line graphs mean differences in run‐time between tgfam‐finder and other tools. The gray bar graph represents genome size of the six plant species.
FAR1, NLR, and CYP450 annotation in plant genomes
We re‐annotated FAR1, NLR and CYP450 genes in 50 plant genomes using tgfam‐finder (Fig. 5). Only 3.5%, 4.3% and 4.0% of the genome sequences (average genome length 1127 Mb) were determined as target regions for the re‐annotation of FAR1, NLR, and CYP450 genes, respectively, indicating that tgfam‐finder used fewer than 50 Mb of genome sequences for efficient annotation (Table S3). On average, 37 FAR1, 328 NLR and 330 CYP450 genes, considering representative loci, were identified in previously annotated gene models of the 50 plant genomes (Table S4). In addition to these, we identified 150, 166 and 86 new FAR1, NLR and CYP450 genes, respectively, using tgfam‐finder (Fig. 5; Table S4). Moreover, we found 1, 13 and 8 intact gene structures in regions containing previously annotated partial FAR1, NLR and CYP450 genes that had no start or stop codon using tgfam‐finder (Table S4). We considered these newly identified and new intact genes as newly annotated genes. In total, 94% of the newly annotated genes had both start and stop codons, indicating that the majority of the newly annotated genes had intact gene structures (Table S5). Moreover, 131 (87%), 116 (65%), and 70 (75%) of the newly annotated FAR1, NLR and CYP450 genes were located in genomic regions that had no genes identified in existing models, respectively (Fig. S7; Table S6).
Fig. 5.
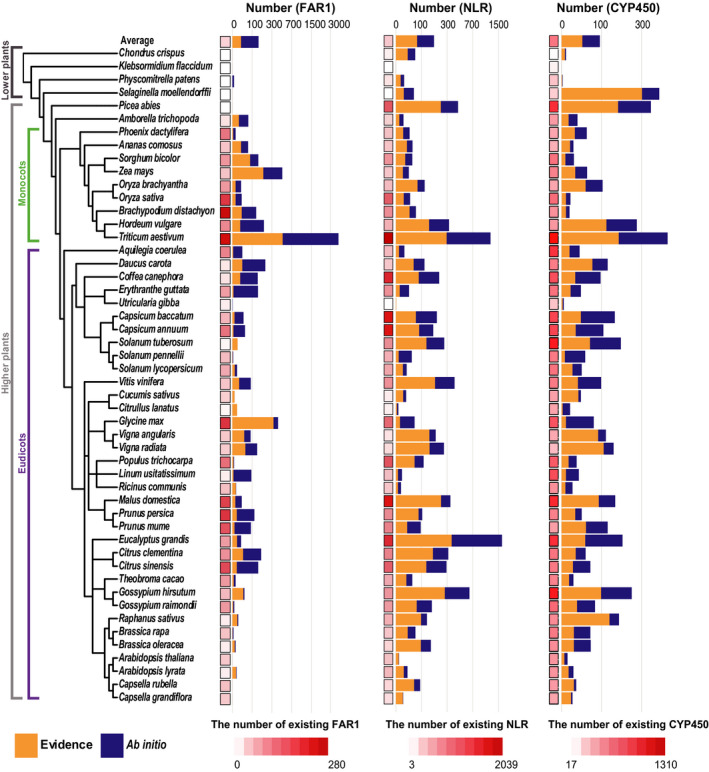
Re‐annotation of FAR1, NLR and CYP450 genes. The heat map indicates the number of existing target genes in representative loci of 50 plant genomes. Bar graphs show the number of newly annotated genes. Colors in the bar represent the number of newly annotated genes from protein or transcriptome evidence (orange) and ab initio model (navy blue).
On average, tgfam‐finder newly annotated 35 FAR1 (24%), 77 NLR (43%) and 43 CYP450 (45%) genes based on protein or transcriptome evidence (Table S7). The newly annotated genes were similar or slightly shorter than those of previously annotated genes, mainly due to the short lengths of genes generated from ab initio prediction (Fig. S8; Table S8). Finally, we constructed new gene models of FAR1, NLR and CYP450 families in the 50 plant genomes including the previously and newly annotated genes (Table S4). The average numbers of FAR1, NLR, and CYP450 genes in the new gene models of the 50 plant genomes were 188, 495 and 418, respectively that were 5‐, 1.5‐ and 1.3‐fold higher than those of the previous annotations (Table S9). This implies that only 20% (37 of 188) of FAR1, 66% (327 of 495) of NLR, and 79% (330 of 417) of CYP450 genes were annotated and used in the previous studies.
Compared to the number of genes in existing gene models, we found a large number of new FAR1 genes in 22 of 50 new gene models, more than a four‐fold higher number, or > 100 new FAR1 genes (Table S9). Specifically, 7 and 19 FAR1 genes were annotated in the existing models of carrot and maize genomes, respectively, but tgfam‐finder identified 218 and 546 new FAR1 genes in carrot and maize, respectively, a > 25‐fold relative to the number of existing genes. In the wheat genome, we detected > 4000 new FAR1 genes. For NLR genes, we found > 200 new genes or more than a two‐fold higher number in new gene models of 17 plant genomes (Table S9). Although only three NLR genes were annotated in the existing model of the Selaginella genome, 63 (> 20‐fold) more NLR genes were detected by tgfam‐finder in the same genome sequence. Furthermore, we identified over 1000 more NLR genes in both Eucalyptus and wheat genome sequences. We also identified a large number of new CYP450 genes in the Selaginella and wheat genomes (Table S9). These results indicate that tgfam‐finder greatly improves the gene models for FAR1, NLR and CYP450 by identifying a number of genes that were omitted in previous annotations.
To study the phylogenetic relationships of FAR1, NLR and CYP450 in the plant genomes, we constructed phylogenetic trees using the new gene models containing the newly annotated and previously annotated genes (Fig. S9). Our analyses revealed that a large number of newly annotated genes were distinctly clustered in specific lineages or comprised expanded groups with the small number of previously annotated genes. This finding suggests that certain gene clades had never been annotated in the earlier studies (Fig. S9).
Proteomic validation of the newly annotated genes
We collected mass spectrometry data from seven plant species in the PRIDE database (Vizcaino et al., 2014) to confirm that the newly annotated genes are indeed translated as proteins (Table S10). We implemented proteome mapping for previously and newly annotated genes in the seven genomes and compared the mapped proteins from both annotations (Figs 6, S10; Datasets S2, S3). In total, we identified mass spectrometry data evidences for 10% (795 of 7591) and 21% (991 of 4764) of previously and newly annotated genes in these plant genomes, respectively, indicating that a significantly higher proportion of the newly annotated genes are indeed translated into proteins (Fisher's exact test, P < 0.0001) (Fig. S10). We detected protein evidence for 15% (CYP450) to 23% (NLR) of newly annotated genes but only 10% (each family) of previously annotated genes (Fig. 6). When we specifically compared 21 newly annotated gene sets to their previously annotated gene sets, evidence from mass spectrometry was significantly enriched in 57% (12 of 21) of the newly annotated gene sets (Fisher's exact test, P < 0.05) (Fig. 6). Taken together, our analyses demonstrate that tgfam‐finder enabled the construction of gene models of target gene families containing previously undiscovered protein‐coding genes as well as potential gene candidates, such as expressed genes and pseudogenes.
Fig. 6.

Proteomic validation of the previously and newly annotated genes. (a–c) Bar graphs represent the percentages of protein‐coding genes in previously (sky blue) and newly (yellow) annotated genes, validated using mass spectrometry in seven plant genomes. Stars on the bar graphs indicate significant differences in protein‐coding gene abundance between the previously and newly annotated genes (Fisher's exact test, P < 0.05).
Genomic features of the newly annotated genes in plant genomes
The genomic positions of the newly annotated genes could be classified into the following three categories: (1) non‐overlapping, (2) overlapping with existing genes without target domain(s), and (3) overlapping with existing partial target genes (Fig. S7). A large portion of non‐overlapping genes overlapped repetitive sequences (Fig. S11A,B). Interestingly, the non‐overlapping FAR1 genes were remarkably resided in the regions containing DNA‐transposons. We observed that a significant number of NLR and CYP450 genes co‐localized with LTR‐retrotransposons (Fig. S11C). Considering previous descriptions of annotation processes (Yandell & Ence, 2012; Bennetzen & Park, 2018), our results suggest that the repeat masking process prior to gene annotation could have a crucial impact in generating imperfect gene models. For newly annotated genes that overlapped existing genes without target domain(s), we observed that newly annotated gene families primarily overlapped with hypothetical genes without known domain(s) (Fig. S12). This result suggests that several newly annotated genes were ignored in previous annotations due to the presence of uncharacterized genes in the same region.
Discussion
Structural gene annotation after genome assembly is an essential prerequisite in genomics and functional gene analyses. To construct an accurate gene model, the correct annotation of true‐positive genes is as critically important as reducing false‐positive genes. However, previous methods have been shown to be insufficient in constructing gene models containing all true‐positive genes, hampered by imperfect methodologies, resources, and knowledge. Here, we describe tgfam‐finder, a highly efficient tool for the automated structural annotation of target‐gene families of interest. We evaluated and demonstrated the competitiveness of tgfam‐finder by re‐annotating FAR1, NLR and CYP450 gene families in plants. We only used publicly available resources and identified large numbers of newly annotated genes that were omitted from existing gene models.
One crucial problem with annotation is the omission of protein‐coding genes as functional gene candidates that can obstruct access to essential information and ultimately generate biased downstream analyses. Our analyses demonstrated a higher annotation accuracy and coverage of tgfam‐finder than the publicly available annotation tools, considering the successful prediction of a large number of newly annotated genes with almost no omissions of previously annotated genes. When we conducted proteomic validation for previously and newly annotated genes using publicly available mass spectrometry data from seven plant species, the outstanding performance of tgfam‐finder was confirmed based on the massive identification of previously undiscovered, true‐positive, protein‐coding genes that were missed in previous annotations. tgfam‐finder is easy to use and requires considerably less run‐time and computing power than full annotation. Compared to the long run‐times and intensive computational power required by the publicly available annotation tools, our results demonstrate that tgfam‐finder enables even novice users to obtain target gene models within a couple of days. This tool is greatly faster especially for the annotation of genomes containing large chromosomal sequences than gemoma and maker2.
In summary, tgfam‐finder enables users to determine their experimental priorities based on annotation evidence and more accurate copy numbers of genes of interest in assembled genomes. Large‐scale comparative studies of gene families will not be biased by missing genes, as frequently noted in previous annotations. Our approach provides an alternative solution for the identification and characterization of target‐gene families, accelerating accurate functional, comparative, and evolutionary studies in plant genomes.
Author contributions
SK and DC conceived the project, designed the content, and organized the manuscript. SK, JP, M‐SK, GYC and KC developed tgfam‐finder and annotated the gene families. SK, M‐SK, Y‐MK, HM and NK collected plant genomic materials. JK, S‐HK, K‐SK, NO, S‐KY, K‐SP, CWM and STK performed experimental validation. SK, M‐KS, K‐TK, JJ, HK, Y‐YL, KHS, HCM and Y‐HL performed phylogenetic analyses and validated the new gene models. SK, MJJ and HK designed and constructed the figures. SK and DC wrote the manuscript. SK and KC contributed equally to this work.
Supporting information
Dataset S1 Generated gene models from tgfam‐finder, gemoma and maker2 for evaluation.
Dataset S2 The list of validated protein‐coding genes in newly annotated gene sets.
Dataset S3 Parameters of proteome analyses in this study.
Dataset S4 The newly annotated genes in plants.
Fig. S1 An automated process of tgfam‐finder pipeline.
Fig. S2 Schematic diagram for annotation evaluation.
Fig. S3 Sensitivity, specificity, PPV and NPV of gene models from tgfam‐finder, gemoma and maker2.
Fig. S4 Average sensitivity, specificity, PPV and NPV of 99 individual gene models from tgfam‐finder, gemoma and maker2.
Fig. S5 Initial gene model structures in genomic regions containing the previously annotated genes of rice genome omitted in annotation of tgfam‐finder.
Fig. S6 Manual inspection of annotated genes from tgfam‐finder, gemoma and maker2.
Fig. S7 Average numbers and proportions of non‐overlapping and overlapping genes in new gene models.
Fig. S8 Length distribution of the previously and newly annotated genes.
Fig. S9 Phylogenetic trees of FAR1 and NLR in plant genomes.
Fig. S10 The total number of previously and newly annotated genes mapped according to mass spectrometry data.
Fig. S11 Percentages and numbers of non‐overlapping genes that share genomic positions with repeat sequences.
Fig. S12 Previously annotated non‐target genes that overlap newly annotated genes.
Table S1 List of 50 plant genomic resources used in this study.
Table S2 The number of expressed genes in gene models grouped by 11 trials.
Table S3 Lengths (ratios) of target regions in plant genomes for re‐annotation.
Table S4 The number of genes generated from re‐annotation using tgfam‐finder in 50 plant genomes.
Table S5 The average number (ratio) of newly annotated genes containing start and stop codons in plants.
Table S6 The number of non‐overlapping and overlapping genes in new gene models.
Table S7 The average number of newly annotated genes from annotation evidences and ab initio prediction.
Table S8 The average length (bp) of previously and newly annotated genes.
Table S9 The number of newly annotated target genes and percentage rate of new gene models compared to existing gene models.
Table S10 Proteome resources and the number of previously and newly annotated genes of seven plant genomes.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
The authors appreciate the assistance from the KOBIC Research Support Program. The authors also acknowledge the following researchers who performed iterative beta testing of tgfam‐finder: Ho‐Sub Shin, Myung‐Shin Kim, and Jun‐Ki Lee at Seoul National University; Namjin Koo at the Korea Research Institute of Bioscience and Biotechnology; and Eunyoung Seo at the University of California, Berkeley. This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF‐2017R1A6A3A04004014) to SK, by a grant from the Agricultural Genome Center of the Next Generation Biogreen 21 Program of RDA (Project no. PJ013153) to DC, and by a NRF grant funded by the Korean Government (no. 2018R1A5A1023599, SRC: Plant Immunity Research Center) to DC. The authors declare that there is no conflict of interest.
Contributor Information
Seungill Kim, Email: ksi2204@uos.ac.kr.
Doil Choi, Email: doil@snu.ac.kr.
References
- Bayer PE, Edwards D, Batley J. 2018. Bias in resistance gene prediction due to repeat masking. Nature Plants 4: 762–765. [DOI] [PubMed] [Google Scholar]
- Bennetzen JL, Park M. 2018. Distinguishing friends, foes, and freeloaders in giant genomes. Current Opinion in Genetics & Development 49: 49–55. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10: 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nature Biotechnology 26: 1367–1372. [DOI] [PubMed] [Google Scholar]
- de Santana Costa MG, Mazzafera P, Balbuena TS. 2017. Insights into temperature modulation of the Eucalyptus globulus and Eucalyptus grandis antioxidant and lignification subproteomes. Phytochemistry 137: 15–23. [DOI] [PubMed] [Google Scholar]
- Feschotte C. 2008. Transposable elements and the evolution of regulatory networks. Nature Reviews Genetics 9: 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J et al 2018. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Research 47: D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Chan CK. 2016. Analysis of RNA‐Seq data using TopHat and Cufflinks. Methods in Molecular Biology 1374: 339–361. [DOI] [PubMed] [Google Scholar]
- Guo J, Wang P, Cheng Q, Sun L, Wang H, Wang Y, Kao L, Li Y, Qiu T, Yang W et al 2017. Proteomic analysis reveals strong mitochondrial involvement in cytoplasmic male sterility of pepper (Capsicum annuum L.). Journal of Proteomics 168: 15–27. [DOI] [PubMed] [Google Scholar]
- Gupta R, Min CW, Kramer K, Agrawal GK, Rakwal R, Park KH, Wang Y, Finkemeier I, Kim ST. 2018. A multi‐omics analysis of Glycine max leaves reveals alteration in flavonoid and isoflavonoid metabolism upon ethylene and abscisic acid treatment. Proteomics 18: 1700366. [DOI] [PubMed] [Google Scholar]
- Holt C, Yandell M. 2011. MAKER2: an annotation pipeline and genome‐database management tool for second‐generation genome projects. BMC Bioinformatics 12: 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson M, Ringli C, Boylan MT, Quail PH. 1999. The FAR1 locus encodes a novel nuclear protein specific to phytochrome A signaling. Genes & Development 13: 2017–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F, Vernaldi S, Maekawa T. 2013. Evolution and conservation of plant NLR functions. Frontiers in Immunology 4: 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G et al 2014. InterProScan 5: genome‐scale protein function classification. Bioinformatics 30: 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SJM. 2006. Prediction of genomic functional elements. Annual Review of Genomics and Human Genetics 7: 315–338. [DOI] [PubMed] [Google Scholar]
- Jupe F, Witek K, Verweij W, Sliwka J, Pritchard L, Etherington GJ, Maclean D, Cock PJ, Leggett RM, Bryan GJ et al 2013. Resistance gene enrichment sequencing (RenSeq) enables reannotation of the NB‐LRR gene family from sequenced plant genomes and rapid mapping of resistance loci in segregating populations. The Plant Journal 76: 530–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambiranda D, Basha SM, Singh R, Snowden J, Mercer R. 2018. Proteome profile of American hybrid grape cv. Blanc du Bois during ripening reveals proteins associated with flavor volatiles and ethylene production. Proteomics 18: e1700305. [DOI] [PubMed] [Google Scholar]
- Keilwagen J, Wenk M, Erickson JL, Schattat MH, Grau J, Hartung F. 2016. Using intron position conservation for homology‐based gene prediction. Nucleic Acids Research 44: e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller O, Odronitz F, Stanke M, Kollmar M, Waack S. 2008. Scipio: using protein sequences to determine the precise exon/intron structures of genes and their orthologs in closely related species. BMC Bioinformatics 9: 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT – the BLAST‐like alignment tool. Genome Research 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nature Biotechnology 37: 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Choi D. 2018. New role of LTR‐retrotransposons for emergence and expansion of disease‐resistance genes and high‐copy gene families in plants. BMB Reports 51: 55–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim MS, Kim YM, Yeom SI, Cheong K, Kim KT, Jeon J, Kim S, Kim DS, Sohn SH et al 2015. Integrative structural annotation of de novo RNA‐Seq provides an accurate reference gene set of the enormous genome of the onion (Allium cepa L.). DNA Research 22: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Park J, Yeom SI, Kim YM, Seo E, Kim KT, Kim MS, Lee JM, Cheong K, Shin HS et al 2017. New reference genome sequences of hot pepper reveal the massive evolution of plant disease‐resistance genes by retroduplication. Genome Biology 18: 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution 33: 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagarde J, Uszczynska‐Ratajczak B, Carbonell S, Perez‐Lluch S, Abad A, Davis C, Gingeras TR, Frankish A, Harrow J, Guigo R et al 2017. High‐throughput annotation of full‐length long noncoding RNAs with capture long‐read sequencing. Nature Genetics 49: 1731–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped‐read alignment with Bowtie 2. Nature Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R et al 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948. [DOI] [PubMed] [Google Scholar]
- Lee D, Redfern O, Orengo C. 2007. Predicting protein function from sequence and structure. Nature Reviews Molecular Cell Biology 8: 995–1005. [DOI] [PubMed] [Google Scholar]
- Li M, Li D, Feng F, Zhang S, Ma F, Cheng L. 2016. Proteomic analysis reveals dynamic regulation of fruit development and sugar and acid accumulation in apple. Journal of Experimental Botany 67: 5145–5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YI, Knowles DA, Humphrey J, Barbeira AN, Dickinson SP, Im HK, Pritchard JK. 2018. Annotation‐free quantification of RNA splicing using LeafCutter. Nature Genetics 50: 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalingam R. 2017. Shotgun proteomics of the barley seed proteome. BMC Genomics 18: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx H, Minogue CE, Jayaraman D, Richards AL, Kwiecien NW, Siahpirani AF, Rajasekar S, Maeda J, Garcia K, Del Valle‐Echevarria AR et al 2016. A proteomic atlas of the legume Medicago truncatula and its nitrogen‐fixing endosymbiont Sinorhizobium meliloti . Nature Biotechnology 34: 1198–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q, Gupta R, Min CW, Kim J, Kramer K, Wang Y, Park SR, Finkemeier I, Kim ST. 2018. A proteomic insight into the MSP1 and flg22 induced signaling in Oryza sativa leaves. Journal of Proteomics 20: 204–209. [DOI] [PubMed] [Google Scholar]
- Min CW, Lee SH, Cheon YE, Han WY, Ko JM, Kang HW, Kim YC, Agrawal GK, Rakwal R, Gupta R et al 2017. In‐depth proteomic analysis of Glycine max seeds during controlled deterioration treatment reveals a shift in seed metabolism. Journal of Proteomics 169: 125–135. [DOI] [PubMed] [Google Scholar]
- Mistry J, Finn RD, Eddy SR, Bateman A, Punta M. 2013. Challenges in homology search: HMMER3 and convergent evolution of coiled‐coil regions. Nucleic Acids Research 41: e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA‐seq reads. Nature Biotechnology 33: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Salzberg SL. 2010. Between a chicken and a grape: estimating the number of human genes. Genome Biology 11: 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Shumate A, Pertea G, Varabyou A, Breitwieser FP, Chang YC, Madugundu AK, Pandey A, Salzberg SL. 2018. CHESS: a new human gene catalog curated from thousands of large‐scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biology 19: 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nature Biotechnology. 29: 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler MA, Werck‐Reichhart D. 2003. Functional genomics of P450s. Annual Review of Plant Biology 54: 629–667. [DOI] [PubMed] [Google Scholar]
- Slater GS, Birney E. 2005. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Tzvetkova A, Morgenstern B. 2006. AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biology 7: S11–S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teer JK, Mullikin JC. 2010. Exome sequencing: the sweet spot before whole genomes. Human Molecular Genetics 19: R145–R151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N et al 2014. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nature Biotechnology 32: 223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H et al 2014. Mass‐spectrometry‐based draft of the human proteome. Nature 509: 582–587. [DOI] [PubMed] [Google Scholar]
- Yandell M, Ence D. 2012. A beginner's guide to eukaryotic genome annotation. Nature Reviews Genetics 13: 329–342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dataset S1 Generated gene models from tgfam‐finder, gemoma and maker2 for evaluation.
Dataset S2 The list of validated protein‐coding genes in newly annotated gene sets.
Dataset S3 Parameters of proteome analyses in this study.
Dataset S4 The newly annotated genes in plants.
Fig. S1 An automated process of tgfam‐finder pipeline.
Fig. S2 Schematic diagram for annotation evaluation.
Fig. S3 Sensitivity, specificity, PPV and NPV of gene models from tgfam‐finder, gemoma and maker2.
Fig. S4 Average sensitivity, specificity, PPV and NPV of 99 individual gene models from tgfam‐finder, gemoma and maker2.
Fig. S5 Initial gene model structures in genomic regions containing the previously annotated genes of rice genome omitted in annotation of tgfam‐finder.
Fig. S6 Manual inspection of annotated genes from tgfam‐finder, gemoma and maker2.
Fig. S7 Average numbers and proportions of non‐overlapping and overlapping genes in new gene models.
Fig. S8 Length distribution of the previously and newly annotated genes.
Fig. S9 Phylogenetic trees of FAR1 and NLR in plant genomes.
Fig. S10 The total number of previously and newly annotated genes mapped according to mass spectrometry data.
Fig. S11 Percentages and numbers of non‐overlapping genes that share genomic positions with repeat sequences.
Fig. S12 Previously annotated non‐target genes that overlap newly annotated genes.
Table S1 List of 50 plant genomic resources used in this study.
Table S2 The number of expressed genes in gene models grouped by 11 trials.
Table S3 Lengths (ratios) of target regions in plant genomes for re‐annotation.
Table S4 The number of genes generated from re‐annotation using tgfam‐finder in 50 plant genomes.
Table S5 The average number (ratio) of newly annotated genes containing start and stop codons in plants.
Table S6 The number of non‐overlapping and overlapping genes in new gene models.
Table S7 The average number of newly annotated genes from annotation evidences and ab initio prediction.
Table S8 The average length (bp) of previously and newly annotated genes.
Table S9 The number of newly annotated target genes and percentage rate of new gene models compared to existing gene models.
Table S10 Proteome resources and the number of previously and newly annotated genes of seven plant genomes.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
The newly annotated gene sequences in plants are deposited in Dataset S4. The new gene models including peptide and coding DNA sequences with gff3 and tsv are accessible at http://tgfam‐finder.snu.ac.kr/.