

Abstract
Autism spectrum disorder (ASD) is characterized by phenotypic heterogeneity and a complex genetic architecture which includes distinctive epigenetic patterns. We report differential DNA methylation patterns associated with ASD in South African children. An exploratory whole‐epigenome methylation screen using the Illumina 450 K MethylationArray identified differentially methylated CpG sites between ASD and controls that mapped to 898 genes (P ≤ 0.05) which were enriched for nine canonical pathways converging on mitochondrial metabolism and protein ubiquitination. Targeted Next Generation Bisulfite Sequencing of 27 genes confirmed differential methylation between ASD and control in our cohort. DNA pyrosequencing of two of these genes, the mitochondrial enzyme Propionyl‐CoA Carboxylase subunit Beta (PCCB) and Protocadherin Alpha 12 (PCDHA12), revealed a wide range of methylation levels (9–49% and 0–54%, respectively) in both ASD and controls. Three CpG loci were differentially methylated in PCCB (P ≤ 0.05), while PCDHA12, previously linked to ASD, had two significantly different CpG sites (P ≤ 0.001) between ASD and control. Differentially methylated CpGs were hypomethylated in ASD. Metabolomic analysis of urinary organic acids revealed that three metabolites, 3‐hydroxy‐3‐methylglutaric acid (P = 0.008), 3‐methyglutaconic acid (P = 0.018), and ethylmalonic acid (P = 0.043) were significantly elevated in individuals with ASD. These metabolites are directly linked to mitochondrial respiratory chain disorders, with a putative link to PCCB, consistent with impaired mitochondrial function. Our data support an association between DNA methylation and mitochondrial dysfunction in the etiology of ASD. Autism Res 2020, 13: 1079‐1093. © 2020 The Authors. Autism Research published by International Society for Autism Research published by Wiley Periodicals, Inc.
Lay Summary
Epigenetic changes are chemical modifications of DNA which can change gene function. DNA methylation, a type of epigenetic modification, is linked to autism. We examined DNA methylation in South African children with autism and identified mitochondrial genes associated with autism. Mitochondria are power‐suppliers in cells and mitochondrial genes are essential to metabolism and energy production, which are important for brain cells during development. Our findings suggest that some individuals with ASD also have mitochondrial dysfunction.
Keywords: DNA methylation, Autism Spectrum Disorder, mitochondrial dysfunction, epigenetics, metabolomic profiles, organic acids, PCCB, PCDHA12
Introduction
Autism spectrum disorder (ASD), which is a highly heterogeneous disorder, has an incidence of 1 in 59 and is four times more prevalent in boys [Baio et al., 2018; Mannion & Leader, 2016; Xu et al., 2019]. ASD is highly heritable [Yip et al., 2018] with a complex genetic architecture and is associated with copy number variants, rare de novo mutations, and Single Nucleotide Polymorphisms (SNPs) [Wiśniowiecka‐Kowalnik & Nowakowska, 2019]. High throughput DNA sequencing identified over 100 genes with a strong link to ASD [De Rubeis et al., 2014; Iossifov et al., 2014]; however, only 49% of ASD liability is reported to be quantitatively associated with common genetic variation [Gaugler et al., 2014]. Despite the heritable nature of ASD, the genetic mechanisms underlying its etiology has not yet been fully elucidated and no molecular marker shows a consistent association with ASD [Hu, 2013].
Epigenetic mechanisms (e.g., chromatin modification, RNA interference and DNA methylation) [Rakyan, Down, Balding, & Beck, 2011] contribute to temporal and spatial gene regulation during cell differentiation and neurodevelopment [Vogel Ciernia & LaSalle, 2016; Xie, Zang, Li, & Shu, 2016; Zahir & Brown, 2011] and are proposed to play a role in ASD etiology [Tremblay & Jiang, 2019]. Global RNA expression differences associated with ASD were initially identified in blood‐derived cell‐lines from ASD patients using mRNA arrays [Hu, 2013], and were subsequently confirmed by RNA sequencing of postmortem brain tissue from ASD individuals [Gandal et al., 2018; Schwede et al., 2018]. Transcriptomic studies identified gene co‐expression modules linked to neuronal activity, synaptic transmission, and receptor signaling associated with ASD [Giovedi, Corradi, Fassio, & Benfenati, 2014; Romero‐Garcia, Warrier, Bullmore, Baron‐Cohen, & Bethlehem, 2019; Wang, Zhao, Lachman, & Zheng, 2018]. Recent transcriptomic studies in brain tissue identified mitochondrial genes that are downregulated in ASD [Gandal et al., 2018; Schwede et al., 2018].
DNA methylation is associated with numerous complex behavioral phenotypes [Feinberg et al., 2015; Hass et al., 2015], including ASD where differential methylation is observed both in single‐gene and genome‐wide studies [Tremblay & Jiang, 2019; Xie et al., 2016]. Loss‐ and gain‐of‐function mutations in genes encoding DNA methylation enzymes (e.g., DNMT3A, TET2, or MECP2) were also identified in ASD [De Rubeis et al., 2014; Iossifov et al., 2014], and differential DNA methylation is found in promoter regions of ASD‐predisposing genes in peripheral tissue and postmortem brain tissue [Kuehner, Bruggeman, Wen, & Yao, 2019; Tremblay & Jiang, 2019]. Epigenome‐wide DNA methylation studies found differentially methylated (DM) regions associated with ASD in twin and case–control studies [Wang et al., 2018; Wong et al., 2014], with DM regions enriched in genes implicated in neurodevelopment and synaptic function canonical pathways [Kumar et al., 2019]. Correlations between genetic variance and DNA methylation reveal several methylation quantitative trait loci (meQTL) associated with ASD [Andrews et al., 2018; Hannon et al., 2018].
Given ASD's numerous comorbidities and the many pleiotropic pathways contributing to its etiology, a paradigm shift from ASD as primarily a brain disorder, to a systems‐level disorder has emerged [Gandal et al., 2018; Hu, 2013]. ASD is now considered to be the result of a combination of overlapping, dysregulated biological networks affecting multiple systems, and this pleiotropy is reflected in the transcriptomic profiles observed in postmortem ASD brain tissue [Chow et al., 2012; Garbett et al., 2008; Voineagu, 2012]. For example, using transcriptomic and GWAS data from five major psychiatric disorders including ASD, Gandal et al. [2018] found shared pathways of transcriptional dysregulation and disease‐specific signatures of gene expression. Genetic variation accounts for only a fraction of the transcriptional dysregulation observed, suggesting a contribution of epigenetic factors to the etiology of psychiatric disorders. In addition, specific gene co‐expression modules involved in mitochondrial function were downregulated in ASD, suggesting a link between ATP metabolism and synaptic transmission [Gandal et al., 2018]. This association between downregulated mitochondrial gene expression and synaptic transmission in postmortem ASD cerebral cortex is reported in other transcriptomic studies [Schwede et al., 2018]. Mitochondrial dysfunction in ASD is also supported by clinical and experimental evidence that demonstrated altered oxidative stress responses in ASD [Frye & Rossignol, 2011; Legido, Jethva, & Goldenthal, 2013; Melnyk et al., 2012; Rose et al., 2017]. Mitochondrial homeostasis is essential for cell function in all tissues [Alston, Rocha, Lax, Turnbull, & Taylor, 2017; Area‐Gomez & Schon, 2014], and mitochondrial dysfunction has been linked to several neurologic disorders [Bose & Beal, 2016; Franco‐Iborra, Vila, & Perier, 2016].
Differential methylation of genes can affect cellular homeostasis and metabolism by altering gene expression in key bioenergetic processes [Elstner & Turnbull, 2012; F. Reinecke, Smeitink, & van der Westhuizen, 2009]. The metabolome represents the functional phenotype of an organism [Roessner & Bowne, 2009] and can provide insight into mitochondrial metabolism. For example, Reinecke et al. [2011] reported that intermediates of several mitochondrial‐based pathways were elevated in the urine of South African children with respiratory chain disorders, with organic acids being most affected. Numerous metabolic markers and mitochondrial metabolites are found to be dysregulated in ASD, with a recent study [Sharon et al., 2019] reporting an altered metabolome associated with the ASD microbiome.
It is predicted that Africa will be home to 10–20 million children with ASD by 2050 [UNICEF, 2014], yet there is a dearth of information regarding the genetics of ASD in African populations with only a handful of publications from Sub‐Saharan Africa [Franz, Chambers, von Isenburg, & de Vries, 2017]. Integrated genomic and transcriptomic ASD studies highlight the importance of epigenetic mechanisms and the environmental component in ASD etiology [Gandal et al., 2018]. In this context, we examine the role of DNA methylation in a South African ASD cohort by performing a pilot screen of 450,000 CpG sites to identify DM genes associated with ASD, and analyze it in a functional pathway framework. We also examine differential methylation using gene‐ and CpG site‐specific methylation methods, and we test for mitochondrial dysfunction by examining urinary metabolites. Our data suggest that DNA methylation contributes to the dysregulation of mitochondrial function in ASD.
Methods
Cohort Design
Our cohort comprised of children with ASD and age‐ and gender‐matched typically developing children as the control group. We recruited males and females (6–17 years), but only prepubertal boys (6–12 years old) were included in the DNA methylation studies [Bell et al., 2012; Lintas, Sacco, & Persico, 2016]. This was done to reduce genetic heterogeneity, since girls recruited generally have a higher ASD genetic load [Levy et al., 2011; Robinson, Lichtenstein, Anckarsater, Happe, & Ronald, 2013] and we had few girls in our cohort (n = 5). To be eligible for our study, children with ASD had a prior, independent diagnosis of ASD, no known comorbidities and attended Autism‐specialist schools in the Western Cape, South Africa. The control group were unrelated children without ASD or other developmental delays. We used the Autism Diagnostic Observation Schedule (ADOS‐2) for ASD phenotyping and to screen children in our control group for the absence of ASD‐defining traits [Lai, Lombardo, Chakrabarti, & Baron‐Cohen, 2013]. ADOS assessments were performed by Research Reliable ADOS‐2 administrators in English, which was the language used at school for all study participants. We obtained informed parental consent, University of Cape Town ethical approval (FSREC076‐2014) and Western Cape Government approval (20141002–37506) for our study.
Study Participants and Tissue Collection
We screened 145 children (93 ASD and 52 controls) using ADOS‐2 over a 4‐year period (2014–2017). DNA was extracted from buccal cells, a recognized proxy for DNA methylation of brain tissue [Berko et al., 2014; Lowe et al., 2013]. Buccal cells were collected using Epicentre Catch‐All™ collection swabs, extracted using a standard protocol [Aljanabi & Martinez, 1997] and quantified using Qubit 2.0 Fluorometer. For urinary organic acid analysis, urine was collected from a subset of our cohort (21 ASD and 13 controls, Table S1). Approximately 20 ml of urine was collected before breakfast into a sterile collection tube and immediately placed on ice, then frozen upon arrival at the laboratory until analysis.
DNA Methylation Beadchip Screen
DNA (500 ng) from 48 children (32 ASD and 16 controls, Table S1) was bisulfite converted using Zymo EZ DNA Methylation‐Gold™ kit, as per manufacturer's instructions. Bisulfite‐converted DNA samples were processed using the Illumina Human Methylation 450 K Beadchip (Illumina, Inc., CA, USA) and scanned using Illumina HiscanSQ at the Core Facility of the Agricultural Research Council (Onderstepoort, South Africa).
Methylation Beadchip Analysis Pipeline
All analyses were performed in R/Bioconductor (R Development Core Team 2011; Huber et al., 2015). Raw beadchip data were preprocessed and normalized using functions from the minfi, missMethyl, and wateRmelon packages [Aryee et al., 2014; Phipson, Maksimovic, & Oshlack, 2016; Pidsley et al., 2013]. Quality control of raw data was completed using wateRmelon::pfilter to remove low quality samples (detection P‐values >0.05). The remaining raw data were used to assess blood cell composition differences (minfi::estimateCellCounts) and normalized using SWAN (missMethyl::SWAN) [Maksimovic, Gordon, & Oshlack, 2012]. A sequence of filters were used to remove probes with a detection P‐value >0.01 in at least one sample, probes with known SNPs (minfi::dropLociWithSnps), and known cross‐reactive probes [Chen et al., 2013]. After filtering, the normalized data (35 samples and 408604 probes remained) and subsequent analyses were performed on M‐values computed from normalized data (with offset 100). Differences in methylation probe levels were obtained using the RUV‐2 method [Gagnon‐Bartsch & Speed, 2012] using 450 K array negative controls and 0.5 false discovery rate (FDR) threshold for the first RUV fit. DM probes (FDR ≤ 0.05) were annotated using Bioconductor annotation package FDb.InfiniumMethylation.hg19 which assigned methylation probes to the gene with the nearest transcription start site. A power simulation was implemented with our sample size setup [Graw, Henn, Thompson, & Koestler, 2019]. A gene was called DM if at least one DM probe was assigned to that gene. To control for gene probe coverage difference, gene‐level P‐values were obtained using 2000 permutations of the sample labels to generate null distributions that retain gene‐specific coverage. P‐values were computed by counting the proportion of times a gene obtained a statistic (maximum of 1 across assigned probes) in the permutated data, compared to the original data.
Pathway Enrichment
We found DM CpG sites across 898 genes in our analysis (Table S2) which were used to identify enriched canonical pathways associated with ASD in our cohort. We identified nine enriched pathways in our dataset (log(P‐value) > 1.3), comprising 32 DM genes (Qiagen's Ingenuity Pathway Analysis, IPA, www.qiagen.com/ingenuity). The 898 genes identified were enriched with genes that have previous association with ASD based on the SFARI gene database (https://gene.sfari.org/database/human-gene/). There were 59 genes in common between our DM and the SFARI gene list. We used IPA to identify enriched molecular networks associated with our ASD DM genes as well as known diseases and functions in our cohort. We found a significant enrichment of mitochondrial metabolic canonical pathways and further explored the enrichment in pathways and functions associated with Mitochondrial Function and Biogenesis.
Enrichment Analysis of Gene Co‐expression Modules and Brain Signatures
Our DM gene list was compared to published transcriptome data from ASD (n = 50) brain tissue [Gandal et al., 2018]. We used the co‐expression modules and 20 top hub gene sets from the transcriptome dataset. Gene symbols were standardized using Bioconductor Human annotation package (org.Hs.eg.db version 3.4.1). Comparison between our DM genes with each module and hub gene sets from the transcriptome data was assessed using a single‐sided Fisher exact‐test. Enrichment of each module in the DM data was computed using preranked GSEA [Subramanian et al., 2005] via the fgsea package with 10,000 permutations. Enrichment analysis in pan‐cortical RNA‐seq dataset was performed in a similar way using the signatures defined in Gandal et al.'s study [Gandal et al., 2018] with standardized gene symbols and genes ordered by decreasing moderated t‐test statistic in each region separately.
Targeted Next Generation Bisulfite Sequencing
EpigenDx, Inc. (MA, USA) performed targeted Next Generation Bisulfite sequencing (tNGBS) on 44 DNA samples (22 ASD and 22 control, Table S1) to validate some of the methylation differences observed in the 450 K screen. Of the samples analyzed, nine ASD and eight control samples had also undergone the 450 K methylation screen. The DM genes sequenced were selected based on previous association with ASD (SFARI gene list) and relevance to our mitochondrial hypothesis. Sixty tNGBS assays were designed to cover 353 CpG sites across 27 genes and the percentage methylation of each CpG site was determined in each sample. Pairwise t‐tests were used to determine the significantly DM CpG sites between ASD and control (P < 0.05) and P‐values were corrected for using the Benjamini–Hochberg adjusted P‐value (FDR) test.
DNA Pyrosequencing
Based on the tNGBS data, two of the genes (PCCB and PCDHA12) that were significantly differentially methylated between ASD and controls were selected for pyrosequencing on a larger sample size (51 ASD and 25 control), including samples previously screened on the 450 K methylation assay and/or the tNGBS assay (Table S1). Three pyrosequencing assays were designed: one for PCCB (intron 1: CpG site 49 to 53) and two for PCDHA12 (exon 1: CpG sites #30–31 and intron 1: CpG sites 248–249). DNA samples were pyrosequenced by EpigenDx, Inc. (MA, USA) and the percentage DNA methylation per sample for each CpG site was quantified. We performed pairwise t‐tests and FDR corrections to determine the significantly DM CpG sites between ASD and controls. Complete gene sequences for PCCB and PCDHA12 (https://www.ensembl.org/index.html) were analyzed in silico using TRANSFAC transcription analysis (http://factor.genexplain.com/cgi-bin/transfac_factor/search.cgi) to identify transcription factor binding sites.
Metabolomic Analysis
Urine samples from 35 children (21 ASD and 13 controls) were collected, of which 18 ASD and all 13 control had been screened in at least one of the three DNA methylation assays (Table S1). Urinary organic acids were extracted and quantified by gas chromatography–mass spectrometry (GC–MS) relative to 3‐phenylbutyric acid and a stable isotope of orotic acid [C. Reinecke et al., 2011]. The GC–MS data were analyzed and deconvoluted using AMDIS32 which was linked to the NIST (National Institute of Standards and Technology) mass spectral search program, as well as an in‐house organic acid specific library. The organic acid concentration was calculated in mmol/mol creatinine. A standard metabolomics‐based data processing workflow was followed [Reinecke et al., 2011]. The data was log 2 transformed before statistical analysis and visualized with principal component analysis (PCA) to investigate the effect of demographic groups on the data. Univariate analysis (Student's t‐test and effect size) was used to identify metabolites that differed significantly between the ASD and control groups.
Results
ASD Study Cohort and Phenotyping
South Africa has no ASD database containing DNA and/or ASD phenotypes, therefore, we built an ASD cohort of 145 participants (93 children with ASD and 52 matched controls). Our cohort was characterized by a range of ASD phenotypes, with autism severity ranging from Moderate to Severe on the ADOS‐2 scale. Seventeen of the 93 children with ASD did not qualify for the epigenetic aspects of our study because they could not provide a DNA sample or had another co‐occurring neurodevelopmental disorder. Of the children with ASD that qualified for our study, ~9% (7 of 76) did not meet the ASD criteria on ADOS‐2 assessment. In addition, ~15.3% (8 of 52) of the children in the control group were excluded from our study cohort because they displayed a behavioral trait that overlaps with “autistic” traits as defined by ADOS‐2 criteria; this highlights the importance of phenotyping controls. In total, 69 children with ASD and 44 typically developing children were retained in our study cohort.
Screening for DM Genes
We completed a preliminary DNA methylation screen using the 450 K beadchip assay on a subset of our cohort (32 ASD and 16 control) and retained 408,604 CpG probes after stringent quality control filtering criteria, which included controlling for contamination from blood cells, batch effects, ethnicity and age. Our power calculation analysis estimated a 20% overall average power to detect 5–20% methylation differences, given our sample size [Graw et al., 2019]. Thus, we expect our differential analysis to have missed many true differentially methylated CpG loci on the whole‐methylome beadchip array, and therefore used this data in combination with the other DNA methylation assays to determine candidate DM genes. We identified DM CpG sites mapped to 898 genes, 39 of which were highly significant (P ≤ 0.005; Table S2). DM genes mapped across all chromosomes, including the Y chromosome. Fifty‐nine of the 898 DM genes have previous association with ASD (https://gene.sfari.org/database/human-gene/; Table S3), including SETD5 and MTR, which are both directly involved in DNA methylation.
Mitochondrial Pathways Implicated in ASD
Using pathway analysis, we identified nine enriched canonical pathways in our DM dataset, of which eight were metabolic pathways occurring primarily in the mitochondria (Table 1). When overlaying canonical pathways and functions associated with Mitochondrial Function and Mitochondrial Biogenesis with our data, additional DM genes were found in the Mitochondrial Dysfunction canonical pathway and the Mitochondrial Biogenesis toxicity list (Table S4), supporting the enrichment of mitochondrial functions in our dataset [Winden et al., 2009]. The Protein Ubiquitination Pathway was the only nonmetabolic enriched pathway and had 17 DM genes (Table 1), many of them crucial for the degradation of damaged mitochondria [Bragoszewski et al., 2017].
Table 1.
Enriched Canonical Pathways in Our Differentially Methylated (DM) Dataset and Associated Genes
| Ingenuity canonical pathway | −Log(P‐value) | Cellular location | DM genes |
|---|---|---|---|
| Methylmalonyl pathway | 2.06 | Mitochondria | MUT, PCCB |
| 2‐Oxobutanoate degradation I | 1.85 | Mitochondria | MUT, PCCB |
| Glutaryl‐CoA degradation | 1.72 | Mitochondria | L3HYPDH, HACD2, EHHADH |
| Triacylglycerol biosynthesis | 1.56 | Endoplasmic Reticulum | LPGAT1, LPCAT2, PLPP5, MOGAT1, AGPAT1 |
| Protein ubiquitination pathway a | 1.51 | Cytoplasm | HSPA2, DNAJC13, PSMB5, DNAJC8, PSMC2, USP51, DNAJC28, USP31, UBB, UBE2C, USP49, MDM2, PSMD10, SMURF1, RBX1, DNAJC5G, PSMA5 |
| Acyl carrier protein metabolism | 1.41 | Mitochondria | AASDHPPT |
| Acetyl‐CoA biosynthesis III (from citrate) | 1.41 | Cytoplasm | ACLY |
| Lipoate salvage and modification | 1.41 | Mitochondria | LIPT1 |
| Superpathway of methionine degradation | 1.33 | Mitochondria | MTR, MUT, PCCB, PRMT1 |
Nonmetabolic pathway.
Our DM enriched pathways were synthesized within a framework of integrated mitochondrial metabolism, given that six canonical pathways were directly linked to ATP production (Fig. 1). In the Lipoate Salvage and Modification pathway, a lipoate moiety is transferred by Lipoyltransferase 1 (LIPT1) onto two enzymes of the tricarboxylic acid cycle (TCA): pyruvate dehydrogenase (PDH) and alpha ketoglutarate dehydrogenase (a‐KGDH). Five other pathways (Fig. 1) all directly involve substrates of the TCA cycle, which produces ATP. We find six DM genes (Mitochondrial Dysfunction; Table S4) that play a direct role in ATP production and reactive oxygen species (ROS); four genes (NDUFA4, NDUFB2, NDUFB4, NDUFB6) that are part of complex I, one gene in complex III (UQCRC2) and one gene in complex IV (COX7B) of the electron transport chain. In addition, the top DM gene on our gene list, STOML2 (P < 0.005), codes for a mitochondrial protein that regulates mitochondrial fusion in response to ROS damage.
Figure 1.
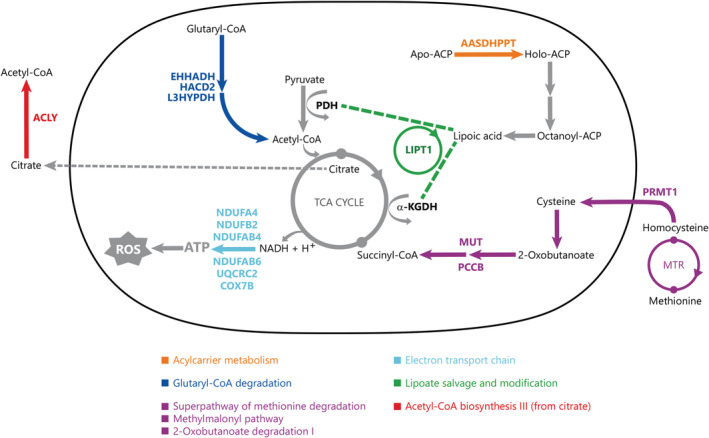
Mitochondrial pathways implicated in Autism Spectrum Disorder (ASD) etiology. Differentially methylated genes in our dataset interact in multiple enriched canonical pathways (Table 1). When examining the enriched pathways in our ASD dataset, we find metabolic pathways which converge on the TCA cycle which could decrease ATP production via the electron transport chain, ETC, and the accumulation of reactive oxygen species, ROS. The differentially methylated genes have a putative association with mitochondrial dysfunction in ASD. Differentially methylated genes are coded in color corresponding to their respective canonical pathways which are indicated in color‐coded boxes. In these pathways, only relevant starting, intermediate and end‐molecules are shown, where arrows can depict multiple reaction steps. Genes shown in color are differentially methylated in our dataset.
Methylated Genes Are Enriched in ASD Coexpression Modules Linked to Mitochondrial Activity
To find the common genes between our DM data and differentially expressed genes in postmortem ASD brain tissue, we computed the enrichment of our genes in the transcriptomic data reported by Gandal et al. [2018]. These authors identified 13 co‐expression modules shared across ASD and other psychiatric disorders in a large cohort, including 50 ASD subjects. These modules were defined using co‐expression network analysis and the core expression pattern of each module was represented by 20 hub genes. We assessed the concordance between co‐expression modules and our DM genes in three ways by calculating the: (a) overlap between our DM genes and the hub genes of each module; (b) overlap between our DM genes and all the genes in each module; and (c) enrichment of each module among the genes associated with the most differential methylation using Gene Set Enrichment Analysis (GSEA). Four of the 13 modules (CD1, CD5, CD7 and CD10) showed a significant overlap with our DM genes in at least one of the analyses (P‐value ≤0.05; Fig. S1). Modules CD5 and CD10 were strongly associated with mitochondrial activity and ASD, while module CD1 was associated with neuron and synapse functions and ASD. Modules CD5, CD10 and CD1 were enriched in neuron‐specific genes and were significantly downregulated in ASD [Gandal et al., 2018].
Downregulated Gene Expression in ASD Occipital Cortex Is Associated with DNA Methylation
To test whether our DM genes were associated with differential expression in different brain regions, we used a published RNA‐seq dataset (Synapse:syn4587609). This data was derived from postmortem brain tissue of 24 ASD and 17 controls, with expression signatures from four cortical regions: frontal, temporal, parietal, and occipital lobes [Gandal et al., 2018]. Using GSEA to test the enrichment of DM genes in each brain region signature, we found a significant negative enrichment in the occipital region. This suggests that methylation differences may be linked to corresponding downregulated gene expression in this region of ASD subjects (Fig. S2).
tNGBS and DNA Pyrosequencing Support the Epigenome‐methylation Screen Results
The tNGBS of 27 DM genes verified the DNA methylation differences seen at CpG loci between ASD and controls. Methylation was measured at 353 CpG loci across 27 genes, with average methylation levels per gene and per CpG locus ranging from 0.2 to 37.78% and 0.07 to 97.44%, respectively (Table S2). Significant differential methylation between ASD and controls was identified at 52 CpG sites across 14 genes (BBS4, CUL3, CYB5B, JMJD1C, LIPT1, MAGT1, MFN1, MTR, PCCB, PCDHA12, PPARGC1A, SIN3A, SRSF1, and STOML2). After FDR correction, 40 CpG sites retained significance (FDR < 0.05, Table S5). In vitro studies have defined dynamic CpG sites based on minimum methylation differences between 10 and 30% [Ziller et al., 2013]. Consequently, a methylation range >10% was used to identify 20 highly variable CpG sites across five genes (LIPT1, PCCB, PCDHA12, PPARGC1A and STOML2) that showed significant (FDR < 0.1) differential methylation in ASD (Table 2).
Table 2.
Highly Variable Differentially Methylated CpG Sites in Autism Spectrum Disorder (ASD) Identified by Targeted Next Generation Bisulphite Sequencing
| Gene | CPG # | Site location | % Mean methylation ASD | % Mean methylation control | FDR |
|---|---|---|---|---|---|
| (% Range) | (% Range) | ||||
| LIPT1 | CpG#‐152 | 5‐Upstream | 14.279 | 10.331 | 0.043 |
| (3.4–26.9) | (1.9–38.2) | ||||
| CpG#‐24 | Intron 2 | 29.896 | 21.721 | 0.038 | |
| (10.9–67.0) | (8.6–56.5) | ||||
| CpG#‐23 | Intron 2 | 27.649 | 17.673 | 0.040 | |
| (5.4–67.7) | (6–59.4) | ||||
| CpG#‐22 | Intron 2 | 30.794 | 20.877 | 0.042 | |
| (8.4–72.2) | (8–60.9) | ||||
| PCCB | CpG#51 | Intron 1 | 18.500 | 23.159 | 0.050 |
| (9.05–26.4) | (14.5–49.78) | ||||
| PCDHA12 | CpG#31 | Exon 1 | 39.745 | 35.559 | 0.107 |
| (14.2–54.93) | (21.23–49.61) | ||||
| CpG#250 | Intron 1 | 7.648 | 5.876 | 0.059 | |
| (2.5–14.89) | (0–10.63) | ||||
| CpG#251 | Intron 1 | 26.538 | 23.233 | 0.096 | |
| (13.92–38.64) | (12–31.78) | ||||
| PPARGC1A | CpG#‐14 | 5‐Upstream | 15.258 | 85.083 | 0.039 |
| (0–42.86) | (0.72–30.74) | ||||
| CpG#‐13 | 5‐Upstream | 20.200 | 12.854 | 0.038 | |
| (4.15–57.36) | (3.76–48.04) | ||||
| CpG#‐12 | 5‐Upstream | 14.258 | 8.402 | 0.040 | |
| (1.88–44.38) | (2.18–33.54) | ||||
| CpG#‐11 | 5‐Upstream | 10.558 | 6.038 | 0.040 | |
| (1.34–34.82) | (1.49–22.45) | ||||
| CpG#‐1 | 5‐Upstream | 25.961 | 19.541 | 0.037 | |
| (7.36–60.76) | (7.54–51.02) | ||||
| CpG#24 | Intron 1 | 9.600 | 6.028 | 0.038 | |
| (1.1–31.1) | (2.0–16.0) | ||||
| CpG#33 | Intron 1 | 21.996 | 13.439 | 0.037 | |
| (2.0–65.1) | (1.0–58.7) | ||||
| CpG#34 | Intron 1 | 25.405 | 14.048 | 0.051 | |
| (1.9–70.1) | (1.0–64.0) | ||||
| CpG#53 | Intron 1 | 89.566 | 96.742 | 0.038 | |
| (91–99.4) | (93.5–99.1) | ||||
| CpG#141 | Intron 2 | 92.673 | 94.828 | 0.051 | |
| (82.65–97.5) | (92.19–98.77) | ||||
| CpG#142 | Intron 2 | 94.998 | 96.609 | 0.053 | |
| (90–98.35) | (91.52–100) | ||||
| STOML2 | CpG#30 | Intron 2 | 201.010 | 16.827 | 0.044 |
| (11.74–35.42) | (9.09–40.08) |
Note. Average % methylation for 22 ASD and 22 controls is shown for 20 highly variable CpG sites (range > 10%, FDR < 0.1).
Both PCCB and PCDHA12 displayed high mean methylation levels (18–39%) and had a wide methylation range across CpG sites (0–54.9%). Given PCCB's central role in our mitochondrial hypothesis and PCDHA12's known role in neurodevelopment, these two genes were further analyzed in a larger cohort of ASD (n = 51) and controls (n = 25). DNA bisulfite pyrosequencing verified the methylation differences seen in both the 450 K array and tNGBS (Table S6). Methylation at all five PCCB CpG sites was significantly different (P = 0.04) between ASD and control, including significant hypomethylation at three sites in ASD (CpG50, CpG51 and CpG53) (Fig. 2). Significant hypomethylation was also identified at two CpG sites in PCDHA12 (CpG31 in Exon 1 and CpG249 in Intron 1) (Fig. 2). Of note, PCCB was identified as DM in all three different DNA methylation assays used. Additionally, in silico transcription analysis of the PCCB gene predicted binding of the transcription factor p53 to CpG51 and CpG52 sites.
Figure 2.
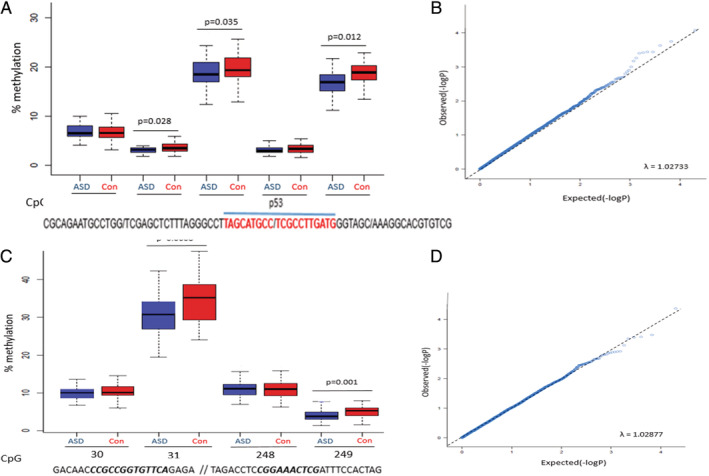
Differential methylation of PCCB and PCHDA12. DNA methylation between Autism Spectrum Disorder (ASD) and control (Con) groups shown as percentage methylation per site. (A) DNA methylation across five PCCB CpG sites (CpG sites 49–53), chromosome location Chr3:136251074–136251144. Transcription factor p53 binding sites shown in red. (B) QQ‐plots with Lambda score for PCCB. (C) DNA methylation across four PCDHA12 CpG sites (CpG sites 30–31 and GpG sites 248–249), chromosomal locations Chr5:140875861–140875870 // Chr5:140882711–140882719. (D) QQ‐plots with Lambda score for PCDHA12. CpG sites with flanking sequences separated by a slash (/); CpG sites in bold italic. Significant FDR‐corrected P‐values are shown for both (A) and (B) and outliers are not shown. Box‐ and QQ‐plots were generated using R version 3.5.3, with the “ggplot” and “car” packages (https://www.statmethods.net/advgraphs/ggplot2.html). Lambda scores, showing association between observed and expected P‐values, were calculated for the QQ‐plots using chi‐squared and Kolmogorov–Smirnov testing.
Urinary Metabolomic Data Are Consistent with Mitochondrial Dysfunction
Urinary organic acids from our ASD cohort were compared to metabolites associated with mitochondrial respiratory chain defects. We confirmed that there was no significant difference in metabolites across different demographic groups (using PCA; data not shown). Three metabolites were markedly elevated (P < 0.05) in the urine of the ASD group: 3‐methylglutaconic acid, 3‐hydroxy‐3‐methylglutaric acid, and ethylmalonic acid (Fig. 3). The magnitude of effect of 3‐methylglutaconic acid and 3‐hydroxy‐3‐methylglutaric acid was significant (d > 0.8), while that of ethylmalonic acid was moderate (d = 0.62).
Figure 3.
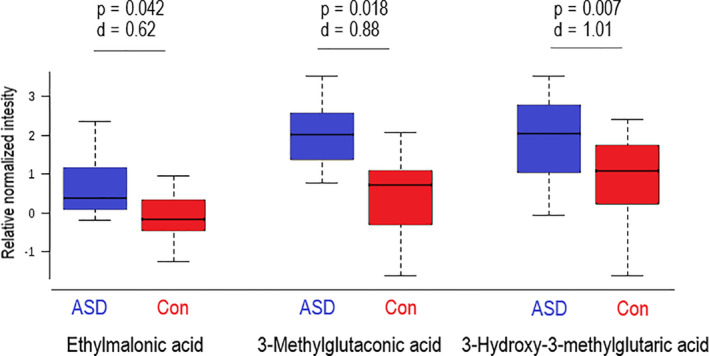
Urinary metabolites in Autism Spectrum Disorder (ASD) and control (Con) groups. Boxplots of three urinary metabolites that differed significantly between ASD and Con. Effect size d‐values (>0.5) are included to show the magnitude by which these metabolites were elevated in the ASD group. Outliers not shown.
Discussion
Autism genetics have been intensively studied since the heritability of ASD was discovered more than 40 years ago. The advent of next generation sequencing technology has resulted in an explosion of ASD genetic data. However, ASD genetic research has primarily been done in Northern‐hemisphere countries, with African populations being understudied [Popejoy & Fullerton, 2016]. This bias [Franz et al., 2017] is exacerbated by the absence of a centralized ASD biobank for African populations. Here, we report epigenetic data for a unique cohort of South African children by investigating DNA methylation as a contributor to ASD etiology. We identify DM genes in our ASD cohort using qualitative and quantitative DNA methylation techniques, in line with published reports that propose a role for DNA methylation in ASD etiology [Tremblay & Jiang, 2019]. In addition, we find DM genes enriched in mitochondrial pathway genes, supporting a role for epigenetic dysregulation of energy metabolism pathways in ASD. We also find a correlation between DM and mitochondrial metabolite concentrations, consistent with a potential mitochondrial dysfunction ASD endophenotype in our cohort.
The limitations of single‐gene analyses are apparent when considering the complicated interplay among multiple genes in complex neurological disorders [Jin et al., 2014]. Transcriptomic studies propose that common pleiotropic pathways underpin numerous neuropsychiatric conditions, including ASD [Gandal et al., 2018; Gupta et al., 2014]. We investigated our DM data within a framework of pathway analysis and identified enrichment of mitochondrial canonical pathways which supports an association between mitochondrial function and ASD [Griffiths & Levy, 2017; Legido et al., 2013; Rossignol & Frye, 2012].
Two DM genes in our cohort, SETD5 and MTR, are directly involved with DNA methylation and have previously been associated with ASD. SETD5 is a methyltransferase within the 3p25 “ASD” microdeletion, with SETD5 mutations previously identified in individuals with ASD [Grozeva et al., 2014]. MTR is a methyltransferase that catalyzes the final step of methionine biosynthesis. Cortical MTR mRNA levels are significantly lower in autistic individuals than controls in postmortem samples within the same age‐range as our AD cohort [Muratore et al., 2013]. The enrichment of the Ubiquitin pathway is also congruent with our mitochondrial hypothesis. Functional mitochondria employ strategies that maintain mitochondrial homeostasis, including fission/fusion events and the ubiquitin‐dependent lysosomal degradation of damaged mitochondria [Bragoszewski, Turek, & Chacinska, 2017]. Taken together, our data suggest a potential role for DNA methylation in the dysregulation of mitochondrial pathways in ASD. This is consistent with a comprehensive meta‐analysis that identified an increased incidence of mitochondrial disease in children with ASD compared to the general population [Rossignol & Frye, 2012]. Since neuronal development depends on efficient oxidative phosphorylation, it is unsurprising that defects in mitochondrial function are associated with a range of neurological disorders [Lin & Beal, 2006; Trushina & McMurray, 2007] including bipolar disorder [Scaini et al., 2016], Alzheimer's disease [Hroudová, Singh, & Fišar, 2014; Salminen et al., 2015] and Parkinson's disease [Bose & Beal, 2016; Franco‐Iborra et al., 2016].
DNA methylation initiates a complex process that includes chromatin remodeling and transcription factor (TF) binding to regulate expression. DNA pyrosequencing validated the DM of two genes, PCDHA12 and PCCB, in our ASD cohort. PCDHA12 codes for a neural cadherin‐like adhesion protein that plays a critical role in cell–cell connections in the brain and is implicated in both ASD [Vaishnavi, Manikandan, & Munirajan, 2014] and other neurodevelopmental disorders [Portales‐Casamar et al., 2016]. PCCB codes for the beta subunit of propionyl‐CoA carboxylase, a mitochondrial enzyme present in three of the nine canonical pathways enriched in our whole‐methylome DM dataset (Table 1, Fig. 1). PCCB is crucial for ATP metabolism, with defects in this gene causing metabolic disorders including propionic acidemia [Wongkittichote, Ah Mew, & Chapman, 2017]. In silico TF analysis revealed two hypomethylated CpG sites in PCCB that overlap with a p53 binding site. Hypomethylation increases TF binding, decondensing normally closed chromatin domains, to allow p53 access to TF binding sites which changes transcription levels [Bao, LoVerso, Fisk, Zhurkin, & Cui, 2017]. Therefore, DM at these p53 binding sites may contribute to aberrant transcription of PCCB. The potential involvement of p53 is noteworthy, as p53 has been linked to ROS accumulation and mitochondrial dysfunction in neurodegenerative disease [Qi et al., 2011]. Mitochondrial accumulation of p53 can result in a cascade of events culminating in mitophagy, p53‐induced apoptosis and cell necrosis [Dai et al., 2016]. The overexpression of p53 in mitochondria has been associated with an increased Ca2+ release into the mitochondrial matrix, leading to mitochondrial dysfunction in Parkinson's disease [Ottolini, Cali, Negro, & Brini, 2013], Huntington's disease [Bae et al., 2005] and Alzheimer's disease [Buizza et al., 2012]. Thus, it is possible that p53 acts in a similar manner in ASD, although we were not able to test whether PCCB DM affects p53 binding, or whether DNA methylation changed mRNA expression levels, due to lack of suitable tissue.
Nevertheless, we show a significant overlap of our DM genes with differentially expressed genes in brain tissue from individuals with ASD from publicly available datasets. First, we find that our DM genes are associated with brain‐region‐specific changes in gene expression, with a significant negative enrichment in the occipital cortex of ASD individuals. This negative enrichment suggests an association between the downregulation of gene expression in this brain region in ASD and differential methylation. Brain imaging data from ASD individuals revealed a decrease in structural connectivity and resting‐state brain activity in the occipital cortex, thought to be associated with the impaired social communication traits typical of ASD, and supporting a role for this brain region in the etiology of ASD [Jung et al., 2019]. Secondly, our DM genes overlapped with genes in the mitochondrial activity and neuron and synapse functions co‐expression modules, which were downregulated in ASD brain tissue [Gandal et al., 2018]. Notably, two DM genes from our dataset, STOML2 and UQCRC2, were both present in a co‐expression module enriched in mitochondrial activity pathways and were downregulated in postmortem ASD brain tissue [Gandal et al., 2018]. STOML2 is the most significantly DM gene in the 450 K dataset, and this DM was also observed using tNGBS. STOML2 codes for a mitochondrial protein that regulates mitochondrial fusion in response to ROS damage, and UQCRC2 is part of mitochondrial Complex III. Additionally, tNGBS identified 20 significant DM CpG sites in PPARGC1A, which acts as a transcriptional regulator of mitochondrial biogenesis [Castora, 2019]. Protein and mRNA levels of genes involved in PPARGC1A signaling have been found to be reduced in ASD patients [Bu et al., 2017]. We have also identified increased mitochondrial copy number in our ASD cohort compared to controls (data not shown), further implicating mitochondrial biogenesis and methylation of PPARGC1A in our ASD endophenotype. Therefore, the differential expression of these three DM genes in ASD is consistent with our hypothesis of an epigenetically driven mitochondrial ASD endophenotype.
Abnormal metabolites are observed in ASD [Griffiths & Levy, 2017] with low serum carnitine levels and elevated lactate, alanine and ammonia levels found in ASD [Filipek, Juranek, Nguyen, Cummings, & Gargus, 2004], which are biomarkers of oxidative phosphorylation and mitochondrial dysfunction. Markers of abnormal ETC function were identified in postmortem brains of ASD children, suggesting a vulnerability of the developing brain [Tang et al., 2013]. Urinary metabolomics represent a powerful tool to analyze multiple mitochondrial metabolites, which can reveal signatures of mitochondrial disease [Reinecke et al., 2011; Wortmann, Kluijtmans, Engelke, Wevers, & Morava, 2012]. Our urinary metabolomics data identified elevated metabolites in ASD, which are elevated in patients with impaired mitochondrial respiration [Reinecke et al., 2011]. Branched‐chain amino acid catabolism occurs in the mitochondria and is sensitive to redox imbalance and perturbations of the TCA cycle. The elevated urinary organic acids identified in ASD are suggestive of impaired ketogenesis and flux of acetyl‐CoA into the TCA cycle [Wortmann et al., 2012], consistent with redox imbalance and impaired mitochondrial function. Importantly, the elevated ethylmalonic acid in our ASD group (P = 0.042) may be linked to a dysregulation of PCCB, since PCCB plays a direct role in the conversion of butyryl‐CoA to ethylmalonic acid [Linster et al., 2011; Nochi, Olsen, & Gregersen, 2017; C. Reinecke et al., 2011]. Thus, the differential methylation observed at PCCB across all three DNA methylation assays performed may lead to altered PCCB expression and enzyme activity, directly affecting ethylmalonic acid concentration. Notably, all three metabolites elevated in our ASD cohort have been previously associated with mitochondrial respiratory chain defects [Roessner & Bowne, 2009]. Our ASD metabolomic profiles are therefore consistent with the epigenetic results and are congruent with an endophenotype of mitochondrial dysfunction in our South African ASD cohort.
The size of our cohort is a clear limitation of our study. However, our samples represent an understudied African population, and therefore add a unique contribution to understanding the epigenetic landscape of ASD in a previously unstudied cohort. The three different methods used to measure DNA methylation all consistently report similar, overlapping trends of differential methylation between ASD and control. Although the 450 K Illumina screen was underpowered given our cohort size [Graw et al., 2019], a clear signature of differential methylation associated with mitochondrial function genes is consistently identified across all three DNA methylation methods used. Furthermore, data from our whole‐epigenome screen significantly overlap with previously published transcriptomic data from ASD brains [Gandal et al., 2018], and the DNA methylation results reported across three methylation assays are supported by our functional metabolomic data.
In summary, our results suggest that DNA methylation is a key epigenetic contributor to ASD etiology and that DM genes associated with ASD in our cohort may play a functional role in mitochondrial homeostasis and dysregulation. Future research investigating the role of mitochondrial mechanisms in ASD etiology may contribute to the identification of novel therapeutic targets that could potentially mitigate ASD symptoms by targeting mitochondrial function. This pharmacologic avenue is currently being explored with promising preliminary results [Legido et al., 2018]. Finally, our study highlights the value of investigating the genetics of an understudied, African ASD cohort for gaining insight into the etiology and endophenotypes of ASD.
Conflict of Interest
The authors declare that no conflict of interest exists.
Supporting information
Figure S1. Differentially methylated genes are enriched with coexpression modules linked to mitochondrial activity and ASD traits. Heatmap of –log 10(P‐value) of the comparisons of coexpression module overlap (“Top 20” and “Module”) and GSEA analysis (“Methylation”) with our differentially methylated ASD genes. Only modules with P‐value ≤0.05 are shown. The color indicates the strength of association, from weak (light blue) to strong (dark blue).
Figure S2. Downregulated genes in the occipital region of ASD brain are enriched with differentially methylated genes. Normalized enrichment scores from GSEA in the ASD signature within each cortical region tested. Color indicates statistical significance from weak (light blue) to strong (dark blue).
Table S1. Samples used in the different DNA methylation and urinary metabolomics assays. A, ASD samples and C, control samples. Participants recruited who were disqualified from the study are not shown.
Table S2. Differentially methylated genes associated with ASD in a South African cohort
Table S3. Genes with known ASD association that are differentially methylated in our South African ASD cohort
Table S4. Differentially methylated mitochondrial genes in our South African ASD cohort as classified by IPA
Table S5. Targeted next generation bisulfite sequencing identified significant differential methylation at 52 CpG sites across 14 genes in ASD (P < 0.05). Genes that are also present on the ASD SFARI list are italicized
Table S6. Pyrosequencing results for PCCB and PCDHA12
Acknowledgments
We thank the National Research Foundation and the Medical Research Council of South Africa for funding this research. EpigenDx, Inc. (USA) cofunded the tNGBS and pyrosequencing experiments. We are extremely grateful to the families and the staff of all the schools that participated in our research.
References
- Aljanabi, S. M. , & Martinez, I. (1997). Universal and rapid salt‐extraction of high quality genomic DNA for PCR‐based techniques. Nucleic Acids Research, 25(22), 4692–4693. 10.1093/nar/25.22.4692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alston, C. L. , Rocha, M. C. , Lax, N. Z. , Turnbull, D. M. , & Taylor, R. W. (2017). The genetics and pathology of mitochondrial disease. The Journal of Pathology, 241(2), 236–250. 10.1002/path.4809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews, S. V. , Sheppard, B. , Windham, G. C. , Schieve, L. A. , Schendel, D. E. , Croen, L. A. , … Ladd‐Acosta, C. (2018). Case–control meta‐analysis of blood DNA methylation and autism spectrum disorder. Molecular Autism, 9, 40 10.1186/s13229-018-0224-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Area‐Gomez, E. , & Schon, E. A. (2014). Mitochondrial genetics and disease. Journal of Child Neurology, 29(9), 1208–1215. 10.1177/0883073814539561 [DOI] [PubMed] [Google Scholar]
- Aryee, M. J. , Jaffe, A. E. , Corrada‐Bravo, H. , Ladd‐Acosta, C. , Feinberg, A. P. , Hansen, K. D. , & Irizarry, R. A. (2014). Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics (Oxford, England), 30(10), 1363–1369. 10.1093/bioinformatics/btu049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae, B.‐I. , Xu, H. , Igarashi, S. , Fujimuro, M. , Agrawal, N. , Taya, Y. , … Sawa, A. (2005). p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron, 47(1), 29–41. 10.1016/j.neuron.2005.06.005 [DOI] [PubMed] [Google Scholar]
- Baio, J. , Wiggins, L. , Christensen, D. L. , Maenner, M. J. , Daniels, J. , Warren, Z. , … Dowling, N. F. (2018). Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. Morbidity and Mortality Weekly Report. Surveillance Summaries (Washington, DC: 2002), 67(6), 1–23. 10.15585/mmwr.ss6706a1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, F. , LoVerso, P. R. , Fisk, J. N. , Zhurkin, V. B. , & Cui, F. (2017). p53 binding sites in normal and cancer cells are characterized by distinct chromatin context. Cell Cycle (Georgetown, TX), 16(21), 2073–2085. 10.1080/15384101.2017.1361064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, J. T. , Tsai, P.‐C. , Yang, T.‐P. , Pidsley, R. , Nisbet, J. , Glass, D. , … Deloukas, P. (2012). Epigenome‐wide scans identify differentially methylated regions for age and age‐related phenotypes in a healthy ageing population. PLoS Genetics, 8(4), e1002629 10.1371/journal.pgen.1002629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berko, E. R. , Suzuki, M. , Beren, F. , Lemetre, C. , Alaimo, C. M. , Calder, R. B. , … Greally, J. M. (2014). Mosaic epigenetic dysregulation of ectodermal cells in autism spectrum disorder. PLoS Genetics, 10(5), e1004402 10.1371/journal.pgen.1004402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose, A. , & Beal, M. F. (2016). Mitochondrial dysfunction in Parkinson's disease. Journal of Neurochemistry, 139(Suppl. 1), 216–231. 10.1111/jnc.13731 [DOI] [PubMed] [Google Scholar]
- Bragoszewski, P. , Turek, M. , & Chacinska, A. (2017). Control of mitochondrial biogenesis and function by the ubiquitin‐proteasome system. Open Biology, 7(4), 170007 10.1098/rsob.170007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu, X. , Wu, D. , Lu, X. , Yang, L. , Xu, X. , Wang, J. , & Tang, J. (2017). Role of SIRT1/PGC‐1alpha in mitochondrial oxidative stress in autistic spectrum disorder. Neuropsychiatric Disease and Treatment, 13, 1633–1645. 10.2147/NDT.S129081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buizza, L. , Cenini, G. , Lanni, C. , Ferrari‐Toninelli, G. , Prandelli, C. , Govoni, S. , … Uberti, D. (2012). Conformational altered p53 as an early marker of oxidative stress in Alzheimer's disease. PLoS One, 7(1), e29789 10.1371/journal.pone.0029789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castora, F. J. (2019). Mitochondrial function and abnormalities implicated in the pathogenesis of ASD. Progress in Neuro‐Psychopharmacology & Biological Psychiatry, 92, 83–108. 10.1016/j.pnpbp.2018.12.015 [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Lemire, M. , Choufani, S. , Butcher, D. T. , Grafodatskaya, D. , Zanke, B. W. , … Weksberg, R. (2013). Discovery of cross‐reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics, 8(2), 203–209. 10.4161/epi.23470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow, M. L. , Pramparo, T. , Winn, M. E. , Barnes, C. C. , Li, H.‐R. , Weiss, L. , … Courchesne, E. (2012). Age‐dependent brain gene expression and copy number anomalies in autism suggest distinct pathological processes at young versus mature ages. PLoS Genetics, 8(3), e1002592 10.1371/journal.pgen.1002592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, C.‐Q. , Luo, T.‐T. , Luo, S.‐C. , Wang, J.‐Q. , Wang, S.‐M. , Bai, Y.‐H. , … Wang, Y.‐Y. (2016). p53 and mitochondrial dysfunction: Novel insight of neurodegenerative diseases. Journal of Bioenergetics and Biomembranes, 48(4), 337–347. 10.1007/s10863-016-9669-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis, S. , He, X. , Goldberg, A. P. , Poultney, C. S. , Samocha, K. , Cicek, A. E. , … Buxbaum, J. D. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature, 515(7526), 209–215. 10.1038/nature13772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elstner, M. , & Turnbull, D. M. (2012). Transcriptome analysis in mitochondrial disorders. Brain Research Bulletin, 88(4), 285–293. 10.1016/j.brainresbull.2011.07.018 [DOI] [PubMed] [Google Scholar]
- Feinberg, J. I. , Bakulski, K. M. , Jaffe, A. E. , Tryggvadottir, R. , Brown, S. C. , Goldman, L. R. , … Feinberg, A. P. (2015). Paternal sperm DNA methylation associated with early signs of autism risk in an autism‐enriched cohort. International Journal of Epidemiology, 44(4), 1199–1210. 10.1093/ije/dyv028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipek, P. A. , Juranek, J. , Nguyen, M. T. , Cummings, C. , & Gargus, J. J. (2004). Relative carnitine deficiency in autism. Journal of Autism and Developmental Disorders, 34(6), 615–623. [DOI] [PubMed] [Google Scholar]
- Franco‐Iborra, S. , Vila, M. , & Perier, C. (2016). The Parkinson disease mitochondrial hypothesis: Where are we at? The Neuroscientist: A Review Journal Bringing Neurobiology, Neurology and Psychiatry, 22(3), 266–277. 10.1177/1073858415574600 [DOI] [PubMed] [Google Scholar]
- Franz, L. , Chambers, N. , von Isenburg, M. , & de Vries, P. J. (2017). Autism spectrum disorder in sub‐saharan africa: A comprehensive scoping review. Autism Research, 10(5), 723–749. 10.1002/aur.1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye, R. E. , & Rossignol, D. A. (2011). Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatric Research, 69(5 Pt 2), 41R–47R. 10.1203/PDR.0b013e318212f16b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon‐Bartsch, J. A. , & Speed, T. P. (2012). Using control genes to correct for unwanted variation in microarray data. Biostatistics (Oxford, England), 13(3), 539–552. 10.1093/biostatistics/kxr034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal, M. J. , Haney, J. R. , Parikshak, N. N. , Leppa, V. , Ramaswami, G. , Hartl, C. , … Geschwind, D. H. (2018). Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science (New York, NY), 359(6376), 693–697. 10.1126/science.aad6469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbett, K. , Ebert, P. J. , Mitchell, A. , Lintas, C. , Manzi, B. , Mirnics, K. , & Persico, A. M. (2008). Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiology of Disease, 30(3), 303–311. 10.1016/j.nbd.2008.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaugler, T. , Klei, L. , Sanders, S. J. , Bodea, C. A. , Goldberg, A. P. , Lee, A. B. , … Buxbaum, J. D. (2014). Most genetic risk for autism resides with common variation. Nature Genetics, 46(8), 881–885. 10.1038/ng.3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovedi, S. , Corradi, A. , Fassio, A. , & Benfenati, F. (2014). Involvement of synaptic genes in the pathogenesis of autism spectrum disorders: The case of synapsins. Frontiers in Pediatrics, 2, 94 10.3389/fped.2014.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graw, S. , Henn, R. , Thompson, J. A. , & Koestler, D. C. (2019). pwrEWAS: A user‐friendly tool for comprehensive power estimation for epigenome wide association studies (EWAS). BMC Bioinformatics, 20(1), 218 10.1186/s12859-019-2804-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, K. K. , & Levy, R. J. (2017). Evidence of mitochondrial dysfunction in autism: Biochemical links, genetic‐based associations, and non‐energy‐related mechanisms. Oxidative Medicine and Cellular Longevity, 2017, 4314025 10.1155/2017/4314025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozeva, D. , Carss, K. , Spasic‐Boskovic, O. , Parker, M. J. , Archer, H. , Firth, H. V. , … Raymond, F. L. (2014). De novo loss‐of‐function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. American Journal of Human Genetics, 94(4), 618–624. 10.1016/j.ajhg.2014.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, S. , Ellis, S. E. , Ashar, F. N. , Moes, A. , Bader, J. S. , Zhan, J. , … Arking, D. E. (2014). Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity‐dependent genes in autism. Nature Communications, 5, 5748 10.1038/ncomms6748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon, E. , Schendel, D. , Ladd‐Acosta, C. , Grove, J. , Agerbo, E. , Als, T. D. , … iPSYCH‐Board ASD Group . (2018). Elevated polygenic burden for autism is associated with differential DNA methylation at birth. Genome Medicine, 10(1), 19 10.1186/s13073-018-0527-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hass, J. , Walton, E. , Wright, C. , Beyer, A. , Scholz, M. , Turner, J. , … Ehrlich, S. (2015). Associations between DNA methylation and schizophrenia‐related intermediate phenotypes—A gene set enrichment analysis. Progress in Neuro‐Psychopharmacology & Biological Psychiatry, 59, 31–39. 10.1016/j.pnpbp.2015.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hroudová, J. , Singh, N. , & Fišar, Z. (2014). Mitochondrial dysfunctions in neurodegenerative diseases: Relevance to Alzheimer's disease. BioMed Research International, 2014, 175062 10.1155/2014/175062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, V. W. (2013). From genes to environment: Using integrative genomics to build a “systems‐level” understanding of autism spectrum disorders. Child Development, 84(1), 89–103. 10.1111/j.1467-8624.2012.01759.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber, W. , Carey, V. J. , Gentleman, R. , Anders, S. , Carlson, M. , Carvalho, B. S. , … Morgan, M. (2015). Orchestrating high‐throughput genomic analysis with bioconductor. Nature Methods, 12(2), 115–121. 10.1038/nmeth.3252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov, I. , O'Roak, B. J. , Sanders, S. J. , Ronemus, M. , Krumm, N. , Levy, D. , … Wigler, M. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature, 515(7526), 216–221. 10.1038/nature13908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, L. , Zuo, X.‐Y. , Su, W.‐Y. , Zhao, X.‐L. , Yuan, M.‐Q. , Han, L.‐Z. , … Rao, S.‐Q. (2014). Pathway‐based analysis tools for complex diseases: A review. Genomics, Proteomics & Bioinformatics, 12(5), 210–220. 10.1016/j.gpb.2014.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, M. , Tu, Y. , Lang, C. A. , Ortiz, A. , Park, J. , Jorgenson, K. , … Kong, J. (2019). Decreased structural connectivity and resting‐state brain activity in the lateral occipital cortex is associated with social communication deficits in boys with autism spectrum disorder. NeuroImage, 190, 205–212. 10.1016/j.neuroimage.2017.09.031 [DOI] [PubMed] [Google Scholar]
- Kuehner, J. N. , Bruggeman, E. C. , Wen, Z. , & Yao, B. (2019). Epigenetic regulations in neuropsychiatric disorders. Frontiers in Genetics, 10, 268 10.3389/fgene.2019.00268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Reynolds, K. , Ji, Y. , Gu, R. , Rai, S. , & Zhou, C. J. (2019). Impaired neurodevelopmental pathways in autism spectrum disorder: A review of signaling mechanisms and crosstalk. Journal of Neurodevelopmental Disorders, 11(1), 10 10.1186/s11689-019-9268-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, M.‐C. , Lombardo, M. V. , Chakrabarti, B. , & Baron‐Cohen, S. (2013). Subgrouping the autism “spectrum”: Reflections on DSM‐5. PLoS Biology, 11(4), 1–7. 10.1371/journal.pbio.1001544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legido, A. , Goldenthal, M. , Garvin, B. , Damle, S. , Corrigan, K. , Connell, J. , … Newschaffer, C. (2018). Effect of a combination of carnitine, coenzyme Q10 and alpha‐lipoic acid (MitoCocktail) on mitochondrial function and neurobehavioral performance in children with autism spectrum disorder (P3.313). Neurology, 90(15 Supplement), P3.313 Retrieved from http://n.neurology.org/content/90/15_Supplement/P3.313.abstract [Google Scholar]
- Legido, A. , Jethva, R. , & Goldenthal, M. J. (2013). Mitochondrial dysfunction in autism. Seminars in Pediatric Neurology, 20(3), 163–175. 10.1016/j.spen.2013.10.008 [DOI] [PubMed] [Google Scholar]
- Levy, D. , Ronemus, M. , Yamrom, B. , Lee, Y. , Leotta, A. , Kendall, J. , … Wigler, M. (2011). Rare de novo and transmitted copy‐number variation in autistic spectrum disorders. Neuron, 70(5), 886–897. 10.1016/j.neuron.2011.05.015 [DOI] [PubMed] [Google Scholar]
- Lin, M. T. , & Beal, M. F. (2006). Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 443(7113), 787–795. 10.1038/nature05292 [DOI] [PubMed] [Google Scholar]
- Linster, C. L. , Noel, G. , Stroobant, V. , Vertommen, D. , Vincent, M.‐F. , Bommer, G. T. , … Van Schaftingen, E. (2011). Ethylmalonyl‐CoA decarboxylase, a new enzyme involved in metabolite proofreading. The Journal of Biological Chemistry, 286(50), 42992–43003. 10.1074/jbc.M111.281527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lintas, C. , Sacco, R. , & Persico, A. M. (2016). Differential methylation at the RELN gene promoter in temporal cortex from autistic and typically developing post‐puberal subjects. Journal of Neurodevelopmental Disorders, 8, 18 10.1186/s11689-016-9151-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe, R. , Gemma, C. , Beyan, H. , Hawa, M. I. , Bazeos, A. , Leslie, R. D. , … Ramagopalan, S. V. (2013). Buccals are likely to be a more informative surrogate tissue than blood for epigenome‐wide association studies. Epigenetics, 8(4), 445–454. 10.4161/epi.24362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksimovic, J. , Gordon, L. , & Oshlack, A. (2012). SWAN: Subset‐quantile within array normalization for Illumina Infinium HumanMethylation450 BeadChips. Genome Biology, 13(6), R44 10.1186/gb-2012-13-6-r44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion, A. , & Leader, G. (2016). An investigation of comorbid psychological disorders, sleep problems, gastrointestinal symptoms and epilepsy in children and adolescents with autism spectrum disorder: A two year follow‐up. Research in Autism Spectrum Disorders, 22, 20–33. 10.1016/j.rasd.2015.11.002 [DOI] [Google Scholar]
- Melnyk, S. , Fuchs, G. J. , Schulz, E. , Lopez, M. , Kahler, S. G. , Fussell, J. J. , … James, S. J. (2012). Metabolic imbalance associated with methylation dysregulation and oxidative damage in children with autism. Journal of Autism and Developmental Disorders, 42(3), 367–377. 10.1007/s10803-011-1260-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratore, C. R. , Hodgson, N. W. , Trivedi, M. S. , Abdolmaleky, H. M. , Persico, A. M. , Lintas, C. , … Deth, R. C. (2013). Age‐dependent decrease and alternative splicing of methionine synthase mRNA in human cerebral cortex and an accelerated decrease in autism. PLoS One, 8(2), e56927 10.1371/journal.pone.0056927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nochi, Z. , Olsen, R. K. J. , & Gregersen, N. (2017). Short‐chain acyl‐CoA dehydrogenase deficiency: From gene to cell pathology and possible disease mechanisms. Journal of Inherited Metabolic Disease, 40(5), 641–655. 10.1007/s10545-017-0047-1 [DOI] [PubMed] [Google Scholar]
- Ottolini, D. , Cali, T. , Negro, A. , & Brini, M. (2013). The Parkinson disease‐related protein DJ‐1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum‐mitochondria tethering. Human Molecular Genetics, 22(11), 2152–2168. 10.1093/hmg/ddt068 [DOI] [PubMed] [Google Scholar]
- Phipson, B. , Maksimovic, J. , & Oshlack, A. (2016). missMethyl: An R package for analyzing data from Illumina's HumanMethylation450 platform. Bioinformatics (Oxford, England), 32(2), 286–288. 10.1093/bioinformatics/btv560 [DOI] [PubMed] [Google Scholar]
- Pidsley, R. , Wong, C. C. Y. , Volta, M. , Lunnon, K. , Mill, J. , & Schalkwyk, L. C. (2013). A data‐driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics, 14(1), 293 10.1186/1471-2164-14-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popejoy, A. B. , & Fullerton, S. M. (2016). Genomics is failing on diversity. Nature, 538(7624), 161–164. 10.1038/538161a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portales‐Casamar, E. , Lussier, A. A. , Jones, M. J. , MacIsaac, J. L. , Edgar, R. D. , Mah, S. M. , … Kobor, M. S. (2016). DNA methylation signature of human fetal alcohol spectrum disorder. Epigenetics & Chromatin, 9, 25 10.1186/s13072-016-0074-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, Z. , He, J. , Su, Y. , He, Q. , Liu, J. , Yu, L. , … Qian, M. (2011). Physical exercise regulates p53 activity targeting SCO2 and increases mitochondrial COX biogenesis in cardiac muscle with age. PLoS One, 6(7), e21140 10.1371/journal.pone.0021140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakyan, V. K. , Down, T. A. , Balding, D. J. , & Beck, S. (2011). Epigenome‐wide association studies for common human diseases. Nature Reviews. Genetics, 12(8), 529–541. 10.1038/nrg3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinecke, C. , Koekemoer, G. , van der Westhuizen, F. , Louw, R. , Lindeque, Z. , Mienie, J. , & Smuts, I. (2011). Metabolomics of urinary organic acids in respiratory chain deficiencies in children. Metabolomics, 8, 264–283. 10.1007/s11306-011-0309-0 [DOI] [Google Scholar]
- Reinecke, F. , Smeitink, J. A. M. , & van der Westhuizen, F. H. (2009). OXPHOS gene expression and control in mitochondrial disorders. Biochimica et Biophysica Acta (BBA)—Molecular Basis of Disease, 1792(12), 1113–1121. 10.1016/j.bbadis.2009.04.003 [DOI] [PubMed] [Google Scholar]
- Robinson, E. B. , Lichtenstein, P. , Anckarsater, H. , Happe, F. , & Ronald, A. (2013). Examining and interpreting the female protective effect against autistic behavior. Proceedings of the National Academy of Sciences of the United States of America, 110(13), 5258–5262. 10.1073/pnas.1211070110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessner, U. , & Bowne, J. (2009). What is metabolomics all about? BioTechniques, 46(5), 363–365. 10.2144/000113133 [DOI] [PubMed] [Google Scholar]
- Romero‐Garcia, R. , Warrier, V. , Bullmore, E. T. , Baron‐Cohen, S. , & Bethlehem, R. A. I. (2019). Synaptic and transcriptionally downregulated genes are associated with cortical thickness differences in autism. Molecular Psychiatry, 24(7), 1053–1064. 10.1038/s41380-018-0023-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, S. , Bennuri, S. C. , Wynne, R. , Melnyk, S. , James, S. J. , & Frye, R. E. (2017). Mitochondrial and redox abnormalities in autism lymphoblastoid cells: A sibling control study. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology, 31(3), 904–909. 10.1096/fj.201601004R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol, D. A. , & Frye, R. E. (2012). Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta‐analysis. Molecular Psychiatry, 17(3), 290–314. 10.1038/mp.2010.136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen, A. , Haapasalo, A. , Kauppinen, A. , Kaarniranta, K. , Soininen, H. , & Hiltunen, M. (2015). Impaired mitochondrial energy metabolism in Alzheimer's disease: Impact on pathogenesis via disturbed epigenetic regulation of chromatin landscape. Progress in Neurobiology, 131, 20 10.1016/j.pneurobio.2015.05.001 [DOI] [PubMed] [Google Scholar]
- Scaini, G. , Rezin, G. T. , Carvalho, A. F. , Streck, E. L. , Berk, M. , & Quevedo, J. (2016). Mitochondrial dysfunction in bipolar disorder: Evidence, pathophysiology and translational implications. Neuroscience and Biobehavioral Reviews, 68, 694–713. 10.1016/j.neubiorev.2016.06.040 [DOI] [PubMed] [Google Scholar]
- Schwede, M. , Nagpal, S. , Gandal, M. J. , Parikshak, N. N. , Mirnics, K. , Geschwind, D. H. , & Morrow, E. M. (2018). Strong correlation of downregulated genes related to synaptic transmission and mitochondria in post‐mortem autism cerebral cortex. Journal of Neurodevelopmental Disorders, 10(1), 18 10.1186/s11689-018-9237-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon, G. , Cruz, N. J. , Kang, D.‐W. , Gandal, M. J. , Wang, B. , Kim, Y.‐M. , … Mazmanian, S. K. (2019). Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell, 177(6), 1600–1618.e17. 10.1016/j.cell.2019.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian, A. , Tamayo, P. , Mootha, V. K. , Mukherjee, S. , Ebert, B. L. , Gillette, M. A. , … Mesirov, J. P. (2005). Gene set enrichment analysis: A knowledge‐based approach for interpreting genome‐wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America, 102(43), 15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, G. , Gutierrez Rios, P. , Kuo, S.‐H. , Akman, H. O. , Rosoklija, G. , Tanji, K. , … Sulzer, D. (2013). Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiology of Disease, 54, 349–361. 10.1016/j.nbd.2013.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay, M. W. , & Jiang, Y.‐H. (2019). DNA methylation and susceptibility to autism spectrum disorder. Annual Review of Medicine, 70, 151–166. 10.1146/annurev-med-120417-091431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushina, E. , & McMurray, C. T. (2007). Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience, 145(4), 1233–1248. 10.1016/j.neuroscience.2006.10.056 [DOI] [PubMed] [Google Scholar]
- UNICEF . (2014). Generation 2030 Africa. UNICEF: Division of Data, Research, and Policy, e_173.
- Vaishnavi, V. , Manikandan, M. , & Munirajan, A. K. (2014). Mining the 3′UTR of autism‐implicated genes for SNPs perturbing microRNA regulation. Genomics, Proteomics & Bioinformatics, 12(2), 92–104. 10.1016/j.gpb.2014.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel Ciernia, A. , & LaSalle, J. (2016). The landscape of DNA methylation amid a perfect storm of autism aetiologies. Nature Reviews. Neuroscience, 17(7), 411–423. 10.1038/nrn.2016.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu, I. (2012). Gene expression studies in autism: Moving from the genome to the transcriptome and beyond. Neurobiology of Disease, 45(1), 69–75. 10.1016/j.nbd.2011.07.017 [DOI] [PubMed] [Google Scholar]
- Wang, P. , Zhao, D. , Lachman, H. M. , & Zheng, D. (2018). Enriched expression of genes associated with autism spectrum disorders in human inhibitory neurons. Translational Psychiatry, 8(1), 13 10.1038/s41398-017-0058-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winden, K. D. , Oldham, M. C. , Mirnics, K. , Ebert, P. J. , Swan, C. H. , Levitt, P. , … Geschwind, D. H. (2009). The organization of the transcriptional network in specific neuronal classes. Molecular Systems Biology, 5, 291 10.1038/msb.2009.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiśniowiecka‐Kowalnik, B. , & Nowakowska, B. A. (2019). Genetics and epigenetics of autism spectrum disorder—Current evidence in the field. Journal of Applied Genetics, 60(1), 37–47. 10.1007/s13353-018-00480-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, C. C. Y. , Meaburn, E. L. , Ronald, A. , Price, T. S. , Jeffries, A. R. , Schalkwyk, L. C. , … Mill, J. (2014). Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Molecular Psychiatry, 19(4), 495–503. 10.1038/mp.2013.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wongkittichote, P. , Ah Mew, N. , & Chapman, K. A. (2017). Propionyl‐CoA carboxylase—A review. Molecular Genetics and Metabolism, 122(4), 145–152. 10.1016/j.ymgme.2017.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wortmann, S. B. , Kluijtmans, L. A. , Engelke, U. F. H. , Wevers, R. A. , & Morava, E. (2012). The 3‐methylglutaconic acidurias: What's new? Journal of Inherited Metabolic Disease, 35(1), 13–22. 10.1007/s10545-010-9210-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, P. , Zang, L.‐Q. , Li, X.‐K. , & Shu, Q. (2016). An epigenetic view of developmental diseases: New targets, new therapies. World Journal of Pediatrics: WJP, 12(3), 291–297. 10.1007/s12519-016-0020-3 [DOI] [PubMed] [Google Scholar]
- Xu, G. , Strathearn, L. , Liu, B. , O'Brien, M. , Kopelman, T. G. , Zhu, J. , … Bao, W. (2019). Prevalence and treatment patterns of autism spectrum disorder in the United States, 2016. JAMA Pediatrics, 173(2), 153–159. 10.1001/jamapediatrics.2018.4208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip, B. H. K. , Bai, D. , Mahjani, B. , Klei, L. , Pawitan, Y. , Hultman, C. M. , … Sandin, S. (2018). Heritable variation, with little or no maternal effect, accounts for recurrence risk to autism spectrum disorder in Sweden. Biological Psychiatry, 83(7), 589–597. 10.1016/j.biopsych.2017.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahir, F. R. , & Brown, C. J. (2011). Epigenetic impacts on neurodevelopment: Pathophysiological mechanisms and genetic modes of action. Pediatric Research, 69(5 Pt. 2), 92R–100R. 10.1203/PDR.0b013e318213565e [DOI] [PubMed] [Google Scholar]
- Ziller, M. J. , Gu, H. , Muller, F. , Donaghey, J. , Tsai, L. T.‐Y. , Kohlbacher, O. , … Meissner, A. (2013). Charting a dynamic DNA methylation landscape of the human genome. Nature, 500(7463), 477–481. 10.1038/nature12433 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Differentially methylated genes are enriched with coexpression modules linked to mitochondrial activity and ASD traits. Heatmap of –log 10(P‐value) of the comparisons of coexpression module overlap (“Top 20” and “Module”) and GSEA analysis (“Methylation”) with our differentially methylated ASD genes. Only modules with P‐value ≤0.05 are shown. The color indicates the strength of association, from weak (light blue) to strong (dark blue).
Figure S2. Downregulated genes in the occipital region of ASD brain are enriched with differentially methylated genes. Normalized enrichment scores from GSEA in the ASD signature within each cortical region tested. Color indicates statistical significance from weak (light blue) to strong (dark blue).
Table S1. Samples used in the different DNA methylation and urinary metabolomics assays. A, ASD samples and C, control samples. Participants recruited who were disqualified from the study are not shown.
Table S2. Differentially methylated genes associated with ASD in a South African cohort
Table S3. Genes with known ASD association that are differentially methylated in our South African ASD cohort
Table S4. Differentially methylated mitochondrial genes in our South African ASD cohort as classified by IPA
Table S5. Targeted next generation bisulfite sequencing identified significant differential methylation at 52 CpG sites across 14 genes in ASD (P < 0.05). Genes that are also present on the ASD SFARI list are italicized
Table S6. Pyrosequencing results for PCCB and PCDHA12