

Abstract
Background
A triple‐secured plasma‐derived von Willebrand factor (pdVWF) almost devoid of factor VIII (FVIII):WILFACTIN®, was approved in France in 2003, and then in other countries for the treatment of patients with von Willebrand disease (VWD).
Objective
To investigate long‐term safety and efficacy of the product in real‐life over the first 5 post‐approval years.
Patients/Methods
This prospective, observational, national post‐marketing study (PMS) enrolled patients of all ages and VWD types. Patients were observed for up to 3 years and treated for one or more occasions. Efficacy was assessed for each major event. Breakthrough bleeding rate 3 days post‐infusion and annualized bleeding rate (ABR) were also evaluated for long‐term prophylaxis.
Results
Overall, 155 of 174 patients enrolled from 31 centers were eligible for efficacy assessment. Most patients (76.8%) were severely affected (VWF:RCo ≤ 15 IU/dL). They were treated for 743 bleeds and 140 surgeries including childbirth. Efficacy outcomes were excellent/good for 98.2% of 56 major surgeries and 94.0% of 67 major bleeds. Approximately 75% of 49 major mucosal bleeds were effectively managed without FVIII co‐administration. In 32 patients receiving prophylaxis, breakthrough bleeding occurred in 1.5% of infusions and median ABR was 1.0 for 20 patients treated ≥ 12 months. Excellent tolerability was confirmed with no safety concerns. No thrombotic events were observed.
Conclusions
Results from this PMS increase the clinical experience of a FVIII‐poor pdVWF in patients of all ages and VWD types including those with thrombotic risk factors and emphasize that giving FVIII is not always mandatory to effectively treat patients with severe VWD.
Keywords: clinical trial, post‐marketing, factor VIII, von Willebrand disease, von Willebrand factor
Essentials.
A post‐approval study on a plasma‐derived von Willebrand factor was completed.
It confirms and extends the pivotal studies’ results for efficacy, safety, and tolerability.
A priming dose of factor VIII was used to tailor therapy in less than half of cases.
No thrombotic events were observed, including in patients with high risk profiles.
1. INTRODUCTION
Von Willebrand disease (VWD) is caused by a deficiency or abnormality of von Willebrand factor (VWF), a multimeric glycoprotein with a high molecular weight. 1 VWF plays a crucial role in primary hemostasis, promoting the adhesion of platelets to subendothelium after vascular injury and in blood coagulation as it carries and protects factor VIII (FVIII) from inactivation and rapid catabolism. 2 , 3 VWD is a heterogeneous disease ranging from moderate to very severe deficiency in VWF and is classified into three different types: type 1 and type 3 are characterized by a partial or (virtually) complete quantitative deficiency of VWF while type 2 results from qualitative defects. 4 The prevalence of VWD in patients referred for clinically relevant bleeding symptoms is about 0.01% although the most severe form type 3 VWD is very rare (0.5‐5 cases/million). 1 , 5 , 6
The goal of VWD therapy is the correction of the VWF defect and, consequently, the correction of abnormal platelet adhesion and the abnormal intrinsic coagulation pathway. 1 Several plasma‐derived VWF/FVIII factor concentrates, initially developed for the treatment of hemophilia A and containing also large amounts of VWF, can be used to treat desmopressin‐unresponsive or intolerant patients with VWD. These VWF/FVIII products vary in VWF/FVIII ratio, depending upon the manufacturing process. 7 , 8 , 9 , 10 , 11 As the FVIII defect in VWD is secondary to that of VWF, the endogenous synthesis of FVIII is normal contrary to hemophilia A. Thus, correction of the VWF defect results also in the correction of FVIII levels, as has been shown in patients with severe VWD. 12 , 13 , 14 , 15 , 16 Repeated administration of dual VWF/FVIII concentrates may induce unnecessary increases in FVIII levels that could promote thrombotic complications when supra‐physiologic in high‐risk patients. 17
A highly purified plasma‐derived von Willebrand factor (pdVWF) concentrate with a very low FVIII content (≤10%) specifically dedicated to the treatment of patients with VWD was therefore developed in France almost 30 years ago. 18 The addition of two further viral inactivation or removal steps to the initial solvent‐detergent treatment led to a highly secured product first approved in France in 2003, with subsequent approvals in more than 30 countries worldwide, known under the names WILFACTIN® or WILLFACT® (LFB, Les Ulis, France). This product is indicated for the treatment of bleeding and prophylaxis (long‐term prophylaxis and surgery) in patients with VWD unresponsive to desmopressin. 19 Compared with VWF/FVIII concentrates, correction of the FVIII defect by this concentrate is not immediate as infused VWF must first stabilize endogenous FVIII. This delay involves specific management of patients with a low FVIII baseline level in some situations; however, this concentrate prevents, in case of repeated administrations, the risk of FVIII accumulation. The safety, efficacy, and tolerability for Wilfactin have been proven in a comprehensive clinical development program. 14 , 20 Here, we report the safety and efficacy results from a post‐marketing study (PMS) conducted to monitor patients over extended periods and assess the product in real‐life practice.
2. DESIGN AND METHODS
2.1. Study design
This open‐label, prospective, observational PMS conducted in 31 centers in France included patients with inherited VWD to be treated with Wilfactin. The study, designed to follow patients for up to 3 years after enrolment, started in September 2004 and ended in December 2009. It was conducted in accordance with international standards and national data protection regulations. The primary objective was to evaluate the safety of Wilfactin in a real‐life setting. The secondary objective was to assess the clinical efficacy of the product in current medical practice and, in particular, during treatment of severe situations (major bleeding and major surgery).
2.2. VWF product
Wilfactin is a pdVWF concentrate characterized by a high VWF activity and a low FVIII content. The residual FVIII:C is ≤ 0.1 International Units (IU) per 1 IU ristocetin cofactor activity of von Willebrand factor (VWF:RCo). The viral inactivation/removal includes three steps: solvent‐detergent treatment, 35 nm nanofiltration, and dry heating of the lyophilized product at 80°C for 72 hours. The other characteristics of the product have been fully described elsewhere. 21
2.3. Patients and treatment
Patients with inherited VWD (unresponsive‐ or with contraindications to desmopressin) who presented for regular appointments or hospitalization were eligible for inclusion and included in a consecutive order. Wilfactin was recommended to be used according to the Summary of Product Characteristics. If an immediate rise in FVIII:C was necessary, it was recommended to co‐administer a FVIII concentrate with the first injection of VWF. For scheduled surgery, it was possible to start VWF treatment 12 to 24 hours before the procedure to allow progressive stabilization of the endogenous FVIII and to administer a second VWF dose just before the procedure (ie, two preoperative injections).
2.4. Data collection
Patients were evaluated at each clinic visit. All collected data were checked for consistency with source data. Historical FVIII and VWF levels recorded in patient files were defined as baseline data. The VWD classification established by each center and based on phenotype and genotype (when available) was also collected at enrolment. Safety endpoints included monitoring of serious adverse events (SAEs), as defined in the Good Clinical Practice guidelines of EU directive 2001/20/EC, and both non‐serious adverse events (AEs) and SAEs considered related to the product. Special attention was given to possible thrombotic events or anaphylactic reactions, development of VWF inhibitors, and modifications in viral status. At each visit, the number of exposure days (EDs), VWF infusions, and VWF:RCo units infused were recorded according to therapeutic situation. Possible associated co‐administration of FVIII as a “priming dose” ie, at the time of first VWF injection or the use of two preoperative infusions, was also recorded. Major bleeds and major surgeries were arbitrarily defined according to the number of EDs (Table S1 in supporting information). Administration of red blood cells (RBC) was collected for major bleedings, surgeries, and childbirths. Plasma VWF:RCo and FVIII:C levels performed at the discretion of the treating physician were not recorded. Investigators were requested to assess the efficacy by using a predefined four‐point scale (excellent, good, moderate, none) at the last infusion for each major surgery and each major bleeding episode without central adjudication (Table S1). Long‐term prophylaxis (LTP), usually defined as at least one infusion per week for at least 45 weeks per year, 22 was evaluated by the record of the number of breakthrough bleeding episodes that occurred within 3 days of the infusion. Annualized bleeding rate (ABR) was also calculated from breakthrough bleeding events occurring within 3 days from infusion for those patients who received at least 12 months of treatment. The prophylaxis duration was defined as the period between the first and the last visit recording LTP infusions. As exact dates of start and end of LTP were not recorded, the frequency of infusions was determined for periods of definite LTP. When the product was administered over relatively brief periods of time to prevent bleedings commonly known to occur in daily life (eg, in advance of physical activity), it was recorded as short‐term prophylaxis. Descriptive statistics (median, ranges, and frequencies) were performed using SAS version 9.1.3 (Cary, NC, USA). Missing data for efficacy endpoint were not included in analyses. Patients with inherited deficiency who had any data on clinical events were included in the efficacy analysis and no distinction was made between patients who completed the study and those lost to follow‐up.
3. RESULTS
A total of 174 patients were enrolled (1 to 20 patients per center). Of these, 158 received at least one study dose (safety population) and 16 did not require replacement therapy. Three patients were excluded from efficacy analysis as they were subsequently diagnosed with acquired or pseudo VWD; the efficacy population thus comprised 155 patients. At the end of the study, 35 patients were prematurely withdrawn: four died and 31 were lost to follow‐up. Patient demographics of the efficacy cohort are summarized in Table 1. Thirty‐three (21.3%) had type 1 VWD, 84 (54.2%) had type 2 (2A = 41, 2B = 14, 2M = 8, 2N = 6, 2A/2N = 1, other type 2 = 14), 33 (21.3%) had type 3, and 5 had a VWD type not clearly classified as type 1 or 2. A total of 119 (76.8%) patients had severe disease defined by VWF:RCo levels ≤ 15 IU/dL according to the European guideline on clinical investigation of human pdVWF product. 23 Regarding the FVIII:C level, 106 (68.4%) patients had ≤ 40 IU/dL, a level considered as the minimum suitable hemostatic cut‐off level, 24 , 25 and 61 (39.4%) had < 20 IU/dL. Pediatric and elderly populations were represented: 14 patients aged < 6 years, 16 between 6 and 11 years, 9 between 12 and 17 years, and 14 ≥ 65 years. At the study entry, 13 children ≤ 10 years of age including one with type 3 VWD had never been exposed to any VWF concentrate.
Table 1.
Patient demographics by VWD type (efficacy population)
| Type 1 | Type 2 | Type 3 | Not classified (N = 5) | All patients (N = 155) | |
|---|---|---|---|---|---|
| (N = 33) | (N = 84) | (N = 33) | |||
| Sex, male/female (N) | 15/18 | 33/51 | 14/19 | 1/4 | 63/92 |
| Age at inclusion (years) | |||||
| Median (range) | 42 (11m.‐78) | 34 (3d.‐83) | 29 (11m.‐58) | 27 (3‐41) | 33 (3d.‐83) |
| <12 years, N (%) | 2 (6.1) | 18 (21.4) | 9 (27.3) | 1 (20.0) | 30 (19.4) |
| 12‐64 years, N (%) | 28 (84.8) | 55 (65.5) | 24 (72.7) | 4 (80.0) | 111 (71.6) |
| ≥65 years, N (%) | 3 (9.1) | 11 (13.1) | 0 ( 0.0) | 0 (0.0) | 14 (9.0) |
| Weight at inclusion (kg) | |||||
| Median (range) | 76 (9.5‐116) | 64 (3.5‐120)* | 56 (8.6‐135) | 58 (15‐67)* | 65.0 (3.5‐135) † |
| BMI in patients ≥ 18 years | |||||
| N with available data/total | 26/31 | 51/60 | 20/21 | 3/4 | 100/116 |
| ≥25.0 kg/m2, N (%) | 17 (65.4) | 25 (49.0) | 8 (40.0) | 0 (0) | 50 (50.0) |
| VWF:RCo plasma levels (IU/dL) | |||||
| Median (range) | 12.5 (1‐40) | 11 (<1‐102 ‡ ) | <3 (<1‐<12) | <10 (2‐12) | <10 (<1‐102 ‡ ) |
| ≤15 IU/dL, N (%) | 21 (63.6) | 60 (71.4) | 33 (100) | 5 (100) | 119 (76.8) |
| VWF:Ag plasma levels (IU/dL) | |||||
| Median (range) | 15 (2‐54) | 31 (9‐182)* | 1 (<1‐<10) | 15 (10‐18) | 19 (<1‐182)* |
| FVIII:C plasma levels (IU/dL) | |||||
| Median (range) | 33 (8‐63) | 38 (2 ‡ ‐102) | 2 (<1‐8) | 16.5 (9‐19) | 27 (<1‐102) |
| ≤40 IU/dL, N (%) | 23 (69.7) | 45 (53.6) | 33 (100) | 5 (100) | 106 (68.4) |
| <20 IU/dL, N (%) | 8 (24.2) | 15 (17.9) | 33 (100) | 5 (100) | 61 (39.4) |
Abbreviations: BMI, body mass index; d., days; FVIII:C, factor VIII coagulant activity; m., months; N, total number of study participants; VWD, von Willebrand disease; VWF:Ag, von Willebrand factor antigen; VWF:RCo, von Willebrand factor ristocetin cofactor activity.
Data missing in 1 patient.
Data missing in 2 patients.
Patient with type 2N.
3.1. Efficacy
The majority of patients (123 of 155) were treated on‐demand with different clinical settings described in Table 2. LTP was used in 32 (20.6%) patients with recurrent bleeding. The median VWF:RCo consumption per patient across all clinical settings (including LTP) was 352 IU/kg per year (range: 15 to 9807) in the subgroup of 106 patients with a follow‐up ≥ 12 months. However, the annual consumption was higher for those under LTP (4239 IU/kg).
Table 2.
Patients’ clinical characteristics by clinical setting (excluding prophylaxis)
| Details cohort | Bleeding episodes | Surgical procedures and childbirths | Invasive procedures | ||
|---|---|---|---|---|---|
| Major | Minor | Major | Minor | ||
| Patients (characteristics at inclusion) | |||||
| Number of patients, N | 83 | 105 | 27 | ||
| 38 | 70 | 50 | 66 | ||
| Male/female | 19/19 | 35/35 | 19/31 | 23/43 | 10/17 |
| Median age (range; years) | 33 (0‐77) | 30 (0‐77) | 32 (2‐83) | 34 (0‐77) | 48 (1‐83) |
| VWF:RCo ≤ 15 IU/dL, N (%) | 35 (92.1) | 66 (94.3) | 36 (72.0) | 45 (68.2) | 23 (85.2) |
| FVIII:C ≤ 40 IU/dL, N (%) | 31 (81.6) | 58 (82.9) | 32 (64.0) | 44 (66.7) | 22 (81.5) |
| FVIII:C < 20 IU/dL, N (%) | 23 (60.5) | 38 (54.3) | 16 (32.0) | 27 (40.9) | 12 (44.4) |
| Events (characteristics at event) | |||||
| Number of clinical events, n | 743 | 140 | 47 | ||
| 67 | 676 | 57 | 83 | ||
| By patient's age | |||||
| <12 years, n (%) | 16 (23.9) | 138 (20.4) | 7 (12.3) | 8 (9.6) | 12 (25.5) |
| 12‐64 years, n (%) | 45 (67.2) | 507 (75.0) | 39 (68.4) | 70 (84.3) | 26 (55.3) |
| ≥65 years, n (%) | 6 (9.0) | 31 (4.6) | 11 (19.3) | 5 (6.0) | 9 (19.1) |
| By VWD type | |||||
| Type 1, n (%) | 5 (7.5) | 49 (7.2) | 12 (21.1) | 21 (25.3) | 8 (17.0) |
| Type 2, n (%) | 36 (53.7) | 298 (44.1) | 34 (59.6) | 47 (56.6) | 21 (44.7) |
| Type 3, n (%) | 25 (37.3) | 328 (48.5) | 10 (17.5) | 12 (14.5) | 18 (38.3) |
| Not classified, n (%) | 1 (1.5) | 1 (0.1) | 1 (1.8) | 3 (3.6) | 0 (0.0) |
| By basal plasma levels | |||||
| VWF:RCo ≤ 15 IU/dL, n (%) | 63 (94.0) | 667 (98.6) | 41 (71.9) | 54 (65.0) | 41 (87.2) |
| FVIII:C ≤ 40 IU/dL, n (%) | 56 (83.6) | 541 (80.0) | 36 (63.1) | 54 (65.0) | 40 (85.1) |
| FVIII:C < 20 IU/dL, n (%) | 40 (59.7) | 429 (63.5) | 17 (29.8) | 31 (37.3) | 22 (46.8) |
| Total number of infusions | 783 | 1462 | 934 | 276 | 103 |
| Total number of exposure days | 608 | 1337 | 676 | 198 | 92 |
Abbreviations: FVIII:C, factor VIII coagulant activity; N, number of patients; n, number of clinical events; VWD, von Willebrand disease; VWF:RCo, von Willebrand factor ristocetin cofactor activity.
3.1.1. Surgery
During the study, 105 patients received treatment to prevent bleeding for 140 surgical procedures including 19 childbirths. Oro‐dental procedures were the most common (37, 26.4%), followed by obstetrical and gynecological procedures (33, 23.6%); see Table S2 in supporting information. In addition, 27 patients received treatment for 47 invasive procedures. Among the 140 surgeries, 57 (40.7%) were major and 83 (59.3%) were minor. Treatment initiation according to the patient's basal FVIII:C is shown in Figure 1. Correction of FVIII:C levels before surgery was considered necessary in 40 of 57 (70.2%) major surgeries compared to 46 of 82 (56.1%) minor surgeries (Figure 1A1). This was achieved more often by giving two VWF preoperative doses 12 to 24 hours apart (20 of 40, 50%) in major surgeries than in minor surgeries (5 of 46, 11%). In the 53 remaining procedures (17 major, 36 minor) FVIII:C levels were considered high enough by the clinician to ensure hemostasis during the procedure: 29 procedures were carried out in patients with basal FVIII:C levels > 40 IU/dL, 15 in patients with FVIII:C 20‐40 IU/dL, and 9 in patients with FVIII:C < 20 IU/dL (Figure 1A2). Regarding these last 9 procedures without apparent excessive bleeding, most patients were under LTP or received a previous VWF infusion within 3 days. Details by type of surgery are shown in Figure S2 in supporting information. During the period covering hospitalization and home treatment for major surgeries, patients received a median number of 13.0 VWF infusions (range 6‐52) for 10.0 EDs (range 6‐45) at a median daily dose of 44.5 VWF:RCo IU/kg (range 17‐128 IU/kg); (Table 3). Overall hemostatic efficacy in 56 major procedures was rated as excellent (n = 40) or good (n = 15) in 98.2% of the cases and moderate in one: a cesarean section complicated with post‐partum hemorrhage (Figure 2A). Percentages of excellent/good responses with or without FVIII correction (100% versus 93.75%) were comparable. All the surgeries performed in children (7 major, 8 minor) were rated as excellent. Red blood cell transfusions were administered on 10 occasions, more commonly in digestive surgery. There were no remarkable differences in the daily dose between major and minor surgeries (Table S2); however, the median treatment duration was shorter in minor (2 EDs) than in major (10 EDs) procedures.
Figure 1.
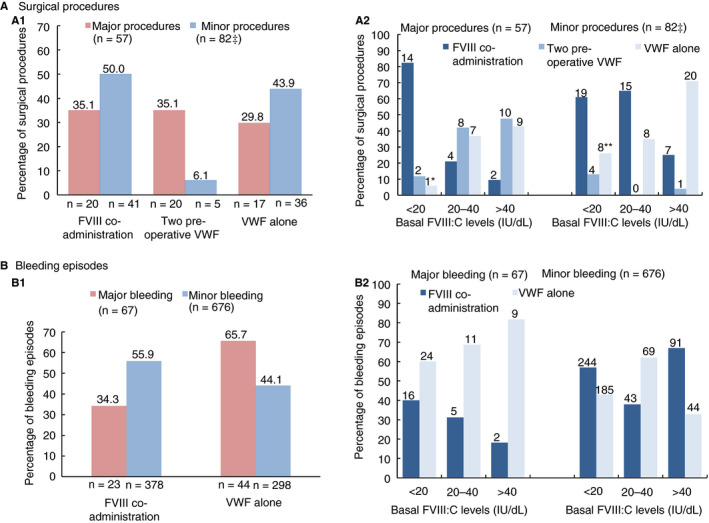
Treatment initiation patterns taking into account the patient's basal factor VIII coagulant activity (FVIII:C) levels for (A) surgical procedures (B) bleeding episodes: (A1) within major and minor procedures; (A2) within the different basal FVIII:C level categories for surgical procedures; (B1) within major and minor bleeding episodes; (B2) within the different basal FVIII:C categories for bleeding episodes. For (A2) and (B2), the number of surgical procedures or bleeding episodes is reported above each bar.
‡Modality to correct FVIII:C level was missing for one patient
* One procedure was performed under long‐term prophylaxis
**Eight procedures were performed either under long‐term prophylaxis (5) or within 3 days following VWF administration for other reason (2), or without factor VIII co‐administration in one patient with type 1 VWD
Table 3.
Summary of treatment (including rehabilitation) for 57 major surgical procedures and 67 major bleeding episodes
| Details cohort | N | n | VWF treatment: median (range) | Red Blood cells (n) | ||||
|---|---|---|---|---|---|---|---|---|
| Number of infusions | Number of exposure days | VWF:RCo dose | ||||||
| IU/kg/day | Total IU/kg | |||||||
| Major procedures | 50 | 57 | 13.0 (6‐52) | 10.0 (6‐45) | 44.5 (17‐128) | 476 (134‐1662) | 10 | |
| Category | Oro‐dental | 5 | 5 | 10.0 (6‐17) | 6.0 (6‐12) | 58.1 (17‐128) | 349 (134‐878) | 0 |
| Obs/Gyn | 16 | 19 | 12.0 (8‐23) | 9.0 (6‐16) | 39.6 (18‐83) | 343 (202‐1325) | 1 | |
| Orthopedic | 10 | 12 | 22.5 (9‐39) | 14.0 (6‐35) | 47.4 (23‐93) | 648 (239‐1662) | 2 | |
| Digestive | 7 | 7 | 12.0 (6‐21) | 10.0 (6‐13) | 39.4 (18‐65) | 329 (220‐849) | 4 | |
| General | 8 | 8 | 17.0 (11‐52) | 9.5 (8‐45) | 69.8 (34‐93) | 725 (560‐1513) | 0 | |
| Urological | 3 | 3 | 8.0 (7‐9) | 7.0 (7‐7) | 55.5 (32‐93) | 389 (226‐651) | 0 | |
| Cardiovascular | 2 | 2 | 21.5 (13‐30) | 12.5 (9‐16) | 46.3 (41‐52) | 598 (369‐827) | 2 | |
| Neurological | 1 | 1 | 28.0 | 27.0 | 41.7 | 1127 | 1 | |
| Age at event | <12 years | 7 | 7 | 13.0 (8‐28) | 9.0 (7‐18) | 70.0 (56‐93) | 644 (389‐1396) | 0 |
| 12 to 64 years | 34 | 39 | 13.0 (6‐52) | 10.0 (6‐45) | 41.0 (17‐128) | 430 (134‐1662) | 5 | |
| ≥65 years | 9 | 11 | 12.0 (6‐34) | 9.0 (6‐25) | 42.7 (26‐93) | 349 (226‐1046) | 5 | |
| Major bleeding episodes | 38 | 67 | 8.5 (1‐50)* | 6.0 (1‐47)* | 56.9 (25‐138)* | 390 (44‐2077) | 29 | |
| Location | Gastrointestinal | 18 | 34 | 8.5 (1‐42) | 5.5 (1‐26) | 54.6 (26‐138) | 369 (44‐2077) | 25 |
| Musculoskeletal | 8 | 13 | 10.0 (4‐24) | 8.0 (4‐17) | 55.9 (31‐100) | 495 (215‐1597) | 0 | |
| Genito‐urinary | 7 | 7 | 13.0 (8‐25)* | 9.5 (5‐24)* | 60.3 (49‐72)* | 406 (154‐1176) | 0 | |
| Nasopharynx | 4 | 7 | 5.0 (4‐21) | 4.0 (4‐12) | 76.5 (42‐107) | 308 (168‐1286) | 3 | |
| Intrabuccal | 1 | 1 | 19.0 | 19.0 | 60.9 | 1158 | 1 | |
| Other | 5 | 5 | 17.0 (3‐50) | 11.0 (3‐47) | 58.0 (25‐78) | 603 (233‐1178) | 0 | |
| Age at event | <12 years | 9 | 16 | 6.5 (2‐24) | 5.5 (2‐19) | 64.7 (50‐100) | 388 (106‐1597) | 1 |
| 12 to 64 years † | 26 | 45 | 9.5 (1‐50)* | 6.5 (1‐47)* | 55.2 (25‐138)* | 390 (44‐2077) | 23 | |
| ≥65 years | 4 | 6 | 8.5 (4‐10) | 5.0 (3‐10) | 58.8 (38‐79) | 350 (113‐566) | 5 | |
Other: other bleeds included “peritoneal hemorrhage” (n = 1), “post‐procedural hemorrhage” (n = 1), “tongue biting” (n = 1), “cerebral hematoma” (n = 1), “hemoptysis” revealing tuberculosis in a 16‐year‐old type 3 patient requiring secondary prophylaxis totaling 47 exposure days (n = 1).
Abbreviations: N, number of patients; n, number of clinical events; Obs/Gyn, Obstetrics/Gynecology; VWD, von Willebrand disease; VWF:RCo, von Willebrand factor ristocetin cofactor activity.
Number of infusions and treatment days were missing for one patient experiencing genito‐urinary bleeding episode, aged between 12 to 64 years and with type 2 VWD (basal VWF:RCo: <10 IU/dL and FVIII:C: 31 IU/dL).
One patient changed age group during the study participation and was present in two age groups: 12 to 64 years and ≥ 65 years.
Figure 2.
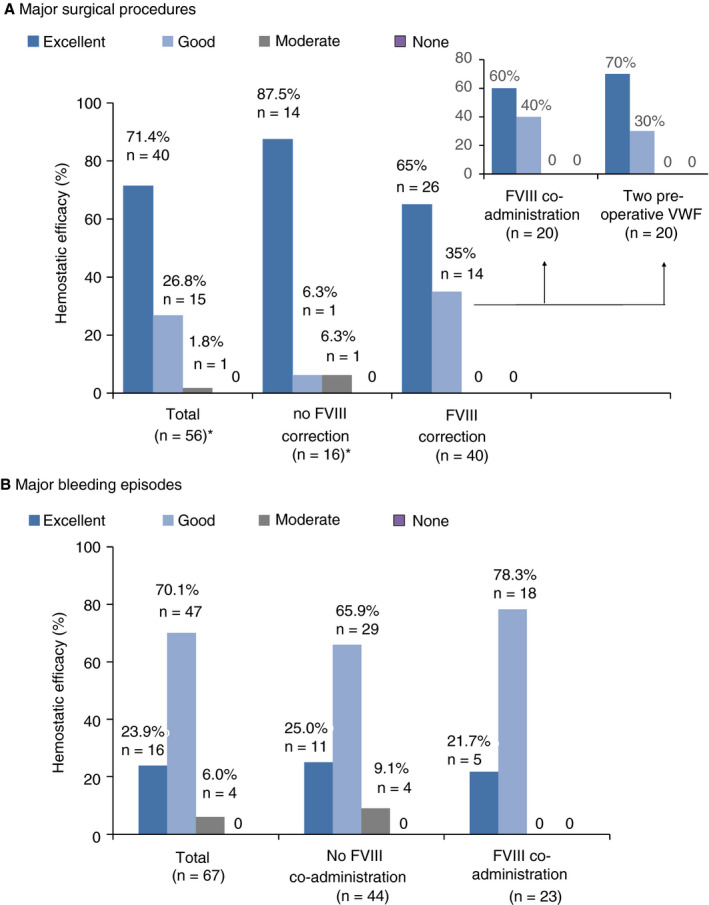
Efficacy of Wilfactin for the treatment of major events according to the factor VIII (FVIII) correction using a four‐point scale (excellent, good, moderate, none): (A) von Willebrand factor (VWF)‐treated major surgical procedures; (B) VWF‐treated major bleeding episodes.
* Efficacy was not assessed for 1 of the 57 major surgeries
Forty‐seven invasive procedures were performed by using a median dose of 44.8 IU/kg/infusion (range 23‐92) and a median number of infusions of 1 (range 1‐8.5). FVIII was co‐administered at the first VWF infusion in 40.4% (19/47) of cases.
3.1.2. Treatment of bleeding episodes
There were 743 bleeding episodes reported in 83 patients (Table 2). Most bleeds (91%) were minor, occurring in 70 patients whereas 67 major bleeding episodes in 38 patients required hospitalization.
The treatment summary for all 67 major bleeding episodes (including 16 experienced by 9 children) is shown in Table 3. Almost all (94%) occurred in patients with VWF:RCo basal levels ≤ 15 IU/dL. The most frequent bleeding locations were gastrointestinal (GI) tract (50.7%) and musculoskeletal (19.4%). At admission, 13 patients with acute anemia received concomitant RBC transfusions on 29 occasions, mainly for GI bleeds (86.2%). Efficacy was assessed as excellent/good in 63 episodes (94.0%) and moderate in 4 (6.0%) (Figure 2B). A moderate assessment was stated for four GI episodes occurring in two adult type 3 patients. Percentages of excellent/good responses with or without factor VIII co‐administration (100% versus 91.0%) were comparable. Median consumption of VWF:RCo from start to complete resolution of the bleed was 390 IU/kg (range 44‐2077). A median of 6.0 EDs (range 1‐47) with a median daily dose of 56.9 IU/kg (range 25‐138) was required. Twenty‐two bleeds required > 10 EDs. A priming dose of factor VIII was administered in combination with Wilfactin at the onset of treatment for 23 of 67 (34.3%) episodes as shown in Figure 1B1. The use of factor VIII co‐administration was more common in musculoskeletal (7/13, 53.8%) than in mucocutaneous bleeds (12/49, 24.5%); (Figure S2 in supporting information). In patients with basal FVIII:C < 20 IU/dL, FVIII was co‐administered in 16 of 40 (40.0%) events (Figure 1B2); in the other cases, patients were judged to have sufficient residual endogenous FVIII due to a previous VWF administration.
Seventy patients were treated for 676 minor bleeding episodes, of which 138 occurred in 17 children < 12 years. Most of these bleeding episodes were treated on an ambulatory basis. The median number of EDs by episode for a given patient was 2.0 (ranging from 1.0 to 12.4 in a patient with severe type 1 repeatedly treated for recurrent hemarthrosis). The median daily dose administered was 49.8 VWF:RCo IU/kg (range 24‐177 IU/kg). Factor VIII was co‐administered at first VWF injection in 55.9% of events (Figure 1B1) and 56.9% (244/429) for those occurring in patients with basal FVIII:C < 20 IU/dL (Figure 1B2).
3.1.3. Long‐term prophylaxis
Long‐term prophylaxis was initiated to prevent recurrent bleeding episodes in 32 VWD patients including five patients below 12 years of age (Table 4). The most frequent reasons for LTP were the prevention of joint bleeding (43.8%) and GI bleeding (40.6%) and mainly concerned type 3 and type 2 VWD patients, respectively. Patients under LTP received 43.5 VWF:RCo IU/kg (median) per infusion administered approximately twice a week, more frequently in patients with a history of GI bleeding compared with those with a history of joint bleeds (median 2.5 versus 1.9 infusions per week). Breakthrough bleeding occurred following 1.5% prophylactic infusions and required additional treatment with Wilfactin. In the subgroup of patients with available data and treated for at least 12 months (n = 20), the median annualized bleeding rate (ABR) was 1.0 (range 0.0 to 11.0). The highest ABR was due to the recurrence of GI bleeding in one patient with type 2A (17 events in 18.6 months). ABR was higher in 8 patients with type 2 than in the 12 type 3 VWD (1.9 versus 0.9). Seven patients did not experience bleeding events.
Table 4.
Treatment summary for long‐term prophylaxis in 32 patients
| Details cohort | Affected patients by type of recurrent bleeding episodes |
Overall N = 32 |
||
|---|---|---|---|---|
| Gastrointestinal | Joint | Other | ||
| N = 13 | N = 14 | N = 5 | ||
| Sex, male/female (N) | 8/5 | 4/10 | 2/3 | 14/18 |
| Age (years) at start of LTP | ||||
| Median (range) | 57.0 (33‐81) | 19.5 (5‐54) | 15.0 (6‐36) | 33.5 (5‐81) |
| <12 years, N (%) | 0 (0.0) | 3 (21.4) | 2 (40.0) | 5 (15.6) |
| 12‐64 years, N (%) | 9 (69.2) | 11 (78.6) | 3 (60.0) | 23 (71.9) |
| ≥65 years, N (%) | 4 (30.8) | 0 (0.0) | 0 (0.0) | 4 (12.5) |
| Type VWD | ||||
| Type 1, N (%) | 0 (0.0) | 0 (0.0) | 1 (20.0) | 1 (3.1) |
| Type 2, N (%) | 9 (69.2) | 2 (14.3) | 2 (40.0) | 13 (40.6) |
| Type 3, N (%) | 4 (30.8) | 12 (85.7) | 2 (40.0) | 18 (56.3) |
| Basal plasma levels | ||||
| VWF:RCo ≤ 15 IU/dL, N (%) | 13 (100) | 14 (100) | 5 (100) | 32 (100) |
| FVIII:C ≤ 40 IU/dL, N (%) | 8 (61.5) | 12 (85.7) | 4 (80.0) | 24 (75.0) |
| FVIII:C < 20 IU/dL, N (%) | 5 (38.5) | 12 (85.7) | 2 (40.0) | 19 (59.4) |
| Prophylaxis duration (months) | ||||
| Median (range) | 37.1 (6.7‐43.4) | 32.5 (6.5‐44.1) | 16.9 (1.1‐38.1) | 35.2 (1.1‐44.1) |
| Infusions for prophylaxis | ||||
| Total number | 4036 | 3341 | 326 | 7703 |
| VWF infusion dose (IU/kg) † | ||||
| Median (range) | 45.2 (22‐55) | 42.2 (26‐76) | 46.6 (27‐53) | 43.5 (22‐76) |
| Number of infusions per week | ||||
| N | 11 | 11 | 1 | 23 |
| Median (range) | 2.5 (1.0‐3.0) | 1.9 (1.2‐3.3) | 0.8 | 2.0 (0.8‐3.3) |
|
Number of bleeding episodes/total infusions † (%) |
56/4036 (1.4%) |
51/3069* (1.7%) |
6/326 (1.8%) |
113/7431* (1.5%) |
| Annualized bleeding rate * | ||||
| N | 10 | 9 | 1 | 20 |
| Median (range) | 1.1 (0.0‐11.0) | 0.8 (0.0‐5.4) | 1.0 | 1.0 (0.0‐11.0) |
Other: Other included menorrhagia (2 patients aged 15 and 36 years), epistaxis (1 patient aged 6 years), hematoma (1 patient aged 6 years), and subchorial hematoma to be prevented during a second pregnancy (1 patient aged 28 years).
Abbreviations: FVIII:C, factor VIII coagulant activity; N, number of patients; VWD, von Willebrand disease; VWF, von Willebrand factor: VWF:RCo, von Willebrand factor ristocetin cofactor activity.
The number of total infusions did not include 272 infusions without knowledge of bleeding occurrence within 3 days.
Bleeding episodes included spontaneous or post‐traumatic bleeds which occurred within 3 days after infusion.
Annualized bleeding rate correspond to the number of bleeding episodes per year in patients with at least 1‐year prophylaxis.
Note that 20 other patients including 5 children were administered 232 short‐term prophylactic infusions at a median dose of 47.3 IU/kg (range 23.7 to 77.7).
3.2. Safety
Clinical safety was assessed in the 158 patients who received Wilfactin. Their follow‐up ranged from 3 days to 49.4 months (median 27.3 months): 108 patients (68.4%) had a follow‐up longer than 1 year and 53 patients (33.5%) longer than 3 years. Patients cumulated 10 928 EDs in the study and received 32.5 million VWF:RCo units. Distribution of EDs by clinical events showed that 71.3% of EDs were related to LTP (Figure S1 in supporting information). No safety issues were identified during the study. A summary of reported AEs is shown in Table S3 in supporting information. SAEs were reported in 21.5% patients and none were considered to be related to the study treatment. Eleven non‐serious AEs considered to be related to Wilfactin occurred after six injections in six patients. All these events were mild or moderate in intensity. The rate of related AEs by patient was 3.8% and 0.05% by injection. Patients showed no clinical signs of inhibitor development: inhibitor testing was performed during the follow‐up for 28 of 158 patients and none of them, including 9 with type 3 VWD, developed an inhibitor to VWF:RCo. No thrombotic complications were reported.
4. DISCUSSION
This PMS including 155 patients with inherited VWD is the largest multicenter prospective survey to date on the clinical use of a VWF concentrate almost devoid of FVIII in real‐life conditions.
With the exception of recombinant VWF (rVWF; Veyvondi), all concentrates available for the treatment of patients with VWD have various amounts of FVIII in addition to VWF with FVIII/VWF ratios from about 0.4 (Haemate P, Voncento) to 0.8 (Fandhi, Alphanate) or 1 (Wilate). 26 The concept of a FVIII‐poor VWF concentrate is based upon the principle that what is missing in VWD patients is VWF only and not FVIII. The first FVIII‐poor VWF concentrate approved in France in 1989 was widely used until the marketing approval of Wilfactin, 13 , 18 the second generation product of this concept incorporating additional safety steps. 21 Previous clinical studies with Wilfactin have shown its particular pharmacodynamic profile, with a progressive post infusion FVIII:C increase raising 40 IU/dL by 6 hours after infusion in type 3 VWD patients. 20 The same results were recently observed with another VWF product without FVIII. 27
This PMS that included 155 patients with inherited VWD confirms and extends the efficacy, safety, and tolerability data of Wilfactin previously demonstrated in previous phase 3 pivotal studies in 50 patients. 14 It was the only VWF concentrate available in France during the study period allowing the description of treatment in a large panel of patients and clinical conditions. Most of the patients were severely affected: 76.8% had basal VWF:RCo ≤ 15 IU/dL and 68% had basal FVIII ≤ 40 IU/dL, a threshold value considered to increase the bleeding risk. 24 , 25 The range in age (3 days to 83 years) shows that the study achieved its goal of enrolling classes of patients poorly represented in clinical trials, notably patients < 6 years or elderly patients (14 patients for both categories). As a whole the patients were treated at a dosage of 40‐50 IU VWF:RCo/kg per day over a period depending upon the clinical setting. The efficacy evaluated only in patients requiring prolonged treatment was assessed as excellent/good in 98.2% of major procedures and in 94.0% of major bleeding episodes. It is noteworthy that there was no remarkable difference in the efficacy rate with respect to the FVIII correction at the treatment initiation.
In this cohort reflecting the current daily practice, 105 patients undergoing 140 surgical procedures were managed with a median daily dose of 51.7 VWF:RCo IU/kg for 4 EDs. The FVIII correction was achieved in 25 of 139 (18%) of surgeries with available data by two preoperative VWF injections with no further factor VIII administration and in 61 (44%) of surgeries by co‐administration of factor VIII at the first VWF injection. In comparison, in pivotal studies 43.5% were managed with two preoperative injections as per protocol and 12.0% with factor VIII co‐administration. These differences reflect the changes in patient management and can be explained by the constraints of the two preoperative injections. Neither of these two options for correcting FVIII was selected in 53 remaining procedures. About half (24) of these procedures were carried out in patients with historical FVIII:C levels ≤ 40 IU/dL, including 9 in patients with levels < 20 IU/dL. Nevertheless, the absence of bleeding complications in the postoperative period suggests the correction of FVIII levels at time of surgery, either due to rapid endogenous increase of FVIII or recent infusion. Based upon these results and overall experience with the use of this concentrate, the presurgical correction of FVIII levels in patients with a preoperative FVIII:C level of < 40 IU/dL has to be recommended for any surgery, taking into account that a rapid rise in endogenous FVIII is observed within 1 hour after VWF injection.
Eighty‐three patients were treated for 743 bleeding episodes including 67 major events. Major bleeds were managed with a median daily dose of 56.9 VWF:RCo IU/kg comparable with the one administered for minor bleeds (49.8 IU/kg). While major bleeding episodes correspond to emergency situations by definition, co‐administration of FVIII was necessary in only one third (34.3%) of cases, mainly for musculoskeletal episodes (53.8%), for which rapid correction of FVIII is essential (as for hemophiliacs). In contrast, 75.5% of mucocutaneous bleeding episodes were effectively treated by only VWF showing that, in this type of situation, the key issue for stopping bleeds is to correct the defective primary hemostasis. Factor VIII co‐administration was slightly more frequent for minor bleeds due to a more cautious approach being taken in the home setting, compared with major bleeds treated at hospital (55.9% versus 34.3%). As a general rule, coadministration of FVIII is highly recommended in cases of soft tissue bleeds or severe bleeds (such as intracranial hemorrhage) in patients whose FVIII:C level at the time of treatment is < 40 IU/dL. However, in cases of mucocutaneous bleeding episodes resulting mainly from defective primary hemostasis, VWF only may be sufficient. Gastrointestinal bleedings are the most challenging bleeds to manage. 28 , 29 , 30 In the present study there were 34 GI major episodes in 18 patients, treated with a median daily dose of 54.6 IU/kg for 5.5 EDs with an efficacy response of 88.2%. This assessment was similar to that reported in other studies based upon the use of VWF/FVIII products: after merging four prospective studies, Berntorp and Windyga described 82% excellent/good responses for 145 GI bleeds including 84 severe in 10 patients. 31 Federici et al provided 84% excellent or good hemostatic efficacy in 19 episodes. 32 Comparable data for rVWF are currently limited to six bleeds with excellent or good efficacy. 27
In this study, the importance of LTP was underlined as a treatment modality for patients with more severe disease, irrespective of VWD type; all 32 patients (18 type 3, 13 type 2, and 1 type 1 VWD) had VWF:RCo ≤15 IU/dL. Clinical effectiveness was suggested by the low rate of bleeding episodes occurring within 3 days of VWF prophylactic infusions and low ABR, especially in type 3. ABR was higher in patients with type 2 than in type 3 VWD (1.9 versus 0.9), which may be expected considering GI as the primary indication in type 2.
Thirty pediatric patients under 12 years of age were treated with Wilfactin for 154 bleeding episodes including 16 major, 15 surgeries including 7 major, and 12 invasive procedures. Five of them received LTP infusions. The study showed that 100% of the major therapeutic situations were treated successfully with excellent/good responses in this population. Results are comparable to those recently reported by Khair et al in 47 children and showed a good to excellent response in 97.7% in bleeding episodes and in 95.1% of the surgical procedures. 33
Prolonged treatment may be needed in major bleeding risk situations. More than 10 EDs were required in 37% of the major bleed cases and major surgeries in this study. It is important to point out that even in such circumstances there were no thrombotic events, including in patients at higher risk: elderly (n = 14), overweight and obese people with body mass index (BMI) ≥25 kg/m2 (n = 50), childbirths (n = 19), orthopedic surgeries (n = 12), or patients with multiple risk factors. Thromboembolic disease is multifactorial, but elevated FVIII levels > 150 IU/dL are considered as an additional risk factor that should be avoided. 34 , 35 , 36 , 37 The infusion of factor VIII in addition to endogenous FVIII production may lead to supraphysiological FVIII levels increasing the thrombotic risk. A few cases of pulmonary embolism and/or venous thrombosis, and superficial thrombophlebitis in postoperative VWD patients receiving dual VWF/FVIII concentrates were reported in VWD patients in the literature. 38 , 39 , 40 , 41 More recently, a deep vein thrombosis was reported in a patient treated with rVWF in the course of a major surgery. 16 Therefore several current therapeutic guidelines recommend to maintain FVIII below 150 IU/dL (Italian guideline, 42 European guideline 43 ) or 250 IU/dL (U.S. guideline 44 ). This objective is certainly easier to achieve with a FVIII‐poor VWF concentrate, which has the potential to raise VWF:RCo to high levels without leading to unwanted high levels of FVIII as recently underlined. 26 , 45
5. CONCLUSION
This study shows that Wilfactin was used in all types of severe inherited VWD across all age groups. This treatment allows a very high efficacy response in surgery to prevent perioperative bleedings, in curative treatment, and in long‐term prophylaxis with no remarkable safety signals. In particular, there were no thrombotic events, including in high‐risk patients. In less than half of the cases a priming dose of factor VIII has to be given to obtain the rapid increase in FVIII:C needed in some situations. This FVIII‐poor pdVWF provides a sound basis for effective and tailored patient treatment across a range of clinical settings.
Conflicts of Interest
This work was funded by LFB, Les Ulis, France. A. Harroche, P. Chamouni have no conflicts of interest to declare. J. Goudemand received support for attending scientific meetings and honoraria from Baxalta‐Shire, Bayer Healthcare, CSL Behring, LFB, Novo‐Nordisk, Roche‐Chugai, and Sobi. S. Claeyssens received support for attending scientific meetings from Baxalta/Shire, Bayer, LFB, Novo‐Nordisk, Octapharma, Sobi, and honoraria from Octapharma and Sobi. N. Itzhar‐Baïkian received travel fees from LFB, Shire‐Takeda, and CSL‐Behring. D. Desprez received research support for her institution and travel fees from Sobi. C. Négrier reports grants and personal fees from Baxalta/Shire, CSL Behring, Octapharma, Sobi, and personal fees from Alnylam Pharmaceuticals, Bayer, LFB, Novo Nordisk, Pfizer, Roche. All are outside the submitted work. H. Chambost has received support for attending scientific meetings and honoraria from Baxalta‐Shire, Bayer Healthcare, CSL Behring, LFB, Novo Nordisk, Octapharma, Pfizer, Roche‐Chugaï, and Sobi; has been an investigator in studies sponsored by Baxter, Bayer, Biomarin, Bioverativ, CSL Behring, LFB, and Octapharma; and has received research support from CSL Behring, LFB, Novo Nordisk, Octapharma, and Sobi (none of these relate to the present study). S. Susen received research support for her institution from LFB and CSL‐Behring, and performed consultancy for LFB, Roche, Sobi, Takeda: fees go to the institution. She is co‐founder and owner of Laelaps Therapeutics. A. Borel‐Derlon received research support for her institution from CSL Behring, Octapharma, Novo Nordisk, Baxalta, Shire: Consultant for Sobi, LFB. F. Bridey and C. Henriet are employees of LFB.
AUTHOR CONTRIBUTIONS
J. Goudemand was a study investigator and supervised and wrote the manuscript. F. Bridey and C. Henriet were involved in the concept and design of the study and the analysis and interpretation of data, and wrote the manuscript. S. Claeyssens, C. Négrier, H. Chambost, N. Itzhar‐Baïkian, A. Harroche, D. Desprez, P. Chamouni were primary investigators for the study and/or contributed to patient enrolment. S. Susen contributed to the interpretation of data. A. Borel‐Derlon was a study investigator and contributed to the design of the study and interpretation of data. All authors reviewed and approved the final manuscript.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the investigators and their team for their contributions to the conduct of the study at the investigative sites in France: C. Berger (Saint‐Etienne), M. A. Bertrand (Besancon), P. Beurrier (Angers), A. Borel‐Derlon (Caen), J. Y. Borg (Rouen), C. Boyer‐Neumann (Clamart), A. Brunot‐Ojeda (Mulhouse), H. Chambost (Marseille), A. Durin (Lyon), A. Faradji (Strasbourg), V. Gay (Chambery), J. Goudemand (Lille), V. Guerin (Bordeaux), C. Guérois/Y. Gruel (Tours), B. Guillet (Rennes), T. Lambert (Le Kremlin‐Bicêtre), M. Laubriat (Annecy), G. Lavigne (Nimes), J. L. Lorenzini (Dijon), A. Marques‐Verdier (Clermont‐Ferrand), P. Moreau (Le Mans), C. Négrier (Lyon), P. Nguyen (Reims), B. Pan‐Petesch (Brest), J. Peynet (Le Chesnay), B. Polack (Grenoble), C. Rothschild (Paris‐Necker), F. Sanderson (Nice), J. F. Schved (Montpellier), P. Sie (Toulouse), A. L. Voyer (Amiens). The authors thank Jane Shepard for her writing assistance and her revision of the manuscript based on input from authors.
Goudemand J, Bridey F, Claeyssens S, et al. Management of von Willebrand disease with a factor VIII‐poor von Willebrand factor concentrate: Results from a prospective observational post‐marketing study. J Thromb Haemost. 2020;18:1922–1933. 10.1111/jth.14928
Manuscript handled by: Flora Peyvandi
Final decision: Flora Peyvandi, 14 May 2020
Funding information
LFB sponsored the Wilfactin observational clinical study.
REFERENCES
- 1. Leebeek FW, Eikenboom JC. Von Willebrand’s disease. N Engl J Med. 2016;375:2067‐2080. [DOI] [PubMed] [Google Scholar]
- 2. Castaman G, Federici AB, Rodeghiero F, Mannucci PM. Von Willebrand’s disease in the year 2003: towards the complete identification of gene defects for correct diagnosis and treatment. Haematologica. 2003;88(1):94‐108. [PubMed] [Google Scholar]
- 3. Lenting PJ, Casari C, Christophe OD, Denis CV. von Willebrand factor: the old, the new and the unknown. J Thromb Haemost. 2012;10:2428‐2437. [DOI] [PubMed] [Google Scholar]
- 4. Sadler JE, Budde U, Eikenboom JC, et al. Working Party on von Willebrand Disease Classification. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4:2103‐2114. [DOI] [PubMed] [Google Scholar]
- 5. Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood. 1987;69:454‐459. [PubMed] [Google Scholar]
- 6. Veyradier A, Boisseau P, Fressinaud E, et al. A laboratory phenotype/genotype correlation of 1167 French patients from 670 families with von Willebrand disease: A new epidemiologic picture. Medicine. 2016;95:e3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mannucci PM, Tenconi PM, Castaman G, Rodeghiero F. Comparison of four virus inactivated plasma concentrates for treatment of severe von Willebrand disease: a cross‐over randomized trial. Blood. 1992;79:3130‐3137. [PubMed] [Google Scholar]
- 8. Mannucci PM, Lattuada A, Ruggeri ZM. Proteolysis of von Willebrand factor in therapeutic plasma concentrates. Blood. 1994;83:3018‐3027. [PubMed] [Google Scholar]
- 9. Lethagen S, Carlson M, Hillarp A. A comparative in vitro evaluation of six von Willebrand factor concentrates. Haemophilia. 2004;10:243‐249. [DOI] [PubMed] [Google Scholar]
- 10. Budde U, Metzner HJ, Müller HG. Comparative analysis and classification of von Willebrand factor/factor VIII concentrates: impact on treatment of patients with von Willebrand disease. Semin Thromb Hemost. 2006;32:626‐635. [DOI] [PubMed] [Google Scholar]
- 11. Riddell A, Vinayagam S, Gomez K, Laffan M, McKinnon T. Evaluation of von Willebrand factor concentrates by platelet adhesion to collagen using an in vitro flow assay. Res Pract Thromb Haemost. 2019;3(1):126‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goudemand J, Mazurier C, Marey A, et al. Clinical and biological evaluation in von Willebrand's disease of a von Willebrand factor concentrate with low factor VIII activity. Br J Haematol. 1992;80:214‐221. [DOI] [PubMed] [Google Scholar]
- 13. Goudemand J, Negrier C, Ounnoughene N, Sultan Y. Clinical management of patients with von Willebrand’s disease with a VHP vWF concentrate: the French experience. Haemophilia. 1998;4:48‐52. [DOI] [PubMed] [Google Scholar]
- 14. Borel‐Derlon A, Federici AB, Roussel‐Robert V, et al. Treatment of severe von Willebrand disease with a high‐purity von Willebrand factor concentrate (Wilfactin): a prospective study of 50 patients. J Thromb Haemost. 2007;5:1115‐1124. [DOI] [PubMed] [Google Scholar]
- 15. Gill JC, Mannucci PM. Thromboembolic incidence with transiently elevated levels of coagulation factors in patients with von Willebrand disease treated with VWF:FVIII concentrate during surgery. Haemophilia. 2014;20:e399‐443. [DOI] [PubMed] [Google Scholar]
- 16. Peyvandi F, Mamaev A, Wang JD, et al. Phase 3 study of recombinant von Willebrand factor in patients with severe von Willebrand disease who are undergoing elective surgery. J Thromb Haemost. 2019;17:52‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mannucci PM. Treatment of von Willebrand's Disease. N Engl J Med. 2004;351:683‐694. [DOI] [PubMed] [Google Scholar]
- 18. Mazurier C, Jorieux S, de Romeuf C, Samor B, Goudemand M. In vitro evaluation of a very‐high‐purity, solvent/detergent‐treated, von Willebrand factor concentrate. Vox Sang. 1991;61:1‐7. [DOI] [PubMed] [Google Scholar]
- 19. LFB, WILFACTIN, WILLFACT .Summary of Product Characteristics; https://www.medicines.org.uk/emc/product/3706/smpc. Accessed December 2019.
- 20. Goudemand J, Scharrer I, Berntorp E, et al. Pharmacokinetic studies on Wilfactin, a von Willebrand factor concentrate with a low factor VIII content treated with three virus‐inactivation/removal methods. J Thromb Haemost. 2005;3:2219‐2227. [DOI] [PubMed] [Google Scholar]
- 21. Mazurier C, Poulle M, Samor B, Hilbert L, Chtourou S. In vitro study of a triple‐secured von Willebrand factor concentrate. Vox Sang. 2004;86:100‐104. [DOI] [PubMed] [Google Scholar]
- 22. Lethagen S. Clinical experience of prophylactic treatment in von Willebrand disease. Thromb Res. 2006;118(Suppl 1):S9‐11. [DOI] [PubMed] [Google Scholar]
- 23. Guideline on the Clinical Investigation of Human Plasma Derived von Willebrand factor Products (CPMP/BPWG/220/02) London, 17 November 2005.
- 24. Guideline on the core SPC for human plasma derived von Willebrand factor (CPMP/BPWG/278/02) London, 17 November 2005.
- 25. Makris M, Oldenburg J, Mauser‐Bunschoten EP, Peerlinck K, Castaman G. Fijnvandraat K, for the subcommittee on Factor VIII, Factor IX and Rare Bleeding Disorders. The definition, diagnosis and management of mild hemophilia A: communication from the SSC of the ISTH. J Thromb Haemost. 2018;16:2530‐2533. [DOI] [PubMed] [Google Scholar]
- 26. Peyvandi F, Kouides P, Turecek PL, Dow E, Berntorp E. Evolution of replacement therapy for von Willebrand disease: from plasma fraction to recombinant von Willebrand factor. Blood Rev. doi: 10.1016/j.blre.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 27. Gill JC, Castaman G, Windyga J, et al. Hemostatic efficacy, safety, and pharmacokinetics of a recombinant von Willebrand factor in severe von Willebrand disease. Blood. 2015;126:2038‐2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Makris M. Gastrointestinal bleeding in von Willebrand disease. Thromb Res. 2006;118(Suppl. 1):S13‐S17. [DOI] [PubMed] [Google Scholar]
- 29. Makris M, Federici AB, Mannucci PM, et al. The natural history of occult or angiodysplastic gastrointestinal bleeding in von Willebrand disease. Haemophilia. 2015;21:338‐342. [DOI] [PubMed] [Google Scholar]
- 30. Franchini M, Mannucci PM. Gastrointestinal angiodysplasia and bleeding in von Willebrand disease. Thromb Haemost. 2014;112:427‐431. [DOI] [PubMed] [Google Scholar]
- 31. Berntorp E, Windyga J. Treatment and prevention of acute bleedings in von Willebrand disease–efficacy and safety of Wilate, a new generation von Willebrand factor/factor VIII concentrate. Haemophilia. 2009;15:122‐130. [DOI] [PubMed] [Google Scholar]
- 32. Federici AB, Barilliari G, Zanon E, et al. Efficacy and safety of highly purified, doubly virus‐inactivated VWF/FVIII concentrates in inherited von Willebrand’s disease: results of an Italian cohort study on 120 patients characterized by bleeding severity score. Haemophilia. 2010;16:101‐110. [DOI] [PubMed] [Google Scholar]
- 33. Khair K, Batty P, Riat R, et al. Wilate use in 47 children with von Willebrand disease: the North London paediatric haemophilia network experience. Haemophilia. 2015;21:e44‐50. [DOI] [PubMed] [Google Scholar]
- 34. Koster T, Blann AD, Briët E, Vandenbroucke JP, Rosendaal FR. Role of clotting factor VIII in effect of von Willebrand factor on occurrence of deep‐vein thrombosis. Lancet. 1995;345:152‐155. [DOI] [PubMed] [Google Scholar]
- 35. Kyrle PA, Minar E, Hirschl M, et al. High plasma levels of factor VIII and the risk of recurrent venous thromboembolism. N Engl J Med. 2000;343:457‐462. [DOI] [PubMed] [Google Scholar]
- 36. Bank I, Libourel EJ, Middeldorp S, et al. Elevated levels of FVIII: C within families are associated with an increased risk for venous and arterial thrombosis. J Thromb Haemost. 2005;3:79‐84. [DOI] [PubMed] [Google Scholar]
- 37. Coppola A, Franchini M, Makris M, Santagostino E, Di Minno G, Mannucci PM. Thrombotic adverse events to coagulation factor concentrates for treatment of patients with haemophilia and von Willebrand disease: a systematic review of prospective studies. Haemophilia. 2012;18:e173‐e187. [DOI] [PubMed] [Google Scholar]
- 38. Miesbach W, Krekeler S, Wolf Z, Seifried E. Clinical use of Haemate® P in von Willebrand disease: a 25‐year retrospective observational study. Thromb Res. 2015;135:479‐484. [DOI] [PubMed] [Google Scholar]
- 39. Mannucci PM. Venous thromboembolism in von Willebrand disease. Thromb Haemost. 2002;88:378‐379. [PubMed] [Google Scholar]
- 40. Franchini M, Targher G, Montagnana M, Lippi G. Antithrombotic prophylaxis in patients with von Willebrand disease undergoing major surgery: when is it necessary? J Thromb Thrombolysis. 2009;28:215‐219. [DOI] [PubMed] [Google Scholar]
- 41. Girolami A, Tasinato V, Sambado L, Peroni E, Casonato A. Venous thrombosis in von Willebrand disease as observed in one centre and as reported in the literature. Blood Coagul Fibrinolysis. 2015;26:54‐58. [DOI] [PubMed] [Google Scholar]
- 42. Mannucci PM, Franchini M, Castaman G, Federici AB. Italian Association of Haemophilia Centres. Evidence‐based recommendations on the treatment of von Willebrand disease in Italy. Blood Transfus. 2009;7:117‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Castaman G, Goodeve A, Eikenboom J. European Group on von Willebrand Disease. Principles of care for the diagnosis and treatment of von Willebrand disease. Haematologica. 2013;98:667‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nichols WL, Hultin MB, James AH, et al. von Willebrand disease (VWD): evidence based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008;14:171‐232. [DOI] [PubMed] [Google Scholar]
- 45. Mannucci PM. New therapies for von Willebrand disease. Blood Adv. 2019;3:3481‐3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material