

Abstract
Archetypal phosphine/borane frustrated Lewis pairs (FLPs) are famed for their ability to activate small molecules. The mechanism is generally believed to involve two‐electron processes. However, the detection of radical intermediates indicates that single‐electron transfer (SET) generating frustrated radical pairs could also play an important role. These highly reactive radical species typically have significantly higher energy than the FLP, which prompted this investigation into their formation. Herein, we provide evidence that the classical phosphine/borane combinations PMes3/B(C6F5)3 and PtBu3/B(C6F5)3 both form an electron donor–acceptor (charge‐transfer) complex that undergoes visible‐light‐induced SET to form the corresponding highly reactive radical‐ion pairs. Subsequently, we show that by tuning the properties of the Lewis acid/base pair, the energy required for SET can be reduced to become thermally accessible.
Keywords: charge transfer, Lewis pairs, photochemistry, radicals, electron transfer
Exciting FLPs! The radical‐ion pairs of the archetypal frustrated Lewis pair (FLP) systems PMes3/B(C6F5)3 and PtBu3/B(C6F5)3 can be accessed by visible‐light‐induced single‐electron transfer. Varying the FLP system shows this process is general for a range of donor–acceptor combinations and can be tuned to proceed thermally.

Introduction
Frustrated Lewis pair (FLP) chemistry is useful for the activation of a variety of small molecules, most notably dihydrogen and carbon dioxide.1 However, the activation of less reactive substrates, such as dinitrogen or methane, remains a challenge. It is generally accepted that FLP reactivity stems from the association of the electron donor (D; Lewis base) and the electron acceptor (A; Lewis acid) to form an encounter complex ([d–A]) that features a reactive pocket in which heterolytic bond cleavage of the substrate occurs.2 Interestingly, recent studies have revealed evidence for the formation of radicals within such systems, which indicates that single‐electron transfer (SET) may also play an important role in Lewis acid/base chemistry.2b, 2c, 3 Such highly reactive radical species could be key to the activation of inert substrates, which we were keen to investigate.
A SET mechanism within FLP chemistry was first suggested by Piers et al.,3b as B(C6F5)3 (BCF) is also known to act as a single‐electron oxidant.4 However, it was noted that the disparity in redox potentials between PtBu3 and BCF would limit the presence of radicals to subnanomolar concentrations for this archetypal FLP.3b When combining PMes3 with BCF, Stephan and co‐workers detected a very weak signal by EPR spectroscopy in chlorobenzene, which they postulated to be the PMes3 ⋅+ radical cation. Switching to the more Lewis acidic Al(C6F5)3 resulted in significantly larger signals accredited to the PMes3 ⋅+ radical cation (Scheme 1 a).2c The absence of radical anions (BCF⋅− and Al(C6F5)3 ⋅−) was attributed to the known rapid degradation of these species through solvolytic pathways.3a, 5, 6 Further indications for a radical pathway were provided by the reaction of PMes3/B(C6F5)3 with Ph3SnH, which afforded [Mes3PH][HB(C6F5)3] and Ph3Sn–SnPh3 instead of the expected [Mes3PH][Ph3SnB(C6F5)3].2c Klare, Müller and co‐workers also observed radical formation when combining trityl (CPh3 +) or silylium cations (SiR3 +) with phosphines (PMes3, P(C6F5)3, P(C6Me5)3 and PTipp3, Tipp=2,4,6‐triisopropylphenyl; Scheme 1 b).7 In the case of trityl, both radical species (⋅CPh3 and R3P+⋅) were persistent, allowing the detection of both components of the radical pair in solution.
Scheme 1.

Literature examples of detected radicals (highlighted in boxes) in cooperative main‐group Lewis acid/base chemistry.2c, 4b, 7 R=Mes, Tipp, C6F5, C6Me5, tBu (only in the case of CPh3 +); SiR′3 +=SiiPr3 +, Si(C6Me5)3 +, SitBuMe2 +, SiEt3 +.
Interestingly, when the analogous reactions of BCF, Al(C6F5)3 and SiR3 + were carried out with PtBu3 as the Lewis base, no radical reactivity was observed. Klare, Müller and co‐workers accredited this to the higher ionisation energy of PtBu3 compared with PMes3 (see Table S1 in the Supporting Information). Another challenge comes from the fact that the PtBu3 ⋅+ radical cation is far less stable than its mesityl analogue, decomposing rapidly at room temperature.7, 8 In addition to phosphines, amines are also potent electron donors, with the amine radical cations, for example, formed by visible‐light photoredox catalysis, having been well explored recently.9 Indeed, Wang et al. reported on the one‐electron oxidation of cyclic triarylamine NbAr3 by B(C6F5)3 and isolated the corresponding radical cation (Scheme 1 c).4b Furthermore, while this manuscript was under revision, Ooi and co‐workers reported on the photoinduced SET between B(C6F5)3 and N,N‐dialkylanilines.10 Again, the BCF⋅− radical anion was not detected in either case.3a, 5
These findings prompted us to focus on understanding SET processes (versus the established concerted, polar pathways) in frustrated Lewis pair chemistry,11 ultimately allowing the rational design of reactive main‐group radical‐ion pairs from the corresponding Lewis acid/base combinations by a single‐electron shift.3c, 12 Mulliken theory describes how interactions between donors and acceptors can lead to the formation of electron donor–acceptor (EDA) complexes [d–A] that exhibit characteristic absorption bands resulting from the promotion of an electron from the donor component to the acceptor component in the EDA complex (Scheme 2).13 It is worth noting that these EDA complexes are analogous to the encounter (or van der Waals) complexes proposed in FLP mechanistic pathways. Kochi and co‐workers showed that such EDA complexes can be formed by mixing tetracyanoethylene and anthracenes, and used picosecond laser spectroscopy to demonstrate that specific irradiation of the charge‐transfer (CT) band indeed induces SET to generate the corresponding radical‐ion pairs [D⋅+–A⋅−].14, 15 Note that these radical species often undergo rapid back‐electron transfer (BET) to regenerate the starting donor–acceptor complex [d–A].
Scheme 2.

Electron donor–acceptor complex formation and subsequent SET to generate the corresponding radical‐ion pair.
Results and Discussion
In applying this knowledge to frustrated Lewis pairs, we first calculated the ionisation energy (IED) and electron affinity (EAA)7 for the archetypal FLP systems PtBu3/BCF and PMes3/BCF at the SCRF/ωB97X‐D/6‐311+G(d,p) level of theory (solvent=toluene).16 This highlighted the fact that there is a large disparity between the EAA of BCF (−3.03 eV) and the IED of PtBu3 (5.95 eV) and PMes3 (5.54 eV), affording energy differences of 2.92 eV (67.2 kcal mol−1) and 2.51 eV (57.8 kcal mol−1), respectively, between the Lewis acid/base pairs and the corresponding radical‐ion pairs (Figure 1). Overcoming such a large energy gap to generate these main‐group radicals by SET is, therefore, unlikely to be a thermal process. This prompted us to further investigate these systems, employing both time‐dependent DFT (TD‐DFT) calculations and a range of spectroscopic techniques, to shed light on the process of radical formation.17
Figure 1.

DFT‐calculated energy needed to access the radical‐ion pairs from the FLP systems PtBu3/PMes3 and BCF.7
It is known that mixing colourless solutions of PMes3 and BCF yields a violet solution,2c, 18 which indicates that interaction with visible light (λ=400–800 nm, ΔE=71.4–35.7 kcal mol−1) is occurring. Although the violet colour has previously been suggested to result from the formation of radical species (PMes3 ⋅+),2c we postulated that this colour change is more likely to result from the formation of a visible‐light‐absorbing EDA complex, generated in the ground state upon association of the electron‐rich phosphine and the electron‐deficient borane. The absorption spectrum of the phosphine–borane encounter complex PMes3–BCF, computed by TD‐DFT at the ωB97X‐D/6‐311++G(d,p) level of theory indeed features an additional absorption band at a longer wavelength than the individual components (λ=439 nm; see Figure S1 in the Supporting Information),19 which is indicative of a CT band.12 Analysis of the frontier molecular orbitals of this encounter complex clearly showed that this CT is from the phosphorus lone pair (HOMO; Figure 2) to the formally vacant p orbital on boron (LUMO; Figure 2).20 UV/Vis spectroscopic analysis of the violet solution of PMes3/BCF in toluene (0.015 m) confirmed this hypothesis and revealed an absorption band in the visible spectrum at λ max=534 nm (Figure 3 a), which corresponds to a vertical excitation energy of 53.5 kcal mol−1 (S0→S1 transition).
Figure 2.
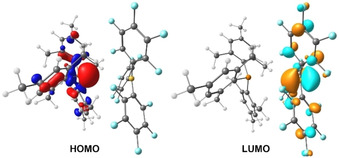
Frontier molecular orbitals of the PMes3/BCF encounter complex, calculated at the ωB97X‐D/6‐311G(d,p) level of theory. Isovalue=0.05.
Figure 3.

a) UV/Vis spectrum of PMes3/BCF (both components: 1.5×10−2 m in toluene) compared with the spectra of the separate components (1.5×10−2 m). b) Experimental EPR spectrum of PMes3/BCF in toluene measured at 30 K during irradiation with visible light (390–500 nm) and simulated EPR spectra of PMes3 ⋅+ and BCF⋅−. c) Transient absorption spectra measured after pulsed excitation of PMes3/BCF with 530 nm light.
To investigate whether irradiation of the coloured EDA complex induces SET, we designed an EPR experiment featuring a light source within the spectrometer. Low‐temperature (30 K) analysis of a violet PMes3/BCF frozen toluene (0.06 m) solution prepared in the dark showed no radical formation, clearly evidencing that the violet colour is not generated by radicals. During irradiation with visible light (390–500 nm) for 90 s, an intense EPR signal corresponding to a superposition of two radical species was observed (Figure 3 b). The broad signal at g=2.005, with a featureless signal at 30 K that shows no resolved hyperfine coupling interactions, is attributed to the BCF⋅− radical anion.15 The other four lines stem from the axially symmetric PMes3 ⋅+ radical cation (g ⊥=2.0055, g ‖=2.0015) exhibiting hyperfine coupling (A ⊥=550 MHz, A ‖=1170 MHz) with the I= phosphorus nucleus; similar spectra have been previously reported in nitrile solvents (A ⊥=427–480 MHz, A ‖=1128–1157 MHz).21 When we stopped irradiating the sample, both radicals remained visible with the signal intensity having only decayed by about 25 % after 6 min at 30 K. We postulate that this relative longevity can be ascribed to the formation of separate PMes3 ⋅+ and BCF⋅− radical ions by electron tunnelling through the frozen solvent.22 Upon removal of the sample from the EPR spectrometer, an intense, dark‐purple colour could be observed, which quickly reverted back to violet as the sample thawed.
Next, we employed transient absorption spectroscopy23 to record the absorption spectra of the PMes3 ⋅+/BCF⋅− radical‐ion pair and determine its lifetime. Using a toluene solution of PMes3/BCF (0.04 m) at room temperature, short laser pulse (<200 fs; λ=530 nm) excitation of the CT band and subsequent time‐resolved picosecond spectroscopy allowed us to detect a broad absorption band (λ max=620 nm; Figure 3 c), which we assign to a superposition of the individual absorption spectra of PMes3 ⋅+ and BCF⋅− (λ≈600 nm in PhCN24 and λ≈600 nm in THF,5 respectively). Decay analysis of this transient absorption showed that the photogenerated radical‐ion pair has a lifetime25 of 237 ps at room temperature, due to rapid BET to the ground‐state EDA complex in solution. Formally, CT‐band excitation produces optical electron transfer, that is, direct population of the CT state ([D⋅+–A⋅−]) within the laser pulse.26 These findings evidence, for the first time, unequivocally that visible light is a necessity to access the PMes3 ⋅+/BCF⋅− radical‐ion pair. Note that the instability of BCF⋅− in combination with the light‐dependence and short lifetime of the radical‐ion pair indicates that the PMes3 ⋅+/BCF⋅− pair will be available in minute quantities and thus is unlikely to account for the follow‐up chemistry reported recently.2c, 3d, 10
Because for a series of donors interacting with a common acceptor, the energies of the CT transitions (hν CT) vary proportionally with the donor ionisation energies (IED; Figure 1),13b we expect the same SET process for the PtBu3/BCF combination to be feasible using light, albeit of higher energy.2c Indeed, mixing the two colourless FLP components PtBu3 and BCF (0.015 m in toluene) resulted directly in a pale‐yellow solution,27 UV/Vis spectroscopic analysis of which also revealed an additional broad absorption band (λ≈400 nm; Figure 4 a) that partly overlaps with the absorptions of the separate species; the CT band of the PtBu3/BCF EDA complex is expected in this region. However, it is also known that PtBu3 and B(C6F5)3 slowly react to form the yellow (λ=372 nm) para‐addition product,26 which, although not visible by 19F NMR spectroscopy, could also be present in low concentrations in this region, hampering unambiguous assignment of the new absorption band. EPR spectroscopy at 30 K of a PtBu3/BCF frozen toluene (6×10−5 m) solution, freshly prepared in the dark, showed no radicals. Yet, to our delight, during irradiation with visible light (390–500 nm) for 90 s, EPR signals corresponding to PtBu3 ⋅+ and BCF⋅− radical ions were observed (Figure 4 b), thereby confirming the existence of a CT band in the visible region. Again, the BCF⋅− radical ion comprises a featureless broad signal at g=2.005, and the remaining four lines result from the axially symmetric PtBu3 ⋅+ radical cation (g ⊥=2.0065, g ‖=2.0012) with hyperfine coupling (A ⊥=580 MHz, A ‖=1365 MHz) with the I= phosphorus nucleus.8 This time, after irradiation, the EPR signals decay notably faster than those of PMes3/BCF, with a 50 % decline in intensity after 6 min at 30 K. Similarly, faster decay was observed when the sample was removed from the EPR spectrometer, as the dark‐purple colour instantly reverted back to pale yellow upon thawing. Ultrafast transient absorption spectroscopy confirmed these observations and showed that excitation of the CT band (<200 fs laser pulse; λ=400 nm) affords the absorption bands of the PtBu3 ⋅+/BCF⋅− radical ions (λ max=680 nm; Figure 4 c) that disappear rapidly (lifetime=6 ps) after the laser pulse. This short lifetime, resulting from facile BET, prevents significant diffusive separation and subsequent chemistry,13c, 22 which accounts for the lack of radical chemistry observed for this FLP system.
Figure 4.

a) UV/Vis spectrum of PtBu3/BCF (both components: 1.5×10−2 m in toluene) compared with the spectra of the separate components (1.5×10−2 m). Inset shows the colour of the solution. b) Experimental EPR spectrum of PtBu3/BCF in toluene measured at 30 K during irradiation with visible light (390–500 nm) and simulated EPR spectra of PtBu3 ⋅+ and BCF⋅−. c) Transient absorption spectra measured after pulsed excitation of PtBu3/BCF with 530 nm light.
Intrigued by the formation of the stable, deep‐blue NbAr3 ⋅+ radical cation when the bridged triphenylamine NbAr3 was treated with BCF in DCM at room temperature for 72 h (Scheme 1 c),4b, 28 we expanded our studies by varying the Lewis base to incorporate triphenylamine (NPh3) and tri‐p‐tolylamine (NpTol3). Calculation of their ionisation energies (IED=5.94 eV for NPh3 and 5.67 eV for NpTol3) at the ωB97X‐D/6‐311+G(d,p) level of theory showed again a large disparity with the EAA of BCF (−3.03 eV), affording energy gaps of 2.91 eV (67.1 kcal mol−1) and 2.64 eV (60.7 kcal mol−1), respectively, between the Lewis acid/base pairs and the corresponding radical ions, which indicates that the needed SET again requires visible light. Indeed, the CT band of the EDA complex was detected by UV/Vis spectroscopy for both NPh3/BCF and NpTol3/BCF in toluene (0.015 m; λ max=500 and 420 nm, respectively; see Figures S17–S19 in the Supporting Information). Interestingly, for NpTol3/BCF, the UV/Vis spectrum changed over time and after 5 h at room temperature the absorption bands of the stable pTol3N⋅+ radical cation were also observed (λ max=590 and 690 nm; see Figure S19).29, 30 EPR analysis confirmed that mixing NPh3 or NpTol3 with BCF yields no SET in the dark, although irradiation with visible light (390–500 nm) promoted photoinduced SET to generate the corresponding radical‐ion pair, the amine radical cations being observed at room temperature (see Figures S27 and S28).31 The absence of BCF⋅− is attributed to its known rapid degradation in solution at room temperature,3a, 5 which is the driving force for the build‐up of the amine radical cation as it prevents regeneration of the ground‐state EDA complex by BET (Scheme 3). Because the computed ionisation energy for the bridged triarylamine NbAr3 (5.67 eV) is equal to that of NpTol3 (5.67 eV),15 these findings strongly suggest that the SET reported4b by Wang et al. between the triarylamine NbAr3 and BCF also proceeds photochemically, and that performing this reaction in broad daylight (or by using a high‐power light‐emitting diode (LED)) will be beneficial.
Scheme 3.

SET to afford the high‐energy radical‐ion pair, which, by decomposition of either the radical cation or radical anion, forms the stable complementary radical ion selectively.
Next, we selected the bulky tris(3,5‐dinitro‐2,4,6‐trimethylphenyl)borane as the Lewis acid (abbreviated as B(NO2‐Mes)3, EAA=−3.04 eV; Figure 5), which has a similar electron affinity to the archetypal borane B(C6F5)3 (EAA=−3.03 eV; Figure 5). The former can be reduced with sodium metal32 to afford the persistent radical anion B(NO2‐Mes)3 ⋅−.33 We postulated that this borane radical anion should also be accessible from an EDA complex by photoinduced SET when a suitable Lewis base is used. For this, we selected PtBu3 (IED=5.95 eV),27 because the energy gap between the corresponding FLP and the PtBu3 ⋅+/B(NO2‐Mes)3 ⋅− radical‐ion pair is 2.91 eV (IED+EAA, 66.9 kcal mol−1). We could not detect a CT band by UV/Vis spectroscopy; the appearance of a small band corresponding to the dark‐red B(NO2‐Mes)3 ⋅− radical anion (510 nm) was the only peak observable that was not attributable to the separate phosphine or borane (see Figure S14 in the Supporting Information). However, we irradiated a pale‐yellow solution of PtBu3/B(NO2‐Mes)3 in DCM (0.03 m) with 455 nm light (2.2 W, LED), avoiding excitation of the separate Lewis acid and base (λ<420 nm), and were delighted to see that facile SET took place within 3 h at room temperature to generate a dark‐red solution of B(NO2‐Mes)3 ⋅− (confirmed by EPR spectroscopy, see Figure S24), facilitated by the degradation of the highly unstable PtBu3 ⋅+ radical cation (70 % conversion, Scheme 3 and Figure S25). This is a proof of principle for the use of Lewis bases as sacrificial one‐electron donors in the photochemical reduction of boranes.
Figure 5.

Ionisation energies (IED, top) and electron affinities (EAA, bottom) of donors and acceptors typical in FLP chemistry calculated at the SCRF/ωB97X‐D/6‐311+G(d,p) level of theory (solvent=toluene).
In light of the foregoing findings, we were also keen to target the generation of radical pairs from Lewis acids and bases through thermal SET by using FLPs for which the energy difference between donor–acceptor and radical pair is smaller, to show that this generates a fundamentally different situation involving equilibria. The combination of PTipp3 or PMes3 with the strongly Lewis acidic trityl cation (+CPh3), previously reported by Klare, Müller and co‐workers, serves as an ideal starting point (Scheme 1 b).7 In both these cases, the radicals obtained by SET at room temperature (PR3 ⋅+ and .CPh3) were detected by EPR spectroscopy,7 and we were keen to elucidate whether this process is general and also occurs in the dark. Note that the high sensitivity of EPR spectroscopy allows for detection of persistent radicals in concentrations as low as 10−8 m. For a 0.06 m solution, this corresponds to an equilibrium with an energy gap (ΔG) of around 9 kcal mol−1 (0.4 eV) between the ground‐state EDA complex and the radical pair, leading to the formation of measurable amounts of radicals (according to its Boltzmann distribution).15 Indeed, combining PMes3 with the trityl source [Ph3C][B(C6F5)4] (IED+EAA=0.3 eV (7.0 kcal mol−1); Figure 5) in toluene in the absence of light resulted in facile SET and detection of the corresponding phosphoniumyl PMes3 ⋅+/⋅CPh3 radical pair by EPR spectroscopy (see Figures S29 and S30).7 As the trityl radical is in equilibrium with its quinoid‐type dimer, Gomberg dimerisation provides an additional driving force towards the radical side of the equilibrium (ΔG=−4.7 kcal mol−1; Figure 6).34
Figure 6.

Energy diagram showing the thermal SET equilibrium for the NpTol3/+CPh3 EDA pair. For computational details, see Figure 5.
Finally, we were curious as to whether we also could achieve the one‐electron oxidation of amines through thermal SET simply by using the trityl cation as the Lewis acid in the dark. Although the energy gap between the EDA pairs formed by combining the arylamines NPh3 or NpTol3 with [Ph3C][B(C6F5)4] and the corresponding radical pairs (ΔG (ΔE)=14.4 (16.3) and 8.9 (10.0) kcal mol−1, respectively; Figure 6) are close to or exceed the threshold for detection by EPR spectroscopy, Gomberg dimerisation drives the equilibrium to the right, allowing for observation of the radical species (Figure 6). As the trityl radical is mostly present as Gomberg's dimer, the EPR spectrum is dominated by the amine radical cations (see Figures S32 and S33).
Conclusion
We have shown that the encounter complexes in FLP chemistry can also be described as EDA complexes, and are susceptible to photoinduced SET to form the corresponding radical pairs. This knowledge has resulted, for the first time, in the controlled generation and detection of the radical‐ion pairs of the archetypal FLPs PMes3/BCF and PtBu3/BCF through visible‐light‐induced SET. This study will allow us to directly probe any FLP‐type reaction facilitated by these systems and determine whether they proceed via radical‐pair formation. Furthermore, we have demonstrated that the energy gap between the EDA complex and the corresponding radical pair can be readily tuned to proceed thermally by changing the Lewis acid and base components. It is therefore possible to predict the nature of the SET between two donor–acceptor species by simple analysis of their ionisation energy and electron affinity. In addition, this work has provided important insights for understanding and controlling the generation of highly reactive radical pairs by photoinduced or thermal SET, which we are currently applying to the design of new radical‐ion pairs with photophysical properties tuned for exploiting radical reactivity. We envision such systems could be highly beneficial for designing FLPs suitable for the activation of inert substrates as well as the development of main‐group photoredox catalysis.35
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Council for Chemical Sciences of The Netherlands Organization for Scientific Research (NWO/CW) through a VIDI grant (J.C.S.), an NWA Idea Generator grant (J.C.S.) and a VENI grant (A.R.J.).
F. Holtrop, A. R. Jupp, N. P. van Leest, M. Paradiz Dominguez, R. M. Williams, A. M. Brouwer, B. de Bruin, A. W. Ehlers, J. C. Slootweg, Chem. Eur. J. 2020, 26, 9005.
Dedicated to Professor Peter Chen on the occasion of his 60th birthday
References
- 1.
- 1a. Welch G. C., San Juan R. R., Masuda J. D., Stephan D. W., Science 2006, 314, 1124–1126; [DOI] [PubMed] [Google Scholar]
- 1b. Paradies J., Coord. Chem. Rev. 2019, 380, 170–183; [Google Scholar]
- 1c. Skara G., De Vleeschouwer F., Geerlings P., De Proft F., Pinter B., Sci. Rep. 2017, 7, 1–15; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Stephan D. W., Science 2016, 354, aaf7229;27940818 [Google Scholar]
- 1e. Stephan D. W., J. Am. Chem. Soc. 2015, 137, 10018–10032; [DOI] [PubMed] [Google Scholar]
- 1f. Stephan D. W., Erker G., Angew. Chem. Int. Ed. 2015, 54, 6400–6441; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6498–6541; [Google Scholar]
- 1g. Jupp A. R., Stephan D. W., Trends Chem. 2019, 1, 35–48. [Google Scholar]
- 2.
- 2a. Paradies J., Eur. J. Org. Chem. 2019, 283–294; [Google Scholar]
- 2b. Liu L., Cao L. L., Zhu D., Zhou J., Stephan D. W., Chem. Commun. 2018, 54, 7431–7434; [DOI] [PubMed] [Google Scholar]
- 2c. Liu L., Cao L. L., Shao Y., Ménard G., Stephan D. W., Chem 2017, 3, 259–267; [Google Scholar]
- 2d. Rocchigiani L., Ciancaleoni G., Zuccaccia C., Macchioni A., J. Am. Chem. Soc. 2014, 136, 112–115; [DOI] [PubMed] [Google Scholar]
- 2e. Rokob T. A., Bakó I., Stirling A., Hamza A., Pápai I., J. Am. Chem. Soc. 2013, 135, 4425–4437; [DOI] [PubMed] [Google Scholar]
- 2f. Grimme S., Kruse H., Goerigk L., Erker G., Angew. Chem. Int. Ed. 2010, 49, 1402–1405; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 1444–1447; [Google Scholar]
- 2g. Daru J., Bakó I., Stirling A., Pápai I., ACS Catal. 2019, 9, 6049–6057. [Google Scholar]
- 3.
- 3a. Lawrence E. J., Oganesyan V. S., Wildgoose G. G., Ashley A. E., Dalton Trans. 2013, 42, 782–789; [DOI] [PubMed] [Google Scholar]
- 3b. Piers W. E., Marwitz A. J. V., Mercier L. G., Inorg. Chem. 2011, 50, 12252–12262; [DOI] [PubMed] [Google Scholar]
- 3c. Dong Z., Cramer H. H., Schmidtmann M., Paul L. A., Siewert I., Müller T., J. Am. Chem. Soc. 2018, 140, 15419–15424; [DOI] [PubMed] [Google Scholar]
- 3d. Soltani Y., Dasgupta A., Gazis T. A., Ould D. M. C., Richards E., Slater B., Stefkova K., Vladimirov V. Y., Wilkins L. C., Willcox D., Melen R. L., Cell. Rep. Phys. Sci. 2020, 1, 100016. [Google Scholar]
- 4.
- 4a. Tao X., Daniliuc C. G., Knitsch R., Hansen M. R., Eckert H., Lübbesmeyer M., Studer A., Kehr G., Erker G., Chem. Sci. 2018, 9, 8011–8018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Zheng X., Wang X., Qiu Y., Li Y., Zhou C., Sui Y., Li Y., Ma J., Wang X., J. Am. Chem. Soc. 2013, 135, 14912–14915; [DOI] [PubMed] [Google Scholar]
- 4c. Harlan C. J., Hascall T., Fujita E., Norton J. R., J. Am. Chem. Soc. 1999, 121, 7274–7275; [Google Scholar]
- 4d. Nakamoto M., Inagaki Y., Ochiai T., Tanaka M., Sekiguchi A., Heteroat. Chem. 2011, 22, 412–416; [Google Scholar]
- 4e. Beddows C. J., Burrows A. D., Connelly N. G., Green M., Lynam J. M., Paget T. J., Organometallics 2001, 20, 231–233; [Google Scholar]
- 4f. Ishida Y., Sekiguchi A., Kobayashi K., Nagase S., Organometallics 2004, 23, 4891–4896; [Google Scholar]
- 4g. Liu L. L., Cao L. L., Shao Y., Stephan D. W., J. Am. Chem. Soc. 2017, 139, 10062–10071; [DOI] [PubMed] [Google Scholar]
- 4h. Liu Y., Solari E., Scopelliti R., Fadaei Tirani F., Severin K., Chem. Eur. J. 2018, 24, 18809–18815. [DOI] [PubMed] [Google Scholar]
- 5. Kwaan R. J., Harlan C. J., Norton J. R., Organometallics 2001, 20, 3818–3820. [Google Scholar]
- 6. Chen J., Chen E. Y.-X., Dalton Trans. 2016, 45, 6105–6110. [DOI] [PubMed] [Google Scholar]
- 7. Merk A., Großekappenberg H., Schmidtmann M., Luecke M.-P., Lorent C., Driess M., Oestreich M., Klare H. F. T., Müller T., Angew. Chem. Int. Ed. 2018, 57, 15267–15271; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15487–15492. [Google Scholar]
- 8. Murugesan R., Subramanian S., Mol. Phys. 1979, 38, 1941–1953. [Google Scholar]
- 9.
- 9a. Hu J., Wang J., Nguyen T. H., Zheng N., Beilstein J. Org. Chem. 2013, 9, 1977–2001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Chen J.-R., Hu X.-Q., Lu L.-Q., Xiao W-J., Chem. Soc. Rev. 2016, 45, 2044; [DOI] [PubMed] [Google Scholar]
- 9c. Morris S. A., Wang J., Zheng N., Acc. Chem. Res. 2016, 49, 1957–1968; [DOI] [PubMed] [Google Scholar]
- 9d. Beatty J. W., Stephenson C. R. J., Acc. Chem. Res. 2015, 48, 1474–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.During the revision of this paper, the Ooi group reported on photoinduced SET between B(C6F5)3 and N,N-dialkylanilines, see: Aramaki Y., Imaizumi N., Hotta M., Kumagai J., Ooi T., Chem. Sci. 2020, 11, 4305–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu L. L., Stephan D. W., Chem. Soc. Rev. 2019, 48, 3454–3463. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Shaik S. S., J. Am. Chem. Soc. 1981, 103, 3692–3701; [Google Scholar]
- 12b. Pross A., Acc. Chem. Res. 1985, 18, 212–219; [Google Scholar]
- 12c. Eberson L., Electron Transfer Reactions in Organic Chemistry (Ed.: L. Eberson), Springer, New York, 1987. [Google Scholar]
- 13.
- 13a. Mulliken R. S., J. Am. Chem. Soc. 1952, 74, 811–824; [Google Scholar]
- 13b. Foster R., J. Phys. Chem. 1980, 84, 2135–2141; [Google Scholar]
- 13c. Rosokha S. V., Kochi J. K., Acc. Chem. Res. 2008, 41, 641–653. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Hilinski E. F., Masnovi J. M., Amatore C., Kochi J. K., Rentzepis P. M., J. Am. Chem. Soc. 1983, 105, 6167–6168; [Google Scholar]
- 14b. Hilinski E. F., Masnovi J. M., Kochi J. K., Rentzepis P. M., J. Am. Chem. Soc. 1984, 106, 8071–8077; [Google Scholar]
- 14c. Kochi J. K., Adv. Phys. Org. Chem. 1994, 29, 185–272; [Google Scholar]
- 14d. Rathore R., Lindeman S. V., Kochi J. K., J. Am. Chem. Soc. 1997, 119, 9393–9404; [Google Scholar]
- 14e.for an ethylene–I2 EDA complex, see: Kalume A., George L., Powell A. D., Dawes R., Reid S. A., J. Phys. Chem. A 2014, 118, 6838–6845. [DOI] [PubMed] [Google Scholar]
- 15.For selected examples of electron donor–acceptor complexes in photocatalysis, see:
- 15a. Arceo E., Jurberg I. D., Álvarez-Fernández A., Melchiorre P., Nat. Chem. 2013, 5, 750–756; [DOI] [PubMed] [Google Scholar]
- 15b. Zhu Y., Zhang L., Luo S., J. Am. Chem. Soc. 2014, 136, 14642–14645; [DOI] [PubMed] [Google Scholar]
- 15c. Nappi M., Bergonzini G., Melchiorre P., Angew. Chem. Int. Ed. 2014, 53, 4921–4925; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5021–5025; [Google Scholar]
- 15d. Woźniak Ł., Murphy J. J., Melchiorre P., J. Am. Chem. Soc. 2015, 137, 5678–5681; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15e. Kandukuri S. R., Bahamonde A., Chatterjee I., Jurberg I. D., Escudero-Adán E. C., Melchiorre P., Angew. Chem. Int. Ed. 2015, 54, 1485–1489; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1505–1509; [Google Scholar]
- 15f. Sandfort F., Strieth-Kalthoff F., Klauck F. J. R., James M. J., Glorius F., Chem. Eur. J. 2018, 24, 17210–17214; [DOI] [PubMed] [Google Scholar]
- 15g. Wu J., Grant P. S., Li X., Noble A., Aggarwal V. K., Angew. Chem. Int. Ed. 2019, 58, 5697–5701; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5753–5757; [Google Scholar]
- 15h.for a recent review, see: Crisenza G. E. M., Mazzarella D., Melchiorre P., J. Am. Chem. Soc. 2020, 142, 5461–5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.See the Supporting Information for further details.
- 17.For an overview of mechanistic studies in photocatalysis, see: Buzzetti L., Crisenza G. E. M., Melchiorre P., Angew. Chem. Int. Ed. 2019, 58, 3730–3747; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3768–3786. [Google Scholar]
- 18. Welch G. C., Stephan D. W., J. Am. Chem. Soc. 2007, 129, 1880–1881. [DOI] [PubMed] [Google Scholar]
- 19.Although TD-DFT calculations provide an excellent basis for the determination of trends, it is notoriously difficult to determine the exact energies for absorption bands in cases of charge transfer, see:
- 19a. Laurent A. D., Jacquemin D., Int. J. Quantum Chem. 2013, 113, 2019–2039; [Google Scholar]
- 19b. Maitra N. T., J. Phys. Condens. Matter 2017, 29, 423001; [DOI] [PubMed] [Google Scholar]
- 19c. Adamo C., Jacquemin D., Chem. Soc. Rev. 2013, 42, 845–856; [DOI] [PubMed] [Google Scholar]
- 19d. Xu P., Zhang C.-R., Wang W., Gong J.-J., Liu Z.-J., Chen H.-S., Int. J. Mol. Sci. 2018, 19, 1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.For charge-transfer emission in oligo(triarylamine/triarylboranes), see: Bonn A. G., Wenger O. S., J. Org. Chem. 2015, 80, 4097–4107. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Il'yasov A. V., Kargin Y. M., Nikitin E. V., Vafina A. A., Romanov G. V., Parakin O. V., Kazakova A. A., Pudovik A. N., Phosphorus Sulfur Relat. Elem. 1980, 8, 259–262; [Google Scholar]
- 21b. Culcasi M., Berchadsky Y., Gronchi G., Tordo P., J. Org. Chem. 1991, 56, 3537–3542. [Google Scholar]
- 22.
- 22a. Wenger O. S., Leigh B. S., Villahermosa R. M., Gray H. B., Winkler J. R., Science 2005, 307, 99–102; [DOI] [PubMed] [Google Scholar]
- 22b. Wenger O. S., Phys. Chem. Chem. Phys. 2013, 15, 10673–10685. [DOI] [PubMed] [Google Scholar]
- 23. Kumpulainen T., Lang B., Rosspeintner A., Vauthey E., Chem. Rev. 2017, 117, 10826–10939. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Tojo S., Yasui S., Fujitsuka M., Majima T., J. Org. Chem. 2006, 71, 8227–8232; [DOI] [PubMed] [Google Scholar]
- 24b. Ménard G., Hatnean J. A., Cowley H. J., Lough A. J., Rawson J. M., Stephan D. W., J. Am. Chem. Soc. 2013, 135, 6446–6449. [DOI] [PubMed] [Google Scholar]
- 25.For the time needed for the concentration of an entity to decrease to 1/e of its initial value by a first-order process, see: Braslavsky S. E., Pure Appl. Chem. 2007, 79, 3, 293–465. [Google Scholar]
- 26. Vân Anh N., Schlosser F., Groeneveld M. M., van Stokkum I. H. M., Würthner F., Williams R. M., J. Phys. Chem. C 2009, 113, 18358–18368. [Google Scholar]
- 27. Marwitz A. J. V., Dutton J. L., Mercier L. G., Piers W. E., J. Am. Chem. Soc. 2011, 133, 10026–10029. [DOI] [PubMed] [Google Scholar]
- 28.Note that [NbAr3]⋅+[Al(ORF)4]− could be isolated when using Ag[Al(ORF)4] (ORF=OC(CF3)3) as one-electron oxidant in the dark.
- 29.For thermal SET between [PhN2][BF4] and tri-p-tolylamine in CH3CN, see: Habraken E. R. M., van Leest N. P., Hooijschuur P., de Bruin B., Ehlers A. W., Lutz M., Slootweg J. C., Angew. Chem. Int. Ed. 2018, 57, 11929–11933; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12105–12109. [Google Scholar]
- 30.
- 30a. Granick S., Michaelis L., J. Am. Chem. Soc. 1940, 62, 2241–2242; [Google Scholar]
- 30b. Seo E. T., Nelson R. F., Fritsch J. M., Marcoux L. S., Leedy D. W., Adams R. N., J. Am. Chem. Soc. 1966, 88, 3498–3503; [Google Scholar]
- 30c. Yurchenko O., Freytag D., zur Borg L., Zentel R., Heinze J., Ludwigs S., J. Phys. Chem. B 2012, 116, 30–39. [DOI] [PubMed] [Google Scholar]
- 31.As the corresponding amine radical cations are known to be persistent at room temperature, an EPR analysis at this temperature is sufficient to prove that SET takes place.
- 32. Connelly N. G., Geiger W. E., Chem. Rev. 1996, 96, 877–910. [DOI] [PubMed] [Google Scholar]
- 33. Bennett E. L., Lawrence E. J., Blagg R. J., Mullen A. S., MacMillan F., Ehlers A. W., Scott D. J., Sapsford J. S., Ashley A. E., Wildgoose G. C., Slootweg J. C., Angew. Chem. Int. Ed. 2019, 58, 8362–8366; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 8450–8454. [Google Scholar]
- 34.For the trityl radical, see:
- 34a. Gomberg M., Ber. Dtsch. Chem. Ges. 1900, 33, 3150–3163; [Google Scholar]
- 34b. Gomberg M., J. Am. Chem. Soc. 1900, 22, 757–771; [Google Scholar]
- 34c. Chesnut D. B., Sloan G. J., J. Chem. Phys. 1960, 33, 637–638; [Google Scholar]
- 34d. Zarkadis A. K., Neumann W. P., Uzick W., Chem. Ber. 1985, 118, 1183–1192; [Google Scholar]
- 34e. Neumann W. P., Uzick W., Zarkadis A. K., J. Am. Chem. Soc. 1986, 108, 3762–3770; [Google Scholar]
- 34f. Fukuzumi S., Kitano T., Ishikawa M., J. Am. Chem. Soc. 1990, 112, 5631–5632. [Google Scholar]
- 35.
- 35a. McAtee R. C., McClain E. J., Stephenson C. R. J., Trends Chem. 2019, 1, 111–125; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35b. Romero N. A., Nicewicz D. A., Chem. Rev. 2016, 116, 10075–10166; [DOI] [PubMed] [Google Scholar]
- 35c. Fu M.-C., Shang R., Zhao B., Wang B., Fu Y., Science 2019, 363, 1429–1434; [DOI] [PubMed] [Google Scholar]
- 35d. Deng Z., Lin J.-H., Xiao J.-C., Nat. Commun. 2016, 7, 10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary