

Abstract
Parkinson's disease (PD) is characterised by selective and severe degeneration of the substantia nigra pars compacta and the locus coeruleus (LC), which underlies the most prominent symptoms. Although α‐synuclein accumulation has long been established to play a causal role in the disease, it alone cannot explain the selective degenerative pattern. Recent evidence shows that the selective vulnerability could arise due to the large presence of cytosolic catecholamines and Ca2+ ions in the substantia nigra pars compacta and LC specifically that can be aberrantly affected by α‐synuclein accumulation. Moreover, each has its own toxic potential, and disturbance of one can exacerbate the toxic effects of the others. This presents a mechanism unique to these areas that can lead to a vicious degenerative cycle. Interestingly, in familial variants of PD, the exact same brain areas are affected, implying the underlying process is likely the same. However, the exact disease mechanisms of many of these genetic variants remain unclear. Here, we review the effects of the PD‐related genes Parkin, PINK1 and DJ‐1. We establish that these mutant varieties can set in motion the same degenerative process involving α‐synuclein, cytosolic catecholamines and Ca2+. Additionally, we show indications that model organisms might not accurately represent all components of this central mechanism, explaining why Parkin, PINK1 and DJ‐1 model organisms often lack a convincing PD‐like phenotype.
Keywords: DJ‐1, Parkin, Parkinson's disease, PINK1, selective vulnerability
Parkinson's disease is characterised by selective and severe degeneration of the substantia nigra pars compacta and the locus coeruleus. The review discusses the effects of the Parkinson's disease‐related genes Parkin, PINK1 and DJ‐1 and establishes that these mutant varieties can set in motion the degenerative process involving α‐synuclein, cytosolic catecholamines and Ca2+.
Abbreviations
- AADC
l‐amino acid decarboxylase
- Ca2+
calcium
- CMA
chaperone‐mediated autophagy
- CNS
central nervous system
- LC
locus coeruleus
- PD
Parkinson's disease
- PQC
protein quality control
- ROS
reactive oxygen species
- SNc
substantia nigra pars compacta
- TH
tyrosine hydroxylase
- VTA
ventral tegmental area
- α‐syn
alpha‐synuclein
1. INTRODUCTION
Parkinson's disease (PD) is a complex age‐related neurodegenerative disease, affecting about 1–2 out of 1,000 people worldwide (Tysnes & Storstein, 2017). It is mainly characterised by a set of motor symptoms, including resting tremor, muscular rigidity, bradykinesia and difficulty with movement initiation (Kalia & Lang, 2015). These main symptoms arise due to progressive degeneration of dopaminergic neurons within the substantia nigra pars compacta (SNc). Loss of these dopaminergic neurons leads to a decreased dopamine output within the basal ganglia, causing an imbalance between the direct and indirect pathways in the basal ganglia–thalamo–cortical motor loop that underlies the motor symptoms (Blandini, Nappi, Tassorelli, & Martignoni, 2000; Przedborski, 2017). It is largely unknown what triggers the disease, although PD is thought to start years or even decades before the appearance of motor symptoms. By this time though, massive neuronal loss in the SNc has already occurred. Definitive diagnosis can generally only be made post‐mortem, through examination of the brain tissue. There are several non‐motor symptoms associated with PD, some of which precede the motor symptoms. These include sleep disturbance, constipation, mood swings and olfactory deficits. However, these are too unspecific for diagnostic use (Visanji & Marras, 2015). Late‐stage disease is often accompanied by cognitive decline and dementia (Kalia & Lang, 2015).
There is currently no treatment available to halt disease progression. The few treatments in use are aimed at symptom alleviation and improving the quality of life. The most common is l‐Dopa, a precursor to dopamine that can cross the blood–brain barrier, which, following uptake into dopaminergic neurons and conversion to dopamine, supplements the dopamine shortage. At later disease stages, when even more of the SNc has been lost, this treatment becomes ineffective. Deep brain stimulation is also used in some cases to restore balance to the basal ganglia–thalamo–cortical motor loop by inhibiting the subthalamic nucleus, normalising the system's output. This therapeutic measure deals with the consequences of the dopamine shortage instead of the shortage itself; however, it also alleviates the motor symptoms and does not halt disease progression.
Parkinson's disease is thus a multifactorial disease, comprising both genetic and environmental risk factors. About 90% of all cases are sporadic, with no determined underlying cause. The remaining 10% of PD cases have been linked to predisposing genes that can be inherited either in an autosomal dominant or recessive manner (Klein & Westenberger, 2012). The first familial variant of PD was discovered in 1997, with a mutation of the SNCA gene, which encodes alpha‐synuclein (α‐syn) (Polymeropoulos et al., 1997). This was a promising discovery, as sporadic PD coincides with progressive intraneuronal accumulation of α‐syn, forming protein aggregates named Lewy bodies throughout the central nervous system (CNS). These findings suggest a causal role for α‐syn in the degenerative process. Moreover, a triplication of wild‐type α‐syn has also been related to familial PD (Singleton et al., 2003), establishing that overexpressing regular α‐syn is sufficient to initiate aggregation and cause disease. Furthermore, this implies a causal role for α‐syn in the degenerative process through a toxic gain‐of‐function mechanism that underlies neuronal death, but what triggers this aggregation in sporadic PD remains elusive.
In recent years, many more disease genes have been identified that encode proteins that seem to be working in many different pathways and have a myriad of functions, though all underlying a similar disease phenotype. This sparked discussion whether PD can be considered one disease or more like a syndrome comprised of a multiple of diseases with a similar phenotype, which may have implications for treatment. However, the distinction between sporadic PD and hereditary forms is not always clear‐cut. Familial varieties often do not have complete penetrance, and sporadic PD patients often do possess some genetic risk factors that include mutations in genes related to familial PD (Schulte & Gasser, 2011). As genetic and sporadic PD share certain characteristics, mainly the specific degeneration of the SNc and locus coeruleus (LC) often accompanied by Lewy bodies, the mechanism underlying the most prominent disease symptoms may very well be the same. Recently, the aberrant interactions between α‐syn and catecholamines have been pointed out as a potential mechanism for the specific degeneration seen in PD (Post, Lieberman, & Mosharov, 2018). This review will discuss how several familial variants of PD may directly facilitate these same aberrant interactions in the specific brain areas involved, providing an explanation for the similar phenotypes of sporadic and familial PD. Identifying such a common mechanism would prove very valuable in terms of potential therapies, especially in these familial varieties where early diagnosis is possible.
2. SPORADIC PARKINSON'S DISEASE AND ALPHA‐SYNUCLEIN
α‐syn is believed to play a causal role in the neurodegeneration seen in PD. It is the primary constituent of Lewy bodies, the protein aggregates accompanying both sporadic PD and most cases of familial PD. Despite it being a high interest topic due to its implications in the disease, surprisingly little is known about its exact endogenous functions. α‐syn is a small, 140 amino acid, evolutionarily conserved protein that is specific to vertebrates (Jakes, Spillantini, & Goedert, 1994; Mor, Ugras, Daniels, & Ischiropoulos, 2016). It consists of three domains: an N‐terminal lipid‐binding domain, an amyloid binding domain and a C‐terminal acidic tail (Emamzadeh, 2016). Indeed, α‐syn often associates with lipid membranes, primarily interacting with membranes with high curvature, like pre‐synaptic vesicles. Coincidentally, α‐syn is largely localised to the pre‐synaptic nerve terminal, where it is believed to play a part in neurotransmitter handling and release (Logan, Bendor, Toupin, Thorn, & Edwards, 2017). It has been shown to promote both vesicle retention in the pre‐synaptic vesicle pool and neurotransmitter release by aiding in the SNARE complex‐mediated fusion of the vesicle with the pre‐synaptic membrane (Bendor, Logan, & Edwards, 2013; Burré, Sharma, & Südhof, 2014; Emamzadeh, 2016). Upon interaction with membranes, the N‐terminal adopts an alpha helix structure (Emamzadeh, 2016). In contrast, α‐syn is a natively largely unstructured protein when localised to the cytosol. These cytosolic, unstructured monomers are more prone to aggregate (Mor et al., 2016).
It is most likely that α‐syn mediates neurodegeneration mainly via a toxic gain‐of‐function mechanism. A loss of function seems an unlikely major contributor as α‐syn KO mice show no symptoms or signs of neurodegeneration. In part, this was attributed to functional redundancy between α‐syn and the closely related β‐ and γ‐synuclein. However, even a triple KO for all synucleins, albeit having some behavioural abnormalities and alterations in neurotransmitter handling, showed no signs of neurodegeneration (Anwar et al., 2011). In contrast, experimental overexpression of α‐syn causes it to aggregate and lead to cell death, which is similar to what is seen in the familial form of PD caused by SNCA gene multiplication. Moreover, several point mutations have been found within the SNCA gene that all lead to autosomal dominant forms of PD: A30P, E46K, H50Q, G51D and A53T. Interestingly, these mutations are clustered in the lipid‐binding domain of α‐syn, affecting its alpha helix structure and lipid affinity. Although they do not behave exactly the same in every respect, the A30P, H50Q and A53T mutants do have in common that they are more unstructured and have an increased propensity to form protofibrils (Narhi et al., 1999; Rutherford, Moore, Golde, & Giasson, 2014). This supports that aggregation of α‐syn likely plays an important role in PD in general, although there may be other factors at play as well. Additionally, α‐syn is a vertebrate‐specific protein, but it can aggregate and mediate specific dopaminergic neurodegeneration in non‐vertebrate organisms like Drosophila melanogaster when experimentally expressed (Feany & Bender, 2000). The dominant manner of inheritance of SNCA‐related PD combined with an intrinsic propensity to aggregate and causing cell death supports a toxic gain‐of‐function mechanism.
The coincidental appearance of Lewy bodies in PD and the toxic properties of aggregating α‐syn indicate that Lewy bodies may play a causal role. Indeed, large aggregates can interfere with cellular processes due to their size. However, aggregation into large aggregates might not be the most harmful aspect of α‐syn aggregation. Smaller, soluble, oligomeric species that generate early on in the aggregation process likely possess the most detrimental properties (Winner et al., 2011). α‐syn oligomers exert their toxic effects by interfering with SNARE complex formation and disrupting membranes, making them more permeable (van Rooijen, Claessens, & Subramaniam, 2010; Winner et al., 2011). This subsequently affects calcium (Ca2+) regulation (Danzer et al., 2007), pre‐synaptic vesicle integrity, neurotransmitter release and mitochondrial function, which are all suspected to be involved in PD pathology. Therefore, favouring large, stable aggregates minimises exposure to these oligomers. Although aggregates themselves are harmful as well, they are likely a lesser of evils when compared to their small oligomeric counterparts.
Most neurodegenerative diseases, including PD, are associated with pathogenic protein aggregation that starts later in life. There are protein quality control (PQC) systems in place within the cell that guard the protein homeostasis and can prevent protein aggregation (Martinez‐Vicente, Sovak, & Cuervo, 2005). Chaperone proteins regulate correct protein folding and work closely together with clearance mechanisms dedicated to degrading misfolded, damaged and even aggregated proteins (Hartl, Bracher, & Hayer‐Hartl, 2011). The main mechanism by which α‐syn is degraded is chaperone‐mediated autophagy (CMA) (Vogiatzi, Xilouri, Vekrellis, & Stefanis, 2008), although the ubiquitin–proteasome system is also involved in regulating α‐syn levels (Webb, Ravikumar, Atkins, Skepper, & Rubinsztein, 2003). The CMA substrate is transported to lysosomes. There, the substrate is recognised by LAMP2A, the lysosomal receptor responsible for transporting CMA substrates into the lysosomal lumen for degradation. The PQC mechanisms, including CMA, are known to decline with age, making cells more susceptible to pathogenic protein accumulation (Hartl et al., 2011). Additionally, the amount of damaged proteins increases with age due to accumulation of somatic mutations in the DNA, as well as increased damage to proteins themselves caused by higher amounts of reactive oxygen species (ROS) due to age‐related decrease in mitochondrial function (Krisko & Radman, 2019). This puts increasing pressure on a declining PQC system, allowing aggregation‐prone proteins to escape this system, which explains why most protein aggregation‐associated neurodegenerative diseases arise at a later age (Martinez‐Vicente et al., 2005).
α‐syn pathology is not strictly cell autonomous, and aggregation‐prone α‐syn may spread from neuron to neuron. PD patients who have received grafted embryonic stem cells to replace lost dopaminergic neurons had Lewy bodies present within these cells upon post‐mortem tissue examination (Li et al., 2008). Mouse models later provided evidence that α‐syn accumulation can spread throughout the brain in a prion‐like manner (Bernis et al., 2015; Planchard, Exley, Morgan, & Rangachari, 2014; Reidy, Sharma, Roberts, & Masison, 2016). Interestingly, Braak and colleagues have reported that, in sporadic PD cases, α‐syn accumulation spreads through the CNS in a specific manner. It first shows up in the enteric nervous system and the olfactory bulb. It then moves up the vagus nerve to the brainstem, subsequently to the basal ganglia, the SNc and LC, and lastly spreading through the cortex (Braak et al., 2003). Interestingly, this pattern correlates to some of the pre‐motor symptoms and the late‐stage dementia. However, the SNc and LC are reached relatively late by this spread of pathological α‐syn.
3. SELECTIVE VULNERABILITY
As PD progresses, α‐syn pathology will spread through most of the CNS and affect many brain areas. However, the pattern of α‐syn accumulation does not correlate well with neuronal cell death (Braak et al., 2003). The SNc and the LC degenerate early on, compared to other degenerating areas, and much more severely, despite being reached by α‐syn pathology relatively late. It has been suggested that these dopaminergic neurons may be more vulnerable because they are large, lack myelin and have a higher energy requirement than other neurons (Bolam & Pissadaki, 2012; Foomt, 1985), resulting in more ROS production and related damage. However, this does not explain why these areas are so vulnerable in PD specifically. Moreover, dopaminergic neurons in the ventral tegmental area (VTA), for example, are relatively spared, despite sharing most of their characteristics with the SNc. The common factor of the SNc and the LC, which is also specific to these areas, is that they both accumulate neuromelanin with age. Neuromelanin is a pigment that gives these areas their characteristic dark colour, after which they are named. Interestingly, neuromelanin mainly consists of iron molecules in conjunction with oxidised catecholamine derivatives native to the respective area, enveloped in a double membrane of autophagic origin (Sulzer et al., 2000; Zucca et al., 2018). Coincidentally, the SNc and LC have elevated levels of cytosolic catecholamines when compared to other catecholaminergic areas. Cytosolic catecholamines can be toxic at a neutral pH, where they can auto‐oxidise and cause damage to the cell. For the mechanisms, this review will focus mostly on the SNc and dopamine, as there are more data available about this area due to its direct link to the motor symptoms of PD, though it is likely that the LC is subjected to similar processes.
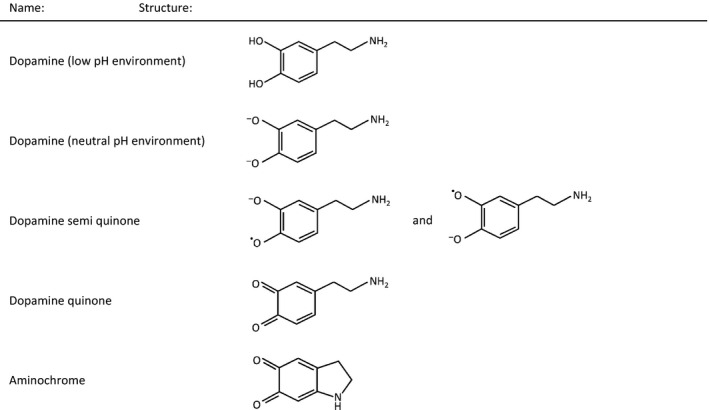
After production, dopamine is immediately stored in pre‐synaptic vesicles by the vesicular dopamine transporter VMAT2. Due to coinciding proton transport required for transporter activity, the vesicle lumen has a low pH value at which dopamine is stable and does not auto‐oxidise. Tyrosine hydroxylase (TH) and l‐amino acid decarboxylase (AADC) have been found to associate with VMAT2, likely to promote dopamine storage in vesicles and limit its presence in the cytosol (Cartier et al., 2010). Given that dopaminergic neurons of the SNc have higher cytosolic dopamine levels compared to other dopaminergic areas, it makes them more susceptible to dopamine oxidation and its toxic consequences. At a neutral pH, dopamine can spontaneously react with oxygen by donating electrons from its O− groups to free oxygen molecules. This results in the formation of dopamine quinones and creates ROS in the process. Dopamine quinones stabilise in the form of aminochrome (Table 1; Muñoz, Huenchuguala, Paris, & Segura‐Aguilar, 2012). While high amounts of ROS are damaging on their own, aminochrome can form adducts with several proteins implicated in PD, including Parkin (LaVoie, Ostaszewski, Weihofen, Schlossmacher, & Selkoe, 2005), DAT (Whitehead, Ferrer, Javitch, & Justice, 2001) and perhaps, most interestingly, α‐syn (Chen & Saez‐Atienzar, 2018; Muñoz et al., 2012). Dopaminergic neurons have mechanisms in place to prevent aminochrome formation, including TH feedback inhibition (Dickson & Briggs, 2013), VMAT2‐mediated storage in vesicles and neuromelanin formation. Indeed, neuromelanin is likely a protective mechanism to sequester reactive dopamine forms to prevent aberrant interactions in the cytosol. This is supported by the fact that neuromelanin is absent at birth and accumulates with age. Neuromelanin formation under normal conditions is almost exclusively seen in humans (Fedorow et al., 2005). However, it can be induced in animal models and cell models in which neuromelanin is normally absent by supplementing them with l‐Dopa, further establishing neuromelanin as a storage mechanism for excess dopamine. This effect can be rescued by overexpression of VMAT2, indicating that it is indeed a consequence of high cytosolic dopamine content (Sulzer et al., 2000).
Table 1.
Structures of dopamine and dopamine quinones. In a low pH environment, dopamine is stable, whereas in a neutral pH environment like the cytosol, it can donate electrons and form dopamine quinones, generating reactive oxygen species (ROS) in the process
Although the human SNc has high levels of cytosolic dopamine, this alone is not sufficient to cause neurodegeneration. However, there is evidence that aggregating α‐syn and cytosolic dopamine can both exacerbate each other's pathogenic properties (Post et al., 2018). Dopamine quinones can stabilise α‐syn oligomers and inhibit aggregate formation. This causes an accumulation of toxic oligomers that subsequently lead to more vesicle damage, causing more dopamine to leak into the cytosol (Conway, Rochet, Bieganski, & Lansbury, 2001). Additionally, dopamine quinones can interact with α‐syn monomers, forming adducts that block LAMP2A and thus CMA. They do so by interacting with LAMP2A, but LAMP2A is not able to transport these adducts to the lysosomal lumen, effectively blocking this transporter (Martinez‐Vicente et al., 2008). The A30P and A53T familial SNCA mutants also block CMA in a similar manner (Cuervo, Wong, & Martinez‐Vicente, 2010; Post et al., 2018). As α‐syn degradation is largely dependent on CMA, it can block its own degradation in the presence of cytosolic dopamine. This can cause an increase of α‐syn levels and consequentially its propensity to aggregate. While overexpression of α‐syn is most toxic in neurons with relatively high cytosolic dopamine content, the opposite is also true. VMAT2‐KD mice have increased cytosolic dopamine levels; however, this only causes degeneration when α‐syn is present as well, and not in α‐syn KO mice. VMAT2‐KD mice with endogenous α‐syn also showed Lewy body accumulation, indicating that increased cytosolic dopamine alone can trigger α‐syn aggregation at normal α‐syn levels (van der Putten et al., 2000). This shows that elevating either α‐syn or cytosolic dopamine levels, with the other present at normal levels, can lead to α‐syn aggregation.
Another common factor of the SNc and LC is that they have high Ca2+ content, due to their intrinsic pacemaking activity. Ca2+ positively regulates catecholamine production (Mosharov et al., 2009), which may contribute to the high catecholamine levels in these areas. Moreover, these areas lack calbindin that binds free Ca2+, while calbindin is present in dopaminergic neurons like the VTA that are relatively spared in PD (Dauer & Przedborski, 2003). Ca2+ is normally translocated to the mitochondrial lumen because of the negative charge generated by proton pump activity. However, additional Ca2+ can leak into the cell through α‐syn oligomer‐mediated permeabilisation of the cell membrane and lysosomal membranes (Bourdenx, Bezard, & Dehay, 2014; Post et al., 2018). High cytosolic Ca2+ levels may contribute to increased catecholamine production (Mosharov et al., 2009) and α‐syn aggregation (Rcom‐H'cheo‐Gauthier, Goodwin, & Pountney, 2014), in addition to being toxic in itself (Orrenius & Nicotera, 1994).
Taken together, the interactions of α‐syn, dopamine and Ca2+ provide a mechanism that can set in motion a vicious cycle of toxicity leading to degeneration (Figure 1). Moreover, the SNc and LC provide the perfect conditions for such a process due to their high cytosolic dopamine and Ca2+ content. More importantly, this mechanism can lead to the same pathological process, regardless of which component triggered it. While familial PD genes are seemingly involved in different processes, it could be that they connect to different parts of this central mechanism and subsequently cause a highly similar degenerative phenotype.
Figure 1.
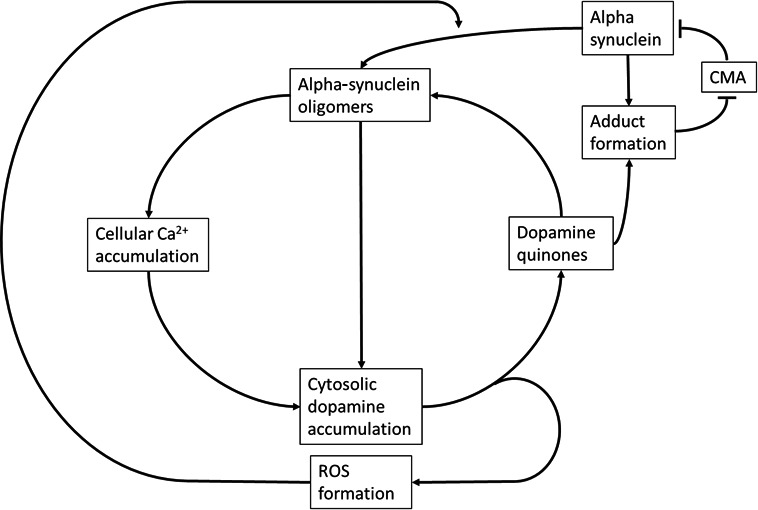
Simplified schematic representation of the toxic cycle underlying the substantia nigra pars compacta degeneration seen in PD. An increase in α‐syn levels can induce α‐syn oligomer formation. These oligomers permeabilise cell and vesicle membranes, causing accumulation of cytosolic dopamine and Ca2+, where the latter further promotes dopamine production. Cytosolic dopamine auto‐oxidises into dopamine quinones, forming reactive oxygen species (ROS) in the process, which in turn promotes α‐syn oligomer formation. Dopamine quinones stabilise α‐syn oligomers as well as forming adducts with α‐syn monomers, which block chaperone‐mediated autophagy (CMA). Subsequently, CMA‐mediated degradation of α‐syn (and other CMA substrates) is impaired leading to further α‐syn accumulation. The factors in this positive feedback cycle all contribute to cellular stress, protein, DNA and lipid damage, eventually leading to cell death
4. FAMILIAL PD—PARKIN AND PINK1
There are many gene variants known to be associated with familial PD. Several are heritable in an autosomal dominant manner, including mutations in SNCA, LRRK2 and VPS35, which all cause late‐onset PD very similar to sporadic PD. Others, namely Parkin, PINK1 and DJ‐1, are heritable in an autosomal recessive manner and result in early‐onset PD (Klein & Westenberger, 2012; Schulte & Gasser, 2011). Many of the PD genes have a myriad of functions and are expressed throughout the CNS or even beyond. Yet, their PD‐associated mutant varieties lead to the same characteristic degeneration of the SNc and LC as seen in sporadic PD. Additionally, the line between sporadic PD and familial PD can be blurry in certain cases. Heterozygosity for recessive variants is a risk factor for sporadic PD, and dominant variants never have complete penetrance (Schulte & Gasser, 2011). The recessive PD variants Parkin, PINK1 and DJ‐1 are interesting for finding a common mechanism linking sporadic and familial PD because homozygotes have 100% penetrance combined with an early disease onset (Schulte & Gasser, 2011). If there is a connection to aberrant α‐syn–dopamine interactions, it must be most clear in these because of this severe phenotype.
Parkin is encoded by the PARK2 gene, and mutations in this gene are known to cause recessive, early‐onset PD (Lücking et al., 2000), where degeneration can occasionally be accompanied by Lewy bodies. Parkin is an E3 ubiquitin ligase and a component of the PQC system, responsible for tagging to be degraded proteins with ubiquitin, which marks them for degradation. There is a large amount of E3 ligases that have their own set of substrates, which provides specificity to the degradation system. Parkin is a ubiquitous protein, but expression is relatively high in the CNS. PD‐associated mutations are spread throughout the whole gene and include truncations, pointing towards a loss of function being responsible for the PD phenotype. Several Parkin KO models have been developed to help identify a neurodegenerative mechanism; however, results remain inconsistent and species‐dependent. In Drosophila melanogaster, Parkin depletion mediated degeneration of the flight muscles, accompanied by mitochondrial damage (Greene et al., 2003). Mouse models showed no clear phenotype but did show behavioural changes (Goldberg et al., 2003) and a general increased cellular sensitivity to apoptosis (Casarejos et al., 2006). Cell models showed morphological changes to mitochondria accompanied by excessive ROS production (Narendra, Tanaka, Suen, & Youle, 2008; Pickrell & Youle, 2015).
Mitochondrial dysfunction and ROS‐related damage had previously been associated with sporadic PD (Schapira et al., 1990). Later research suggested the involvement of α‐syn, which can be imported into mitochondria where it blocks complex 1 of the mitochondrial respiration chain, leading to mitochondrial depolarisation and ROS production (Martínez et al., 2018; Reeve et al., 2015). This sparked the idea that mitochondrial deficits may play a causal role in PD. Interestingly, Parkin was found to be involved in mitophagy, the clearance of mitochondria through macro‐autophagy. Moreover, the signalling pathway regulating this process also includes PINK1, another gene involved in familial PD (Pickrell & Youle, 2015). Coincidentally, PINK1 mutations also cause recessive, early‐onset PD, which is generally associated with Lewy bodies (Maria Valente et al., 2004). Experimentally, a PINK1 KO can be rescued through Parkin overexpression, whereas a Parkin KO cannot be rescued by PINK1 overexpression, indicating that PINK1 functions upstream of Parkin. Aged or damaged mitochondria lose their membrane potential, which causes PINK1 to translocate to the outer mitochondrial membrane. Here, PINK1 recruits Parkin and mediates its activation by phosphorylation of Parkin, relieving Parkin's auto‐inhibited state in which it is normally present. Parkin then mediates clustering of these mitochondria and subsequent clearance via mitophagy (Matsuda et al., 2010; Pickrell & Youle, 2015). PD‐associated mutations in the PINK1 gene are all localised in or very near to its kinase domain, indicating a loss of function of this kinase domain as the culprit. Indeed, this is the kinase domain that phosphorylates Parkin in the mitophagy process and mutant varieties in this region fail to recruit Parkin (Song et al., 2013).
It is still debated whether Parkin and PINK1 deficiency‐mediated mitophagy impairment is sufficient to cause a PD phenotype. Indeed, Parkin and PINK1 are not the only means of mitophagy regulation. For example, the MUL1 pathway can compensate for Parkin/PINK1 deficiency and removing both pathways aggravates the phenotype beyond the loss of a single pathway (Yun et al., 2014). Additionally, in mouse myocytes, Parkin can still be recruited to defective mitochondria in the absence of PINK1 (Kubli et al., 2015). However, specifically under high ATP demand, PINK1 KO does lead to mitochondrial issues. Subsequently, cytosolic Ca2+ levels rise as Ca2+ export pumps require a lot of ATP. The SNc and LC coincidentally have a high ATP demand due to their intrinsic pacemaking activity; thus, PINK1 deficiency could potentially lead to issues in these areas specifically (Heeman et al., 2011). Additionally, the increase in cytosolic Ca2+ positively regulates catecholamine production (Mosharov et al., 2009), while the ATP deficiency can impair VMAT2 functioning, both of which cause an increase in cytosolic dopamine levels.
Aside from its role in mitochondrial quality control, Parkin has other interesting functions with potential implications in PD. For example, Parkin can protect against dopamine toxicity and apoptosis in human neuroblastoma cells. It specifically suppressed dopamine‐induced ROS production and subsequent apoptosis, while having no effect on the consequences of ROS in general. Moreover, this protective effect was not seen in Parkin with PD‐linked mutations (Jiang, Ren, Zhao, & Feng, 2004). Lastly, in a rat model for PD expressing aggregation‐prone A30P α‐syn, Parkin overexpression protected from neurodegeneration while mediating an increase in α‐syn aggregation (Lo Bianco et al., 2004). This is in line with the notion that α‐syn aggregates are potentially protective when compared to small oligomeric species. Thus, Parkin could potentially antagonise the oligomer‐stabilising effect of cytosolic dopamine.
Taken together, Parkin and PINK1 work closely together in regulating mitochondrial quality control, an effect that is dependent on Parkin functionality. A loss of Parkin function due to mutation or lack of activation by the upstream PINK1 may lead to mitochondrial issues, increased oxidative stress and Ca2+ disturbances that the SNc and LC are specifically vulnerable to due to their high energy demand. These conditions subsequently favour dopamine oxidation and α‐syn accumulation, while Parkin's protective effects against dopamine toxicity and its ability to deal with the consequences of pathogenic α‐syn accumulation are impaired.
5. FAMILIAL PD—DJ‐1
Several mutations in the PARK7 gene, encoding DJ‐1, have been shown to cause recessive, early‐onset PD (Bonifati et al., 2003; Malgieri & Eliezer, 2008) that may be associated with Lewy body accumulation (Taipa et al., 2016). Similar to Parkin and PINK1 mutations, DJ‐1 homozygosity is accompanied by a 100% disease penetrance with an early onset (Schulte & Gasser, 2011). DJ‐1 is a small protein with a myriad of functions, including ROS scavenging (Andres‐Mateos et al., 2007), metal ion binding (Björkblom et al., 2013), chaperone activity (Shendelman, Jonason, Martinat, Leete, & Abeliovich, 2004), CMA regulation(Xu et al., 2017) and transcriptional regulation. The PD‐related mutations in DJ‐1 cause a loss of structure, increased degradation and/or decreased dimerisation (Malgieri & Eliezer, 2008). Together with heterozygote susceptibility to sporadic PD, this also indicates a loss of function being the underlying cause. Due to its wide range of functions, it has proven very difficult to elucidate how it mediates specific the catecholaminergic neuronal degeneration leading to PD. However, there are several connections that can be made to mechanisms involved in PD.
One such mechanism is that DJ‐1 protects against ROS through direct interaction with mitochondria. DJ‐1 deficiency leads to altered mitochondrial morphology, likely causing slight alterations in mitophagy and a subsequent increase of ROS production (McCoy & Cookson, 2011; Trempe & Fon, 2013). This effect can be rescued by Parkin and/or PINK1 overexpression, as these respond to depolarised mitochondria and regulate their subsequent clearance (Trempe & Fon, 2013). Moreover, DJ‐1 can be oxidised at Cys‐106 to function as a ROS scavenger (Andres‐Mateos et al., 2007), while DJ‐1 levels also increase in response to oxidative stress caused by dopamine to suppress ROS accumulation (Lev et al., 2013). Thus, a lack of DJ‐1 can contribute to increased oxidative stress.
Additionally, DJ‐1 interacts with α‐syn and can influence the aggregation process of pathological α‐syn. DJ‐1 can directly inhibit the early aggregation steps of α‐syn by acting as a redox‐dependent molecular chaperone. Oxidising conditions make proteins, including α‐syn, more prone to aggregate; thus, it makes sense to have a condition‐dependent defence mechanism. The chaperone activity of DJ‐1 requires oxidation of Cys‐53 caused by the presence of ROS. DJ‐1 was shown to suppress the formation of α‐syn oligomers, and subsequently their toxic effects (Shendelman et al., 2004). Additionally, DJ‐1 deficiency caused a decrease in LAMP2A expression, the receptor required for CMA‐mediated α‐syn degradation, and subsequent α‐syn accumulation (Xu et al., 2017). Taken together, the lack of DJ‐1 may contribute to α‐syn aggregation through impaired chaperoning activity and decreased α‐syn degradation.
Most intriguing though are the effects of DJ‐1 deficiency on dopamine homeostasis. In Dictyostelium discoideum, DJ‐1 KD did not result in the expected mitochondrial dysfunction‐related phenotype. However, decreased DJ‐1 expression caused a slower phagocytosis rate, while increased expression caused a higher phagocytosis rate (Chen et al., 2017). Interestingly, this effect has been found to extend to neurons, where DJ‐1 KO in primary cultured mouse neurons severely impaired synaptic vesicle endocytosis. The effect was completely rescued after transfection with WT DJ‐1, but not after transfection with PD‐related mutant varieties of DJ‐1. Moreover, the exocytosis process was unaffected by DJ‐1 KO. Altogether, this caused a severe depletion of the pre‐synaptic vesicle pool upon neuronal stimulation (Kyung et al., 2018). Additionally, DJ‐1 regulates the expression of VMAT2, the sole transporter responsible for sequestering dopamine inside pre‐synaptic vesicles. DJ‐1 overexpression boosts VMAT2 expression, while DJ‐1 KD resulted in decreased VMAT2 expression. As DJ‐1 levels increase under (dopamine‐induced) oxidative stress, this likely serves as a feedback mechanism where DJ‐1 increases VMAT2 in response to increased cytosolic dopamine levels (Lev et al., 2013). Conversely, DJ‐1 depletion can severely impair the dopamine storage ability of dopaminergic neurons.
Interestingly, DJ‐1 KO alone does not lead to a neurodegenerative phenotype in mice, while it leads to early‐onset PD in humans. DJ‐1‐deficient mice do exhibit alterations in dopamine metabolism compared to WT mice. Young DJ‐1‐deficient mice show an increase in dopamine turnover accompanied by increased ROS production. In aged DJ‐1‐deficient mice, this turnover is decreased compared to WT, while there is an increase in dopamine re‐uptake via DAT, causing an accumulation of striatal dopamine (Raman et al., 2013). While cytosolic dopamine normally interacts with and inhibits TH as a feedback mechanism, the presence of oxidised dopamine species can increase TH activity on the long term (Acheson, Zigmond, & Stricker, 1980). Although there are signs of cellular damage, DJ‐1 KO mice do not show signs of neurodegeneration and cell death (Raman et al., 2013). The discrepancy is likely due to human SNc neurons having higher overall and oxidised dopamine levels than their mouse counterparts, making them more susceptible to dopamine oxidation. This is reflected by neuromelanin formation in the human SNc, while mice lack this pigmentation (Burbulla et al., 2017). Experimentally increasing dopamine levels in WT mouse iPSC have no significant effect on oxidised dopamine levels. However, doing the same thing in DJ‐1 KO mouse iPSC caused a dramatic increase in oxidised dopamine levels. In vivo experiments in mice also showed that combining DJ‐1 KO with l‐Dopa treatment caused SNc neurodegeneration with α‐syn aggregation. Additionally, they demonstrated that combining DJ‐1 deficiency with aggregation‐prone α‐syn A53T showed more severe neurodegeneration and increased oxidised dopamine levels than in α‐syn A53T mice with functional DJ‐1 (Burbulla et al., 2017). This corroborates that aggregating α‐syn can increase oxidised dopamine levels and vice versa.
Taken together, DJ‐1 deficiency can cause major disturbances in dopamine storage through depletion of the pre‐synaptic vesicle pool and decreased VMAT2 expression. In addition, in mice it mediates dopamine accumulation at a later age through increased dopamine re‐uptake. Lastly, DJ‐1 deficiency leads also to increased oxidative stress, which it could normally protect against. Combined, this provides the perfect conditions for dopamine oxidation and quinone formation, which in turn can mediate α‐syn aggregation and cellular damage leading to neurodegeneration.
6. DISCUSSION
Genetic cases of a disease can offer critical insight into the disease mechanism and pathways involved. However, genes related to familial variants of PD are often involved in multiple cellular functions and pathways, making it difficult to distinguish exactly what functionality underlies the PD‐related symptoms. Due to the highly similar degeneration of the SNc and LC between sporadic and familial PD, it is likely that the mechanism involved in these areas is the same as in sporadic PD, although the trigger may be different. The recessive familial PD gene variants of Parkin, PINK1 and DJ‐1 are shown to be capable of causing disruptions in both dopamine and Ca2+ homeostasis, leading to dopamine quinone formation, mitochondrial issues and related oxidative stress. In turn, these conditions trigger α‐syn aggregation and subsequently set in motion a positive feedback cycle towards neuronal death. In contrast to sporadic PD, α‐syn aggregation should start in the SNc and LC in these specific genetic cases, as it is triggered by conditions unique to these areas. It would be very interesting to see Braak staging of α‐syn propagation in these cases specifically if possible. Taken together, the mutant varieties of PD‐related genes, although operating in different pathways, may all disrupt at least one of the three key players central to the neurodegeneration seen in PD, leading to disease. However, this review only discussed the known recessive variants and it remains to be elucidated whether similar connections can be made for the autosomal dominant familial variants of PD.
The disease mechanisms of DJ‐1 and Parkin dysfunction specifically could also have implications for PD in general. DJ‐1 forms conjugates with dopamine quinones, with modifications by dopamine on both Cys‐53 and Cys‐106. The former causes the formation of covalent dimers, while the latter makes DJ‐1 more thermodynamically unstable and causes the formation of dopamine‐modified high molecular weight species (Girotto et al., 2012). Normally, oxidation of these specific Cys residues is required for DJ‐1 to function as a chaperone and ROS scavenger, respectively (Andres‐Mateos et al., 2007; Shendelman et al., 2004). Although the exact consequences of these dopamine quinone interactions remain to be elucidated, it almost certainly interferes with its normal functioning. Thus, the protective functions of DJ‐1 may also be impaired to some extent in PD cases unrelated to DJ‐1 mutation. Additionally, dopamine quinones can covalently bind Parkin, forming adducts that functionally inactivate it and cause Parkin to cluster together in insoluble aggregates (LaVoie et al., 2005). This presents another potential loop, where dopamine toxicity can disable a mechanism that would normally protect against it. Taken together, in PD cases unrelated to DJ‐1 or Parkin mutation, accumulation of dopamine quinones can cause impairment of these two proteins, which in turn could contribute to the degenerative process. This may provide an example why the distinction between familial and sporadic PD is not always clear‐cut. Impairment of the same proteins may play a part in the disease process, even though dysfunction can arise at both the genetic and the protein level depending on the case.
Further research using model organisms is required to unravel the specifics of the disease mechanisms and find the best therapeutic avenue per case. However, the importance of both dopamine and α‐syn in SNc degeneration stands out, as it is seen in many model organisms for PD, including the commonly used MPTP‐induced models. MPTP is a toxin that mediates specific PD‐like neurodegeneration, with prominent neuronal death in the SNc and LC. Since its discovery in the 70s, it has been widely used to replicate the consequences of PD‐related neurodegeneration in model organisms, in addition to providing evidence that PD symptoms can occur due to environmental toxin exposure. MPTP makes use of dopamine transporters like DAT and VMAT2 to cross membranes. Once in the cell, it exerts its toxic effects by blocking complex 1 of the mitochondrial respiration chain, causing mitochondrial dysfunction, high levels of ROS and eventually cell death (Langston, 2017). Indeed, mitochondrial deficits are also observed in Parkin/PINK1 deficiency, in which it causes disturbances that the SNc and LC are particularly vulnerable to. Interestingly, in MPTP‐induced models, other dopaminergic areas like the VTA are relatively spared, just as is seen in PD. Moreover, prolonged exposure to MPTP causes α‐syn accumulation, indicating that the selective sensitivity to MPTP may involve similar mechanisms implicated in PD (Meredith & Rademacher, 2011). This is supported by the finding that α‐syn KO mice are resistant to MPTP toxicity (Dauer et al., 2002), and increased cytosolic dopamine levels may enhance MPTP toxicity in mice. However, the latter is difficult to assess as increased dopamine levels were induced through dopamine transporter manipulation (Lohr et al., 2016; Masoud et al., 2015; Takahashi et al., 1997). As MPTP makes use of these same transporters, this would potentially alter cellular MPTP levels as well, yielding inaccurate results. However, it would be interesting to see whether MPTP exposure after l‐Dopa treatment also enhances MPTP toxicity, as this would likely have a lesser effect on transporter presence. Altogether, the specificity of MPTP‐mediated toxicity cannot be attributed only to its specific use of dopamine transporters but may also involve the presence of α‐syn and dopamine. This provides additional insight into how MPTP‐induced models emulate PD‐related degeneration so accurately. Additionally, it indicates that modulating the presence of α‐syn and dopamine could indeed be effective in preventing degeneration of the SNc.
Research into specific familial variants of PD has been particularly difficult because animal models often do not exhibit any PD‐related symptoms. Recent findings in a DJ‐1 KO mouse model suggest that this may be because the human SNc contains significantly higher amounts of cytosolic dopamine than any model organism (Burbulla et al., 2017). Indeed, it is becoming clear that in familial PD variants like Parkin, PINK1 and DJ‐1, α‐syn aggregation with subsequent neurodegeneration is likely a secondary effect that is induced by disruption of dopamine and Ca2+ homeostasis. Due to the lower dopamine levels in these model organisms, it is conceivable that there is not enough dopamine to significantly disrupt, resulting in a lack of phenotype. Elevating dopamine levels through l‐Dopa administration in the case of these DJ‐1 KO mice caused them to develop PD‐like phenotype, which either condition alone was unable to accomplish. Thus, model organisms in general may not accurately reflect the human situation in terms of SNc dopamine homeostasis, resulting in potential disease mechanisms being overlooked. Indeed, Parkin KO mice also show no clear PD phenotype on their own, which could very well be due to the same reason. The mechanism of Parkin deficiency‐related degeneration is partly dependent on the dopamine toxicity it causes through mitochondrial issues and increased cytosolic dopamine levels (Mosharov et al., 2009). It is likely that in mice, this effect is not dramatic enough to trigger excessive dopamine toxicity due to the lower dopamine levels in this species. Subsequently, the adverse effects on α‐syn, for example, remain absent, and neurodegeneration does not occur. Taken together, increasing neuronal dopamine levels in concert with a familial PD mutation may be required to achieve a correct phenotype in models for other genetic PD variants as well. Indeed, the conditions for developing PD are almost exclusively seen in humans. To understand the complex interactions underlying the degenerative process, future studies should be aimed at updating PD models where necessary to accurately reflect the human situation. Interestingly, if cytosolic dopamine levels can be the determining factor whether neurodegeneration occurs in the presence of DJ‐1 mutation, it may present an interesting potential therapeutic avenue for these cases.
Elucidating a degenerative mechanism may open new avenues for therapeutic intervention. It now seems that the most prominent degeneration in sporadic PD as well as familial PD variants involves α‐syn accumulation, cytosolic catecholamines and Ca2+. Each can be the potential trigger, depending on the underlying disease variant, and all can exacerbate each other's toxic effects. Although there is a central mechanism, each PD‐related mutation can disrupt this mechanism via a different route. In terms of therapy, this means that the required treatment will likely need to be case‐specific, dependent on the underlying genetic variant and pathway affected. Given the many genetic varieties underlying PD, many different therapeutic strategies may have to be developed. On the other hand, the central degenerative mechanism also provides a potential bottleneck where all different pathways intersect. It has been suggested that modulation of one or more of α‐syn, cytosolic catecholamines and Ca2+ could protect SNc and LC neurons from PD‐related degeneration in general (Post et al., 2018), given that these are the primary contributors to toxicity. When the disruption of either of these is caused by an underlying genetic mutation, modulation of this specific component affected may prove sufficient to prevent or delay symptomatic disease onset by preventing excessive toxicity in the SNc and LC. This could prove particularly effective in variants where the pathology originates in these areas. For example, if the primary disease mechanism of DJ‐1 mutation‐related degeneration is the formation of dopamine quinones through disruption of dopamine homeostasis, preventing excessive dopamine oxidation by modulating cytosolic dopamine levels could suffice as a therapy. Moreover, this same therapy may work in other variants as well, albeit by slowing down disease progression or preventing a selection of symptoms. In sporadic PD where α‐syn is the primary culprit, it may prove less effective as the pathological spread of α‐syn continues and will still cause further degeneration, dementia and other (non‐motor) symptoms; however, it may slow degeneration of the areas where catecholamines play a contributing role. Taken together, a central degenerative mechanism provides an opportunity for a general therapeutic strategy. Nevertheless, the effectiveness may be dependent on the underlying cause, and in certain variants, case‐dependent treatment may be necessary.
Although there are promising results regarding the mechanisms underlying PD, the main challenge for application of therapeutic measures, however, is diagnostics. By the time patients exhibit the motor symptoms by which they are diagnosed, they have already lost over 50% of their SNc neurons, and it is not possible to regain what has been lost. Additionally, upon extensive neuronal death, other factors come in to play that start contributing to the degenerative process, complicating the potential for therapeutic measures even further and making them less effective. For example, disruption of the basal ganglia–thalamo–cortical motor loop due to SNc degeneration causes increased activity of the subthalamic nucleus, which feeds back to the SNc. The increase in activity mediates increased glutamate release at the SNc, potentially leading to excitotoxicity that further contributes to neuronal degeneration (Rodriguez, Obeso, & Olanow, 1998). Additionally, the death of neuromelanin‐containing neurons causes neuromelanin, and thus iron and catecholamine quinones, to be released in the extracellular environment. This triggers microglia and causes a state of chronic neuroinflammation that results in further neuronal damage (Zhang et al., 2011). For effective treatment of PD, and neurodegenerative diseases in general, therapeutic measures should ideally be taken before the onset of degeneration, which makes early diagnosis critical. While timely diagnosis of sporadic PD is a major hurdle due to a lack of symptoms and disease markers, it is possible for variants caused by known genetic mutations, like Parkin, PINK1 or DJ‐1. In these cases, therapies aimed at modulating catecholamine oxidation, cytosolic‐free Ca2+ levels and/or α‐syn accumulation may be able to prevent or at least delay disease onset as they could be applied before the onset of degeneration.
It is very likely that due to the many disease‐related genes and pathways, there is no single cause for PD. However, this review shows that familial variants related to Parkin, PINK1 and DJ‐1 can set in motion a chain of events that triggers the same degenerative mechanism that is present in sporadic PD. The aberrant interactions between α‐syn, cytosolic catecholamines and Ca2+ ions, where disruption of one will disrupt the others, can set in motion a vicious toxic cycle leading to neurodegeneration. Moreover, this mechanism is specific to the SNc and LC, which explains why these areas are so selectively and severely affected in sporadic and familial cases alike. While there are many genetic triggers that may require as many different therapies to prevent the disease, the convergence of pathways on a single mechanism also provides us with a strategic bottleneck for potential intervention in disease progression. The usage of accurate models is extremely important to elucidate this topic, as there is evidence that most model organisms do not have high enough cytosolic catecholamine levels to reflect the human situation. However, future research utilising solid model systems can provide us with new potential therapeutic avenues to pursue.
CONFLICT OF INTEREST
The authors have none to declare.
AUTHOR CONTRIBUTIONS
All authors have equally contributed.
van der Vlag M, Havekes R, Heckman PRA. The contribution of Parkin, PINK1 and DJ‐1 genes to selective neuronal degeneration in Parkinson's disease. Eur J Neurosci. 2020;52:3256–3268. 10.1111/ejn.14689
Edited by Eilis Dowd.
The peer review history for this article is available at https://publons.com/publon/10.1111/ejn.14689
DATA AVAILABILITY STATEMENT
No data are available as this is a review.
REFERENCES
- Acheson, A. L. , Zigmond, M. J. , & Stricker, E. M. (1980). Compensatory increase in tyrosine hydroxylase activity in rat brain after intraventricular injections of 6‐hydroxydopamine. Science, 207, 537–540. 10.1126/science.6101509 [DOI] [PubMed] [Google Scholar]
- Andres‐Mateos, E. , Perier, C. , Zhang, L. , Blanchard‐Fillion, B. , Greco, T. M. , Thomas, B. , … Dawson, V. L. (2007). DJ‐1 gene deletion reveals that DJ‐1 is an atypical peroxiredoxin‐like peroxidase. Proceedings of the National Academy of Sciences USA, 104, 14807–14812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar, S. , Peters, O. , Millership, S. , Ninkina, N. , Doig, N. , Connor‐Robson, N. , … Buchman, V. L. (2011). Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. Journal of Neuroscience, 31, 7264–7274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendor, J. T. , Logan, T. P. , & Edwards, R. H. (2013). The function of α‐synuclein. Neuron, 79, 1044–1066. 10.1016/j.neuron.2013.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernis, M. E. , Babila, J. T. , Breid, S. , Wüsten, K. A. , Wüllner, U. , & Tamgüney, G. (2015). Prion‐like propagation of human brain‐derived alpha‐synuclein in transgenic mice expressing human wild‐type alpha‐synuclein. Acta Neuropathologica Communications, 3, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkblom, B. , Adilbayeva, A. , Maple‐Grødem, J. , Piston, D. , Ökvist, M. , Xu, X. M. , … Møller, S. G. (2013). Parkinson disease protein DJ‐1 binds metals and protects against metal‐induced cytotoxicity. Journal of Biological Chemistry, 288, 22809–22820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blandini, F. , Nappi, G. , Tassorelli, C. , & Martignoni, E. (2000). Functional changes of the basal ganglia circuitry in Parkinson's disease. Progress in Neurobiology, 62, 63–88. [DOI] [PubMed] [Google Scholar]
- Bolam, J. P. , & Pissadaki, E. K. (2012). Living on the edge with too many mouths to feed: Why dopamine neurons die. Movement Disorders, 27, 1478–1483. 10.1002/mds.25135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati, V. , Rizzu, P. , van Baren, M. J. , Schaap, O. , Breedveld, G. J. , Krieger, E. , … Heutink, P. (2003). Mutations in the DJ‐1 gene associated with autosomal recessive early‐onset Parkinsonism. Science, 299, 256–259. 10.1126/science.1077209 [DOI] [PubMed] [Google Scholar]
- Bourdenx, M. , Bezard, E. , & Dehay, B. (2014). Lysosomes and α‐synuclein form a dangerous duet leading to neuronal cell death. Frontiers in Neuroanatomy, 8, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak, H. , Tredici, K. D. , Rüb, U. , de Vos, R. A. , Jansen Steur, E. N. , & Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson's disease. Neurobiology of Aging, 24, 197–211. [DOI] [PubMed] [Google Scholar]
- Burbulla, L. F. , Song, P. , Mazzulli, J. R. , Zampese, E. , Wong, Y. C. , Jeon, S. , … Krainc, D. (2017). Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science, 357, 1255–1261. 10.1126/science.aam9080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré, J. , Sharma, M. , & Südhof, T. C. (2014). α‐Synuclein assembles into higher‐order multimers upon membrane binding to promote SNARE complex formation. Proceedings of the National Academy of Sciences USA, 111, E4274–E4283. 10.1073/pnas.1416598111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier, E. A. , Parra, L. A. , Baust, T. B. , Quiroz, M. , Salazar, G. , Faundez, V. , … Torres, G. E. (2010). A biochemical and functional protein complex involving dopamine synthesis and transport into synaptic vesicles. Journal of Biological Chemistry, 285, 1957–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarejos, M. J. , Menendez, J. , Solano, R. M. , Rodriguez‐Navarro, J. A. , Garcia de Yebenes, J. , & Mena, M. A. (2006). Susceptibility to rotenone is increased in neurons from parkin null mice and is reduced by minocycline. Journal of Neurochemistry, 97, 934–946. [DOI] [PubMed] [Google Scholar]
- Chen, S. , Annesley, S. J. , Jasim, R. A. F. , Musco, V. J. , Sanislav, O. , & Fisher, P. R. (2017). The Parkinson's disease‐associated protein DJ‐1 plays a positive nonmitochondrial role in endocytosis in Dictyostelium cells. Disease Models & Mechanisms, 10, 1261–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, V. , & Saez‐Atienzar, S. (2018). A tango for two: Dopamine and α‐synuclein synergy may explain nigrostriatal degeneration in Parkinson's disease. Movement Disorders, 33, 249–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway, K. A. , Rochet, J. C. , Bieganski, R. M. , & Lansbury, P. T. (2001). Kinetic stabilization of the alpha ‐synuclein protofibril by a dopamine‐alpha‐synuclein adduct. Science, 294, 1346–1349. [DOI] [PubMed] [Google Scholar]
- Cuervo, A. M. , Wong, E. S. P. , & Martinez‐Vicente, M. (2010). Protein degradation, aggregation, and misfolding. Movement Disorders, 25, S49–S54. [DOI] [PubMed] [Google Scholar]
- Danzer, K. M. , Haasen, D. , Karow, A. R. , Moussaud, S. , Habeck, M. , Giese, A. , … Kostka, M. (2007). Different species of alpha‐synuclein oligomers induce calcium influx and seeding. Journal of Neuroscience, 27, 9220–9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer, W. , Kholodilov, N. , Vila, M. , Trillat, A.‐C. , Goodchild, R. , Larsen, K. E. , … Hen, R. (2002). Resistance of alpha ‐synuclein null mice to the Parkinsonian neurotoxin MPTP. Proceedings of the National Academy of Sciences USA, 99, 14524–14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer, W. , & Przedborski, S. (2003). Parkinson's disease: Mechanisms and models. Neuron, 39, 889–909. 10.1016/S0896-6273(03)00568-3 [DOI] [PubMed] [Google Scholar]
- Dickson, P. W. , & Briggs, G. D. (2013). Tyrosine hydroxylase: Regulation by feedback inhibition and phosphorylation. Advances in Pharmacology, 68, 13–21. [DOI] [PubMed] [Google Scholar]
- Emamzadeh, F. N. (2016). Alpha‐synuclein structure, functions, and interactions. Journal of Research in Medical Sciences, 21(1), 29 10.4103/1735-1995.181989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feany, M. B. , & Bender, W. W. (2000). A Drosophila model of Parkinson's disease. Nature, 404, 394–398. 10.1038/35006074 [DOI] [PubMed] [Google Scholar]
- Fedorow, H. , Tribl, F. , Halliday, G. , Gerlach, M. , Riederer, P. , & Double, K. L. (2005). Neuromelanin in human dopamine neurons: Comparison with peripheral melanins and relevance to Parkinson's disease. Progress in Neurobiology, 75, 109–124. 10.1016/j.pneurobio.2005.02.001 [DOI] [PubMed] [Google Scholar]
- Foomt, S. L. (1985). Impulse conduction properties of noradrenergic locus coeruleus axons projecting to monkey cerebrocortex. Neuroscience, 15(3), 765–777. 10.1016/0306-4522(85)90077-6 [DOI] [PubMed] [Google Scholar]
- Girotto, S. , Sturlese, M. , Bellanda, M. , Tessari, I. , Cappellini, R. , Bisaglia, M. , … Mammi, S. (2012). Dopamine‐derived quinones affect the structure of the redox sensor DJ‐1 through modifications at Cys‐106 and Cys‐53. Journal of Biological Chemistry, 287, 18738–18749. 10.1074/jbc.M111.311589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg, M. S. , Fleming, S. M. , Palacino, J. J. , Cepeda, C. , Lam, H. A. , Bhatnagar, A. , … Shen, J. (2003). Parkin‐deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. Journal of Biological Chemistry, 278, 43628–43635. [DOI] [PubMed] [Google Scholar]
- Greene, J. C. , Whitworth, A. J. , Kuo, I. , Andrews, L. A. , Feany, M. B. , & Pallanck, L. J. (2003). Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proceedings of the National Academy of Sciences USA, 100, 4078–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl, F. U. , Bracher, A. , & Hayer‐Hartl, M. (2011). Molecular chaperones in protein folding and proteostasis. Nature, 475, 324–332. 10.1038/nature10317 [DOI] [PubMed] [Google Scholar]
- Heeman, B. , Van den Haute, C. , Aelvoet, S.‐A. , Valsecchi, F. , Rodenburg, R. J. , Reumers, V. , … Baekelandt, V. (2011). Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. Journal of Cell Science, 124, 1115–1125. [DOI] [PubMed] [Google Scholar]
- Jakes, R. , Spillantini, M. G. , & Goedert, M. (1994). Identification of two distinct synucleins from human brain. FEBS Letters, 345(1), 27–32. 10.1016/0014-5793(94)00395-5 [DOI] [PubMed] [Google Scholar]
- Jiang, H. , Ren, Y. , Zhao, J. , & Feng, J. (2004). Parkin protects human dopaminergic neuroblastoma cells against dopamine‐induced apoptosis. Human Molecular Genetics, 13, 1745–1754. [DOI] [PubMed] [Google Scholar]
- Kalia, L. V. , & Lang, A. E. (2015). Parkinson's disease. The Lancet, 386(9996), 896–912. 10.1016/S0140-6736(14)61393-3. [DOI] [PubMed] [Google Scholar]
- Klein, C. , & Westenberger, A. (2012). Genetics of Parkinson's disease. Cold Spring Harbor Perspectives in Medicine, 2, a008888 10.1101/cshperspect.a008888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krisko, A. , & Radman, M. (2019). Protein damage, ageing and age‐related diseases. Open Biology, 9(3), 180249 10.1098/rsob.180249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli, D. A. , Cortez, M. Q. , Moyzis, A. G. , Najor, R. H. , Lee, Y. , & Gustafsson, Å. B. (2015). PINK1 is dispensable for mitochondrial recruitment of Parkin and activation of mitophagy in cardiac myocytes. PLoS ONE, 10, e0130707 10.1371/journal.pone.0130707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyung, J. W. , Kim, J.‐M. , Lee, W. , Ha, T.‐Y. , Cha, S.‐H. , Chung, K.‐H. , … Park, S. M. (2018). DJ‐1 deficiency impairs synaptic vesicle endocytosis and reavailability at nerve terminals. Proceedings of the National Academy of Sciences USA, 115, 1629–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston, J. W. (2017). The MPTP story. Journal of Parkinson's Disease, 7, S11–S19. 10.3233/JPD-179006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVoie, M. J. , Ostaszewski, B. L. , Weihofen, A. , Schlossmacher, M. G. , & Selkoe, D. J. (2005). Dopamine covalently modifies and functionally inactivates parkin. Nature Medicine, 11, 1214–1221. [DOI] [PubMed] [Google Scholar]
- Lev, N. , Barhum, Y. , Pilosof, N. S. , Ickowicz, D. , Cohen, H. Y. , Melamed, E. , & Offen, D. (2013). DJ‐1 protects against dopamine toxicity: Implications for Parkinson's disease and aging. The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences, 68, 215–225. [DOI] [PubMed] [Google Scholar]
- Li, J.‐Y. , Englund, E. , Holton, J. L. , Soulet, D. , Hagell, P. , Lees, A. J. , … Brundin, P. (2008). Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host‐to‐graft disease propagation. Nature Medicine, 14, 501–503. [DOI] [PubMed] [Google Scholar]
- Lo Bianco, C. , Schneider, B. L. , Bauer, M. , Sajadi, A. , Brice, A. , Iwatsubo, T. , & Aebischer, P. (2004). Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha‐synuclein rat model of Parkinson's disease. Proceedings of the National Academy of Sciences USA, 101, 17510–17515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan, T. , Bendor, J. , Toupin, C. , Thorn, K. , & Edwards, R. H. (2017). α‐Synuclein promotes dilation of the exocytotic fusion pore. Nature Neuroscience, 20, 681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr, K. M. , Chen, M. , Hoffman, C. A. , McDaniel, M. J. , Stout, K. A. , Dunn, A. R. , … Miller, G. W. (2016). Vesicular Monoamine transporter 2 (VMAT2) level regulates MPTP vulnerability and clearance of excess dopamine in mouse striatal terminals. Toxicological Sciences, 153, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lücking, C. B. , Dürr, A. , Bonifati, V. , Vaughan, J. , De Michele, G. , Gasser, T. , … Brice, A. (2000). Association between early‐onset Parkinson's disease and mutations in the Parkin gene. New England Journal of Medicine, 342, 1560–1567. [DOI] [PubMed] [Google Scholar]
- Malgieri, G. , & Eliezer, D. (2008). Structural effects of Parkinson's disease linked DJ‐1 mutations. Protein Science, 17, 855–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maria Valente, E. , Abou‐Sleiman, P. M. , Caputo, V. , Muqit, K. M. M. , Harvey, K. , Gispert, S. , … Wood, N. W. (2004). Hereditary early‐onset Parkinson's disease caused by mutations in PINK1. Science, 304, 1158–1160. [DOI] [PubMed] [Google Scholar]
- Martínez, J. H. , Fuentes, F. , Vanasco, V. , Alvarez, S. , Alaimo, A. , Cassina, A. , … Velazquez, F. (2018). Alpha‐synuclein mitochondrial interaction leads to irreversible translocation and complex I impairment. Archives of Biochemistry and Biophysics, 651, 1–12. [DOI] [PubMed] [Google Scholar]
- Martinez‐Vicente, M. , Sovak, G. , & Cuervo, A. M. (2005). Protein degradation and aging. Experimental Gerontology, 40, 622–633. [DOI] [PubMed] [Google Scholar]
- Martinez‐Vicente, M. , Talloczy, Z. , Kaushik, S. , Massey, A. C. , Mazzulli, J. , Mosharov, E. V. , … Cuervo, A. M. (2008). Dopamine‐modified alpha‐synuclein blocks chaperone‐mediated autophagy. J. Clin. Invest., 118, 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoud, S. T. , Vecchio, L. M. , Bergeron, Y. , Hossain, M. M. , Nguyen, L. T. , Bermejo, M. K. , … Salahpour, A. (2015). Increased expression of the dopamine transporter leads to loss of dopamine neurons, oxidative stress and l‐DOPA reversible motor deficits. Neurobiology of Diseases, 74, 66–75. 10.1016/j.nbd.2014.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda, N. , Sato, S. , Shiba, K. , Okatsu, K. , Saisho, K. , Gautier, C. A. , … Tanaka, K. (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. Journal of Cell Biology, 189, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy, M. K. , & Cookson, M. R. (2011). DJ‐1 regulation of mitochondrial function and autophagy through oxidative stress. Autophagy, 7, 531–532. 10.4161/auto.7.5.14684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith, G. E. , & Rademacher, D. J. (2011). MPTP mouse models of Parkinson's disease: An update. J. Parkinsons. Dis., 1, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mor, D. E. , Ugras, S. E. , Daniels, M. J. , & Ischiropoulos, H. (2016). Dynamic structural flexibility of α‐synuclein. Neurobiology of Diseases, 88, 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov, E. V. , Larsen, K. E. , Kanter, E. , Phillips, K. A. , Wilson, K. , Schmitz, Y. , … Sulzer, D. (2009). Interplay between cytosolic dopamine, calcium, and α‐synuclein causes selective death of substantia nigra neurons. Neuron, 62, 218–229. 10.1016/j.neuron.2009.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz, P. , Huenchuguala, S. , Paris, I. , & Segura‐Aguilar, J. (2012). Dopamine oxidation and autophagy. Parkinson's Disease, 2012, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra, D. , Tanaka, A. , Suen, D.‐F. , & Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. Journal of Cell Biology, 183, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narhi, L. , Wood, S. J. , Steavenson, S. , Jiang, Y. , Wu, G. M. , Anafi, D. , … Citron, M. (1999). Both familial Parkinson's disease mutations accelerate alpha‐synuclein aggregation. Journal of Biological Chemistry, 274, 9843–9846. [DOI] [PubMed] [Google Scholar]
- Orrenius, S. , & Nicotera, P. (1994). The calcium ion and cell death. Journal of Neural Transmission. Supplementum, 43, 1–11. [PubMed] [Google Scholar]
- Pickrell, A. M. , & Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron, 85, 257–273. 10.1016/j.neuron.2014.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planchard, M. S. , Exley, S. E. , Morgan, S. E. , & Rangachari, V. (2014). Dopamine‐induced α‐synuclein oligomers show self‐ and cross‐propagation properties. Protein Science, 23, 1369–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos, M. H. , Lavedan, C. , Leroy, E. , Ide, S. E. , Dehejia, A. , Dutra, A. , … Nussbaum, R. L. (1997). Mutation in the alpha‐synuclein gene identified in families with Parkinson's disease. Science, 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Post, M. R. , Lieberman, O. J. , & Mosharov, E. V. (2018). Can interactions between α‐synuclein, dopamine and calcium explain selective neurodegeneration in Parkinson's disease? Frontiers in Neuroscience, 12, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski, S. (2017). The two‐century journey of Parkinson disease research. Nature Reviews Neuroscience, 18, 251–259. [DOI] [PubMed] [Google Scholar]
- Raman, A. V. , Chou, V. P. , Atienza‐Duyanen, J. , Di Monte, D. A. , Bellinger, F. P. , & Manning‐Boğ, A. B. (2013). Evidence of oxidative stress in young and aged DJ‐1‐deficient mice. FEBS Letters, 587, 1562–1570. [DOI] [PubMed] [Google Scholar]
- Rcom‐H'cheo‐Gauthier, A. , Goodwin, J. , & Pountney, D. (2014). Interactions between calcium and alpha‐synuclein in neurodegeneration. Biomolecules, 4, 795–811. 10.3390/biom4030795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve, A. K. , Ludtmann, M. H. R. , Angelova, P. R. , Simcox, E. M. , Horrocks, M. H. , Klenerman, D. , … Abramov, A. Y. (2015). Aggregated α‐synuclein and complex I deficiency: Exploration of their relationship in differentiated neurons. Cell Death & Disease, 6, e1820 10.1038/cddis.2015.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidy, M. , Sharma, R. , Roberts, B. L. , & Masison, D. C. (2016). Human J‐protein DnaJB6b cures a subset of saccharomyces cerevisiae prions and selectively blocks assembly of structurally related amyloids. Journal of Biological Chemistry, 291, 4035–4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez, M. C. , Obeso, J. A. , & Olanow, C. W. (1998). Subthalamic nucleus‐mediated excitotoxicity in Parkinson's disease: A target for neuroprotection. Annals of Neurology, 44, S175–S188. 10.1002/ana.410440726 [DOI] [PubMed] [Google Scholar]
- Rutherford, N. J. , Moore, B. D. , Golde, T. E. , & Giasson, B. I. (2014). Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of α‐synuclein. Journal of Neurochemistry, 131, 859–867. [DOI] [PubMed] [Google Scholar]
- Schapira, A. H. V. , Cooper, J. M. , Dexter, D. , Clark, J. B. , Jenner, P. , & Marsden, C. D. (1990). Mitochondrial complex I deficiency in Parkinson's disease. Journal of Neurochemistry, 54, 823–827. [DOI] [PubMed] [Google Scholar]
- Schulte, C. , & Gasser, T. (2011). Genetic basis of Parkinson's disease: Inheritance, penetrance, and expression. Application of Clinical Genetics, 4, 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendelman, S. , Jonason, A. , Martinat, C. , Leete, T. , & Abeliovich, A. (2004). DJ‐1 is a redox‐dependent molecular chaperone that inhibits α‐synuclein aggregate formation. PLoS Biology, 2, e362 10.1371/journal.pbio.0020362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton, A. B. , Farrer, M. , Johnson, J. , Singleton, A. , Hague, S. , Kachergus, J. , … Gwinn‐Hardy, K. (2003). alpha‐Synuclein locus triplication causes Parkinson's disease. Science, 302, 841 10.1126/science.1090278 [DOI] [PubMed] [Google Scholar]
- Song, S. , Jang, S. , Park, J. , Bang, S. , Choi, S. , Kwon, K.‐Y. , … Chung, J. (2013). Characterization of PINK1 (PTEN‐induced putative kinase 1) mutations associated with Parkinson disease in mammalian cells and Drosophila. Journal of Biological Chemistry, 288, 5660–5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer, D. , Bogulavsky, J. , Larsen, K. E. , Behr, G. , Karatekin, E. , Kleinman, M. H. , … Zecca, L. (2000). Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proceedings of the National Academy of Sciences USA, 97, 11869–11874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipa, R. , Pereira, C. , Reis, I. , Alonso, I. , Bastos‐Lima, A. , Melo‐Pires, M. , & Magalhães, M. (2016). DJ‐1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain, 139, 1680–1687. 10.1093/brain/aww080 [DOI] [PubMed] [Google Scholar]
- Takahashi, N. , Miner, L. L. , Sora, I. , Ujike, H. , Revay, R. S. , Kostic, V. , … Uhl, G. R. (1997). VMAT2 knockout mice: Heterozygotes display reduced amphetamine‐conditioned reward, enhanced amphetamine locomotion, and enhanced MPTP toxicity. Proceedings of the National Academy of Sciences USA, 94, 9938–9943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trempe, J.‐F. , & Fon, E. A. (2013). Structure and function of parkin, PINK1, and DJ‐1, the three musketeers of neuroprotection. Frontiers in Neurology, 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tysnes, O.‐B. , & Storstein, A. (2017). Epidemiology of Parkinson's disease. Journal of Neural Transmission, 124, 901–905. [DOI] [PubMed] [Google Scholar]
- van der Putten, H. , Wiederhold, K. H. , Probst, A. , Barbieri, S. , Mistl, C. , Danner, S. , … Bilbe, G. (2000). Neuropathology in mice expressing human alpha‐synuclein. Journal of Neuroscience, 20, 6021–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooijen, B. D. , Claessens, M. M. A. E. , & Subramaniam, V. (2010). Membrane permeabilization by oligomeric α‐synuclein. In search of the mechanism. PLoS ONE, 5, e14292 10.1371/journal.pone.0014292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visanji, N. , & Marras, C. (2015). The relevance of pre‐motor symptoms in Parkinson's disease. Expert Review of Neurotherapeutics, 15, 1205–1217. [DOI] [PubMed] [Google Scholar]
- Vogiatzi, T. , Xilouri, M. , Vekrellis, K. , & Stefanis, L. (2008). Wild type alpha‐synuclein is degraded by chaperone‐mediated autophagy and macroautophagy in neuronal cells. Journal of Biological Chemistry, 283, 23542–23556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb, J. L. , Ravikumar, B. , Atkins, J. , Skepper, J. N. , & Rubinsztein, D. C. (2003). Alpha‐Synuclein is degraded by both autophagy and the proteasome. Journal of Biological Chemistry, 278, 25009–25013. [DOI] [PubMed] [Google Scholar]
- Whitehead, R. E. , Ferrer, J. V. , Javitch, J. A. , & Justice, J. B. (2001). Reaction of oxidized dopamine with endogenous cysteine residues in the human dopamine transporter. Journal of Neurochemistry, 76, 1242–1251. [DOI] [PubMed] [Google Scholar]
- Winner, B. , Jappelli, R. , Maji, S. K. , Desplats, P. A. , Boyer, L. , Aigner, S. , … Riek, R. (2011). In vivo demonstration that alpha‐synuclein oligomers are toxic. Proceedings of the National Academy of Sciences USA, 108, 4194–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, C.‐Y. , Kang, W.‐Y. , Chen, Y.‐M. , Jiang, T.‐F. , Zhang, J. , Zhang, L.‐N. , … Chen, S.‐D. (2017). DJ‐1 inhibits α‐synuclein aggregation by regulating chaperone‐mediated autophagy. Frontiers in Aging Neuroscience, 9, 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun, J. , Puri, R. , Yang, H. , Lizzio, M. A. , Wu, C. , Sheng, Z.‐H. , & Guo, M. (2014). MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. eLife, 3, e01958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , Phillips, K. , Wielgus, A. R. , Liu, J. , Albertini, A. , Zucca, F. A. , … Zecca, L. (2011). Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: Implications for progression of Parkinson's disease. Neurotoxicity Research, 19, 63–72. 10.1007/s12640-009-9140-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucca, F. A. , Vanna, R. , Cupaioli, F. A. , Bellei, C. , De Palma, A. , Di Silvestre, D. , … Zecca, L. (2018). Neuromelanin organelles are specialized autolysosomes that accumulate undegraded proteins and lipids in aging human brain and are likely involved in Parkinson's disease. NPJ Parkinson's Disease, 4, 17 10.1038/s41531-018-0050-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data are available as this is a review.