

Abstract
Readily prepared tetraarylborates undergo selective (cross)‐coupling through oxidation with Bobbitt's salt to give symmetric and unsymmetric biaryls. The organic oxoammonium salt can be used either as a stoichiometric oxidant or as a catalyst in combination with in situ generated NO2 and molecular oxygen as the terminal oxidant. For selected cases, oxidative coupling is also possible with NO2/O2 without any additional nitroxide‐based cocatalyst. Transition‐metal‐free catalytic oxidative ligand cross‐coupling of tetraarylborates is unprecedented and the introduced method provides access to various biaryl and heterobiaryl systems.
Keywords: biaryls, oxidative coupling, oxoammonium salts, tetraarylborates, transition-metal-free
From B‐aryl to biaryl: Various biaryl and heterobiaryl systems are accessible via selective (cross)‐coupling of readily prepared tetraarylborates through oxidation with Bobbitt's salt. The oxoammonium salt can either be used as a stoichiometric oxidant or as a catalyst in combination with in situ generated NO2 as a cocatalyst and O2 as the terminal oxidant. For reactive borate salts, coupling can also be achieved without the Bobbitt salt by using NO2 as the sole catalyst.
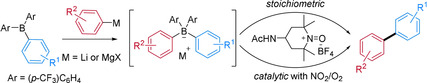
Biaryls play an important role as structural motifs in biologically active compounds, organic materials, and pharmaceuticals, rendering the development of novel methods for aryl–aryl bond formation a topic of ongoing interest.1 Since the first reported biaryl synthesis by Ullmann in 1901,2 different synthetic strategies have been introduced, including transition‐metal‐catalyzed coupling,3 direct4 and dehydrogenative4d, 5 C−H arylation, and decarboxylative coupling.6 In addition, significant efforts have been devoted to the development of transition‐metal‐free alternatives.7, 8, 9 As in the classical Suzuki–Miyaura coupling,10 many of these transition‐metal‐free variants use organoboron compounds as aryl donors.7a, 9 Along these lines, it has been reported that tetraphenylborate undergoes coupling to biphenyl via electrochemical11 or photochemical12 oxidation. Still, only a few methods for the direct oxidative biaryl synthesis via intramolecular ligand coupling of borate complexes with chemical oxidants have been presented. In these transformations iridium(IV),13 vanadium(V),14 a ZnII 4L6 cage,15 and chlorosilanes in the presence of dioxygen16 were chosen as oxidants (Scheme 1 A,B). However, mainly the preparation of symmetric biaryls was achieved by using these methods, whereas the oxidative cross‐coupling of unsymmetric tetraarylborates still remains challenging and is limited to a few examples.14, 16 During the preparation of this manuscript, Didier and co‐workers reported an electrochemical oxidative coupling of unsymmetric tetraarylborates to give the corresponding mixed biaryls.17
Scheme 1.
Oxidative coupling of tetraarylborates (A and B) and transition‐metal‐free selective cross‐coupling of in situ generated unsymmetric tetraarylborates, with a stoichiometric oxoammonium salt as oxidant or by using a catalytic process with O2 as the terminal oxidant (C).
We envisioned that a nitroxide‐derived oxoammonium salt, such as the commercial Bobbitt salt 1,18 could be used as a mild and cheap oxidant for the transition‐metal‐free coupling of tetraarylborates. Herein we disclose the realization of that idea and show that this approach can be used not only for the preparation of symmetric biaryls, but also for the selective cross‐coupling of in situ generated unsymmetric tetraarylborates. Unsymmetric tetraarylborates can be readily formed by addition of an aryl Grignard or aryllithium species to a triaryl borane (Scheme 1 C). It will be shown that the electron‐poor para‐trifluoromethylphenyl group mainly acts as a dummy ligand in these borates, opening the door to selective cross‐coupling reactions.
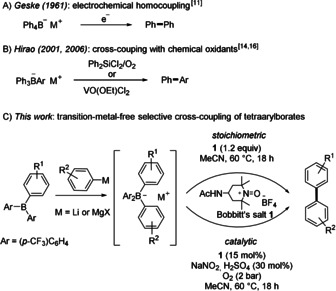
We commenced our studies by exploring the applicability of TEMPO+ salts as oxidants in the coupling of tetraarylborates 2. (p‐Tolyl)4BNa (2 a) was chosen as the test substrate. To our delight, formation of the biaryl 3 a was achieved in 81 % yield by using 1.2 equivalents of TEMPO+BF4 − in MeCN at 60 °C. Application of the cheaper Bobbitt salt 1 (4‐(acetylamino)‐2,2,6,6‐tetramethyl‐1‐oxo‐piperidinium tetrafluoroborate),19 which is a slightly stronger oxidant, led to a further improvement of the result and 3 a was isolated in 88 % yield. The robustness of the homocoupling was shown by running the reaction at 2 mmol scale, after which a comparable yield of 84 % was obtained.
After we had identified an efficient organic oxidant, we tested various commercially available or readily accessible tetraarylborates 2 b–i in the oxidative homocoupling (Scheme 2, Method A). The countercation M+ of the borate salt (Na+ or K+) does not influence the coupling process and is therefore not further specified here (see the Supporting Information). Smooth coupling was observed for the tetraphenylborate (2 b), tetraarylated borates bearing alkyl‐substituted aryl moieties (2 c and 2 d), and also for the tetra‐β‐naphthylborate salt (2 e) and the corresponding products 3 b–e were isolated in excellent yields (88–90 %). A good yield of 75 % was also achieved for the para‐methoxy analogue 3 f, while the oxidative coupling of tetrakis(2‐thienyl)borate 2 h to bithienyl 3 h occurred with slightly lower efficiency (50 %). Although the homocoupling of the para‐chlorophenyl‐substituted borate salt could be realized in high yield (3 h, 85 %), the presence of more electron‐withdrawing CF3 groups almost completely suppressed the transformation, probably due to the increased oxidation potential of 2 i, 11c and only traces of 3 i could be detected by GC‐MS analysis. Importantly, the reluctance of trifluoromethylphenyl groups in such borate salts to engage in oxidative C−C coupling can be harnessed for selective cross‐coupling processes (see below).
Scheme 2.
Transition‐metal‐free oxidative homocoupling of tetraarylborates by three different methods (A, B and C). Yields provided represent isolated yields. Conditions: Method A: 2 (0.30 mmol), 1 (0.36 mmol) in MeCN (3.0 mL); Method B: 2 (0.2 mmol), 1 (0.03 mmol,), NaNO2 (0.06 mmol), H2SO4 (0.06 mmol), 2 bar O2 in MeCN (2.0 mL); Method C: 2 (0.1 mmol), NaNO2 (0.03 mmol), H2SO4 (0.03 mmol), 2 bar O2 in MeCN (1.0 mL). a 84 % yield when conducted on a 2 mmol scale. b 20 mol % of NaNO2 and H2SO4 were used. c conducted on a 0.1 mmol scale d detected by GC‐MS analysis.
TEMPO+BF4 − is a two‐electron oxidant with TEMPOBAr2 being formed as the byproduct in these couplings. In principle, TEMPOBAr2 can be reoxidized to the starting oxoammonium salt ex situ. Despite this option, we were looking for a cheaper and more straightforward variant that uses the nitroxide component as a catalyst. Oxoammonium salts derived from TEMPO have found widespread application as cocatalysts in transition‐metal‐catalyzed aerobic oxidations of alcohols.20 Since 2004, transition‐metal‐free processes using TEMPO as a catalyst, nitric oxide as a cocatalyst, and molecular oxygen as the terminal oxidant have been developed.21 Motivated by these reports, we tested different cocatalytic systems to regenerate the oxoammonium salt in situ.
We were very pleased to find that by addition of 30 mol % NaNO2 along with H2SO4 under an O2 atmosphere, biaryls 3 a–h could be obtained under catalytic conditions using 15 mol % of Bobbitt's salt (Scheme 2, method B). In the catalytic cycle, in situ generated NO2 is suggested to reoxidize 4‐AcNH‐TEMPOBAr2 to the corresponding oxoammonium salt and NO2 is regenerated by O2.21 Notably, for this protocol an oxygen pressure of 2 bar was sufficient, which could be applied using a normal Schlenk tube and an autoclave was not required. Biaryls 3 a–3 e were obtained in good to very good yields (up to 85 %) using the nitroxide/NO2 cocatalysis protocol. Compared to the stoichiometric variant (method A), method B generally provided slightly lower yields, as documented by the preparation of the biaryls 3 f, 3 h, and the bithienyl 3 g. In these cases, the ate complexes were fully converted and the corresponding phenol derivatives were observed as main side products, explaining the lower yields. Surprisingly, control experiments revealed that in the absence of the oxoammonium salt 1, coupling of the tetraarylborates 2 a–h still occurs. Obviously, NO2 is able to directly oxidize a tetraarylborate salt, further lowering the costs of our homocoupling process. Through direct oxidation with catalytic NO2 under O2 atmosphere, yields of 24–85 % were achieved showing that, at least for the readily oxidized, more electron‐rich tetraarylborate salts, method C provides satisfying results (Scheme 2). Methods B and C, which are based on simple and cheap catalysts, represent to our knowledge the first transition‐metal‐free catalytic protocols for the oxidative ligand coupling of tetraarylborates.
We next turned our attention to the more challenging cross‐coupling using unsymmetric tetraarylborate salts as substrates. Preliminary studies revealed that oxidation of tetraarylborates, derived from Ph3B and p‐tolyllithium or (4‐methoxyphenyl)lithium, with TEMPO+BF4 − almost exclusively led to the formation of the cross‐coupling products Ph‐p‐CH3C6H4 (53 %) and Ph‐p‐CH3OC6H4 (52 %, see the Supporting Information). However, triphenyl(4‐(trifluoromethyl)phenyl)borate gave biphenyl as the main product (52 %) and only a trace amount of the trifluoromethylated biaryl was observed (see the Supporting Information). These findings and the failed oxidative homocoupling of borate 2 i (see above) indicate that electron‐withdrawing substituents at the aryl moiety lower the ligand's coupling tendency which should enable selective coupling of more electron‐ rich arenes in mixed borate salts. Notably, (para‐tolyl)pheB(pin)Li and also the more reactive (para‐tolyl)pheB(cat)Li did not react with TEMPO+BF4 − to 4‐methylbiphenyl (see the Supporting Information, pin=pinacolato, cat=catecholato), indicating that the coupling is restricted to tetraarylborates. Moreover, alkyl/aryl coupling did not work, as tested for the oxidation of lithium triethyl(6‐methoxynaphthalen‐2‐yl)borate with the Bobbitt salt (see the Supporting Information).
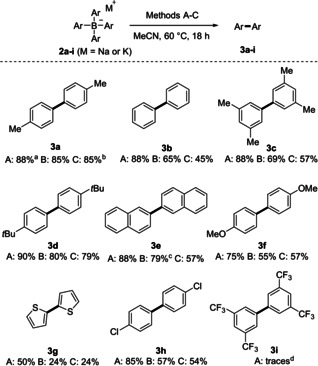
We therefore designed a synthetic strategy for the oxidative cross‐coupling of in situ generated mixed tetraarylborates containing two para‐trifluoromethylated phenyl groups as dummy ligands (Scheme 3). Bench‐stable aryl(p‐CF3C6H4)2B‐NH3 adducts 4 were chosen as starting materials that could be readily transformed in situ to the corresponding triarylboranes upon treatment with HCl (in Et2O). Addition of aryllithium or aryl Grignard reagents resulted in the formation of the corresponding mixed tetraarylborate salts (aryl1aryl2(p‐CF3C6H4)2BM with M=Li or MgX).
Scheme 3.
Transition‐metal‐free oxidative cross‐coupling of mixed tetraarylborates using methods A and B. Yields provided represent isolated yields of the overall sequence. Conditions: Method A: 4 (0.22 mmol), HCl (0.24 mmol), Ar′‐M (0.20 mmol) 1 (0.24 mmol) in MeCN (2.0 mL); Method B: 4 (0.22 mmol), HCl (0.24 mmol), Ar′‐M (0.20 mmol) 1 (30 μmol), NaNO2 (60 μmol), H2SO4 (60 μmol), 2 bar O2 in MeCN (2.0 mL). a Isolated as an inseparable mixture with 4‐(trifluoromethyl)‐1,1′‐biphenyl.
After removal of the solvent, the crude borate complex was redissolved in acetonitrile and subsequently oxidized using either stoichiometric or catalytic amounts of Bobbitt's salt (1), applying methods A or B. For example, reaction of 4 a and p‐tolyllithium, followed by 1‐mediated oxidative coupling (1.2 equivalents) gave the unsymmetric biaryl 5 a in 74 % yield. Notably, a comparatively good yield of 68 % was achieved using catalytic amounts of 1, NaNO2, H2SO4, and O2 (method B), demonstrating the synthetic value of this “catalytic” procedure in cross‐coupling reactions as well. With both methods the undesired trifluoromethylated byproduct was detected only in a trace amount by GC‐MS analysis and could not be isolated.
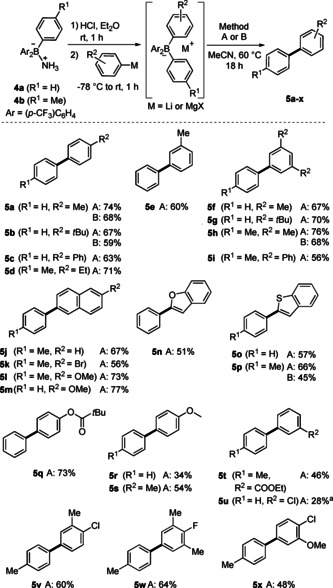
As depicted in Scheme 3, the sequence was applicable to the synthesis of various unsymmetric (hetero)biaryls 5 a–x. Moderate to good yields (56–76 %) were obtained for the cross‐coupling of alkyl‐ and aryl‐substituted aryl groups (5 b–5 i). Method B was applied to the synthesis of tert‐butyl‐ and trimethyl‐substituted biaryls 5 b and 5 h, which were isolated in 59 % and 68 % yield, respectively. Naphthyl groups engaged in the cross‐coupling and both electron‐withdrawing and ‐donating substituents were tolerated at the bicyclic core without affecting the selectivity (5 j–m). Heteroaryl cross‐coupling worked as well, shown by the synthesis of 2‐phenylbenzofuran (5 n) and 2‐phenylbenzothiophene derivatives 5 o and 5 p obtained in yields of 51–66 %. When we applied the catalytic variant (method B), 5 p was formed in 45 % yield. The pivaloyl‐substituted biphenyl 5 q was generated in 73 % yield. The coupling comprising more electron‐rich methoxy‐substituted aryl groups occurred with good overall yields, although a low selectivity was noted in these cases. Thus, while 4‐methoxy‐1,1′‐biphenyl (5 r) was isolated in 34 % yield, the undesired 4‐methoxy‐4′‐(trifluoromethyl)‐1,1′‐biphenyl was also obtained in comparable yield (35 %). However, after we switched to the methylated analogue, a better cross‐selectivity was observed and 5 s was obtained in 54 % yield, along with 22 % yield of the para‐trifluoromethylated biaryl side product. Biaryl 5 t, bearing an ester moiety, was obtained in 46 % yield. The chloro‐substituted biaryl 5 u was also successfully prepared, albeit with a lower yield. For both cases, lower yields were mainly caused by reduced cross‐coupling selectivities resulting in the formation of biaryl side products derived from the dummy ligand. Selective cross‐coupling of halogenated aryl groups with the p‐tolyl substituent was achieved and biaryls 5 v–5 x were isolated in moderate to good yields.
Finally, we developed a modular approach starting with bis(4‐(trifluoromethyl)phenyl)isopropoxyborane in which two different aryl groups can be added sequentially and subsequently coupled to the respective biaryls. The overall sequence comprises the selective cross‐coupling of two different aryllithium compounds and the readily accessed bis(4‐(trifluoromethyl)phenyl)isopropoxy borane acts as a coupling reagent (Scheme 4). We applied this sequence, and unsymmetric biaryls 5 a, 5 f, 5 y, and 5 z were successfully prepared in overall yields of 43–56 %.
Scheme 4.
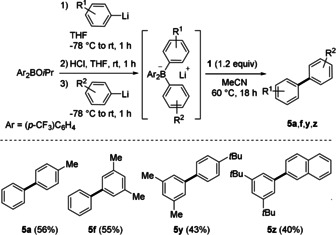
Modular strategy for the transition‐metal‐free oxidative cross‐coupling of two different aryllithium compounds. Yields provided represent isolated yields. Conditions: 4 (0.22 mmol), Aryl1Li (0.22 mmol), HCl (0.24 mmol), Aryl2Li (0.20 mmol), 1 (0.24 mmol) in MeCN (2.0 mL).
Based on our observations and earlier reports,13, 22 we propose the following mechanism for the ligand cross‐coupling in tetraarylborates (Scheme 5). One‐electron oxidation of borate 4 by Bobbitt's salt occurs most likely selectively at the most electron‐rich aryl moiety, while the two dummy substituents remain untouched. The thus generated intermediate I undergoes an intramolecular 1,2‐aryl shift23 to afford cyclohexadienyl radical II. Migration of the more electron‐rich aryl group is favored over migration of the two trifluoromethylated phenyl groups.24 As supported by crossover experiments of Hirao et al., the ligand coupling likely proceeds intramolecularly.14 Intermediate II could further react via two slightly different pathways. One‐electron oxidation of II to cation III by 4‐AcNH‐TEMPO, followed by nucleophilic attack of the thus generated 4‐AcNH‐TEMPO− at the boron atom affords biaryl 5 along with the byproduct V. Alternatively, radical/radical cross‐coupling of II with 4‐AcNH‐TEMPO, steered by the persistent radical effect,25 leads to intermediate IV which can then heterolyze to III.
Scheme 5.
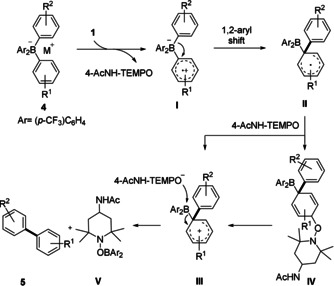
Proposed mechanism for the oxidative cross‐coupling of tetraarylborates with Bobbitt's salt.
In summary, we have reported a novel transition‐metal‐free oxidative ligand coupling in tetraarylborates for the synthesis of various biaryls using an oxoammonium salt as an inexpensive and mild oxidant. This strategy could also be applied to selective cross‐couplings for the preparation of unsymmetric biaryls. Tetraarylborates bearing different aryl groups were formed in situ and the cross‐selectivity was controlled by installation of unreactive trifluoromethylated phenyl groups as dummy ligands. In addition to the standard method that uses a stoichiometric amount of Bobbitt's salt 1, ligand coupling in tetraarylborate salts could also be achieved using oxidant 1 as a catalyst in combination with NO2 as a cocatalyst and O2 as the terminal oxidant. For reactive borate salts, coupling worked even in the absence of 1 with NO2 as the sole catalyst. Transition‐metal‐free, catalytic processes for the oxidative ligand coupling in tetraarylborates are unprecedented.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the European Research Council ERC (Advanced Grant agreement No. 692640) and the Fonds der Chemischen Industrie (fellowship to C.G.) for supporting this work. We thank Dr. Kazuhiro Okamoto (Kyoto University) for conducting initial experiments.
C. Gerleve, A. Studer, Angew. Chem. Int. Ed. 2020, 59, 15468.
References
- 1.
- 1a. Horton D. A., Bourne G. T., Smythe M. L., Chem. Rev. 2003, 103, 893–930; [DOI] [PubMed] [Google Scholar]
- 1b. Hajduk P. J., Bures M., Praestgaard J., Fesik S. W., J. Med. Chem. 2000, 43, 3443–3447; [DOI] [PubMed] [Google Scholar]
- 1c. Klekota J., Roth F. P., Bioinformatics 2008, 24, 2518–2525; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Cepanec I. in Synthesis of Biaryls, Elsevier, New York, 2004; [Google Scholar]
- 1e. Hassan J., Sévignon M., Gozzi C., Schulz E., Lemaire M., Chem. Rev. 2002, 102, 1359–1470; [DOI] [PubMed] [Google Scholar]
- 1f. Wencel-Delord J., Panossian A., Leroux F. R., Colobert F., Chem. Soc. Rev. 2015, 44, 3418–3430. [DOI] [PubMed] [Google Scholar]
- 2. Ullmann F., Bielecki J., Chem. Ber. 1901, 34, 2174–2185. [Google Scholar]
- 3.
- 3a. Stanforth S. P., Tetrahedron 1998, 54, 263–303; [Google Scholar]
- 3b. Johansson Seechurn C. C. C., Kitching M. O., Colacot T. J., Snieckus V., Angew. Chem. Int. Ed. 2012, 51, 5062–5085; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5150–5174; [Google Scholar]
- 3c. Hajipour A. R., Rafiee F., Appl. Organomet. Chem. 2013, 27, 412–418. [Google Scholar]
- 4.
- 4a. Alberico D., Scott M. E., Lautens M., Chem. Rev. 2007, 107, 174–238; [DOI] [PubMed] [Google Scholar]
- 4b. McGlacken G. P., Bateman L. M., Chem. Soc. Rev. 2009, 38, 2447–2464; [DOI] [PubMed] [Google Scholar]
- 4c. Ackermann L., Vincente R., Kapdi A. R., Angew. Chem. Int. Ed. 2009, 48, 9792–9826; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9976–10011; [Google Scholar]
- 4d. Kuhl N., Hopkinson M. N., Wencel-Delord J., Glorius F., Angew. Chem. Int. Ed. 2012, 51, 10236–10254; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10382–10401. [Google Scholar]
- 5.
- 5a. Stuart D. R., Fagnou K., Science 2007, 316, 1172–1175; [DOI] [PubMed] [Google Scholar]
- 5b. Yang Y., Lan J., You J., Chem. Rev. 2017, 117, 8787–8863; [DOI] [PubMed] [Google Scholar]
- 5c. Zhang Y.-F., Shi Z.-J., Acc. Chem. Res. 2019, 52, 161–169; [DOI] [PubMed] [Google Scholar]
- 5d. Röckl J. L., Pollok D., Franke R., Waldvogel S. R., Acc. Chem. Res. 2020, 53, 45–61. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Gooßen L. J., Deng G., Levy L. M., Science 2006, 313, 662–664; [DOI] [PubMed] [Google Scholar]
- 6b. Baudoin O., Angew. Chem. Int. Ed. 2007, 46, 1373–1375; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 1395–1397; [Google Scholar]
- 6c. Perry G. J. P., Larrosa I., Eur. J. Org. Chem. 2017, 3517–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For reviews see
- 7a. Sun C.-L., Shi Z.-J., Chem. Rev. 2014, 114, 9219–9280; [DOI] [PubMed] [Google Scholar]
- 7b. Piazzolla F., Colognese F., Temperini A., Curr. Org. Chem. 2018, 22, 2537–2554. [Google Scholar]
- 8.For selected examples see:
- 8a. Krasovskiy A., Tishkov A., del Amo V., Mayr H., Knochel P., Angew. Chem. Int. Ed. 2006, 45, 5010–5014; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5132–5136; [Google Scholar]
- 8b. Maji M. S., Pfeiffer T., Studer A., Angew. Chem. Int. Ed. 2008, 47, 9547–9550; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 9690–9692; [Google Scholar]
- 8c. Dohi T., Ito M., Yamaoka N., Morimoto K., Fujioka H., Kita Y., Angew. Chem. Int. Ed. 2010, 49, 3334–3337; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3406–3409; [Google Scholar]
- 8d. Sun C.-L., Li H., Yu D.-G., Yu M., Zhou X., Lu X.-Y., Huang K., Zheng S.-F., Li B.-J., Shi Z.-J., Nat. Chem. 2010, 2, 1044–1049; [DOI] [PubMed] [Google Scholar]
- 8e. Shirakawa E., Hayashi Y., Itoh K.-I., Watabe R., Uchiyama N., Konagaya W., Masui S., Hayashi T., Angew. Chem. Int. Ed. 2012, 51, 218–221; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 222–225; [Google Scholar]
- 8f. Dewanji A., Murarka S., Curran D. P., Studer A., Org. Lett. 2013, 15, 6102–6105; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8g. Kloss F., Neuwirth T., Haensch V. G., Hertweck C., Angew. Chem. Int. Ed. 2018, 57, 14476–14481; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14684–14689; [Google Scholar]
- 8h. Hilton M. C., Zhang X., Boyle B. T., Alegre-Requena J. V., Paton R. S., McNally A., Science 2018, 362, 799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Wang Y., Noble A., Sandford C., Aggarwal V. K., Angew. Chem. Int. Ed. 2017, 56, 1810–1814; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1836–1840; [Google Scholar]
- 9b. Liu W., Li J., Querard P., Li C.-J., J. Am. Chem. Soc. 2019, 141, 6755–6764; [DOI] [PubMed] [Google Scholar]
- 9c. Paul S., Das K. K., Manna S., Panda S., Chem. Eur. J. 2020, 26, 1922–1927. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Miyaura N., Yamada K., Suzuki A., Tetrahedron Lett. 1979, 20, 3437–3440; [Google Scholar]
- 10b. Miyaura N., Suzuki A., Chem. Rev. 1995, 95, 2457–2483. [Google Scholar]
- 11.
- 11a. Geske D. H., J. Phys. Chem. 1959, 63, 1062–1070; [Google Scholar]
- 11b. Geske D. H., J. Phys. Chem. 1962, 66, 1743–1744; [Google Scholar]
- 11c. Beil S. B., Möhle S., Enders P., Waldvogel S. R., Chem. Commun. 2018, 54, 6128–6131. [DOI] [PubMed] [Google Scholar]
- 12. Doty J. C., Grisdale P. J., Evans T. R., Williams J. L. R., J. Organomet. Chem. 1971, 32, C35–C37. [Google Scholar]
- 13. Abley P., Halpern J., J. Chem. Soc. D 1971, 1238–1239. [Google Scholar]
- 14. Mizuno H., Sakurai H., Amaya T., Hirao T., Chem. Commun. 2006, 5042–5044. [DOI] [PubMed] [Google Scholar]
- 15. Lu Z., Lavendomme R., Burghaus O., Nitschke J. R., Angew. Chem. Int. Ed. 2019, 58, 9073–9077; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9171–9175. [Google Scholar]
- 16. Sakurai H., Morimito C., Hirao T., Chem. Lett. 2001, 30, 1084–1085. [Google Scholar]
- 17. Music A., Baumann A. N., Spieß P., Plantefol A., Jagau T.-C., Didier D., J. Am. Chem. Soc. 2020, 10.1021/jacs.9b12300. [DOI] [PubMed] [Google Scholar]
- 18. Merbouh N., Bobbitt J. M., Brückner C., Org. Prep. Proced. Int. 2004, 36, 1–31. [Google Scholar]
- 19.
- 19a. Bobbitt J. M., J. Org. Chem. 1998, 63, 9367–9374; [Google Scholar]
- 19b. Mercadante M. A., Kelly C. B., Bobbitt J. M., Tilley L. J., Leadbeater N. E., Nat. Protoc. 2013, 8, 666–676; [DOI] [PubMed] [Google Scholar]
- 19c. Tilley L. J., Bobbitt J. M., Murray S. A., Camire C. E., Eddy N. A., Synthesis 2013, 45, 326–329; [Google Scholar]
- 19d. Hickey D. P., Schiedler D. A., Matanovic I., Doan P. V., Atanassov P., Minteer S. D., Sigman M. S., J. Am. Chem. Soc. 2015, 137, 16179–16186. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Vogler T., Studer A., Synthesis 2008, 1979–1993; [Google Scholar]
- 20b. Wertz S., Studer A., Green Chem. 2013, 15, 3116–3134. [Google Scholar]
- 21.For selected examples see:
- 21a. Liu R., Liang X., Dong C., Hu X., J. Am. Chem. Soc. 2004, 126, 4112–4113; [DOI] [PubMed] [Google Scholar]
- 21b. Wang X. L., Liu R. H., Jin Y., Liang X. M., Chem. Eur. J. 2008, 14, 2679–2685; [DOI] [PubMed] [Google Scholar]
- 21c. He X., Shen Z., Mo W., Sun N., Hu B., Hu X., Adv. Synth. Catal. 2009, 351, 89–92; [Google Scholar]
- 21d. Wertz S., Studer A., Adv. Synth. Catal. 2011, 353, 69–72. [Google Scholar]
- 22. Dhital R. N., Sakurai H., Asian J. Org. Chem. 2014, 3, 668–684. [Google Scholar]
- 23.For recent reviews on 1,2-migrations of boronate complexes see:
- 23a. Namirembe S., Morken J. P., Chem. Soc. Rev. 2019, 48, 3464–3474; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Kischkewitz M., Friese F. W., Studer A., Adv. Synth. Catal. 2020, 362, 10.1002/adsc.201901503; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23c. Tappin N. D. C., Renaud P., Chimia 2020, 74, 33–38. [DOI] [PubMed] [Google Scholar]
- 24. Pachuau Z., Lyngdoh R. H. D., J. Chem. Sci. 2004, 116, 83–91. [Google Scholar]
- 25. Leifert D., Studer A., Angew. Chem. Int. Ed. 2020, 59, 74–108; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 74–110. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary