

Abstract
Introduction
We sought to determine whether survival motor neuron (SMN) protein blood levels correlate with denervation and SMN2 copies in spinal muscular atrophy (SMA).
Methods
Using a mixed‐effect model, we tested associations between SMN levels, compound muscle action potential (CMAP), and SMN2 copies in a cohort of 74 patients with SMA. We analyzed a subset of 19 of these patients plus four additional patients who had been treated with received gene therapy to examine SMN trajectories early in life.
Results
Patients with SMA who had lower CMAP values had lower circulating SMN levels (P = .04). Survival motor neuron protein levels were different between patients with two and three SMN2 copies (P < .0001) and between symptomatic and presymptomatic patients (P < .0001), with the highest levels after birth and progressive decline over the first 3 years. Neither nusinersen nor gene therapy clearly altered SMN levels.
Discussion
These data provide evidence that whole blood SMN levels correlate with SMN2 copy number and severity of denervation.
Keywords: compound muscle action potential, denervation, SMN protein levels, spinal muscular atrophy, survival motor neuron
Abbreviations
- BforSMA
biomarkers for spinal muscular atrophy
- CMAP
compound muscle action potential
- ECL
electrochemiluminescence
- FDA
US Food and Drug Administration
- GLMM
generalized linear mixed model
- ICC
intraclass correlation coefficient
- LPDR
longitudinal pediatric data repositories
- SMA
spinal muscular atrophy
- SMN
survival motor neuron
- SMN1
survival motor neuron 1 gene
- SMN2
survival motor neuron 2 gene
1. INTRODUCTION
Spinal muscular atrophy (SMA) is a devastating neuromuscular disease, but novel targeted molecular therapies are transforming our current approach to diagnosis and management. Identifying biomarkers to track denervation in patients with SMA early in life is a high priority for SMA clinicians as newborn screening for SMA becomes increasingly common, providing the opportunity for early treatment intervention. Survival motor neuron 2 gene (SMN2) copy number is a primary disease modifier in SMA in which the number of SMN2 copies inversely correlates with phenotypic severity. 1 , 2 , 3 Recently, we developed and validated an assay to accurately measure SMN protein levels in whole blood. 4 However, the usefulness of this assay in the clinical setting remains uncertain. Thus, we sought to determine whether patients with SMA with two or three SMN2 copies, which are expected to present with symptoms in the first year of life, could be distinguished according to their circulating SMN protein levels to help further stratify risk for infants with SMA diagnosed in the presymptomatic setting.
Molecular therapies have demonstrated improvements in morbidity and ventilator‐free survival in patients with early infantile‐onset SMA. 5 , 6 , 7 , 8 The US Food and Drug Administration (FDA) has approved the use of two therapies for patients with SMA, the antisense oligonucleotide nusinersen that modulates pre–messenger RNA splicing of the SMN2 gene and the SMN1 gene replacement therapy onasemnogene abeparvovec. No longitudinal studies have yet examined whole blood SMN protein levels to determine whether these new therapies have any impact on peripheral SMN protein levels. Here, we first tested the hypothesis that circulating SMN protein levels correlate with age and severity of denervation in patients with SMA. Then, we analyzed the impact of intervention in infants treated with onasemnogene abeparvovec or nusinersen to determine whether these therapies alter circulating SMN protein levels early in life.
2. MATERIALS AND METHODS
2.1. Study approval
Written informed consent was obtained from parents for all participants under approved institutional ethics review board protocols at the Massachusetts General Hospital (No. 2016P000469) and the University of Utah (Protocol No. 8751).
2.2. Participants and experimental design
We queried the Project Cure SMA and the SPOT SMA longitudinal pediatric data repositories (LPDR) for whole‐blood samples suitable for determining SMN protein levels. The Project Cure SMA and SPOT SMA LPDRs are housed within the research electronic data capture web application at the Newborn Screening Translational Research Network, and contain natural history data and treatment data, respectively. Inclusion criteria included patients with SMA with two, three, or four SMN2 copies, including some who had received nusinersen therapy at some point during the course of longitudinal follow‐up. Exclusion criteria for this initial analysis included patients receiving other available therapies including gene therapy with onasemnogene abeparvovec because of limited numbers of samples. This query resulted in 209 blood samples from 74 patients with SMA. Whole blood SMN protein levels were determined in all these samples at PharmOptima (Portage, Michigan). PharmOptima coauthors (R.P., R.S., P.Z.) who were involved in sample analysis and provision of data for subsequent statistical analysis were blinded to information regarding age, sex, SMA type, SMN genotype, and treatment exposure. Maximum ulnar compound muscle action potential (CMAP) data available at the same visit for which blood samples were obtained and recorded.
We next sought to analyze SMN levels for patients with data available in the first 3 years of life to determine whether SMN protein levels were associated with SMN2 copy number early in life because recent data from fetal and infant autopsies had provided evidence that SMN levels were highest in tissues during fetal development and rapidly declined postbirth. We identified 19 of these 74 patients with available data for this age range and included data from four additional patients who received gene therapy starting in early 2019 in Massachusetts General Hospital.
2.3. Survival motor neuron protein levels, maximum CMAP amplitude, and SMN2 copy number
Survival motor neuron protein levels in whole blood were determined by using an electrochemiluminescence (ECL) immunoassay protocol recently developed and validated by our groups. 4 Maximum ulnar CMAP amplitude was obtained by recording from the abductor digiti minimi muscle after ulnar nerve stimulation at the wrist, as previously described. 9 SMN1 and SMN2 copy numbers were determined by using quantitative polymerase chain reaction. 10
2.4. Statistical analysis
Generalized linear mixed model (GLMM) with random intercept was applied in SAS 9.4 (SAS Institute, Cary, North Carolina). Log transformation was performed to the age variable, and data were controlled for age. Sex and treatment status (receiving or not receiving therapy with nusinersen) were not significant confounding factors in our model. As prerequisites of modeling random effects, the intraclass correlation coefficient (ICC) estimated from random intercept form was 0.785, and the design effect was estimated to be 2.358. If we used simple random sampling, we would inflate the variance by more than twofold. We used the rule of thumb when applying the mixed‐effect model, which is appropriate when ICC is estimated to be greater than 0.1. To test a potential association between SMN protein levels and CMAP values, we included data collected at the same time point, and multiple imputation was performed to handle missing CMAP values. 11 SAS default using the Markov chain Monte Carlo method was used to impute values. There was no statistically significant difference between preimputed and postimputed CMAP data. For the second set with 23 patients between 0 and 3 years of age, SMN protein levels were compared among patients with two, three, and four SMN2 copies and also compared on the basis of clinical symptoms, including presymptomatic, type 1, type 2, or type 3 patients with SMA, by using GLMM. Differences in SMN protein levels were reported as mean difference in picograms per milliliter, 95% CI, and P values. Critical value was set as P < .05.
3. RESULTS
3.1. Survival motor neuron protein levels in whole blood correlate with severity of denervation
Length of longitudinal follow‐up varied and ranged from a single data point in one patient to a patient with follow‐up of 42.9 months, with a median of 1.1 months for the 74 patients. Table 1 presents the distribution of sex, age, SMA type, and SMN2 copies for this cohort. Among these 74 patients with SMA, 31 received nusinersen at some point, but further statistical analysis revealed that nusinersen treatment was not a confounding factor in our model. To analyze SMN levels for patients with data available in the first 3 years of life, a second data set included 86 observations from a total of 23 patients with SMA. Table 1 presents the distribution of sex, age, SMA type, and SMN2 copies for this second cohort. Within this second cohort of 23 patients with SMA, 11 received no therapy, eight received nusinersen, three received onasemnogene abeparvovec, and one received both therapies.
TABLE 1.
Characteristics of patients with SMA
| Variables | Patients with SMA, n = 74 | Set with 0–3‐y‐old patients, n = 23 |
|---|---|---|
| Sex, n (%) | ||
| Boys | 32 (43.2) | 9 (39) |
| Girls | 42 (56.8) | 14 (61) |
| Age, SD (range), mo | 11.1 ± 13.0 (0–57.5) | 8.96 ± 10.09 (0–34.1) |
| SMN protein levels, SD (range), pg/mL | 7940 ± 4408 (1167–2045) | 10 765 ± 6021 (2749–32 045) |
| Maximum CMAP, SD (range), mV | 4.9 ± 3.6 (0.2–16.1) | 4.6 ± 3.3 (0.2–12.2) |
| SMN2 copies, n (%) | ||
| 2 | 11 (14.9) | 9 (39.1) |
| 3 | 37 (50.0) | 13 (56.5) |
| 4+ | 26 (35.1) | 1 (4.3) |
| SMA type, n (%) | ||
| 1 | 8 (10.8) | 6 (26.1) |
| 2 | 21 (28.4) | 6 (26.1) |
| 3a | 22 (29.7) | 2 (8.7) |
| 3b | 13 (17.6) | 0 (0) |
| 4 | 1 (1.3) | 0 (0) |
| Presymptomatic | 9 (12.2) | 9 (39.1) |
Abbreviations: CMAP, compound muscle action potential; SMA, spinal muscular atrophy; SMN, survival motor neuron; SMN2, survival motor neuron 2 gene.
To determine whether SMN protein levels in whole blood are associated with the severity of denervation in patients with SMA, we used 209 observations from 74 patients with SMA to test the association between SMN protein levels and maximum ulnar CMAP. The distribution of sex, age, SMA type, and SMN2 copies among patients with SMA are presented in Table 1. We found significant associations between SMN protein levels and maximum ulnar CMAP after correcting for age and SMN2 copy number (P = .04; 95% CI = 4.1, 374.1). On average, 1 mV increase in CMAP values was associated with 189.08 pg/mL increase in whole blood SMN protein levels after controlling for age and SMN2 copy number. Thus, these initial findings provided evidence that lower whole blood SMN protein levels are associated with more severe denervation in patients with SMA.
3.2. Survival motor neuron protein levels in whole blood correlate with SMN2 copy number early in life
We next investigated a cohort of 23 infants between 0 and 36 months of age with available data to determine whether SMN protein levels in whole blood were associated with SMN2 copy number early in life. Only one of these patients had four SMN2 copies, while 22 infants had two or three SMN2 copies. We found significant differences in SMN protein levels between patients with two and three SMN2 copies after correcting for age (P < .0001; 95% CI = 5476.9, 9671.1; Figure 1A). On average, patients with SMA with three SMN2 copies had 7514.2 pg/mL higher SMN protein levels compared with patients with SMA with two SMN2 copies after controlling for age. In addition, when analyzing this cohort based on SMA subtype, we found that patients with SMA type 1 had a 7699.6 pg/mL decrease (P < .0001, 95% CI = 4538.7, 10 861.0), and patients with SMA type 2 had a 4802.0 pg/mL decrease (P = .01, 95% CI = 1112.4, 8491.7) in SMN protein levels compared with presymptomatic patients after controlling for age (Figure 1B). Among these 23 patients, 11 received no treatment intervention, eight received nusinersen, three received onasemnogene abeparvovec, and one received both therapies. However, these treatment interventions were not a confounding factor for the mixed‐effect model. These data provide evidence that SMN protein levels measured by the ECL immunoassay in whole blood correlate with SMN2 copy number early in life.
FIGURE 1.
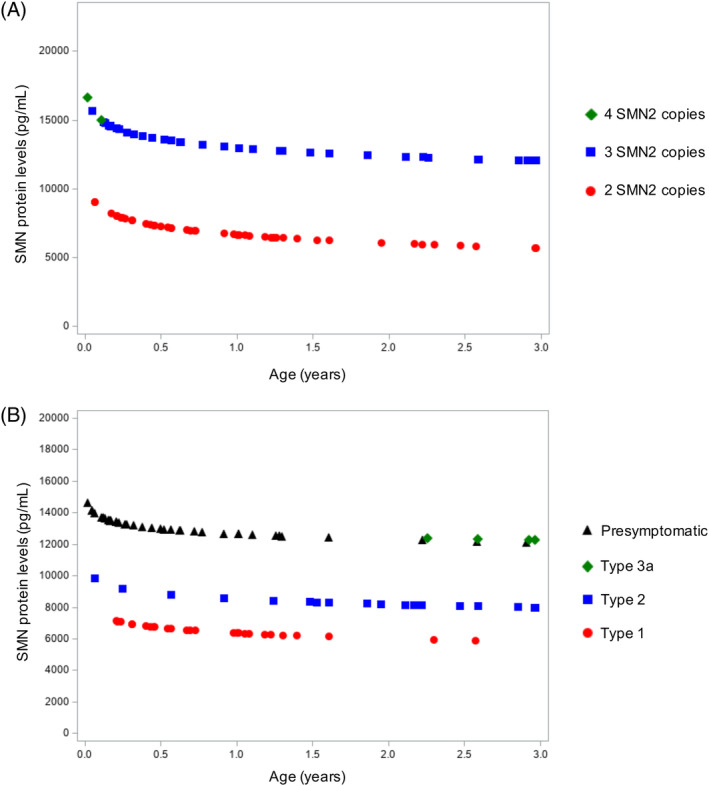
Mixed‐effect model comparing SMN protein levels in whole blood among patients with SMA with two, three, and four SMN2 copies (A) and among different SMA types (B) early in life. A, Patients with SMA were divided according to SMN2 copies, including nine patients with two SMN2 copies, 13 patients with three SMN2 copies, and one patient with four SMN2 copies. B, Patients with SMA were divided according to clinical symptoms, including nine patients who were presymptomatic, six patients with SMA type 1, six patients with SMA type 2, and two patients with SMA type 3. Among the nine patients with SMA who were presymptomatic, one patient had four SMN2 copies, one patient had three SMN2 copies, and seven patients had three SMN2 copies. SMA, spinal muscular atrophy; SMN, survival motor neuron; SMN2, survival motor neuron 2 gene [Color figure can be viewed at wileyonlinelibrary.com]
3.3. Effects of nusinersen and onasemnogene abeparvovec treatment on SMN levels in whole blood
To provide data regarding the potential effect of nusinersen or onasemnogene abeparvovec on whole blood SMN protein levels, we plotted SMN protein levels over time in individual cases treated with onasemnogene abeparvovec or nusinersen. Here, we report data from three patients who received onasemnogene abeparvovec therapy (Figure 2A), one patient who received cotherapy with onasemnogene abeparvovec and nusinersen (Figure 2B), and eight patients who received only nusinersen (Figure 2C) between the ages of 0 and 36 months. Neither onasemnogene abeparvovec nor nusinersen appear to increase SMN protein levels in whole blood, providing evidence that neither of these therapies significantly alter red cell or platelet levels of SMN, which account for the majority of the SMN detected in the ECL assay.
FIGURE 2.
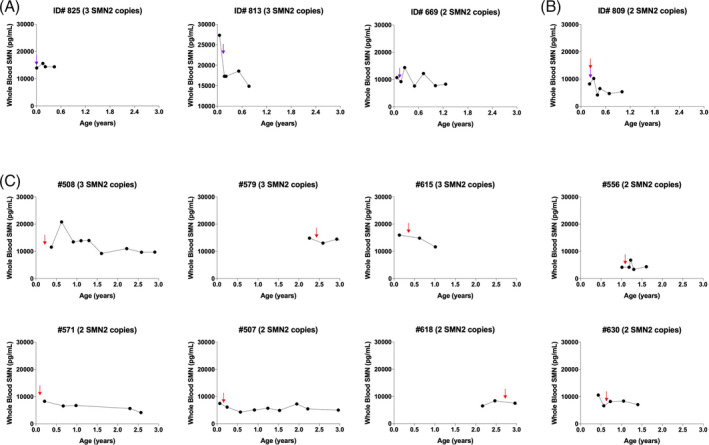
Nusinersen and onasemnogene abeparvovec therapies do not increase SMN protein levels in whole blood early in life. Case series for patients treated with onasemnogene abeparvovec (A), onasemnogene abeparvovec plus nusinersen (B), or only nusinersen (C). Blue arrows indicate the start of onasemnogene abeparvovec therapy, while red arrows indicate the start of nusinersen treatment. SMN, survival motor neuron; SMN2, survival motor neuron 2 gene [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
We demonstrate that SMN protein levels in whole blood are clearly associated with SMN2 copy number and severity of denervation and that infants with two or three SMN2 copies have declining SMN levels with differing trajectories in the first years of life according to their SMN2 dosage. Therefore, whole blood SMN protein levels could prove a useful adjunct to SMN2 copy number data in helping to guide the urgency of therapeutic intervention in newborns and young infants with two compared with three or more SMN2 copies. Recent data in newborns with two SMN2 copies from the NURTURE (ClinicalTrials.gov identifier: NCT02386553) and SPRINT (ClinicalTrials.gov identifier: NCT03505099) trials who received treatment initiation with either nusinersen or onasemnogene abeparvovec early in infancy provide evidence of a divergence in outcomes from the earliest timepoints measured compared with those with three SMN2 copies, and these newborns with two SMN2 copies appear to lag in motor milestone development relative to their peers with higher SMN2 dosage. 12 While experts agree that treatment should be initiated as early as possible in newborns with SMA who have two or three copies to ensure the best outcomes, there are challenges that can delay initiation of treatment with currently available molecular therapies, including the local approval of these therapies because of the high costs, the logistics involving insurance approval, and dosing. In addition, controversy remains about whether an infant with four or more SMN2 copies should be treated as early as possible and what additional factors should influence this decision. In this context, whole blood SMN protein level is a simple readout and, with further validation, could prove a potential additional tool in helping to determine the most ideal timing for treatment. For example, if an infant with SMA with four SMN2 copies identified on newborn screening is clinically presymptomatic but has lower SMN protein levels (eg, <10 000 pg/mL) more typically seen in infants with two or three SMN2 copies, these data may support the premise that early intervention rather than watchful waiting is indeed warranted.
Data on infants with four SMN2 copies are limited in the current study. However, the increasing implementation of newborn screening should provide the opportunity to identify many more such babies, and their data will be critically important. Survival motor neuron protein levels could prove complementary to other biomarkers currently showing promise in heralding the onset of acute denervation, such as plasma neurofilament heavy chain levels. 3 At this time, additional prospective data on newborns with four or more SMN2 copies for all biomarker candidates are required because data currently are limited primarily to those with two or three SMN2 copies. 12 , 13
Survival motor neuron protein levels decrease over time from birth. Researchers in a recent study of autopsy tissues from patients with SMA found that SMN protein levels were higher during fetal development and decreased over time after birth in the spinal cord and cortex. 14 These findings provide evidence that the role of SMN is more critical in the prenatal period and early in life, but the molecular mechanisms underlying the decrease with age remains unclear. Our mixed effect model corroborated these findings by detecting an age effect, providing evidence of a decrease in whole‐blood SMN levels over time in patients with SMA, independent of the number of SMN2 copies. A main limitation of our study is the lack of longitudinal data from healthy controls to determine the age effects in a healthy population. In a previous cross‐sectional study, 4 we showed that whole blood SMN protein concentrations in control patients are higher than 30 000 pg/mL, while all patients with SMA analyzed in that study and in the current study had concentrations <20 000 pg/mL. However, additional longitudinal data from age‐matched control patients are required to better determine normal SMN protein levels for each specific age cohort. In this context, researchers in a previous study did not find a strong correlation between SMN2 copies and circulating SMN protein levels in patients with SMA aged 2 to 13 years, 3 providing evidence that there may be more significant modulation of SMN at the posttranscriptional level during early development.
According to the data presented here, nusinersen or onasemnogene abeparvovec therapies do not seem to result in sustained increases in SMN in whole blood, indicating that this assay is not a useful predictor of response to currently available therapies. Nusinersen status was not a confounding factor, nor did it present significant associations with CMAP when we corrected for age, providing evidence that nusinersen does not affect SMN levels. Intrathecal administration most likely increases SMN levels only in the central nervous system but not in blood cells. For gene therapy, we have available data from only four patients, and we observed a fluctuation in SMN levels in these specific cases. Gene therapy was not a confounding factor in our statistical analysis, but, because of the small sample, we conclude that additional studies are required to confirm these findings in larger longitudinal cohorts. Gene therapy may not increase and sustain whole blood SMN levels in the long term because blood cells are dividing, and the adenoassociated vector 9 is not designed to effectively integrate the DNA. Therefore, testing blood SMN protein levels is not a useful predictor of response to currently FDA‐approved therapies. However, understanding the full impact of SMN deficiency outside of the central nervous system is important because SMN regulates the assembly of spliceosome machinery, which is critical for the functionality of all cell types. 15 , 16 This assay might be useful to predict the response of other therapies that may increase systemic SMN protein levels.
A main challenge in the SMA field is to identify predictive and treatment responsive biomarkers to track disease status. The BforSMA (biomarkers for spinal muscular atrophy) study 3 , 17 was conducted to identify novel biomarkers for SMA. In addition to testing and reporting that patients with SMA had lower circulating SMN protein levels compared with controls, 3 the BforSMA study screened plasma from a cohort of SMA type 1, 2, and 3 patients and age‐ and sex‐matched controls, and identified 97 proteins and 59 metabolites that correlated with SMA phenotypes across a wide spectrum of disease severity. 17 The effects of therapies in these molecules must be further determined. Moreover, two additional candidate biomarkers to track disease severity in patients with SMA include circulating levels of light or heavy chain neurofilaments 13 and serum creatinine. 18 Motor neurons are selectively vulnerable to SMN deficiency, and, when motor neurons are degraded, they are broken up and secreted into the cerebrospinal fluid and serum/plasma. While circulating neurofilament levels decrease over time, nusinersen treatment in newborns in the NURTURE study was demonstrated to be associated with a faster and greater decline in its levels. 13 Finally, we recently reported that patients with SMA not only have low serum creatinine concentrations compared with age‐matched healthy controls but that the severity of denervation is associated with serum creatinine levels, which can be considered an indicator of muscle wasting. 18 The effects of available therapies on serum creatinine levels also requires additional study. In future research, it will be critical to have a more comprehensive longitudinal data set combining all these potential biomarkers to determine the best predictors of treatment response to these molecular and gene therapies.
In summary, nusinersen and onasemnogene abeparvovec therapies do not increase SMN protein levels in whole blood in patients with SMA. However, SMN protein levels in whole blood reflect the SMN genotype and the severity of denervation and may provide added information to help determine when and where to deliver the most effective therapies. These novel observations are clinically relevant because patients with SMA with variable SMN2 dosage can be further distinguished according to their circulating SMN protein levels to help stratify risk for infants whose diagnosis is in the presymptomatic setting. This is the first longitudinal cohort study to evaluate the potential usefulness of whole blood SMN protein levels measurements as a potential adjunct biomarker in predicting outcomes in patients with SMA. We propose that whole blood SMN protein measurements should be included as an exploratory biomarker in the prospective assessment of all newborn patients with SMA in conjunction with other promising candidate blood biomarkers, such as neurofilament levels. Such data, when it is combined with other important molecular, neurophysiological, metabolic and imaging biomarkers, will considerably improve our understanding about what factors are most important in maximizing long‐term outcomes.
CONFLICT OF INTEREST
Kathryn J. Swoboda is a consultant for Biogen and AveXis, and receives clinical trial funding from AveXis and Biogen. All other authors report no conflicts of interest.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
ACKNOWLEDGMENTS
The authors thank all the patients and families who participated in this study. Kathryn J. Swoboda received financial support from NICHD R01HD054599, Biogen, and Cure SMA; Rebekah Poxson, Rachel Schwartz, and Phillip Zaworski received support in the form of salaries from PharmOptima.
Alves CRR, Zhang R, Johnstone AJ, et al. Whole blood survival motor neuron protein levels correlate with severity of denervation in spinal muscular atrophy. Muscle & Nerve. 2020;62:351–357. 10.1002/mus.26995
Funding information Biogen, Grant/Award Number: Biogen; Cure SMA; Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), Grant/Award Number: R01HD054599; Cure SMA, Grant/Award Number: Cure SMA; National Institute of Child Health and Human Development
REFERENCES
- 1. Mailman MD, Heinz JW, Papp AC, et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2 . Genet Med. 2002;4(1):20‐26. [DOI] [PubMed] [Google Scholar]
- 2. Feldkötter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real‐time LightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70(2):358‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Crawford TO , Paushkin SV, Kobayashi DT, et al. Evaluation of SMN protein, transcript, and copy number in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS One. 2012;7(4):e33572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zaworski P, Von Herrmann KM, Taylor S, et al. SMN protein can be reliably measured in whole blood with an electrochemiluminescence (ECL) immunoassay: implications for clinical trials. PLoS One. 2016;11(3):e0150640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Corey DR. Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat Neurosci. 2017;20(4):497‐499. [DOI] [PubMed] [Google Scholar]
- 6. Wood MJA, Talbot K, Bowerman M. Spinal muscular atrophy: antisense oligonucleotide therapy opens the door to an integrated therapeutic landscape. Hum Mol Genet. 2017;26(R2):R151‐R159. [DOI] [PubMed] [Google Scholar]
- 7. Mendell JR, Al‐Zaidy S, Shell R, et al. Single‐dose gene‐replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713‐1722. [DOI] [PubMed] [Google Scholar]
- 8. Mendell J, Al‐Zaidy S, Shell R, et al. AVXS‐101 phase 1 gene therapy clinical trial in SMA type 1: end‐of‐study event free survival and achievement of developmental milestones. Neuromuscul Disord. 2017;27(Suppl 2):S208. [Google Scholar]
- 9. Lewelt A, Krosschell KJ, Scott C, et al. Compound muscle action potential and motor function in children with spinal muscular atrophy. Muscle Nerve. 2010;42(5):703‐708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Prior TW, Krainer AR, Hua Y, et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet. 2009;85(3):408‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Young R, Johnson DR. Handling missing values in longitudinal panel data with multiple imputation. J Marriage Fam. 2015;77(1):277‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Vivo DC, Bertini E, Swoboda KJ, et al. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: interim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019;29(11):842‐856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Darras BT, Crawford TO , Finkel RS, et al. Neurofilament as a potential biomarker for spinal muscular atrophy. Ann Clin Transl Neurol. 2019;6(5):932‐944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ramos DM, d’Ydewalle C, Gabbeta V, et al. Age‐dependent SMN expression in disease‐relevant tissue and implications for SMA treatment. J Clin Invest. 2019;129(11):4817‐4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Swoboda KJ. Romancing the spliceosome to fight spinal muscular atrophy. N Engl J Med. 2014;371(18):1752‐1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nery FC, Siranosian JJ, Rosales I, et al. Impaired kidney structure and function in spinal muscular atrophy. Neurol Genet. 2019;5(5):e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Finkel RS, Crawford TO , Swoboda KJ, et al. Candidate proteins, metabolites and transcripts in the biomarkers for spinal muscular atrophy (BforSMA) clinical study. PLoS One. 2012;7(4):e35462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alves CRR, Zhang R, Johnstone AJ, et al. Serum creatinine is a biomarker of progressive denervation in spinal muscular atrophy. Neurology. 2020;94(9):e921‐e931. [DOI] [PMC free article] [PubMed] [Google Scholar]