

Abstract
BACKGROUND
Blood group A and B antigens are synthesized by glycosyltransferases regulated by a complex molecular genetic background. A multibase deletion in the ABO gene was identified in two related blood donors. To define its hereditary character and to evaluate genotype–phenotype associations, a detailed study including 30 family members was conducted.
METHODS AND MATERIALS
ABO phenotyping was performed with agglutination techniques and adsorption‐elution tests. The secretor status was determined. Allele‐specific sequencing of ABO and genotyping of family members by a mutation‐specific polymerase chain reaction were carried out. Functional analysis included cloning of complementary DNA and transfection experiments in HeLa cells. The antigen expression was investigated by flow cytometry and adsorption‐elution method.
RESULTS
Sequencing analysis revealed a 24‐bp deletion in Exon 5 and the adjacent intronic region of ABO. The alteration was inherited by 16 family members. Nine of them being heterozygous for the mutated allele failed to express A antigen on their erythrocytes as found by routine typing. In particular samples, however, adsorption‐elution studies indicated inconclusive results. HeLa cells transfected with aberrant gene transcripts did not express blood group antigen A.
CONCLUSION
The variation causes defects in messenger RNA splicing, most likely inactivating the transferase as observed by serological typing and in vitro expression analysis. These data suggest a novel mechanism associated with blood group O and extend the knowledge of exceptionally rare ABO splice site mutations and deletions. With increased understanding of the molecular bases of ABO, the diagnostics may be further enhanced to ensure the safest possible use of the blood supply.
ABBREVIATIONS
- AAs
amino acids
- cDNA
complementary DNA
- GT
glycosyltransferase
- mRNA
messenger RNA
- PCR
polymerase chain reaction
- SNPs
single‐nucleotide polymorphisms
- SSP
sequence‐specific priming
- V1
Variant 1
- V2
Variant 2
- V3
Variant 3
The ABO blood group is defined by the presence or absence of specific carbohydrate structures on the red blood cell (RBC) membrane and corresponding antibodies in the serum. 1 To prevent acute hemolytic transfusion reaction, ABO‐compatible blood products have to be provided. Inconclusive serologic ABO phenotyping results of patients and blood donors need to be clarified by genetic testing when weak A or B phenotypes are suspected.
The ABO gene consists of seven exons spanning about 20 kilobase pairs and encodes glycosyltransferases synthesizing the immunodominant terminal glycan structures of glycoproteins and glycolipids on the cell surface and in body fluids. 2 , 3 The coding region consists of 1065 bases. Exons 6 and 7 encode for the catalytic active domain of the protein, which is composed of 354 amino acids (AAs) in its functional form. Glycosyltransferase (GT) A, encoded by ABO*A1.01 (A 1 ) allele and GT B, encoded by ABO*B.01 (B 1 ), add N‐acetylgalactosamine and D‐galactose to the common precursor H antigen, generating ABO A and B antigen, respectively. These transferases are type II transmembrane proteins with an amino (N)‐terminal cytoplasmic tail and a carboxy (C)‐terminal domain in the lumen of the Golgi apparatus. 4
The difference between A and B transferase is based on four different AA exchanges in Exon 7 determining the specificity of the enzyme's nucleotide sugar‐binding sites. 2 Any polymorphism or mutation that changes the activity or specificity of the encoded enzyme may alter the ABO phenotype.
The homozygous presence of unfunctional O alleles results in the absence of A or B antigens and therefore manifests as ABO blood group O. The most common inactive ABO*O.01 (O 1 ) alleles are caused by a frameshift deletion of guanine in Exon 6 (c.261delG, p.Thr88Profs*31) in a sequence that otherwise is identical to the consensus A1 sequence. 5 Another well‐known inactivating polymorphism is c.802G > A (p.Gly268Arg), characterizing the ABO*O.02 (O 2) alleles. 6 However, other mechanisms for blood group O have rarely been described. 7 , 8
In some cases variants of FUT1 and FUT2 genes encoding fucosyltransferases may be the reason for discrepant serologic ABO grouping results, based on reduced or absent precursor H antigen. Fucosyltransferase 2 (FUT2) regulates the expression of soluble ABH antigens in saliva and bodily secretions (secretors). 9 , 10 Since the synthesis of Lewis blood group Antigen b depends on the activity of fucosyltransferase 2, the presence of Lewis antigen is associated with the individuals' secretor status, finding not only Lewis b but also ABH antigen in saliva of secretors. 11
Beyond that, numerous known single‐nucleotide polymorphisms (SNPs), insertion‐ or deletion‐based mutations are spread throughout the seven exons of the ABO gene or located in regulatory regions influencing the transferase activity. 12 , 13 , 14 , 15 , 16 These ABO subgroup alleles and unusual O alleles, containing alterations other than the well‐known guanine deletion in Exon 6, are often associated with ABO typing discrepancies. 7 , 8 , 17 , 18 , 19 Based on characteristic reactivity with different antisera, the presence of anti‐A1 in the individual's serum and the secretor status, weak A subgroup phenotypes may be differentiated. 20
Here, we characterize a novel ABO subgroup allele, defined by a complex deletion generating aberrant splicing variants. Initially found in two related Styrian blood donors (Caucasians) with an unusual ABO phenotype, the mutation turned out to span three generations of family members.
MATERIAL AND METHODS
ABO blood group phenotyping
Routine ABO typing of the two index blood donors was performed on the Olympus automated blood group testing system (Olympus PK7300, Beckman Coulter, Hamburg, Germany). ABO phenotyping and Lewis antigen testing of other family members were done with standard serologic and gel matrix techniques (MicroTyping system, Bio‐Rad). ABO antigens were tested with monoclonal and human antiserum. ABO isoagglutinins were investigated at room temperature, and at 4°C enhanced by bromelin.
Adsorption‐elution studies of the individuals' RBCs serologically typed as blood group O (n = 9) were carried out. Therefore, monoclonal anti‐A and anti‐A,B as well as human anti‐A and human anti‐A,B sera were used, followed by acid elution technique (Gamma ELU‐Kit II, Immucor, Inc.).
ABO antigens in the saliva of selected samples (n = 9) were tested by the hemagglutination inhibition method according to the AABB Technical Manual. 21 Human plasma of ABO blood group A and B, as well as murine monoclonal anti‐A (clones MH04 und 3D3 and anti‐A [Blend]) and monoclonal anti‐B (a mix of NB1.19, NB10.5A5 and NB10.3B4; and anti‐B [Blend]) were used. Saliva from people known to be ABO A or ABO B secretors, and nonsecretors were tested as positive and negative controls, respectively.
All the monoclonal antibodies used for ABO phenotyping of RBCs and for antigen testing in saliva originated from Bioclone (Ortho Clinical Diagnostics).
Molecular genetic analysis
Genomic DNA was prepared from peripheral white blood cells and buccal swabs by magnetic particle technology with a DNA isolation system (GenoM‐6, Qiagen). Genotyping was performed with the ABO type variant sequence‐specific primers (SSP) PCR kit (BAGene, BAG Health Care, Lich Germany). Allele‐specific amplification of the ABO gene, based on the heterozygous SNP 437C > T in Intron 2, was performed by long‐range polymerase chain reaction (PCR) in two fragments (Peqlab Biotechnologie). Oligonucleotide primers were designed with the online primer design tool BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast) and synthesized by MWG Synthesis (Eurofins Genomics). Sanger sequencing was done in forward and reverse direction (Table 1). ABO's regulatory regions (promotor, enhancer, +5.8‐kb site in Intron 1) were analyzed by genomic PCR and direct sequencing with a cycle sequencing kit (BigDye Terminator, V3.1, Life Technologies, Thermo Fisher). 18 Thermal cycling was carried out in a thermal cycler (Veriti or Proflex, Applied Biosystems, Thermo Fisher). For defining secretor status FUT1:H and FUT2:Secretor were analyzed as previously described. 22
TABLE 1.
Primer sequences for allele‐specific amplification and sequencing of the mutant ABO gene
| Primer name | Sequence (5′ to 3′) | Purpose | Size of PCR product |
|---|---|---|---|
| ABO_In1_12797_Amp_F*5 | GATCTGGACTGGGTTTGGAG | Allele‐specific amplification and sequencing of Fragment 1 | 655 bp |
| ABO_In2_437C/T_R* | CGCCACCAGTGCCTTGG/A | Allele‐specific amplification and sequencing of Fragment 1 | |
| ABO_In2_437C/T_F† | CCTCAGGGACTGCACTGAC/T | Allele‐specific amplification and sequencing of Fragment 2 | 6152 bp |
| ABO_Ex7_Amp_R† 5 | CCTAGGCTTCAGTTACTCAC | Allele‐specific amplification and sequencing of Fragment 2 | |
| ABO_Ex3_R | GGTCAAGGCTGACTCCAG | Sequencing of Fragment 2 | |
| ABO_Int3_88F | CCGCTCTCTATGCTCTGCCC | Sequencing of Fragment 2 | |
| ABO_Int3_1243R | GTTGCAGTCTGAGGTCTACGTTC | Sequencing of Fragment 2 | |
| ABO_Ex4_F | TTCGACGTGTCTGGTGAATGTGT | Sequencing of Fragment 2 | |
| ABO_Ex4_R | TCAAAACTGAAGCTCCAGCTCCAT | Sequencing of Fragment 2 | |
| ABO_Int4_138F | CAGTCTCTACCCTGACTTGGC | Sequencing of Fragment 2 | |
| ABO_Int4_778R | GCTGCACAGCAATGTGAATGGACT | Sequencing of Fragment 2 | |
| ABO_Int4_1447R | GCCATTGCTTTCCACTTGACTT | Sequencing of Fragment 2 | |
| ABO_Ex5_F1 | TGAGACACAACCCCTTACGTCCC | Sequencing of Fragment 2 | |
| ABO_In5_193R | AAGAGACGCAAGTCAGAGAAAG | Sequencing of Fragment 2 | |
| ABO_Ex6_F | TGAGTGGAGTTTCCAGGTGGG | Sequencing of Fragment 2 | |
| ABO_In6_223R | GCCTCTGGAGAAGGAGCT | Sequencing of Fragment 2 | |
| ABO_In6_942R | CAGATGCACCACGTTCTCC | Sequencing of Fragment 2 | |
| ABO_Ex7_F1 | CATCGCTGGGAAGAGGATGAAGTG | Sequencing of Fragment 2 | |
| ABO_Ex7_720R | GCTGCTTCCGTAGAAG | Sequencing of Fragment 2 | |
| ABO_Ex7_506F | AGCTGTCAGTGCTGGAG | Sequencing of Fragment 2 | |
| ABO_Ex1_Pro_F‡ | GGCGCCGTCCCTTCCTAG | Amplification and sequencing | 266 bp |
| ABO_Ex1_Pro_R‡ | ATTCCCTGCGGTAGCGGCT | Amplification and sequencing |
The ABO gene region including Exon 2 to Exon 7 was analyzed by allele‐specific amplification and direct sequencing of two fragments. Exon 1 and the promotor region were investigated by sequencing of genomic amplification products. Employed forward (F) and reverse (R) primers, and the size of PCR products with control template (genotype A1/O1) using the respective primer pairs (*, †, ‡) are described. The primers used for the allele‐specific amplifications differ by polymorph bases on the last nucleotide position at the 3′ end (indicated in bold print). bp = base pairs.
To screen the genomic DNA for the 24‐bp deletion a mutation‐specific PCR was developed. The primer pair (5′‐TCCATGCAAACTCACGTGGC‐ 3′ and 5′‐GGAAACCGCCCTCTAATACCT‐3′) was used to amplify PCR fragments covering the mutated ABO gene region in Exon 5 and Intron 5. PCR products were visualized on a 2% agarose gel prestained with gelred (GelRed Dropper Bottle, Olerup SSP AB).
Translation of the complementary DNA (cDNA) sequence to protein sequence was done by using the in‐silico translation tool of SIB ExPASy Bioformatics resources portal. 23 The newly identified ABO allele (LK022841) was submitted to the European Nucleotide Archive on May 29, 2014.
In this paper, allele designation according to ISBT nomenclature 24 is used.
All blood donors confirmed in the questionnaire for donation eligibility, that sample material may be used for scientific exploration. Additionally, written consent was obtained from all studied individuals. All the data were kept pseudonymized.
Cloning and gene expression analysis
ABO gene transcript synthesis was performed as described 8 with the primer pair hABO_BamHI_F (5′‐CTCGGATCCATGGCCGAGGTGTTGCGGACG‐3′) and hABO_XhoI_R (5′‐CCGCTCGAGGAAACAGAGTTTACCCGTTCTGC‐3′). cDNA was cloned into mammalian expression vector pcDNA3.1, kindly provided by Axel Seltsam, German Red Cross Blood Service NSTOB Institute. 25 The restriction sites BamHI and XhoI were used.
Prepared plasmid constructs were transformed into competent Escherichia coli (NEB 5‐alpha, New England Biolabs) via heat shock. The plasmid DNA was isolated (PureLink HiPure Plasmid Midiprep Kit, Invitrogen, Thermo Fisher) and validated by sequencing analysis using the primers T7_R (5′‐GTAATACGACTCACTATAGGGCG‐3′) and hABO_XhoI_R. Hela cells (ACC 57, DSMZ), homozygous for O 1 alleles, were transfected with 2.5‐μg plasmid DNA containing the two different mutation specific ABO transcript variants A1‐like mut (V1 and V2). The A 1 wild‐type and O 1 cDNA were tested as positive and negative expression controls, respectively. A pcDNA3.1 (+) plasmid expressing green fluorescence protein (GFP) was used as the internal expression control (LifeTech Austria, Thermo Fisher).
Approximately 1 × 106 HeLa cells per well were transiently transfected with the reagent Lipofectamine 3000 (Invitrogen, Life Technologies, Thermo Fisher) according to the manufacturer's instruction. Forty‐eight hours later, the cells were harvested and analyzed for ABO A antigen expression by flow cytometry (Cytoflex, Beckman Coulter) with a mouse anti‐human blood group A monoclonal antibody conjugated with BV421 (BD Horizon). BV421 was excited by violet laser and detected in the standard Pacific Blue filter set. To monitor the transfection efficiency, GFP expression was measured by using the fluorescein isothiocyanate channel.
To exclude dead cells, 1 × 105 cells were incubated for 20 minutes at room temperature with the antibody and costained with 4′,6‐diamidino‐2‐phenylindole (Invitrogen, Thermo Fisher). Due to the use of two different plasmids, GFP expression was not used as a gate, although the positive control demonstrated a coexpression of GFP and A antigen. Unstained HeLa cells were used as control cells.
Adsorption‐elution tests using 2 × 106 to 4 × 106 transfected cells and human anti‐A,B plasma (blood group O) were performed in anti‐human globulin and neutral gel cards (Bio‐Rad) at 37°C.
RESULTS
Allele‐specific ABO sequencing of two related blood donors with ABO discrepancy revealed a variant ABO allele. The ABO*A1‐ like allele was defined by a deletion of 4 bps (c.236‐239delCGTG) at the 3′ end of Exon 5, continuing in a deletion of 20 bps downstream in Intron 5 (Fig. 1).
Fig. 1.
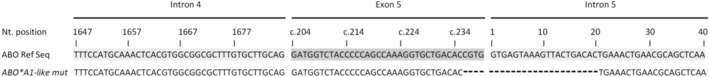
Partial nucleotide sequences of ABO Exon 5 and flanking 5′ and 3′ intronic regions. The identified variation in Exon 5 (c.236‐239del) and Intron 5 (del 1‐20) in the ABO*A1‐like mut allele is shown. The deletion of nucleotides compared to the ABO*A1.01 Reference sequence (NG_006669.1) is indicated by hyphens. The numbering of nucleotides in Exon 5 refers to consecutive numbering according to the coding sequence (c.). Nucleotides of Intron 5 are numbered starting from the first base of the intron. Nt = Nucleotide.
In‐silico protein analyses predicted the disruption of the reading frame at AA 79, resulting in an altered sequence and a premature stop codon after AA 158.
Analysis of individuals who inherited the mutated allele (designated as ABO*A1‐like mut) together with an ABO*B or a common nonfunctional ABO*O allele in trans, indicated blood group B or O by serologic antigen typing. By commercial SSP PCR these samples were erroneously genotyped as A 1 B 1 or A 1 O 1, respectively.
Reverse blood typing indicated weakly reacting anti‐A (1+ to 3+), and several samples lacked the expected serum reactivity against A2 test cells.
Sixteen of 30 investigated family members had the deletion in their ABO gene (Fig. 2) with consistent results for peripheral blood white cells and buccal cells.
Fig. 2.
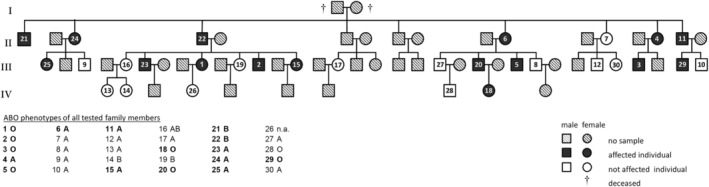
Pedigree of family members tested for the multibase deletion in their ABO gene. The ABO phenotypes as determined by serologic testing are listed, the members typed positive for the genetic alteration are indicated in bold print. Sixteen individuals, spanning three generations (II, III, IV), were found to have shared the deletion in their ABO gene. The mutation, when present in trans with a common ABO*O or B allele was reflected in the absence of A antigen expression in the affected family members (1, 2, 3, 5, 18, 20, 21, 22, and 29). n.a. = not available.
Adsorption‐elution studies of four individuals' RBCs carrying the ABO*A1‐like mut allele indicated trace amounts of A antigen on their RBCs. However, inconsistent results were observed when using different antisera. Repeated analyses with fresh peripheral blood samples of two individuals did not confirm the previous findings.
Twenty individuals were defined to be secretors by being Le (a‐b+), confirmed by the results of FUT2 gene analysis (data not shown).
Eight of them were genotyped as ABO*A1‐like mut/ABO*O (n = 6) or ABO*A1‐like mut/ABO*B (n = 2) and tested negative for the secretion of A antigen in the saliva. The index blood donor sample 2 was phenotyped Le (a + b‐), consistent with the detected inactivating polymorphism c.428G > A (p.Trp143Ter) in FUT2.
Sequence analysis of the ABO's regulatory regions including promotor, enhancer, and + 5.8 kb GATA site in Intron 1 as well as FUT1 did not reveal any deleterious variation.
ABO blood typing test results are summarized in Table 2.
TABLE 2.
Aberrant ABO phenotypes and genotypes of family members tested positive for the 24‐bp deletion
| ABO phenotyping | Genotyping | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| RBCs | Reactivity of serum | Eluate | Secretor status | ABO alleles | |||||
| Sample ID | ABO antigen | A1 cells | A2 cells | B cells | Sample 1 | Sample 2 | Saliva | ||
| 1* | O |
3+ (RT) 3+ (4°C) |
1+ (RT) 3+ (4°C) |
4+ |
A AG neg B AG neg |
n.a. | Secretor |
A AG neg B AG neg |
ABO * O.01.02 ABO * A1‐like mut |
| 2* | O |
2+ (RT) 3+ (4°C) |
Neg RT Neg (4°C) |
4+ |
A AG neg B AG neg |
A AG neg B AG neg |
Non‐Secretor |
A AG neg B AG neg |
ABO * O.01.02 ABO * A1‐like mut |
| 3 | O |
1+ (RT) 2+ (4°C) |
Neg RT Neg (4°C |
4+ |
A AG neg B AG neg |
A AG pos† B AG neg |
Secretor |
A AG neg B AG neg |
ABO * O1.01.01 ABO * A1‐like mut |
| 5 | O |
2+ (RT) 3+ (4°C) |
Neg (RT) 2+ (4°C) |
4+ |
B AG neg |
A AG neg B AG neg |
Secretor |
A AG neg B AG neg |
ABO * O.01.68 ABO * A1‐like mut |
| 18 | O |
1+ (RT) 2+ (4°C). |
Neg (RT) Neg (4°C) |
4+ |
A AG neg B AG neg |
n.a. | Secretor |
A AG neg B AG neg |
ABO * O1.01.01 ABO * A1‐like mut |
| 20 | O |
2+ (RT) 3+ (4°C) |
Neg (RT) 1+ (4°C) |
4+ |
B AG neg |
n.a. | Secretor |
A AG neg B AG neg |
ABO * O.01.68 ABO * A1‐like mut |
| 21 | B |
3+ (RT) 3+ (4°C) |
Neg (RT) 2+ (4°C) |
Neg |
A AG neg B AG pos |
n.a. | Secretor |
A AG neg B AG pos |
ABO * B.01 ABO * A1‐like mut |
| 22 | B |
2+ (RT) 3+ (4°C) |
Neg (RT) Neg (4°C) |
Neg |
A AG neg B AG pos |
n.a. | Secretor |
A AG neg B AG pos |
ABO * B.01 ABO * A1‐like mut |
| 29 | O |
2+ (RT) 3+ (4°C) |
Neg (RT) Neg (4°C) |
3+ |
B AG neg |
n.a. | Secretor |
A AG neg B AG neg |
ABO * O.01.02 ABO * A1‐like mut |
ABO typing results of individuals heterozygous for the mutation and a non‐A encoding O or B allele in trans are summarized. Isoagglutinin testing with A1, A2, and B test cells at room temperature or at 4°C is shown. Positive reactions are indicated by 1+ to 4+. No agglutination is indicated by “Neg.” Adsorption‐elution studies were performed using monoclonal and human anti‐A and anti‐A,B antibodies. The reactive antisera are displayed in the table. Samples 3, 5, 20, and 29 revealed inconclusive adsorption‐elution test results. As expected for the nonsecretor (Sample 2), no antigen expression was detected in the saliva.
Index blood donor Samples 1 and 2.
With monoclonal anti‐A.
With human anti‐AB.
With monoclonal anti‐A,B.
n.a. = not available.
Samples 5, 18, 20, and 21 (Table 2) were subject to reverse transcription PCR. Further, RNA of samples 11 and 15 with the blood group phenotype A were reverse transcribed.
cDNA variants with varying product sizes ranging from about 500 to 1100 bps were obtained. As validated by Sanger sequencing, the amplicons larger than 800 bps contained at least nucleotide sequences of Exon 6 and/or Exon 7 of the ABO gene. Transcripts of the control sample homozygous for wild‐type ABO*A1.01 indicated a single fragment of around 1100 bps after gel electrophoresis.
Six different ABO*A1‐like mut containing cDNAs consisting of at least 800 bps were cloned into pcDNA3.1 vector. Seventy‐eight clones were investigated in total. Thereby, alternative and aberrant splicing variants (Fig. 3), but also full‐length messenger RNA (mRNA) of ABO*A1.01, ABO*B.01, ABO*A2.01 and ABO*O.01.01 alleles were identified. The results are summarized in Table 3.
Fig. 3.
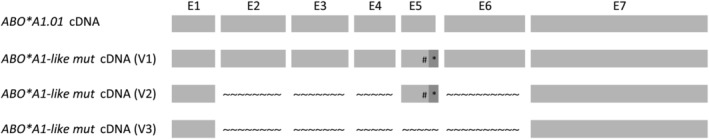
Schematic representation of selected splicing variants identified by subcloning and sequencing of ABO cDNA. ABO*A1.01 cDNA indicates the normal full‐length ABO mRNA transcript, as identified in the ABO*A.01 wild type control. The splicing variants V1 to V3 were predicted to result in frameshift mutations and truncation of the encoded transferase. The V1 and V2 cDNAs were used for transfection of HeLa cells. E1‐E7 Exons 1‐7; # Exon 5 c.236‐239del; * Intron 5 Ins 21‐55; Skipped exons are indicated by the wavy line.
TABLE 3.
ABO transcript variants detected by cloning of mutation containing ABO cDNAs
| Sample ID (cDNA) | Genotype | Transcript variants | Full‐length ABO transcripts |
|---|---|---|---|
| 5 | ABO*O.01.68/ABO*A1‐like mut |
ABO*O.01.68 ABO*A1‐like mut (V1) |
‐ |
| 11 | ABO*A1.01/ABO*A1‐like mut |
ABO*A1.01 ABO*A1‐like mut (V1) |
ABO*A1.01 |
| 15 | ABO*A2.01/ABO*A1‐like mut |
ABO*A2.01 ABO*A1‐like mut (V2) |
ABO*A2.01 |
| 18 | ABO*O.01.01/ABO*A1‐like mut |
ABO*O.01.01 ABO*A1‐like mut (V1 + V3) |
ABO*O.01.01 |
| 20 | ABO*O.01.68/ABO*A1‐like mut | ABO*O.01.68 | ‐ |
| 21 | ABO*B.01/ABO*A1‐like mut | ABO*B.01 | ABO*B.01 |
Aberrant transcript variants associated with the ABO*A1‐like mut allele (V1‐V3) and alternative spliced ABO mRNAs of the alleles in trans are indicated. No full‐length ABO transcript was observed in cDNAs of Samples 5 and 20.
cDNA = complementary DNA.
Transcript variant 1 (V1) harbored a deletion in Exon 5 and retention of nucleotides 21 to 55 of Intron 5. The in‐silico translation to protein sequence predicted a reading frame shift at AA 79 and a premature stop at AA 158 resulting in an enzyme with a defective catalytic domain.
In addition to the variation in V1, transcript Variant 2 (V2) exhibited skipping of Exon 2, 3, 4 and 6. An early frame shift at AA 11 with a premature stop at AA 19 was predicted. The presence of an alternative initial methionine downstream theoretically may encode a transferase consistent with AA 142 to 354 of the wild‐type enzyme.
In transcript Variant 3 (V3) Exon 1 was directly joined to Exon 7 as previously observed by Chen et al. 16 when investigating alternative splicing variants for individuals with Ael phenotype (ABO*AEL.04). Translation to protein sequence predicted an early frame shift and the termination after AA 14. The possible use of an alternative start codon downstream at position 35 would translate in an entirely different protein sequence.
HeLa cells were transfected with the two ABO*A1‐like mut cDNAs (Fig. 3, V1 and V2), where encoding of a transferase with residual enzymatic activity could not be ruled out.
V1‐ and V2‐transfected cells were negative for the expression of A antigen as determined by flow cytometry (Fig. 4: V1 = 0.68 ± 0.5%; V2 = 0.61 ± 0.33%; ABO*A1.01 [wild type] 79.17 ± 5.39%, ABO*O.01.01 [negative control] 0.69 ± 0.3%, each n = 3) and adsorption‐elution tests (n = 3).
Fig. 4.
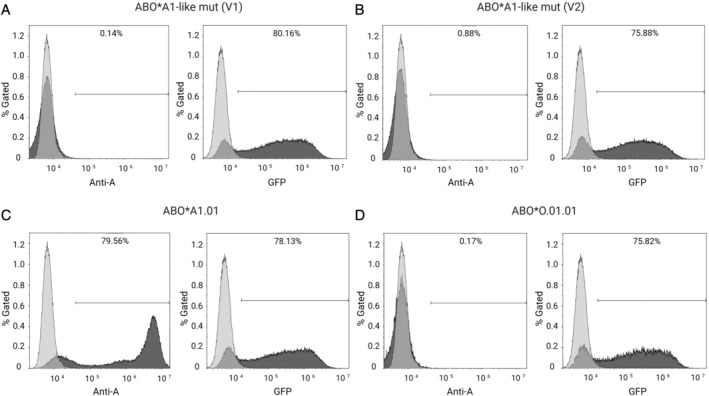
Representative flow cytometry analyses of transfected HeLa cells for blood group A antigen and GFP expression. The used anti‐human blood group A monoclonal antibody binds specifically to blood group A expressed on transfected HeLa cells. A or GFP expression is indicated (dark gray filled) and untreated HeLa cells were used as control cells (light gray, respective control). (A) ABO*A1‐like mut (V1) transfected cells with 80% GFP expression, indicating positive transfection. (B) ABO*A1‐like mut (V2) transfected cells with 75% GFP expression. Despite good transfection efficiencies, V1 and V2 did not result in expression of A antigen. (C) Positive control, ABO*A1.01 (wild type) transfectants, indicating 79% A and 78% GFP expression. (D) Negative control, ABO*O.01.01 transfectants, demonstrating a lack of A expression but indicating 75% of GFP expressing cells.
V3 was predicted to result in a very short protein sequence lacking the stem region and entire catalytic domain of the encoded transferase. Therefore, this variant was not investigated in the transfection experiments.
DISCUSSION
ABO blood typing ambiguity in two related blood donors led to the identification of an ABO*A1‐ like allele with a multibase deletion at the Exon 5/Intron 5 junction of the gene, affecting the 5′donor splice site.
While routine phenotyping did not detect A antigen on erythrocytes and in the saliva (secretors) of the affected individuals, adsorption‐elution tests of RBCs indicated trace amounts of A antigen expression in some samples. However, adsorption‐elution results were not conclusive, depending on the respective antiserum used.
The potency and efficiency of an antiserum relies on the presence of polyclonal or monoclonal antibodies in human‐ or animal‐source reagents. The use of different clones of the same specificity and the use of various diluents to formulate reagents may influence the reproducibility of the tests. 26 In relation to phenotyping of ABO subgroup RBCs, discrepant results obtained with different antibodies have previously been described. 27 , 28
The weakened presence of anti‐A1 and anti‐A observed in the serum of some samples may count for low A antigen expression. Based on the possible existence of varying sequences and varying allele copy numbers, a residual A transferase activity may occur in some of the individuals. The lack of the determination of ABO mRNA copy numbers is a limitation of our study. However, the detected transcript variants (V1 and V2) were defined as being inactive by our further investigations.
As suggested by others who investigated samples genotyped homozygous for deletional as well as nondeletional O alleles, the weak anti‐A activity observed on reverse phenotyping may be due to causes not related to the ABO genotype. 29 , 30
Basically, the presence of transcripts encoding a functional A transferase in other tissues cannot be excluded.
Previously, it has been shown for other GTs that one defective enzyme can be rescued by another and thereby regain its function. 31 , 32 In our study, extended ABO phenotyping including adsorption‐elution tests of the samples heterozygous for the mutated allele and ABO*B.01 (Samples 21 and 22) did not indicate the presence of ABO A antigen, with consistently negative results (Table 2).
Therefore, we conclude that the obtained inconsistent and not reproducible adsorption‐elution test results of some individuals' samples may be due to unspecific reactions and likely to be false positive.
By investigating ABO mRNA, we found erroneous gene transcripts. As known for aberrant mRNA splicing owing to splice junction mutations, exon skipping and intron retention have been observed. 33 , 34
Six splice‐site mutations in A or B alleles have previously been correlated with weak A or B subgroups, respectively, and are listed in the International Society of Blood Transfusion database. Five of them are attributed to single‐nucleotide substitutions, and only one sequence variant results from a single‐nucleotide deletion. 16 , 35 , 36 , 37 , 38
We detected in different samples the aberrant ABO transcript V1 that leads to a frame shift at codon position 79. It was predicted to result in the translation of an entirely different protein with a premature termination of translation at AA Residue 158.
Previously, Hult et al. 39 described a weak A allele that is based on an ABO*A2.01 backbone (c.467C > T and c.1061delC) containing the additional SNP c.236C > T (p.Pro79Leu) in Exon 5 of the gene. The prolin to leucin substitution is located in the stem region of the protein and is suggested to perturb dimerization and enzyme function, causing the very low A antigen levels detected on the erythrocytes. 6
The common single base deletion c.261delG characterizing O 1 alleles results in a frameshift at AA 88 and finally leads to a premature truncation 31 AAs downstream. In relation to that, we hypothesize similar effects on the transferase activity founded by our multibase deletion causing a frameshift at Position 79.
Routine ABO genotyping relies on the well‐known variants to determine whether an inactive O allele is present. Thus, the applied SSP‐based PCR failed to detect the previously unknown mutation and in cases like ours may not be reliable. 40
Finally, in this study, 16 family members were identified to carry the genetic alteration causing a defect in mRNA splicing. Because of consistent results of antigen expression studies with peripheral blood and saliva as well as the in vitro transfection experiments the variant may be classified as being inactivating.
Even considering some residual A antigen expression that is not detected by our routine agglutination tests, the bedside test would be negative.
RBCs carrying very low levels of A antigen are thought to be relatively unsusceptible to damage by ABO antibodies, and thus suggested not to be of clinical relevance when transfused to blood group O and B patients. 15 , 29 , 41 Retrospective analysis going backwards until 2012 revealed that RBCs of two of the donors investigated in this study (Samples 2 and 3) were transfused to several patients with blood group O. No adverse transfusion reactions of the blood‐receiving patients have been reported to our hemovigilance system to date, as also described by Yazer et al. 42
Recently, Möller et al. 43 mentioned an unorthodox 24‐bp deletion while analyzing the data set from the 1000 Genomes project. In fact, it is the same deletion that we are reporting here. The deletion was detected by next‐generation sequencing analyses in nine individuals, showing great diversity, not only regarding ethnic distribution but also regarding the allelic background. The individuals were mainly East Asian, European, and mixed American with Mexican ancestry. Interestingly, within the specifically resequenced samples, the assigned deletion could not be determined, and the variant could be confirmed by neither in‐silico nor in vitro experiments. Finally, the authors suggested this variation to be based on false prediction by next‐generation sequencing data analysis.
However, in our study we were able to characterize the deletion in detail, and therefore we could verify a novel, putative ABO O allele not relying on c.261delG. With respect to the observations hinted by the 1000 Genomes data, the mutation seems to be more common and not only passed on within a single family as we anticipated initially.
The enormous genetic heterogeneity underlying the defined ABO subgroups has become apparent. We augment the ABO allele database by the only multibase deletion resulting in an ABO mRNA splicing defect described to date. With increased knowledge on the ABO blood group system the ABO typing strategies may be improved for safe transfusion therapy.
WEB‐BASED RESOURCES
ExPASy: SIB bioinformatics resource portal. http://web.expasy.org/translate/
European Nucleotide Archive. http://www.ebi.ac.uk/ena/data/view/LK022841
ISBT database: http://www.isbtweb.org/fileadmin/user_upload/Working_parties/WP_on_Red_Cell_Immunogenetics_and/001_ABO_Alleles_v1.2.pdf
CONFLICT OF INTEREST
The authors have disclosed no conflicts of interest.
ACKNOWLEDGMENTS
Anja Stoisser, Lisa Rohrhofer, Marie‐Therese Frisch, Silvia Sorantin, Katharina Sebauer and Maria Stubenrauch are acknowledged for their technical assistance. The authors thank Axel Seltsam for kindly providing the ABO wild‐type vector construct.
REFERENCES
- 1. Laura D. Blood groups and red cell antigens. Bethesda, MD: National Center for Biotechnology Information (US); 2005. [Google Scholar]
- 2. Yamamoto F, Clausen H, White T, et al. Molecular genetic basis of the histo‐blood group ABO system. Nature 1990;345:229‐33. [DOI] [PubMed] [Google Scholar]
- 3. Hakomori S. Antigen structure and genetic basis of histo‐blood groups A, B and O: their changes associated with human cancer. Biochim Biophys Acta 1999;1473:247‐66. [DOI] [PubMed] [Google Scholar]
- 4. Paulson JC, Colley KJ. Glycosyltransferases. Structure, localization, and control of cell type‐specific glycosylation. J Biol Chem 1989;264:17615‐8. [PubMed] [Google Scholar]
- 5. Seltsam A, Hallensleben M, Kollmann A, et al. The nature of diversity and diversification at the ABO locus. Blood 2003;102:3035‐42. [DOI] [PubMed] [Google Scholar]
- 6. Lee HJ, Barry CH, Borisova SN, et al. Structural basis for the inactivity of human blood group O2 glycosyltransferase. J Biol Chem 2005;280:525‐9. [DOI] [PubMed] [Google Scholar]
- 7. Hosseini‐Maaf B, Irshaid NM, Hellberg A, et al. New and unusual O alleles at the ABO locus are implicated in unexpected blood group phenotypes. Transfusion 2005;45:70‐81. [DOI] [PubMed] [Google Scholar]
- 8. Matzhold EM, Drexler C, Wagner T. A novel ABO O allele caused by a large deletion covering two exons of the ABO gene identified in a Caucasian family showing discrepant ABO blood typing results. Transfusion 2016;56:2739‐43. [DOI] [PubMed] [Google Scholar]
- 9. Kelly RJ, Rouquier S, Giorgi D, et al. Sequence and expression of a candidate for the human secretor blood group alpha(1,2)fucosyltransferase gene (FUT2). Homozygosity for an enzyme‐inactivating nonsense mutation commonly correlates with the non‐secretor phenotype. J Biol Chem 1995;270:4640‐9. [DOI] [PubMed] [Google Scholar]
- 10. Larsen RD, Ernst LK, Nair RP, et al. Molecular cloning, sequence, and expression of a human GDP‐L‐fucose:beta‐D‐galactoside 2‐alpha‐L‐fucosyltransferase cDNA that can form the H blood group antigen. Proc Natl Acad Sci U S A 1990;87:6674‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Watkins WM. Biochemistry and genetics of the ABO, Lewis, and P blood group systems. Adv Hum Genet 1980;10:1‐136, 379‐85. [DOI] [PubMed] [Google Scholar]
- 12. Seltsam A, Hallensleben M, Kollmann A, et al. Systematic analysis of the ABO gene diversity within Exons 6 and 7 by PCR screening reveals new ABO alleles. Transfusion 2003;43:428‐39. [DOI] [PubMed] [Google Scholar]
- 13. Patnaik SK, Helmberg W, Blumenfeld OO. BGMUT: NCBI dbRBC database of allelic variations of genes encoding antigens of blood group systems. Nucleic Acids Res 2012;40:D1023‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ogasawara K, Yabe R, Uchikawa M, et al. Molecular genetic analysis of variant phenotypes of the ABO blood group system. Blood 1996;88:2732‐7. [PubMed] [Google Scholar]
- 15. Seltsam A, Das Gupta C, Wagner FF, et al. Nondeletional ABO*O alleles express weak blood group A phenotypes. Transfusion 2005;45:359‐65. [DOI] [PubMed] [Google Scholar]
- 16. Chen DP, Sun CF, Ning HC, et al. Genetic and mechanistic evaluation for the weak A phenotype in Ael blood type with IVS6 + 5G>A ABO gene mutation. Vox Sang 2015;108:64‐71. [DOI] [PubMed] [Google Scholar]
- 17. Olsson ML, Irshaid NM, Hosseini‐Maaf B, et al. Genomic analysis of clinical samples with serologic ABO blood grouping discrepancies: identification of 15 novel A and B subgroup alleles. Blood 2001;98:1585‐93. [DOI] [PubMed] [Google Scholar]
- 18. Matzhold EM, Wagner A, Drexler C, et al. Novel ABO gene variants caused by missense mutations in Exon 7 leading to discrepant ABO blood typing results. Transfusion 2015;55:1589‐90. [DOI] [PubMed] [Google Scholar]
- 19. Matzhold EM, Drexler C, Staudacher E, et al. A novel variant B allele at the ABO gene locus characterized by a duplication‐based insertion of 27 nucleotides identified in an Iraqi male with a weak B subgroup phenotype. Transfusion 2018;58:1318‐9. [DOI] [PubMed] [Google Scholar]
- 20. Storry JR, Olsson ML. The ABO blood group system revisited: a review and update. Immunohematology 2009;25:48‐59. [PubMed] [Google Scholar]
- 21. Brecher I. Technical manual. 15th ed. AABB: Bethesda, MD; 2005. [Google Scholar]
- 22. Matzhold EM, Helmberg W, Wagner T, et al. Identification of 14 new alleles at the fucosyltransferase 1, 2, and 3 loci in Styrian blood donors, Austria. Transfusion 2009;49:2097‐108. [DOI] [PubMed] [Google Scholar]
- 23. Artimo P, Jonnalagedda M, Arnold K, et al. ExPASy: SIB Bioinformatics resources portal. Nucleic Acids Res 2012;40:W597‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reid ME, Lomas‐Francis C, Olsson ML. The blood group antigen FactsBook. 3rd ed. New York, NY: Elsevier Academic Press; 2012. [Google Scholar]
- 25. Seltsam A, Das Gupta C, Bade‐Doeding C, et al. A weak blood group A phenotype caused by a translation‐initiator mutation in the ABO gene. Transfusion 2006;46:434‐40. [DOI] [PubMed] [Google Scholar]
- 26. Moulds MK. Review: monoclonal reagents and detection of unusual or rare phenotypes or antibodies. Immunohematology 2006;22:52‐63. [PubMed] [Google Scholar]
- 27. Moore S, Chirnside A, Micklem LR, et al. A mouse monoclonal antibody with anti‐A,(B) specificity which agglutinates Ax cells. Vox Sang 1984;47:427‐34. [DOI] [PubMed] [Google Scholar]
- 28. McGowan A, Tod A, Chirnside A, et al. Stability of murine monoclonal anti‐A, anti‐B and anti‐A,B ABO grouping reagents and a multi‐centre evaluation of their performance in routine use. Vox Sang 1989;56:122‐30. [DOI] [PubMed] [Google Scholar]
- 29. Wagner FF, Blasczyk R, Seltsam A. Nondeletional ABO*O alleles frequently cause blood donor typing problems. Transfusion 2005;45:1331‐4. [DOI] [PubMed] [Google Scholar]
- 30. Yazer MH, Olsson ML. The O2 allele: questioning the phenotypic definition of an ABO allele. Immunohematology 2008;24:138‐47. [PubMed] [Google Scholar]
- 31. Storry JR, Condon J, Hult AK, et al. An age‐dependent ABO discrepancy between mother and baby reveals a novel A(weak) allele. Transfusion 2015;55:422‐6. [DOI] [PubMed] [Google Scholar]
- 32. Salmon C, Cartron JP. Le renforcement allelique [Allelic enhancement]. Rev Fr Transfus Immunohematol 1976;19:145‐55. [DOI] [PubMed] [Google Scholar]
- 33. Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base‐pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet 1992;90:41‐54. [DOI] [PubMed] [Google Scholar]
- 34. Nakai K, Sakamoto H. Construction of a novel database containing aberrant splicing mutations of mammalian genes. Gene 1994;141:171‐7. [DOI] [PubMed] [Google Scholar]
- 35. Yu LC, Twu YC, Chou ML, et al. Molecular genetic analysis for the B(3) allele. Blood 2002;100:1490‐2. [DOI] [PubMed] [Google Scholar]
- 36. Olsson M. A splice‐site mutation defines the Afinn allele at the blood group ABO locus (abstract). Transfusion 2000;40(10S):13S. [Google Scholar]
- 37. Hosseini‐Maaf B, Smart E, Chester MA, et al. The Abantu phenotype in the ABO blood group system is due to a splice‐site mutation in a hybrid between a new O1‐like allelic lineage and the A2 allele. Vox Sang 2005;88:256‐64. [DOI] [PubMed] [Google Scholar]
- 38. Chen DP, Tseng CP, Wang WT, et al. Genetic and mechanistic evaluation for the mixed‐field agglutination in B3 blood type with IVS3+5G>A ABO gene mutation. PLoS One 2012;7:e37272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hult AK, Yazer MH, Jorgensen R, et al. Weak A phenotypes associated with novel ABO alleles carrying the A2‐related 1061C deletion and various missense substitutions. Transfusion 2010;50:1471‐86. [DOI] [PubMed] [Google Scholar]
- 40. Olsson ML, Chester MA. Polymorphism and recombination events at the ABO locus: a major challenge for genomic ABO blood grouping strategies. Transfus Med 2001;11:295‐313. [DOI] [PubMed] [Google Scholar]
- 41. Mollison PL. Blood transfusion in clinical medicine. 6th ed. Oxford: Blackwell Scientific; 1979. [Google Scholar]
- 42. Yazer MH, Hult AK, Hellberg A, et al. Investigation into A antigen expression on O2 heterozygous group O‐labeled red blood cell units. Transfusion 2008;48:1650‐7. [DOI] [PubMed] [Google Scholar]
- 43. Moller M, Hellberg A, Olsson ML. Thorough analysis of unorthodox ABO deletions called by the 1000 Genomes project. Vox Sang 2018;113:185‐97. [DOI] [PubMed] [Google Scholar]