

Abstract
Glenzocimab (ACT017) is a humanized monoclonal antigen‐binding fragment (Fab) directed against the human platelet glycoprotein VI, a key receptor for collagen and fibrin that plays a major role in thrombus growth and stability. Glenzocimab is being developed as an antiplatelet agent to treat the acute phase of ischemic stroke. During a phase I study in healthy volunteers, the population pharmacokinetics (PK) and pharmacodynamics (PD) of glenzocimab were modeled using Monolix software. The PK/PD model thus described glenzocimab plasma concentrations and its effects on ex vivo collagen‐induced platelet aggregation. Glenzocimab was found to have dose‐proportional, 2‐compartmental PK with a central distribution volume of 4.1 L, and first and second half‐lives of 0.84 and 9.6 hours. Interindividual variability in clearance in healthy volunteers was mainly explained by its dependence on body weight. The glenzocimab effect was described using an immediate effect model with a dose‐dependent half maximal inhibitory concentration: Larger doses resulted in a stronger effect at the same glenzocimab plasma concentration. The mechanism of the overproportional concentration effect at higher doses remained unexplained. PK/PD simulations predicted that 1000‐mg glenzocimab given as a 6‐hour infusion reduced platelet aggregation to 20% in 100% of subjects at 6 hours and in 60% of subjects at 12 hours after dosing. Simulations revealed a limited impact of creatinine clearance on exposure, suggesting that no dose adjustments were required with respect to renal function. Future studies in patients with ischemic stroke are now needed to establish the relationship between ex vivo platelet aggregation and the clinical effect.
Keywords: ACT017, acute phase of ischemic stroke, anti‐GPVI Fab, glenzocimab, population PK/PD
A thrombotic ischemic stroke occurs when a clot forms inside an artery, blocking blood flow to the brain. A critical treatment goal during the acute phase is to resolve the clot and inhibit new clot formation, thus restoring and maintaining blood supply. The only 2 therapeutic options available at present are thrombolysis within the 3 (United States) or 4.5 (European Union) hours following the initial symptoms and/or thrombectomy that can be performed within 6 hours. However, no treatment options are available to cover platelet aggregation during the first 24 hours after a stroke has occurred and when the patient is receiving emergency care. After 24 hours, aspirin is typically administered to inhibit platelet aggregation. 1 However, this is already after irreversible neurological damage may have occurred due to oxygen deprivation. Treatment with glenzocimab (ACT017) aims to close this important treatment gap during the first 24 hours after a stroke has occurred. It is initially planned that glenzocimab should be administered no later than 1 hour after the end of alteplase administration, thus concomitantly in most cases. However, a recent amendment to the current clinical phase 2 study (NCT03803007) has provided for a wider time window that enables administration up to 7.5 hours after the start of symptoms with a dosage regimen that would ensure the inhibition of platelet activation and aggregation with a rapid onset of effect and a duration lasting at least 6 or 12 hours. Because of our limited understanding of potential interactions between glenzocimab and other anticoagulants, the duration of this effect should not last beyond 24 hours, thus avoiding any overlap with the actions of another anticoagulant.
Glenzocimab is a humanized Fab that blocks platelet glycoprotein VI (GPVI) function and is currently in clinical development for the acute treatment of ischemic stroke. Because activated platelets initiate hemostatic clot formation and provide a scaffold for the activation of coagulation, 2 the purported mechanism of action of glenzocimab is to prevent clot growth, inhibit downstream clotting in microvessels, and inhibit the cellular processes that involve platelet‐neutrophil interactions and the release of oxygen radicals. Surface plasmon resonance measurements have shown that glenzocimab binds to GPVI with a dissociation constant of 4.1 nM. In vitro experiments have shown that glenzocimab inhibits collagen‐induced platelet aggregation after preincubation with human platelet‐rich plasma in a concentration‐dependent manner and a half maximal inhibitory concentration (IC50) of 3.2 μg/mL. Studies in cynomolgus monkeys to investigate the ex vivo effects of glenzocimab on platelet aggregation found efficient and reversible inhibition of GPVI function without any effects on the platelet count or bleeding time. 3 The safety and tolerability as well as the pharmacokinetics (PK) and pharmacodynamics (PD) of glenzocimab were investigated during a phase I study in healthy volunteers. 4 In an assay using collagen‐induced platelet aggregation, ex vivo aggregation was used as a PD marker. The objective of the present study was to develop a population PK/PD model for glenzocimab in healthy volunteers and to determine the required dose and infusion regimen necessary to reduce ex vivo platelet aggregation to 20% at 6 or 12 hours after administration in 95% of individuals. It is currently not known how ex vivo platelet aggregation is related to clinical efficacy, and it remains to be seen in the upcoming phase II study whether this 20% target is a valid level for a clinical effect. The treatment goal was motivated by the need to bridge the treatment gap after the initiation of emergency treatment and to procure maximal effect in almost all individuals. Simulations were also performed to investigate the degree to which platelet aggregation returned to normal at 24 hours when other anticoagulants are typically being administered. Further, this study tried to investigate individual factors influencing the PK and PD in order to determine any possible dose adjustments required to maintain the therapeutic goal. Because the molecular weight of glenzocimab (48 kDa) is lower than the glomerular filtration limit of 70 kDa, it was expected that glenzocimab would undergo renal clearance, which would be sensitive to renal function. In an emergency setting, the estimated glomerular filtration rate (eGFR) will not be available, so plasma creatinine concentration was used as a proxy. In many cases, targeted cell surface binding is known to induce target‐mediated drug disposition (TMDD) 5 where drug binds to a cell surface receptor and is eliminated by receptor internalization. Therefore, as a measure of the abundance of GPVI, the platelet count was investigated as a possible effect on PK.
Materials and Methods
Study and Data Description
PK and PD data came from a phase I, first‐in‐human, randomized, double‐blind, placebo‐controlled study with a single‐ascending‐dose design in healthy volunteers. The study included 48 healthy volunteers, 36 of whom received glenzocimab (ACT017) and 12 placebo. The study was conducted by QPS (in the Netherlands) in compliance with the current revision of the Declaration of Helsinki, International Council for Harmonization guidelines for Good Clinical Practice, and current regulations (Medical Research Human Subjects Act). The clinical trial protocol, information for subjects, and informed consent form were all approved by the Independent Ethics Committee for the trial center. The competent authority in the Netherlands was notified, and a nonobjection statement was issued. All subjects provided their written informed consent before any screening procedures were carried out.
Glenzocimab was administered as 6‐hour intravenous (IV) infusion at a volume of 500 mL with 0.9% NaCl. Twenty‐five percent of the total dose was delivered within the first 15 minutes and 75% over the remainder of the 6‐hour infusion. There were 6 active drug‐treated and 2 placebo‐treated individuals in each of the 6 dose groups: 62.5, 125, 250, 500, 1000, and 2000 mg. Glenzocimab concentrations (PK) and ex vivo platelet aggregation (PD) were determined in each individual both before dosing and then 15 minutes and 1, 4, 6, 8, 10, 14, 18, 24, and 48 hours after the start of the infusion. Glenzocimab concentrations in all individuals, as well as ex vivo platelet aggregation in the individuals from the 500‐ and 2000‐mg dose groups, were also determined at 144 ± 48 hours after the start of infusion. A total of 390 PK observations and 404 PD observations were included in the population PK/PD analysis.
Analysis of Glenzocimab Concentrations in Blood Samples
Glenzocimab concentrations were measured in platelet‐free plasma using a validated liquid chromatography–tandem mass spectrometry assay. The extraction method consisted of immunocapture, reduction, alkylation, and digestion, followed by solid‐phase extraction. The lower limit of quantification was 0.3 μg/mL. Concentrations were calculated by interpolation from a calibration curve. 4
Ex Vivo Platelet Aggregation Assay
The effect of glenzocimab on collagen‐induced platelet aggregation was measured by light transmission aggregometry. The principle is to record changes to light transmission through a sample containing platelets. The more inhibited the platelet aggregation, the less light is transmitted. Platelet aggregation was triggered by adding 2.5 μg/mL collagen to platelet‐rich plasma obtained by centrifuging citrated blood. Aggregation was determined 250 seconds after the addition of collagen. 4 The maximal intensity of collagen‐induced platelet aggregation was approximately 80%. An aggregation of 15% to 20% corresponded to an almost maximal biological efficacy and was obtained at saturation of GPVI sites on platelets. 3 Below 15% to 20% of intensity, the measurement of platelet aggregation lost precision and reproducibility.
Population PK/PD Modeling
Population PK/PD parameters were estimated using the stochastic approximation of expectation and maximization algorithm implemented under the Monolix Suite 2018R1. 6 Observations that were below the limit of quantitation (BLQ) were included in the estimation of population parameters using the M3 method. 7 Standard errors of parameter estimates were derived from the Fisher information matrix using stochastic approximation. For individual parameters, conditional means and standard deviations were computed. The –2 log‐likelihood (–2LL) was computed using importance sampling. Rsmlx 1.1.0 was used to build the covariate model. 8
Population PK and PD Base Structural Model
The structural model for the pharmacokinetics of glenzocimab had a central compartment with volume V1 and a peripheral compartment with volume V2. Glenzocimab was eliminated via first‐order clearance Cl from the central compartment and the exchange between the central and peripheral compartments was described using an exchange coefficient Q. The IV administration of glenzocimab was modeled with 2 fixed infusion rates, one for the first 15 minutes (rate = 25% of total amount / 15 minutes) and one for the remainder of the total 6‐hour infusion period (rate = 75% of total amount / 5 hours 45 minutes). To model platelet aggregation, an immediate sigmoid response model with the plasma glenzocimab concentration as driver was used:
| (1) |
where PPA(t) was the ex vivo platelet aggregation as a percentage at time t, and CACT017(t) was the glenzocimab plasma concentration at time t. Base_PPA was the baseline ex vivo platelet aggregation as a percentage prior to treatment with glenzocimab, Imax the maximal reduction in platelet aggregation, and IC50 the glenzocimab concentration for half‐maximal inhibition.
Population PK/PD Stochastic Model
To determine the interindividual variability (IIV) of PK and PD parameters, individual parameters were modeled using log‐normal distributions. The equation for an individual parameter was:
| (2) |
where was the population typical parameter, and η was a random variable with mean 0 and standard deviation ω. The effect of continuous covariates on PK parameters was modeled with the equation:
| (3) |
where was the covariate of the subject i, was the reference value, and β was the estimated covariate coefficient. The effect of categorical covariates was modeled with the equation:
| (4) |
where if the individual covariate was within the category and otherwise, and β was the estimated covariate coefficient. The reference values for the covariates were 70 kg for body weight, 50 years for age, 0.79 mg/dL for creatinine, 220 × 109/L for platelet count (PLT), and female for sex. A combined error model inclusive of additive and proportional errors was used to model the glenzocimab observations:
| (5) |
where yobs was the observed glenzocimab concentration, ypred the model prediction, ε was an independent random variable normally distributed with mean 0 and variance 1, and a1 and b1 the parameters of the error model. PD observations were logit‐normally transformed between 0 and 100 and a constant error model was used to fit the transformed PD observations:
| (6) |
where yobs was the observed platelet aggregation, ypred the model prediction, both between 0 and 100%, ε was an independent random variable, normally distributed with mean 0 and variance 1, and a2 the parameter of the error model. This residual error model was selected to take account of the fact that PD observations were all between 0 and 100% with a smaller residual error toward both ends of the range.
Model Development and Covariate Model Building
The population PK model with the covariate effects was developed independently of the PD data. In a second step, the PK/PD data were combined, and a full PK/PD model was developed while maintaining the previously determined structural and statistical properties of the PK model. To build the PK covariate model, conditional sampling for the stepwise approach based on correlation tests (COSSAC) with the Rsmlx library was used. 9 Model selection with COSSAC was based on a statistically significant difference in the –2LL (chi‐squared test) at a level of significance of P = .01. To build the PD covariate model, the selection of covariates was based on a statistically significant difference in the –2LL and a significant covariate coefficient (Wald test) at a level of significance of P = .01. For the covariate search, typical PK covariates (age, sex, and body weight) were tested. The creatinine concentration (CRE) was tested as a covariate proxy for eGFR, because the latter cannot be determined in the emergency setting when glenzocimab will be administered. The PLT was tested as a covariate because GPVI, the target of glenzocimab, is located on platelets. Age, sex, body weight, CRE, and PLT were tested for all PK parameters: Cl, V1, V2, and Q. The dose was not tested as a covariate because visual inspection of the concentration profiles indicated that the PK of glenzocimab was dose proportional. Different covariance matrix structures were then tested after building the covariate model to avoid the identification of false correlations that might result from latent covariate effects. Pearson's correlation test between sampled random effects from conditional distributions was used to select the parameter pairs for testing.
Model Evaluation
To evaluate the model, visual predictive checks (VPCs), goodness‐of‐fit (GoF) plots, and the relative standard error of the parameter estimates were used as generated by Monolix. The VPCs compared the median and the 10th and 90th percentiles of the observed data with the corresponding 90% prediction interval of the simulated percentiles. GoF plots were constructed using random sampling from the conditional distributions of the individual parameters. 10 BLQ data were included in the diagnostic plots by sampling the BLQ predictions from the conditional distribution. 7 Plots showing individual parameter estimates vs covariates were generated with R and the mode of conditional distributions of the individual parameter estimates from Monolix.
Pharmacokinetic/Pharmacodynamic Simulations
Simulations were performed in R using the mlxR library. 11 These simulations included IIV, random effect correlations, and the covariate effects. Covariates for each simulated individual were sampled from a random distribution with means and standard deviations from the distributions observed in phase I healthy volunteers, truncated at the minimum and maximum observed values. Simulations were performed without the residual error of the model prediction. For each simulation scenario, 1000 individuals were simulated, and the percentages of individuals achieving 20% platelet aggregation at 6 or 12 hours were computed.
Results
Demographic Data
Of the 36 subjects treated who were included in the PK/PD analysis, 36% were women and 64% men; 92% of the population was white. The median age was 56 years and the median body weight 74 kg (Table 1).
Table 1.
Demographic Features of the Study Group
| Baseline Characteristics | Statistics |
|---|---|
| Sex (female/male), N (%) | 13/23 (36.1/63.9) |
| Race (white/other), N (%) | 33/3 (91.7/8.3) |
| eGFR (normal/mild renal impairment), N (%) | 29/7 (81/19) |
| Creatinine (mg/dL), median (range) | 0.77 (0.46‐1.2) |
| Age, y, median (range) | 56 (22‐63) |
| Body weight, kg, median (range) | 74 (52‐107) |
| Platelet counts, 109/L, median (range) | 207 (159‐323) |
eGFR indicates estimated glomerular filtration rate.
Normal >90 mL/min/1.73m2. 90 mL/min/1.73m2 ≥ Mild > 60 mL/min/1.73m2. No individual ≤60 mL/min/1.73m2.
Pharmacokinetic/Pharmacodynamic Observations
The plot of dose‐normalized glenzocimab plasma concentrations showed that glenzocimab PK was dose proportional in the observed range of concentrations (Figure S1). This indicated that there was no TMDD. When the dose was increased, the effect on platelet aggregation increased in degree and duration. Glenzocimab 1000 mg and 2000 mg appeared to have maxed out the inhibitory effect up to the 18‐hour time point. However, the duration of the effect with the 2000‐mg dose was longer when compared to the 1000‐mg dose (Figure 1, left). For the same plasma concentration of glenzocimab, the effect on platelet aggregation was greater with the higher doses (Figure 1, right).
Figure 1.
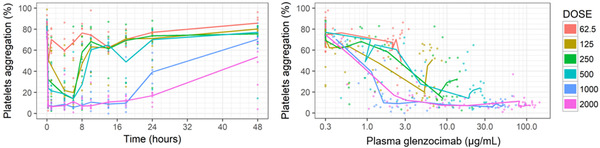
Individual and median ex vivo platelet aggregation vs time (left). Individual and median ex vivo platelet aggregation vs glenzocimab plasma concentrations (right). Doses in milligrams.
Population PK Modeling
Glenzocimab displayed 2‐compartmental plasma PK, with linear clearance from the central compartment. The final PK model included correlations between Cl, V1, and Q. The relative standard errors (RSEs) were all <8% for fixed effects, <22% for random effects, <20% for correlations, and <30% for covariate coefficients. These RSE values were low, indicating an accurate estimation of parameter values. The GoF plot showed no bias, and data pairs in the population predicted vs observed GoF plot clustered tightly around the diagonal. The larger spread around the diagonal at low concentrations that was visible on this log‐log plot reflected the contribution of the additive error from the combined error model (Figure S2). The VPC demonstrated a good model prediction for all dose concentrations (Figure 2).
Figure 2.
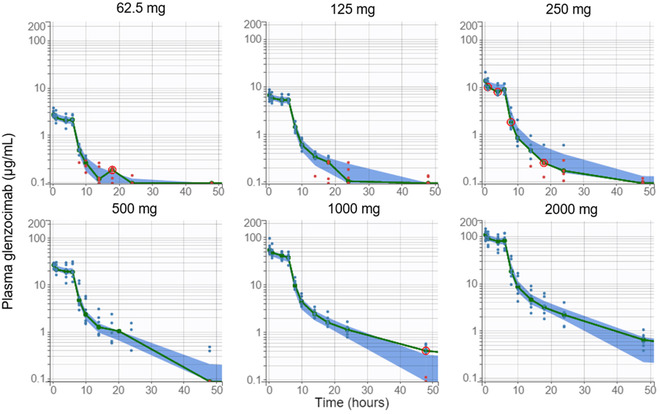
Visual predictive check for glenzocimab plasma concentrations by dose. Empirical median (green line) and 90% prediction interval of the model for the median (blue area). Red circles indicate empirical medians outside of the model prediction interval. Observations as blue dots; observations below the limit of quantitation as red dots.
For a typical 50‐year old subject weighing 70 kg, and 0.79 mg/dL CRE, a clearance of 2.7 L/h, a central volume of 4.1 L, and a peripheral volume of 6.9 L were estimated (Table 2). From these parameters, an initial half‐life of 0.84 hours and a terminal plasma half‐life of 9.6 hours were calculated. Between‐subject variability in the PK parameters was small, with a coefficient of variation (%CV) within the range of 14% to 27% and with 18% for the clearance of glenzocimab. Body weight was found to be a statistically significant covariate regarding glenzocimab clearance (βCl_tBW = 1.1), central distribution volume (βV1_tBW = 0.69), and intercompartmental exchange coefficient (βQ_tBW = 0.81). Heavier subjects had a larger central distribution volume, as well as a more rapid clearance and intercompartmental exchange. Age was found to be a statistically significant covariate with respect to clearance (βCl_tAGE = –0.30) and the intercompartmental exchange coefficient (βQ_tAGE = –0.32). Older subjects displayed slower clearance and intercompartmental exchange. CRE was found to be a statistically significant covariate for clearance (βCl_tCRE = –0.57). Subjects with higher CRE concentrations had slower clearance (Figure S3). The addition of age, body weight, and CRE covariates on clearance helped to lower the %CV from 28% to 18%. The body weight covariate was able to explain a large part of the total between‐subject variability of clearance (Figure 3).
Table 2.
Population PK/PD Parameter Estimates
| Description | Parameter (Unit) | Estimation (%CVa) | RSEb (%) | P Value |
|---|---|---|---|---|
| Fixed effects | ||||
| Clearance | Cl (L/h) | 2.67 (18) | 3.6 | |
| Central distribution volume | V1 (L) | 4.1 (15) | 2.9 | |
| Intercompartmental exchange | Q (L/h) | 0.626 (14) | 3.5 | |
| Peripheral distribution volume | V2 (L) | 6.89 (27) | 7.5 | |
| Baseline platelet aggregation | Base_PPA (%) | 79.8 | 1.6 | |
| ACT017 concentration required for 50% inhibition | IC50 (μg/mL) | 0.924 (228) | 27 | |
| Maximum inhibition | Imax (%) | 72.9 | 1.9 | |
| Standard deviations | ||||
| Clearance | ωCl | 0.182 | 12 | |
| Central distribution volume | ωV1 | 0.148 | 13 | |
| Intercompartmental exchange | ωQ | 0.144 | 20 | |
| Peripheral distribution volume | ωV2 | 0.265 | 22 | |
| ACT017 concentration required for 50% inhibition | ωIC50 | 1.35 | 15 | |
| Correlations | ||||
| Intercompartmental exchange and clearance | rQ_Cl | 0.796 | 14 | |
| Central volume and clearance | rV1_Cl | 0.626 | 18 | |
| Central volume and intercompartmental exchange | rV1_Q | 0.84 | 13 | |
| Covariate coefficients | ||||
| Age on clearancec | βCl_tAGE | −0.304 | 29 | .0005 |
| BW on clearanced | βCl_tBW | 1.09 | 18 | 2.4e‐08 |
| CRE on clearancee | βCl_tCRE | −0.566 | 22 | 5.1e‐06 |
| BW on central distribution volumef | βV1_tBW | 0.694 | 22 | 6.1e‐06 |
| Age on intercompartmental exchangeg | βQ_tAGE | −0.318 | 29 | .00051 |
| BW on intercompartmental exchangeh | βQ_tBW | 0.812 | 23 | 1.5e‐05 |
| Dose on inhibition potencyi | βIC50_tDOSE | −0.989 | 22 | 7.4e‐06 |
| PLT on maximum inhibitionj | βImax_tPLT | 0.17 | 35 | .0038 |
| Observational error | ||||
| Constant PK error | a1 (μg/mL) | 0.0869 | 16 | |
| Proportional coefficient of PK error model | b1 (‐) | 0.0514 | 9.4 | |
| Constant PD error | a2 (‐) | 0.778 | 3.7 | |
BW indicates body weight; CRE, creatinine concentration; PD, pharmacodynamic; PK, pharmacokinetic; PLT, platelet count; RSE, relative standard error.
%CV computed as 100*sqrt(exp(SD^2)‐1.
Stochastic approximation used for estimation of RSE.
Covariate effect: .
Covariate effect: .
Covariate effect: .
Covariate effect: .
Covariate effect: .
Covariate effect: .
Covariate effect: .
Covariate effect: .
Figure 3.
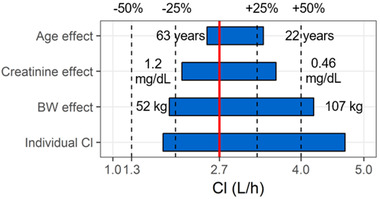
Covariate effects on glenzocimab clearance for the observed range of age, creatinine, and body weight in the phase I study. Solid red line represents clearance for a typical individual, and dotted lines 50%, 75%, 125%, and 150% of this value. Numbers show range. The full range of individual clearance estimates is shown in the lowest row.
Population PK/PD Modeling
The initial PD model described an immediate effect of plasma glenzocimab on platelet aggregation. The immediate effect was modeled as a sigmoid with the half‐effect at the IC50 and the maximal effect Imax. The initial model had a constant residual error, IIV on IC50, and no covariates. This model did not reproduce the observation that the effect on platelet aggregation at the same glenzocimab plasma concentration was stronger at higher doses. To reproduce this behavior, various PD models were tested and included turnover models, effect compartment models, and kinetic binding models, but none of them was able to reproduce the dose‐dependent potency. The immediate response model was therefore taken forward, and a dose covariate on IC50 was added. Indeed, dose was found to be a statistically significant covariate (βIC50_tDOSE = –0.99), and this model successfully reproduced the dose‐dependent effect on potency. The inhibition potency was greater at higher doses and consistent with the stronger inhibition of platelet aggregation observed with higher doses at a given glenzocimab concentration (Figure 4). The dose covariate explained a large part of the total between‐subject variability of IC50 (Figure S4). The baseline PLT was found to be a statistically significant covariate on Imax (βImax_tPLT = 0.17). Subjects with higher PLT had a larger Imax, that is, a stronger inhibition of platelet aggregation at saturating glenzocimab concentrations. However, the effect was small, with Imax varying from 69% to 78% over the range of observed PLT, without any additional between‐subject variability on this parameter. Baseline platelet counts were not found to be a statistically significant covariate on IC50. No correlation was found between IC50 and glenzocimab clearance. The final PD model was of the immediate response type, with the dose as a covariate on IC50 and the baseline platelet concentration PLT as a covariate on Imax. The PD observations were logit‐normally transformed between 0 and 100% and a constant residual error model was used. IIV was included on the IC50. The RSE values for fixed effects were lower than 2% for Imax and Base_PPA, and 27% for the IC50. The RSE for the IC50 random effect was 15%, and for covariate coefficients at most 35%, respectively. These RSE values were low, indicating accurately estimated parameter values. The GoF plot displayed no bias, and the 90% prediction interval from the residual error model for the logit‐normally transformed observations matched well with the shape of the plot (Figure S5). The VPC showed that the median for the observed platelet aggregation was within the predicted interval, except on a few occasions (Figure 5). These deviant values were seen at different dose levels and time points, suggesting that this was due to outlier observations rather than a biased PD model. Similarly, the 10th and 90th percentiles of observed platelet aggregation were within the predicted intervals, except for a few outliers (Figure S6). It was concluded that the onset of inhibition and the return to baseline were well characterized. Overall, the VPCs indicated that the model appropriately characterized the observations without any systematic bias.
Figure 4.
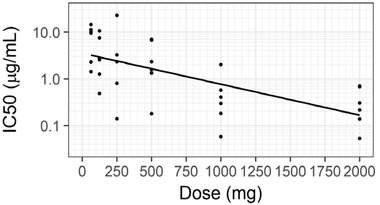
Individual half maximal inhibitory concentration (IC50) estimates (mode of conditional distribution) on log10 scale vs dose with regression line.
Figure 5.
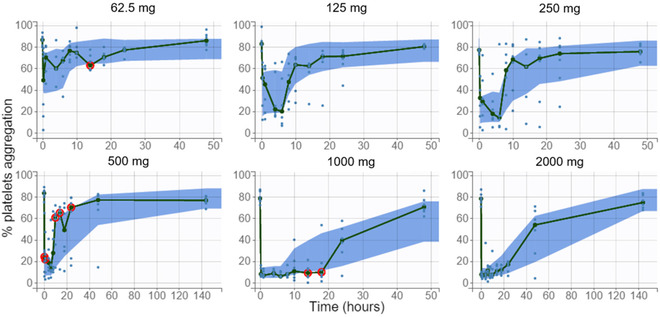
Visual predictive check for ex vivo platelet aggregation by dose. Empirical median (green line) and 90% prediction interval of the model for the median (blue area). Red circles indicate empirical medians outside of the model prediction interval. Observations as blue dots.
Pharmacokinetic/Pharmacodynamic Simulations
PK/PD simulations were run for a population with a covariate distribution similar to that of the phase I healthy volunteer population (Table 1). Simulations were run for a 6‐hour infusion, with 25% of the amount being delivered during the first 15 minutes. The simulations predicted that a dose of 750 mg resulted in ≤20% residual platelet aggregation at 6 hours in 95% of individuals, and that 2750 mg would be required to achieve the same target at 12 hours (Figure 6A).
Figure 6.
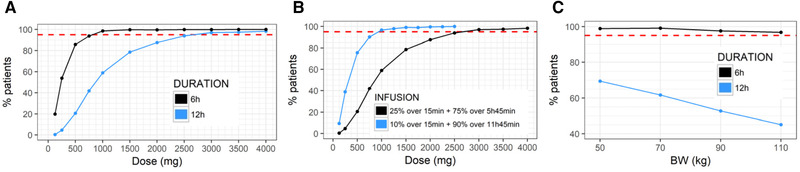
Dashed red line indicates the target of 95% patients. A, Percentage of individuals achieving 20% platelet aggregation at 6 hours or 12 hours vs dose with the phase I infusion scheme. B, Percentage of individuals achieving 20% platelet aggregation at 12 hours vs dose with a 6‐hour vs 12‐hour infusion. C, Percentage of individuals achieving 20% platelet aggregation at 6 hours or 12 hours vs body weight for a 1000‐mg dose with the phase I infusion scheme.
Slower Infusion Rates
Under the original infusion scheme, 25% of the dose was given during the first 15 minutes. Under this scheme, an overshoot in concentration was observed at 15 minutes. An alternative infusion scheme with a flat plasma concentration profile was also tested, with 17.5% of the dose being delivered during the first 15 minutes. Further, an infusion with a constant rate over the entire 6‐hour period was tested. This approach resulted in a longer time being required to reach steady state (Figure S7). For the constant 6‐hour infusion of 1000 mg, it was predicted that it would take 45 minutes to reduce platelet aggregation to 20%. With the 2 options at an increased infusion rate during the first 15 minutes, it took 5 minutes to reduce platelet aggregation to 20% (Figure S8).
12‐Hour Infusion
A 12‐hour infusion was compared as an alternative to the 6‐hour infusion. PK/PD simulations were run for glenzocimab doses of 500, 1000, 1500, or 2000 mg, given over either 6 or 12 hours. Because the same dose was administered over a longer period, the steady‐state concentration dropped by about 50% but was maintained for 12 rather than 6 hours (Figure S9). The dose necessary to ensure that at 12 hours 95% of individuals achieved reduction to 20% platelet aggregation was reduced from ∼2750 mg for a 6‐hour infusion to ∼1000 mg for a 12‐hour infusion (Figure 6B).
Recovery at 24 Hours
The effect on platelets up to 24 hours was simulated for the 6‐ and 12‐hour infusions to assess recovery (Figure S10). For doses of 500, 750, or 1000 mg given as a 6‐hour infusion, platelet aggregation at 24 hours was back at 55%, 40%, and 30%, respectively. With the same doses being given as a 12‐hour infusion, platelet aggregation at 24 hours reached 50%, 35%, and 25%. It should be noted that 80% aggregation was the baseline. However, when selecting equivalent doses on the basis of the effect at 12 hours, the dose for a 12‐hour infusion could then be reduced when compared to a 6‐hour infusion, resulting in a weaker effect at 24 hours.
Impact of body weight, Creatinine, and Age on Glenzocimab Exposure and Platelet Aggregation
The impacts of body weight, eGFR, and age on exposure to glenzocimab were tested in 3 different groups for each individual factor. The eGFRs tested were 45, 75, and 105 mL/min, while those of body weight were 40, 70, and 100 kg, and those of age were 25, 50, and 75 years. Simulated PK profiles revealed a moderate impact of eGFR and age on the concentrations during the first 6 hours when compared to the impact of body weight (Figure S11). For this reason, the body weight effect was further tested on platelet aggregation. Indeed, the larger glenzocimab distribution volume and more rapid clearance of heavier individuals that resulted in lower glenzocimab plasma exposure also caused a weaker effect (Figure 6C). However, sensitivity to body weight differed at 6 vs 12 hours for the 1000‐mg dose. At 6 hours, >95% of individuals were predicted to achieve reduction to 20% platelet aggregation, regardless of the body weight. At 12 hours, the percentage of individuals achieving 20% platelet aggregation fell from 70% for individuals with a body weight of 50 kg to 45% for individuals with a body weight of 110 kg.
Discussion
Glenzocimab clearance was estimated at 2.7 L/h, the central distribution volume at 4.1 L and the peripheral distribution volume at 6.9 L for a 50‐year‐old individual weighing 70 kg. The initial and terminal half‐lives were estimated at 0.84 and 9.6 hours, respectively. These parameter values were typical of a Fab with a distribution limited to plasma and interstitial space. 12 The clearance, central distribution volume, and intercompartmental exchange parameter were body weight dependent with coefficients of βCl_tBW = 1.1, βV1_tBW = 0.69, and βQ_tBW = 0.81, indicating a strong dependence. Indeed, the IIV in the clearance of %CV = 18% could largely be explained by the differences in body weight between individuals. PK simulations demonstrated that heavier individuals displayed lower concentrations during the infusion and a more rapid clearance of glenzocimab after its completion. Because glenzocimab is planned as a treatment in an emergency setting where eGFR values will not be available, the creatinine concentration was used as a marker of renal function. Creatinine concentration was found to be a covariate of glenzocimab clearance with a coefficient βCl_tCRE = –0.57. This was in accordance with the molecular weight of glenzocimab of 48 kDa, which is below the glomerular filtration limit. However, molecular size is not the only parameter that determines the rate of renal elimination and tubular reabsorption of macromolecules, and protection from elimination can occur. 13 Glenzocimab was therefore expected to be at least partially cleared by the kidneys. The sensitivity of glenzocimab to creatinine concentrations was less marked than to body weight. Simulations revealed few differences in glenzocimab concentrations up to 12 hours for an eGFR of 45, 75, or 105 mL/min/1.73 m2, corresponding to individuals with moderate, mild, or no renal function impairment. It should be noted that the creatinine concentration range that was tested during the phase I study was 0.46 to 1.2 mg/dL, corresponding to an eGFR of about 60 to 140 mL/min/1.73 m2, which constitutes the range for which the PK model is valid. Therefore, the predicted exposure in individuals with moderate renal impairment was an extrapolation from the model and still needs to be confirmed. Age was a statistically significant factor on clearance, but the simulations showed few differences during the first 12 hours between the concentration profiles for ages of 25 to 75 years. The baseline platelet count was not found to be a covariate of glenzocimab clearance or central distribution volume, suggesting that the binding of glenzocimab to GPVI, which is expressed on platelets, did not affect glenzocimab PK in the tested dose range.
The effect of glenzocimab on ex vivo platelet aggregation was found to be nonlinear with dose. Higher doses produced a stronger effect for the same glenzocimab plasma concentration. This phenomenon was modeled with a direct response relationship with a dose covariate on inhibition potency (IC50). An attempt was made to determine whether a dynamic glenzocimab‐to‐GPVI binding model could reproduce this observation. This model described the interaction between glenzocimab, GPVI, and the binding complex as a dynamic equilibrium using the surface plasmon resonance determined kon and koff rates for the formation and dissociation of the complex. In this model, a higher dose results in higher concentrations of glenzocimab bound to GPVI. Because of the fast plasma elimination, the plasma glenzocimab will not reflect GPVI‐bound glenzocimab that will be retained because of the slow koff rate. However, this model failed to describe the observed effect on aggregation, and there is currently no explanation for this nonlinear effect of glenzocimab on platelet aggregation, suggesting that the full mechanism of action of GPVI and glenzocimab are not yet understood.
Baseline ex vivo platelet aggregation (Base_PPA) and maximal inhibition of ex vivo platelet aggregation (Imax) were found to be 80% and 73 %, respectively. The IC50 for a dose of 1000 mg was estimated at 0.47 μg/mL. The baseline platelet concentration was found to be a covariate of Imax. However, the effect was small, with a βImax_tPLT = 0.17, and likely not clinically relevant. Because of the limited number of subjects available for this analysis, the covariates thus identified will need to be confirmed using data from larger studies.
For the simulations, the treatment goal was set at 20% platelet aggregation. It remains to be seen whether this is a valid level for a clinical effect and how this translates from healthy subjects to patients suffering from ischemic stroke. Below 15% to 20% of intensity the measurement of platelet aggregation loses its precision and reproducibility. Thus, a 10% or 15% treatment goal would not be expected to achieve a dramatic improvement in efficacy. There have been no reports comparing the reactivity of platelets to collagen in patients suffering from ischemic stroke vs healthy subjects. However, it is possible to state that the inhibition of collagen‐induced (2.5 μg/mL) platelet aggregation to 20% corresponds to an almost maximum biological efficacy obtained at the saturation of GPVI sites on platelets. 3 Platelet activity after an ischemic stroke is increased, 14 protein expression is broadly changed, 15 and GPVI expression was shown to be upregulated in one report. 16 , 17 Other reports have mentioned a shedding of the extracellular domain of GPVI to which glenzocimab and collagen bind. 18 , 19 Thus, ischemic stroke patients may have platelets that are less prone to inhibition by glenzocimab. Even if the reactivity of platelets to collagen were greater in patients than in healthy subjects, the decrease in the biological efficacy of glenzocimab should be limited. For a comparison, the biological efficacy of clopidogrel, an antiplatelet drug that targets the adenosine diphosphate receptor P2Y12 and is widely used in cardiovascular pathologies is considered to be satisfactory when the inhibition of platelet aggregation in response to adenosine diphosphate is >30%. 20 Interestingly, platelet function–guided modifications to antiplatelet therapy (clopidogrel and/or aspirin) in patients with ischemic stroke or a transient ischemic attack have been shown to be associated with significantly higher rates of adverse clinical outcomes (death from ischemic events or bleeding). 21 Other factors, such as fibrin may make a significant contribution to the thrombogenic effect, and the activity of glenzocimab on fibrin may be an important component in the resulting clinical activity.
The effect on platelet aggregation was evaluated 6 or 12 hours after the start of the infusion, as well as at 24 hours. With 1000‐mg glenzocimab the treatment goal was achieved by nearly 100% of individuals at 6 hours and by 60% of individuals at 12 hours. In order to obtain a 95% response rate at 12 hours, a dose of 2750 mg was predicted. For the 1000‐mg dose and the proposed infusion of 25% of the dose being delivered within the first 15 minutes, a reduction to 20% platelet aggregation would be obtained within 5 minutes of starting the infusion. If the infusion rate were kept constant for the entire 6‐hour infusion time, 20% platelet aggregation would be obtained 45 minutes after the start of the infusion. However, for doses ≥2000 mg the onset of an effect was reduced to <10 minutes and the difference between the infusion rates became progressively smaller. Thus, for doses <2000 mg the high rate of infusion during the first 15 minutes is justified to enable a rapid onset of effect. Doses >2000 mg are higher than those tested during the phase I study. While simulations for doses >2000 mg extrapolated beyond the doses tested, they were justified based on the PK properties of glenzocimab. Glenzocimab was likely eliminated via glomerular filtration with a catabolic contribution via endocytosis to the general capillary endothelium. This is well established for proteins with a molecular size of 48 kDa. 22 There may also be specific elimination via receptor uptake, but the present data did not suggest that this occurred with glenzocimab: PK was dose proportional and there was no characteristic bend in the PK curve suggestive of TMDD. Elimination therefore occurred via nonsaturable pathways, so it can be expected that the dose‐proportional PK within the range of 62.5 to 2000 mg would extend to 4000 mg.
As an alternative, a 12‐hour infusion was tested with the same total doses as the 6‐hour infusion. It was found that the 12‐hour infusion required a lower dose to ensure that 95% of individuals would achieve 20% platelet aggregation at 12 hours. This could be explained by the relatively short half‐life of glenzocimab; to sustain the effect at 12 hours with a 6‐hour infusion, the dose had to be increased overproportionally to compensate for the rapid elimination after completion of the infusion. It was found that with the same doses, the 6‐ and 12‐hour infusions produced similar effects at 24 hours. However, with the 12‐hour infusion, lower doses could be given to obtain the same effect at 12 hours compared to the 6‐hour infusion; a 12‐hour scheme might therefore offer a better option to balance a strong effect at 12 hours vs a recovery of aggregation at 24 hours. However, so long as there is no clinically established link between PK, PD, and clinical outcomes (regarding both efficacy and safety), it remains highly speculative to predict which administration scheme would be the most appropriate.
Body weight was identified as a statistically significant covariate on glenzocimab PK. Simulations showed that with the 1000‐mg dose the effect on platelet aggregation at 6 hours was not sensitive to body weight, but at 12 hours there was a dependence on body weight. This body weight dependence decreased with higher doses. Without any patient data on the relationship among PK, PD, and clinical effect, it was unclear whether such body weight dependence might require any dose adjustment. The eGFR dependence of the glenzocimab PK in individuals with mild or moderate renal impairment was weak, and no dose adjustment was required to maintain the effect. No clinically relevant dependence of the PK/PD relationship on the platelet baseline count was identified. Similarly, while there was an age dependence of glenzocimab PK, it was not found to be clinically relevant; therefore, it did not support a dose adjustment for age in the target population that could be up to 85 years old.
Conclusions
The glenzocimab population PK/PD model adequately described the PK and PD data observed in healthy individuals and can be used to reliably predict glenzocimab plasma concentrations and the effect on ex vivo platelet aggregation in individuals aged 22 to 63 years, with a body weight of 52 to 107 kg and an eGFR of 60 to 140 mL/min/1.73 m2.
The goal for treatment with glenzocimab was to obtain a rapid onset of effect that would last for 6 or 12 hours and a return to close‐to‐normal platelet aggregation at 24 hours. The principal pharmacological property of glenzocimab that dictates its posology is its short plasma half‐life, which requires that it should be infused intravenously for 6 or 12 hours in order to maintain the necessary duration of effect. From the PK/PD simulations it was concluded that with 750 mg of glenzocimab given as a 6‐hour infusion with 25% of the dose being delivered during the first 15 minutes the 6‐hour goal would be achieved: between 5 minutes and 6 hours after the start of the infusion 95% of individuals would achieve ≤20% platelet aggregation. At 24 hours the remaining platelet aggregation would be about 40%. A dose of 2750 mg was predicted for the 12‐hour goal, with close to a full effect at 24 hours. Alternatively, to meet the 12‐hour goal, the infusion was prolonged to 12 hours and this reduced the required dose to 1000 mg and increased residual platelet aggregation at 24 hours to about 30%. From this it could be concluded that to avoid any possible drug‐drug interactions with other antithrombotic drugs that would be introduced at 24 hours, a 6‐hour IV infusion and the 6‐hour goal offered a better balance between the effect desired at 6 hours and a return to normal platelet aggregation at 24 hours.
While body weight was identified as a statistically significant covariate on clearance, the predicted effect on platelets at 6 hours was weak within the relevant body weight range, and no dosage adjustment to body weight was proposed. eGFR was also identified as a statistically significant covariate on clearance. PK/PD simulations predicted via extrapolation that the eGFR effect in individuals with mild or moderate renal impairment was weak and would not require any dose adjustment. These findings will need to be confirmed by including data from individuals with moderate renal impairment in the PK/PD analysis.
The information gained from this study was valuable in terms of guiding the choice of dose‐infusion regimen for glenzocimab in future studies on patients and will help to establish the link between exposure, the effect on ex vivo platelet aggregation, and clinical end points once these data become available.
Conflicts of Interest
M.M., L.R., Y.P. are consultants to Acticor. M.J.‐P. and P.B. are founders and consultants to Acticor. K.L. is an employee of Acticor‐Biotech.
Funding
Funding was provided by Acticor Biotech SAS.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding authors and from Kristell Lebozec (kristell.lebozec@acticor-biotech.com) on reasonable request.
Supporting information
Supporting information
Acknowledgments
The authors thank Victoria Hawken for kindly reviewing and greatly improving the language of this manuscript.
References
- 1. Powers WJ, Rabinstein AA, Ackerson T, et al. Guidelines for the early management of patients with acute ischemic stroke, a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2018;49:e46‐e99. [DOI] [PubMed] [Google Scholar]
- 2. Ivanciu L, Stalker TJ. Spatiotemporal regulation of coagulation and platelet activation during the hemostatic response in vivo. J Thromb Haemost. 2015;13:1949‐1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lebozec K, Jandrot‐Perrus M, Avenard G, Favre‐Bulle O, Billiald P. Design, development and characterization of ACT017, a humanized Fab that blocks platelet's glycoprotein VI function without causing bleeding risks. MAbs. 2017;9(6):945‐958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Voors C, Lebozec K, Dogterom P, et al. Safety and tolerability, pharmacokinetics and pharmacodynamics of ACT017, an anti‐platelet glycoprotein VI Fab: first in human healthy volunteer trial. Arterioscler Thromb Vasc Biol. 2019;39:956‐964. [DOI] [PubMed] [Google Scholar]
- 5. Peletier LA, Gabrielsson J. Dynamics of target‐mediated drug disposition: characteristic profiles and parameter identification. J Pharmacokinet Pharmacodyn. 2012;39:429‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Monolix version 2018R1 . Antony, France: Lixoft SAS; 2018.
- 7. Samson A, Lavielle M, Mentre F. Extension of the SAEM algorithm to left‐censored data in nonlinear mixed‐effects model: Application to HIV dynamics model. Comput Stat Data Anal. 2006;51(3):1562‐1574. [Google Scholar]
- 8. Lavielle M. Rsmlx: Running Monolix from R. Version 1.1.0 . http://rsmlx.webpopix.org. Published 2018.
- 9. Chauvin J, Ayral G, Traynard P. COSSAC for efficient covariate selection. 27th Population Approach Group Europe (PAGE) Meeting; Montreux, Switzerland; May 29–June 1, 2018.
- 10. Lavielle M, Ribba B. Enhanced method for diagnosing pharmacometric models: random sampling from conditional distributions. Pharmaceutical Res. 2016;33:2979‐2988. [DOI] [PubMed] [Google Scholar]
- 11. Lavielle M. mlxR: simulation of longitudinal data. Version 3.3.0. http://simulx.webpopix.org. Published 2018.
- 12. Renard C, Grene‐Lerouge N, Beau N, Baud F, Scherrmann JM. Pharmacokinetics of digoxin‐specific Fab: effects of decreased renal function and age. Br J Clin Pharmacol. 1997;44:135‐138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lawrence MG, Altenburg MK, Sanford R, et al. Permeation of macromolecules into the renal glomerular basement membrane and capture by the tubules. Proc Natl Acad Sci. 2017;114(11):2958‐2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yip HK, Chen SS, Liu JS, et al. Serial changes in platelet activation in patients after ischemic stroke: role of pharmacodynamic modulation. Stroke. 2004;35:1683‐1687. [DOI] [PubMed] [Google Scholar]
- 15. Cevik O, Baykal AT, Sener A. Platelets proteomic profiles of acute ischemic sroke patients. PLoS One. 2016;11(6):e0158287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bigalke B, Lindemann S, Ehlers R, et al. Expression of platelet collagen receptor glycoprotein VI is associated with acute coronary syndrome. Eur Heart J. 2006;27:2165‐2169. [DOI] [PubMed] [Google Scholar]
- 17. Bigalke B, Stellos K, Geisler T, et al. Expression of platelet glycoprotein VI is associated with transient ischemic attack and stroke. Eur J Neurol. 2010;17:111‐117. [DOI] [PubMed] [Google Scholar]
- 18. Al‐Tamini M, Gardiner EE, Thom JY, et al. Soluble glycoprotein VI is raised in the plasma of patients with acute ischemic stroke. Stroke. 2011;42(2):498‐500. [DOI] [PubMed] [Google Scholar]
- 19. Wurster T, Poetz O, Stellos K, et al. Plasma levels of soluble glycoprotein VI (sGPVI) are associated with ischemic stroke. Platelets. 2013;24(7):560‐565. [DOI] [PubMed] [Google Scholar]
- 20. Mallouk N, Labruyère C, Reny JL, et al. Prevalence of poor biological response to clopidogrel. Thromb Haemost. 2012;107:494‐506. [DOI] [PubMed] [Google Scholar]
- 21. Depta JP, Fowler J, Novak E, et al. Clinical outcomes using a platelet function‐guided approach for secondary prevention in patients with ischemic stroke or transient ischemic attack. Stroke. 2012;43:2376‐2381. [DOI] [PubMed] [Google Scholar]
- 22. Li Z, Shah DK. Two‐pore physiologically based pharmacokinetic model with de novo derived parameters for predicting plasma PK of different size protein therapeutics. J Pharmacokinet Pharmacodyn. 2019;46:305‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information