

Abstract
Amyotrophic lateral sclerosis (ALS) is a debilitating neurodegenerative disorder with complex biology and significant clinical heterogeneity. Many preclinical and early phase ALS clinical trials have yielded promising results that could not be replicated in larger phase 3 confirmatory trials. One reason for the lack of reproducibility may be ALS biological and clinical heterogeneity. Therefore, in this review, we explore sources of ALS heterogeneity that may reduce statistical power to evaluate efficacy in ALS trials. We also review efforts to manage clinical heterogeneity, including use of validated disease outcome measures, predictive biomarkers of disease progression, and individual clinical risk stratification. We propose that personalized prognostic models with use of predictive biomarkers may identify patients with ALS for whom a specific therapeutic strategy may be expected to be more successful. Finally, the rapid application of emerging clinical and biomarker strategies may reduce heterogeneity, increase trial efficiency, and, in turn, accelerate ALS drug development.
Keywords: amyotrophic lateral sclerosis, biomarkers, clinical trials, disease heterogeneity, enrichment strategies, outcome measures
Abbreviations
- ALS
amyotrophic lateral sclerosis
- ALSFRS‐R
ALS Functional Rating Scale‐Revised
- AMPA
α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic
- BEST
Beta‐Blocker Evaluation of Survival Trial
- CAFS
Combined Analysis of Function and Survival
- CRP
C‐reactive protein
- CSF
cerebral spinal fluid
- fALS
familial ALS
- FVC
forced vital capacity
- miRNA
micro‐RNA
- MSC
mesenchymal stromal cell
- MSC‐NTF
MSC‐neurotrophic factor
- ncRNA
noncoding RNA
- PRO‐ACT
Pooled Resource Open‐Access ALS Clinical Trials
- sALS
sporadic ALS
- SOD1
superoxide dismutase 1
- SVC
slow vital capacity
- TDP‐43
transactive response DNA binding protein 43 kDa
1. INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease primarily characterized by the progressive deterioration of cortical and spinal motor neurons. Substantial clinical heterogeneity among patients with ALS is seen, especially in terms of site of onset (bulbar vs spinal), relative degree of upper and lower motor neuron involvement, and progression rate.1 There is also known genetic heterogeneity, with more than 40 identified ALS genes.2 Many of these confer susceptibility, not necessarily causation, thus further contributing to biological heterogeneity. Disease heterogeneity has implications for the efficient development of innovative therapeutics. Because clinical and biological heterogeneity are not fully understood, individual risk assessment, participant stratification in clinical trials, and timing of treatment interventions may involve considerable challenges and uncertainties with respect to therapeutic strategies. Riluzole was approved for the treatment of ALS in the 1990s, but there has been limited success with the development of additional treatment options. According to information registered at ClinicalTrials.gov (https://clinicaltrials.gov/), since 2007, when new US Food and Drug Administration registration requirements were established for drug trials, over 80 phase 2 or phase 3 ALS trials have been completed, terminated, or suspended. These studies have yielded only one additional approved treatment (Edaravone) to slow the progression of ALS. Part of the reason for the low success rate of ALS trials, in addition to uncertain biological targets or late intervention, may be the lack of strategies to address clinical heterogeneity.3
The goals of this review are to better describe the sources of heterogeneity in ALS; to propose how this heterogeneity can be managed by using validated disease measures and outcomes, biomarkers, and prognostic models; and to examine the use of these strategies in clinical trials.
2. SOURCES OF HETEROGENEITY
The complex biological heterogeneity of ALS includes an expanding list of genetic factors that interacts across neuronal and noncell autonomous pathology.2, 4 More recently, spatiotemporal analysis of the molecular pathology in ALS has provided evidence of a complex interplay of several distinct neuronal and nonneuronal cell types.5
From a genetics perspective, genome‐wide association studies may be consistent with a multigenic process in both familial ALS (fALS) and sporadic ALS (sALS).6 Sporadic ALS occasionally arises from spontaneous mutations in some of the same genes that are known to cause fALS. While genetic mutations can be predictive of disease characteristics (eg, C9orf72 expression may be associated with cognitive and behavioral changes7), clinical features and survival may be highly variable for a given mutation (eg, patients with a mutation in superoxide dismutase 1 [SOD1] show differences in phenotype and in speed of disease progression8), even within families. This variability likely results from the specific allelic mutation from additional risk‐associated modifier genes6 as well as from putative environmental factors.
A broad range of cellular and molecular abnormalities has been noted in both sALS and fALS. Cytoskeletal abnormalities9 and intracellular protein aggregates; alterations in DNA and RNA processing, transport, and function; mitochondrial dysfunction; and disrupted oxidative homeostasis have all been implicated.10, 11 Central nervous system inflammation also appears to play a key role in the progression of motor neuron degeneration, irrespective of the specific upstream disease pathophysiology.12
Prior to the current state of understanding of ALS biological heterogeneity, drug development in ALS primarily focused on ALS as a single disease with proposed common pathophysiology and a predictably relentless clinical course. Recently, this view has changed with an increased recognition of the variability in rates of progression, heterogeneity of phenotypes (degree of upper and lower motor neuron involvement), and differences in allelic mutations of known genetic forms as larger clinical data sets such as the Pooled Resource Open‐Access ALS Clinical Trials (PRO‐ACT) database13 have become available.
3. ADDRESSING CLINICAL HETEROGENEITY
In ALS, heterogeneity in the rate of functional decline and overall rate of disease progression and survival may be related to initial clinical manifestations.14 For example, upper limb (P = .010) or bulbar (P = .005) weakness may be associated with more rapid rate of functional decline,14 an observation recently confirmed by using the PRO‐ACT database.15 To address these challenges, standardized disease outcome measures, prognostic models, and biomarkers have been used to characterize the ALS clinical trial population, with varying success (Figure 1).
Figure 1.
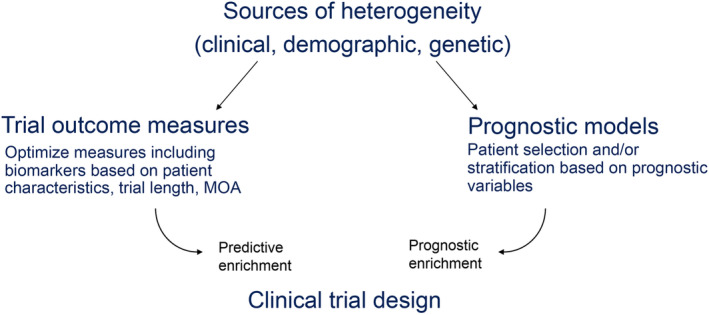
Sources of heterogeneity in amyotrophic lateral sclerosis clinical trials. MOA, mode of action
4. DISEASE OUTCOME MEASURES
Disease outcome measures in ALS include measures of function, survival, and quality of life. Several tools are available for measuring different types of function, and each is associated with benefits and limitations for addressing the biologic and clinical heterogeneity of the study population (Tables 1 and 2). When survival is used as an endpoint, there are intrinsic and extrinsic factors that may affect interpretation of the data,16 and combining survival with ventilatory endpoints (eg, tracheostomy or noninvasive ventilation) must also be considered.17 Riluzole appears to have a modest effect by prolonging survival by approximately 3 months in patients with ALS and, possibly, in the last clinical stage of ALS, as reported in a recent retrospective study.18, 19 Thus, riluzole may be a confounder affecting the survival endpoint in trials when there is an imbalance of the number of patients on riluzole or when the duration of exposure to riluzole differs between the varying treatment allocation arms.
Table 1.
ALS disease outcome measures of physical function and muscle strength
| Outcome | Heterogeneity | Tools | Benefits | Limitations |
|---|---|---|---|---|
| Physical function |
Domain with greatest subscore deterioration51 Rate of decline33 |
ALSFRS‐R52 |
Effective measure of progression Strong predictor of survival53, 54 Reflects clinically meaningful change over time55 Progression of group ALSFRS‐R trajectories form a linear model56 |
Subjective Individual ALSFRS‐R trajectories display variable curvilinearity56 |
| Muscle strength |
Variability in overall rates of decline for different muscle groups60 Large interpatient variability in rates of decline60, |
HHD60 | ||
| ATLIS61 | Additional research required |
Abbreviations: ALS, amyotrophic lateral sclerosis; ALSFRS‐R, ALS Functional Rating Scale‐Revised; ATLIS, Accurate Test of Limb Isometric Strength; FVC, forced vital capacity; HHD, hand‐held dynamometry; MMT, manual muscle testing.
Table 2.
ALS disease outcome measures of respiratory function, muscle integrity, cortical function, and speech function
| Outcome | Heterogeneity | Tools | Benefits | Limitations |
|---|---|---|---|---|
| Respiratory function | Baseline measurement and rate of decline57 | FVC/SVC57 |
Commonly used to assess disease status and outcomes in clinical trials |
Only moderate correlation with ALSFRS‐R respiratory subscale59 Nondiaphragm muscles or obstructive causes may affect results59 |
| Muscle integrity | Rate of decline of muscle integrity62 | EIM62 |
Less variable than HHD and ALSFRS‐R Correlates with survival |
To date, little has been published on using EIM in clinical trials |
| Cortical function | SICI amplitude reduced and MEP amplitude increased in early ALS, precede neurodegeneration63 | TMS64 |
Noninvasive Measures several cortical outputs Discriminates early and late disease stages |
Difficult to determine precisely which cortical neurons and the extent of the cortical area affected with each TMS pulse/stimulation Surface regions of the cortex more likely than the subcortical regions are targeted |
| Speech function | Decline in speech intelligibility during disease progression | Wave65 | Automatic estimation less time intensive and causes less patient fatigue than standard clinical examination of oral motor function | Data are preliminary and sample size is small |
Abbreviations: ALS, amyotrophic lateral sclerosis; ALSFRS‐R, ALS Functional Rating Scale‐Revised; EIM, electrical impedance myography; FVC, forced vital capacity; HHD, hand‐held dynamometry; SICI, short‐interval intracortical inhibition; SVC, slow vital capacity; TMS, transcranial magnetic stimulation.
Survival over a longer time period may help to convince neurologists and patients that a drug or treatment is effective; however, the feasibility of the long study length and placebo controls in survival endpoint studies is a challenge for study participants and their caregivers. The Combined Analysis of Function and Survival (CAFS) was developed to overcome the limitations of measuring function or survival alone as a primary outcome; this method allows a treatment effect on either outcome to be detected when there is a strong effect on one but not the other.20
The revised Airlie House consensus guidelines have recently provided recommendations about how disease outcome measures can account for biologic and clinical heterogeneity in ALS,21 and efforts to define appropriate endpoints are ongoing. Several statistical methods were recently assessed for simultaneous analysis of function and survival by using the PRO‐ACT database. A joint model, in which the ALS Functional Rating Scale‐Revised (ALSFRS‐R) was incorporated into the survival model as a covariate, was found to provide the most consistency among treatment scenarios and had a greater ability to detect smaller treatment effects compared with other statistical methods, including CAFS.22 Moreover, additional power might be achieved with analyses of the ALSFRS‐R subdomain scores rather than total score or by incorporating other outcomes into the model, such as respiratory or muscle strength measures.
5. BIOMARKERS
Biomarkers can be diagnostic, prognostic, or predictive, and each may potentially provide insights into the reasons for heterogeneity in the clinical trial population in different ways. There are currently no individual or group of biomarkers that accurately serve in a diagnostic, prognostic, or predictive capacity in ALS. Diagnostic biomarkers may define different biotypes of the ALS syndrome that may theoretically respond to different therapeutic strategies, could allow the exclusion of cases that are not “true” ALS, or could select for patients at a specific disease stage; prognostic biomarkers could allow for the selection of patients with a specific disease stage and/or rate of progression; and predictive biomarkers could identify patients most likely to respond to a treatment. Validated biomarker development is, therefore, one of the most important needs among ALS researchers.23
Several biomarker candidates are being explored, including “wet” (tissue based), digital (eg, imaging or neurophysiology), and inflammatory and genetic varieties and micro‐RNAs (miRNA; Tables 3 and 4). One candidate is cerebral spinal fluid (CSF) and blood neurofilament (Table 3), which shows promise as an objective measure of disease progression that could provide an early indication of whether a treatment is effective in a particular subgroup of patients.24 Another example is transactive response DNA binding protein 43 kDa (TDP‐43), which is higher in the CSF of patients with frontotemporal dementia‐ALS spectrum disorder than in the CSF of controls; accumulation of TDP‐43 is the most significant pathological finding in approximately 95% of ALS cases25 (Table 4). Furthermore, miRNAs26, 27, 28 associated with neuroprotective or neuroinflammatory pathways were assessed as potential biomarkers because they may be relevant to the mode of action of mesenchymal stromal cells (MSC) transplantation. These cells produce elevated levels of neurotrophic factors after collection and in vitro propagation in preparation for autologous transplant.29 Ongoing research with RNA‐seq technology is being used to investigate miRNA and other short noncoding RNA (ncRNA) species as potential biomarkers (Table 4). Specific ncRNAs were shown to vary between patients with ALS and non‐ALS controls.30
Table 3.
Potential neurophysiological, imaging, and tissue‐based biomarkers for use in ALS clinical trials
| Biomarker | Prognostic value |
|---|---|
| Neurophysiological | |
| MUNE/MUNIX, EIM, TMS |
Markers of disease progression and predictors of survival Some can detect changes prior to symptom onset63 |
| Imaging/MRI | |
| Diffusion tensor imaging, functional MRI, iron‐sensitive sequences, voxel‐based morphometry | Detect changes that correlate with other measures of disease66, 67 |
| Tissue‐based/“wet” | |
| Serum creatinine |
Correlates with ALSFRS‐R, muscle strength, and survival13 Loss correlates with progression and is reduced in dexpramipexole‐treated patients68 |
| Uric acid level |
Correlates with ALSFRS‐R, muscle strength, and survival13 Independent beneficial affect associated with higher urate levels in dexpramipexole‐treated patients69 |
| Urinary extracellular cleavage domain of neurotrophin receptor p75 | Inversely related to ALSFRS‐R scores at first visit, increase with disease progression, and baseline values predict survival70, 71 |
| Plasma light and heavy chain neurofilament proteins |
Light chain levels Correlate well with rate of disease progression72 Higher in fast vs slow progressors, and remain relatively constant during progression heavy chain levels73 Levels declined with progression in rapid progressors Overall levels in ALS not significantly different from controls |
| CSF light and heavy chain neurofilament proteins | Both neurofilament types in the CSF correlate with rate of progression74 |
Abbreviations: ALS, amyotrophic lateral sclerosis; ALSFRS‐R, ALS Functional Rating Scale‐Revised; CSF, cerebrospinal fluid; C9orf72 = EIM, electrical impedance myography; MUNE, motor unit number estimation; MUNIX, motor unit number index; TMS, transcranial magnetic stimulation.
Table 4.
Potential inflammatory, genetic, and miRNA biomarkers for use in ALS clinical trials
| Biomarker | Prognostic value |
|---|---|
| Inflammatory | |
| CHIT1 |
Elevated in the CSF of patients with ALS vs controls/other neurological diseases Levels correlate with rate of progression75 |
| FoxP3 |
Tregs that downmodulate inflammatory responses are inversely correlated with rate of ALS progression Transcription factor FoxP3 is required for Treg suppressive function Early reduced FoxP3 levels predict rapid future progression and shortened survival76 |
| Genetic | |
|
LILRA2, ITGB2, and CEBPD expressed in peripheral lymphocytes |
Expression levels were predictive of rate of ALS progression in microglia and lymphoblastoid cell lines77 |
| SOD1 mutations |
Correlate with disease severity; mutant SOD1 proteins in intracellular inclusions and CSF are cytotoxic78 A4V (SOD1A4V) has an exceptionally aggressive disease course, with shorter disease duration and lower median survival than other SOD1 mutations79 |
| TDP‐43 mutations | Higher in the CSF of patients with FTD‐ALS spectrum disorder compared with controls25 |
| miRNA | |
| CSF miRNA | Used as biomarkers for many cancers and neurodegenerative disorders26, 27, 28 |
| miR‐132 |
Binds TDP‐43 and modulates development and maturation of axons and dendrites Neuroprotective in tauopathies via a caspase 3‐mediated mechanism |
| miR‐146a |
Negatively regulates innate immunity in astrocytes, glia, and Tregs82, 83, 84 Abundantly expressed in human CSF27 |
Abbreviations: ALS, amyotrophic lateral sclerosis; CEBPD, CCAAT/enhancer‐binding protein δ; CHIT1, chitotriosidase; CSF, cerebrospinal fluid; fALS, familial ALS; FTD‐ALS, frontotemporal dementia‐ALS; FUS, fused in sarcoma; ITGB2, integrin subunit β 2; LILRA2, Leukocyte immunoglobulin‐like receptor subfamily A member 2; miRNA, micro‐RNA; sALS, sporadic ALS; SOD1, superoxide dismutase 1; TDP‐43, transactive response DNA binding protein 43 kDa; Treg, T regulatory cell.
Novel methods to identify potential biomarkers are also being developed, including an exploratory platform in which proteomic workflows were applied to study the cross‐phenotype variance of peripheral blood mononuclear cells and plasma/brain proteins. This method provided more sensitivity compared with conventional case–control studies in a single matrix and provided a rationale for identification of biomarkers to aid phenotypic stratification prior to trial enrollment.31
6. PROGNOSTIC MODELS
Prognostic models for disease progression or survival continue to evolve and incorporate additional features, including biomarkers, that may ultimately improve clinical trial design1, 32 and may potentially reduce sample size requirements through the use of several prognostic variables. Such models can be practically applied to enrich a clinical trial with patients who are expected to progress to advanced disease more slowly/quickly or who are likely to have a longer/shorter survival time (Figure 1), and they can also be used as covariates in analysis. One example is the evaluation of ALS progression and individual risk stratification in a 3‐month run‐in period to both reduce clinical heterogeneity and improve estimation of pretreatment‐posttreatment effects.33 Inclusion/exclusion criteria can be based on key variables (such as age and rate of disease progression) to enrich for patients with a greater likelihood of responding to a treatment while reducing interpatient variability.
To demonstrate how biomarker prognostic modeling can be used to address biological heterogeneity, researchers examined demographic, clinical, and laboratory data— including 15 blood chemistry values—to determine whether these metrics were predictive of the ALSFRS‐R rate of decline.13 Higher baseline levels of creatinine or uric acid were associated with slower declines in ALSFRS‐R and vital capacity and with longer survival. Higher body mass index was predictive of longer survival, whereas bulbar onset, older age at onset, and decreased time from onset to diagnosis independently predicted shorter survival.13 In response to the Prize4Life Challenge,34 algorithms were developed to accurately predict the ALSFRS‐R slope of decline by using a subset of the PRO‐ACT database. Two algorithms were identified that outperformed both a baseline model developed by challenge organizers and predictions by ALS clinicians.34 In addition to confirming the variables previously identified as predictors of decline in PRO‐ACT, the new algorithms identified creatine kinase, phosphorous, pulse, and blood pressure. It was estimated that use of the aggregated predictions from the two models could reduce trial sample size by 20%.34
Additional insights came from Pfohl et al,35 who performed a single‐site retrospective analysis of 38 clinical variables for over 800 deceased patients. Prognostic variables for survival were analyzed for changes in predictive ability during disease progression. Time variables, such as patient age, time from onset, time from diagnosis, and disease duration, were the dominant predictors for survival beyond 1 year and changed over time or with disease progression. The authors concluded that the ALSFRS‐R rate of decline is more clinically significant for individual patients than it is for groups and that it should not be the only measure in population‐based models.35 It is not clear whether this model, based on clinic patient records, is applicable to clinical trial populations.
However, a random forest model for predicting ALSFRS‐R score was developed by using baseline clinical trial data and was found to be applicable to a real‐world data set from a single clinic.32 Compared with preslope or generalized linear models, the random forest model performed significantly better at predicting ALSFRS‐R scores and had less error over longer time intervals (eg, 18–36 months). In trials in which ALSFRS‐R is the main outcome measure, this model could be used to assess treatment effects by comparing predicted vs observed ALSFRS‐R rates of decline.
In addition, Westeneng et al1 recently described and validated a model for prediction of survival at the individual level based on eight predefined sources of heterogeneity: age at onset, forced vital capacity (FVC), diagnostic delay, ALSFRS‐R slope, bulbar onset, definite ALS, presence of frontotemporal dementia, and the C9orf72 repeat expansion. This survival model initially was based on data from a population‐based study in the Netherlands and was validated with an external data set from 13 centers in Europe.36 The authors applied the model to distinguish five groups on the basis of time from symptom onset to the composite survival outcome (use of noninvasive ventilation for more than 23 hours per day, tracheostomy, or death). The median predicted/observed times ranged from very short (17.7/16.5 months) to very long (91/85.6 months). The authors suggested that the model could be applied to patient care and clinical trial design but cautioned that the predictions of the model should be used only to guide decisions in the medical community and should not be shared with patients.
Furthermore, prognostic models can also be used to assess observed vs predicted outcomes (Figure 2) and therefore provide important information about the effects of heterogeneity within the trial population. This approach represents an improvement over use of natural history controls, in which contemporaneous data sets must be selected to avoid confounding due to improvements in supportive care. Trials assessing observed vs predicted outcomes could also obviate a placebo arm and might be appropriate for phase 2 studies. In fact, prognostic methods may improve trials that do not enrich enrollment on the basis of progression or survival rates. When investigators applied a predictive survival algorithm to trial stratification during simulated randomizations, it was found to reduce the randomization failure rate and sample size required for sufficient statistical power compared with a standard randomization scheme.37
Figure 2.
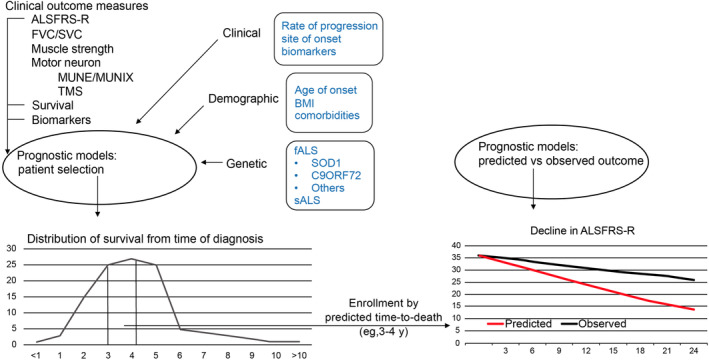
Use of prognostic models to assess the effects of heterogeneity and guide appropriate trial designs. ALS, amyotrophic lateral sclerosis; ALSFRS‐R, ALS Functional Rating Scale‐Revised; BMI, body mass index; fALS, familial ALS; FVC, forced vital capacity; MUNE, motor unit number estimation; MUNIX, motor unit number index; sALS, sporadic ALS; SOD1, superoxide dismutase 1; SVC, slow vital capacity; TMS, transcranial magnetic stimulation
Applying prognostic models in this way has shown that efforts to reduce heterogeneity through baseline inclusion criteria rather than individual risk estimates may not always achieve the desired outcome.37 In addition, stricter inclusion criteria that select for patients with more homogeneous disease could slow the rate of trial enrollment by reducing the pool of eligible patients.36 In an evaluation of the effects of restrictive patient selection in ALS trials, van Eijk et al36 found that more stringent eligibility criteria did not necessarily translate to changes in survival time or functional decline, although these criteria do tend to enrich for younger men with milder disease and may also slow down recruitment. Instead, the authors determined individual risk profiles on the basis of the European Network for the Cure of ALS survival model.37 This approach optimized sample size and eligibility rate better than any of 38 trials based on eligibility criteria alone.37 The investigators noted that enrolling patients on the basis of an individual risk estimate offers the promise of achievable statistical power with a manageable sample size, although researchers still must strike a balance between trial efficacy and generalizability of findings. The authors demonstrated that, on average, approximately 60% of patients with ALS are deemed ineligible for clinical trials at diagnosis,36, 37 so generalizability of findings is a concern that must be addressed.
Predictive enrichment is an alternative approach for reducing heterogeneity that selects patients most likely to respond to a treatment on the basis of either empirical evidence (eg, response to previous treatment) or mode of action‐based data (Figure 1). Li et al38 found that they could identify an “enrichable subgroup” of responders by retrospectively applying their scoring systems to data from the Beta‐Blocker Evaluation of Survival Trial (BEST)39 by incorporating baseline characteristics of the patients enrolled in BEST into their model. This retrospective use of data could be employed in future trials to enrich for likely responders.
7. CURRENT USE OF CLINICAL TRIAL ENRICHMENT STRATEGIES
Both prognostic and predictive enrichment strategies have been applied to ALS trial designs. In the clinical trial process that ultimately led to the approval of the free radical scavenger edaravone, initial phase 3 clinical trial results failed to show treatment benefits.40 Results of post hoc analyses indicated that wide variations in the range of changes in ALSFRS‐R scores may have obscured treatment effects, and a subpopulation of patients in early disease did experience a treatment benefit.41 In a subsequent phase 3 study, researchers prospectively enrolled patients on the basis of criteria identified in the post hoc analysis of the initial phase 3 study: scores of 2 or more on all ALSFRS‐R items, FVC ≥80% predicted, duration of disease up to 2 years, and disease progression characterized by a decrease of 1–4 ALSFRS‐R points during the 12‐week observation period before randomization. The results demonstrated a significantly mitigated decline in ALSFRS‐R scores (approximately 33% over the 24‐week treatment period) compared with placebo.42
A similar approach was not successful in demonstrating a benefit in response to NP001, a negative modulator of macrophage/monocyte activation. A phase 2 study appeared to identify a group of responders with higher baseline levels of the inflammatory marker C‐reactive protein (CRP).43 Investigators subsequently enrolled patients with elevated CRP levels in a second phase 2 study, but the findings did not demonstrate any treatment effect in this enriched population. Modifications to the study design were also made in the phase 3 trial for the fast‐skeletal troponin muscle activator tirasemtiv to offset for tolerability to increasing doses observed in a phase 2B study. The phase 3 trial involved a greater number of patients to account for dropouts resulting from drug‐related adverse events44 and included a longer open‐label run‐in period to establish a titrated (rather than fixed) dose that would be tolerated for the trial duration. The primary endpoint for the phase 3 trial was chosen on the basis of phase 2B results, in which significant benefits were observed for changes in slow vital capacity (SVC) but not in ALSFRS‐R.44 Unfortunately, none of these changes led to a positive trial outcome.
Generally, heterogeneity in the ALS patient population is currently being addressed by focusing on either biological or clinical heterogeneity.45, 46, 47 In the latter approach, some trials are selecting for patients who are more likely to progress to advanced disease quickly. Another example of an enrichment strategy is the trial examining pharmacodynamic effects of retigabine and riluzole in patients with ALS, in which selection of patients was based on transcranial magnetic stimulation data.48 Other representative ongoing or recently completed trials in which one or more of these enrichment approaches are reported are presented in Table 5. Most enrichment criteria are for earlier stages of disease, which suggests a predictive approach in which patients with earlier disease are believed more likely to respond compared with those with more advanced disease. This predictive approach relies heavily on “actionable” biomarkers for clinical decision making. Four of these trials also included or excluded patients on the basis of biomarkers, such as SOD1 mutations (or other monogenic causes of ALS), inflammatory gene expression, and serum urate levels. Although low urate is associated with poorer prognosis, other prognostic criteria were not applied when serum urate levels were used to exclude patients (NCT03168711). Thus, urate level appears to be a predictive criterion rather than a prognostic approach for identifying likely responders.
Table 5.
Ongoing or recently completed clinical trials using enrichment criteria
| Abbreviated Title (NCT No.) | Phase | Inclusion Criteria | Enrichment Type |
|---|---|---|---|
| A Biomarker Study to Evaluate Ibudilast in ALS (NCT02714036) | 1/2 | UMNB 25; FVC > 50% | Disease stage |
| A Trial of Tocilizumab in ALS (NCT02469896) | 2 | High expression of inflammatory genes and UMNB 25 |
Biomarker Disease stage |
| AMX0035 in ALS (NCT03127514) | 2 | Disease onset ≤18 months; SVC >60% | Disease stage |
| Arimoclomol in ALS (NCT03491462) | 3 |
Disease onset ≤18 months ALSFRS‐R 35; SVC >80% |
Disease stage |
| Conservative Iron Chelation as a Disease‐ modifying Strategy in ALS (NCT03293069) | 2/3 | Disease onset ≤18 months, <6 months since the diagnosis; ALSFRS‐R 36; SVC >70% inspiratory pressure >60 | Disease stage |
| Dual Treatment With Lithium and Valproate in ALS (NCT03204500) | 2 | Disease onset between 6 and18 mo; SVC >60% | Disease stage |
| Efficacy and Safety of Plasma Exchange with Albutein 5% in Patients With ALS (NCT02872142) | 2 | Disease onset ≤18 mo; FVC >70% | Disease stage |
| IC14 for Treatment of ALS (NCT03508453) | 2 | ALSFRS‐R decline 3 points in previous 3 mo; seated FVC >65% | Prognostic Disease stage |
|
Intrathecal Autologous Adipose‐derived MSC for ALS (NCT03268603) |
2 | Disease onset <2 y; SVC >65% | Disease stage |
| Perampanel for Sporadic ALS (NCT03019419) | 2 | Disease onset <2 y; ALSFRS‐R decrease between −2 and − 5 at 12 w; ALSFRS‐R respiratory subscale 12 | Prognostic Disease stage |
| Pimozide in Patients With Neuromuscular Junction Transmission Dysfunction Due to ALS (NCT02463825) | 2 | Decremental response 5.0% in at least 1 nerve‐muscle pair | Disease stage |
| Rapamycin Treatment for ALS (NCT03359538) | 2 |
Non‐SOD1; symptom onset ≤18 mo; FVC > 70% |
Biomarker Disease stage |
| Rasagiline in ALS (NCT01786603) | 2 | Disease onset <2 y; SVC >75% | Disease stage |
|
Safety and Tolerability of Antiretroviral (Triumeq) in Patients With ALS (NCT02868580) |
2 | Nonmonogenic ALS | Biomarker |
| Safety of Urate Elevation in ALS (NCT03168711) | 2 | Serum urate <5.5 mg/dL | Biomarker |
| Safety and Efficacy of Repeated Administrations of NurOwn in ALS (NCT03280056) | 3 | Rapid progressors; disease onset <2 y; ALSFRS‐R > 25; SVC >65%; age <60 y | Prognostic Disease stage |
| The Effect of RNS60 on ALS Biomarkers (RNS60) (NCT03456882) | 2 | ALSFRS‐R bulbar and spinal score 3 for swallowing, cutting food, handling utensils, and walking; FVC >80% | Disease stage |
| Transplantation of Astrocytes, Derived From Human Embryonic Stem Cells, in ALS (NCT03482050) | 1/2 | ALSFRS >30 and diagnosis <2 y | Disease stage |
| Transplantation of Human Glial Restricted Progenitor Cells in ALS (NCT02478450) | 1/2 | FVC >65% | Disease stage |
| Two Intrathecal Doses of Autologous MSC for ALS (NCT02917681) | 1/2 | ALSFRS >30; FVC >65% | Disease stage |
Abbreviations: ALS, amyotrophic lateral sclerosis; ALSFRS‐R, ALS Functional Rating Scale‐Revised; FVC, forced vital capacity; MSC, mesenchymal stem cells; NCT, ClinicalTrials.gov identifier; SOD1, superoxide dismutase 1; SVC, slow vital capacity; UMNB, upper motor neuron burden.
Despite the considerable effort that has gone into identifying variables that predict progression or survival, only three trials (two phase 2 trials and one phase 3 trial) used prognostic criteria including the rate of ALSFRS‐R decline as well as earlier stage disease (Table 5). In the ongoing phase 3 trial evaluating repeat intrathecal administration of autologous MSC‐neurotrophic factor (MSC‐NTF) cells (NCT03280056), investigators are recruiting patients with more rapid disease progression (decline of 1 ALSFRS‐R point/month) in the 3‐month run‐in period. Additional prognostic factors in this trial include younger age (60 years) and earlier disease (ALSFRS‐R ≥ 25, SVC ≥65%, and disease onset within 2 years). The trial design is based on results of previous MSC‐NTF phase 2 trial data showing that a prespecified group of patients with more rapidly progressive disease experienced greater improvement in the rate of ALSFRS‐R score decline.49 The primary efficacy endpoint is the change in slope of the ALSFRS‐R compared with placebo by using the run‐in period ALSFRS‐R slope to estimate pretreatment‐posttreatment effects for each study participant. Secondary endpoints include levels of CSF neurotrophic factors, miRNAs (miR‐132 and miR‐146a), and inflammatory markers previously shown to change in response to MSC‐NTF treatment.28, 47 These biomarker measures are potentially valuable for assessing correlations with outcomes and may generate data that are useful for predicting responders.
Binding of extracellular TDP‐43 to CD14 has been implicated in microglial activation with resultant motor neuron toxicity in ALS.50 In an ongoing phase 2 trial, investigators are evaluating the therapeutic potential of IC14, an anti‐CD14 monoclonal antibody (Table 5). Patients included in this trial have rapidly progressive disease, defined as declines of 3 points in the 3 months prior to enrollment as well as seated FVC 65% of the predicted value. The study investigators will measure treatment‐related changes in several disease biomarkers: neurofilament, urinary p75, neurotrophin receptor, cytokines, and soluble CD14. Clinical outcomes, including changes in ALSFRS‐R, seated FVC, and quality of life, will also be determined.
In another phase 2 trial, investigators are evaluating perampanel, an α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic (AMPA) receptor antagonist and approved antiepileptic drug (Table 5). Eligible patients are those with relatively early disease; onset must have been within 2 years, FVC must be >80%, and respiratory ALSFRS‐R subscores must total at least 12. In addition, disease progression must be intermediate to rapid (ALSFRS‐R total declines of −2 to −5 over a 12‐week run‐in period). However, it appears that only functional/clinical measures (ALSFRS‐R, manual muscle test, and FVC) will be assessed in this trial. Biomarker analysis is not included in the trial record and, without a positive outcome on one or more functional measures, it is unclear whether mechanistic information will be obtained relative to the effects of Perampanel on AMPA‐mediated pathology.
In conclusion, recent advances have improved our understanding of the complex biological mechanisms of ALS, and it is hoped that further progress may ultimately explain how these mechanisms contribute to the clinical heterogeneity that remains a challenge in the design and interpretation of ALS trials. The application of individual risk stratification or prognostic modeling may reduce the clinical heterogeneity of the populations studied in trials and increase clinical trial efficiency. The use of predictive biomarkers may identify patients with ALS for whom a specific therapeutic strategy may be expected to be more successful. Finally, the application of these emerging clinical and biomarker strategies within platform trials in which several targeted therapies can be evaluated simultaneously may accelerate ALS drug development.
CONFLICT OF INTEREST
Namita Goyal has received research support from Amylyx Therapeutics, Brainstorm Cell Therapeutics, Cytokinetics, Orphazyme, and Orion and provided advisory board support for Cytokinetics, Biogen, Acceleron, and MT Pharma. James Berry has been a consultant for Clene Nanomedicine, Orion Pharmaceuticals, and Denali Therapeutics and has received research support from Anelixis Therapeutics, Amylyx Therapeutics, Brainstorm Cell Therapeutics, Biogen, Cytokinetics, MT Pharma of America, and Neuraltus Pharmaceuticals. Anthony Windebank has received support for conduct of clinical trials from BrainStorm Cell Therapeutics. Nathan Staff serves as site investigator for clinical trials funded by Brainstorm Cell Therapeutics and Orion Pharmaceuticals. Nicholas Maragakis serves on scientific advisory boards for Brainstorm Cell Therapeutics, Clene Nanomedicine and Orion Pharma. Leonard H. van den Berg serves on scientific advisory boards for the Biogen, Cytokinetics, Orion, and Sarepta. Angela Genge serves on the advisory boards of Avexis, Alexion, AL‐S Pharma, Biogen, Brainstorm, Akcea, Cytokinetics, Sanofi, Mitsubishi, and Novartis. Robert Miller has received research support from BrainStorm Cell Therapeutics, Cytokinetics, Amylyx, Neuraltus, Bioelectron and provided advisory board support for Cytokinetics, AveXis, Neuraltus and MT Pharma. Merit Cudkowicz has been a consultant for Biohaven, Biogen, Takeda, Avexis, and Revalesio and chaired DSMB for Lilly. Ralph Kern, Yael Gothelf and Chaim Lebovits are Brainstorm Cell Therapeutics employees. Robert Baloh reports no conflict of interest.
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
ACKNOWLEDGMENTS
Brainstorm Cell Therapeutics provided funding to Porterhouse Medical, a medical writing company, to assist in manuscript preparation and to coordinate editorial review by the authors. Brainstorm employees contributed to the conception and review of the manuscript along with the other authors.
Goyal NA, Berry JD, Windebank A, et al. Addressing heterogeneity in amyotrophic lateral sclerosis CLINICAL TRIALS. Muscle Nerve. 2020;62:156–166. 10.1002/mus.26801
REFERENCES
- 1. Westeneng HJ, Debray TPA, Visser AE, et al. Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol. 2018;17:423‐433. [DOI] [PubMed] [Google Scholar]
- 2. Ghasemi M, Brown RH Jr. Genetics of amyotrophic lateral sclerosis. Cold Spring Harbor Perspect Med. 2018;8(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goyal NA, Cudkowicz ME, Berry JD, et al. A systematic review of enrichment strategies for current clinical trials. In: Proceedings from the ALS 29th International Symposium on ALS/MND; December 7‐9, 2018; Glasgow. United Kingdom.. [Google Scholar]
- 4. Taylor JP, Brown RH Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maniatis S, Aijo T, Vickovic S, et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science. 2019;364:89‐93. [DOI] [PubMed] [Google Scholar]
- 6. van Rheenen W, Shatunov A, Dekker AM, et al. Genome‐wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet. 2016;48:1043‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Floeter MK, Gendron TF. Biomarkers for amyotrophic lateral sclerosis and frontotemporal dementia associated with hexanucleotide expansion mutations in C9orf72 . Front Neurol. 2018;9:1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cudkowicz ME, McKenna‐Yasek D, Sapp PE, et al. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann Neurol. 1997;41:210‐221. [DOI] [PubMed] [Google Scholar]
- 9. Nicolas A, Kenna KP, Renton AE, et al. Genome‐wide analyses identify KIF5A as a novel ALS gene. Neuron. 2018;97:1268‐1283. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vijayakumar UG, Milla V, Cynthia Stafford MY, Bjourson AJ, Duddy W, Duguez SM. A systematic review of suggested molecular strata, biomarkers and their tissue sources in ALS. Front Neurol. 2019;10:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Recabarren‐Leiva D, Alarcon M. New insights into the gene expression associated to amyotrophic lateral sclerosis. Life Sci. 2018;193:110‐123. [DOI] [PubMed] [Google Scholar]
- 12. Chiot A, Lobsiger CS, Boillee S. New insights on the disease contribution of neuroinflammation in amyotrophic lateral sclerosis. Curr Opin Neurol. 2019;32:764‐770. [DOI] [PubMed] [Google Scholar]
- 13. Atassi N, Berry J, Shui A, et al. The PRO‐ACT database: design, initial analyses, and predictive features. Neurology. 2014;83:1719‐1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Watanabe H, Atsuta N, Nakamura R, et al. Factors affecting longitudinal functional decline and survival in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:230‐236. [DOI] [PubMed] [Google Scholar]
- 15. Daghlas I, Lever TE, Leary E. A retrospective investigation of the relationship between baseline covariates and rate of ALSFRS‐R decline in ALS clinical trials. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:206‐211. [DOI] [PubMed] [Google Scholar]
- 16. Lee CT, Chiu YW, Wang KC, et al. Riluzole and prognostic factors in amyotrophic lateral sclerosis long‐term and short‐term survival: a population‐based study of 1149 cases in Taiwan. J Epidemiol. 2013;23:35‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gordon PH, Corcia P, Lacomblez L, et al. Defining survival as an outcome measure in amyotrophic lateral sclerosis. Arch Neurol. 2009;66:758‐761. [DOI] [PubMed] [Google Scholar]
- 18. Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012;CD001447. [DOI] [PubMed] [Google Scholar]
- 19. Fang T, Al Khleifat A, Meurgey JH, et al. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: a retrospective analysis of data from a dose‐ranging study. Lancet Neurol. 2018;17:416‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berry JD, Miller R, Moore DH, et al. The combined assessment of function and survival (cafs): a new endpoint for ALS clinical trials. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:162‐168. [DOI] [PubMed] [Google Scholar]
- 21. van den Berg LH, Sorenson E, Gronseth G, et al. Revised Airlie House consensus guidelines for design and implementation of ALS clinical trials. Neurology. 2019;92:e1610‐e1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Eijk RP, Eijkemans MJ, Rizopoulos D, van den Berg LH, Nikolakopoulos S. Comparing methods to combine functional loss and mortality in clinical trials for amyotrophic lateral sclerosis. Clin Epidemiol. 2018;10:333‐341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Taga A, Maragakis NJ. Current and emerging ALS biomarkers: utility and potential in clinical trials. Expert Rev Neurother. 2018;18:871‐886. [DOI] [PubMed] [Google Scholar]
- 24. Turner MR. Progress and new frontiers in biomarkers for amyotrophic lateral sclerosis. Biomark Med. 2018;12:693‐696. [DOI] [PubMed] [Google Scholar]
- 25. Majumder V, Gregory JM, Barria MA, Green A, Pal S. TDP‐43 as a potential biomarker for amyotrophic lateral sclerosis: a systematic review and meta‐analysis. BMC Neurol. 2018;18:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Waller R, Goodall EF, Milo M, et al. Serum miRNAs miR‐206, 143‐3p and 374b‐5p as potential biomarkers for amyotrophic lateral sclerosis (ALS). Neurobiol Aging. 2017;55:123‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Waller R, Wyles M, Heath PR, et al. Small RNA sequencing of sporadic amyotrophic lateral sclerosis cerebrospinal fluid reveals differentially expressed mirnas related to neural and glial activity. Front Neurosci. 2017;11:731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berry J, Aricha R, Kaspi H, et al. MicroRNA changes in the NurOwn phase 2 ALS randomized trial: relationship to neuroprotection and innate immunity. Presented at: The 17th Annual Northeast ALS Consortium Meeting; October 2‐4, 2018; Clearwater Beach. Florida.. [Google Scholar]
- 29. Petrou P, Gothelf Y, Argov Z, et al. Safety and clinical effects of mesenchymal stem cells secreting neurotrophic factor transplantation in patients with amyotrophic lateral sclerosis: results of phase 1/2 and 2a clinical trials. JAMA Neurol. 2016;73:337‐344. [DOI] [PubMed] [Google Scholar]
- 30. Joilin G, Leigh PN, Newbury SF, Hafezparast M. An overview of MicroRNAs as biomarkers of ALS. Front Neurol. 2019;10:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leoni E, Bremang M, Mitra V, et al. Combined tissue‐fluid proteomics to unravel phenotypic variability in amyotrophic lateral sclerosis. Sci Rep. 2019;9:4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Taylor AA, Fournier C, Polak M, et al. Predicting disease progression in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2016;3:866‐875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thakore NJ, Lapin BR, Pioro EP, Pooled Resource Open‐Access ALSCTC. Trajectories of impairment in amyotrophic lateral sclerosis: insights from the Pooled Resource Open‐Access ALS Clinical Trials cohort. Muscle Nerve. 2018;57:937‐945. [DOI] [PubMed] [Google Scholar]
- 34. Kuffner R, Zach N, Norel R, et al. Crowdsourced analysis of clinical trial data to predict amyotrophic lateral sclerosis progression. Nat Biotechnol. 2015;33:51‐57. [DOI] [PubMed] [Google Scholar]
- 35. Pfohl SR, Kim RB, Coan GS, Mitchell CS. Unraveling the complexity of amyotrophic lateral sclerosis survival prediction. Front Neuroinform. 2018;12:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Eijk RPA, Westeneng HJ, Nikolakopoulos S, et al. Refining eligibility criteria for amyotrophic lateral sclerosis clinical trials. Neurology. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berry JD, Taylor AA, Beaulieu D, et al. Improved stratification of ALS clinical trials using predicted survival. Ann Clin Transl Neurol. 2018;5:474‐485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li J, Zhao L, Tian L, et al. A predictive enrichment procedure to identify potential responders to a new therapy for randomized, comparative controlled clinical studies. Biometrics. 2016;72:877‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Beta‐Blocker Evaluation of Survival Trial I , Eichhorn EJ, Domanski MJ, Krause‐Steinrauf H, Bristow MR, Lavori PW. A trial of the beta‐blocker bucindolol in patients with advanced chronic heart failure. N Engl J Med. 2001;344:1659‐1667. [DOI] [PubMed] [Google Scholar]
- 40. Abe K, Itoyama Y, Sobue G, et al. Confirmatory double‐blind, parallel‐group, placebo‐controlled study of efficacy and safety of edaravone (MCI‐186) in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:610‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Takahashi F, Takei K, Tsuda K, Palumbo J. Post hoc analysis of MCI186‐17, the extension study to MCI186‐16, the confirmatory double‐blind, parallel‐group, placebo‐controlled study of edaravone in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:32‐39. [DOI] [PubMed] [Google Scholar]
- 42. Writing G, Edaravone ALSSG. Safety and efficacy of edaravone in well‐defined patients with amyotrophic lateral sclerosis: a randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2017;16:505‐512. [DOI] [PubMed] [Google Scholar]
- 43. Miller RG, Block G, Katz JS, et al. Randomized phase 2 trial of NP001‐a novel immune regulator: safety and early efficacy in ALS. Neurol Neuroimmunol Neuroinflamm. 2015;2:e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Andrews JA, Cudkowicz ME, Hardiman O, et al. VITALITY‐ALS, a phase III trial of tirasemtiv, a selective fast skeletal muscle troponin activator, as a potential treatment for patients with amyotrophic lateral sclerosis: study design and baseline characteristics. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:259‐266. [DOI] [PubMed] [Google Scholar]
- 45. Nicholson KA, Cudkowicz ME, Berry JD. Clinical trial designs in amyotrophic lateral sclerosis: does one design fit all? Neurotherapeutics. 2015;12:376‐383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lunetta C, Lizio A, Maestri E, et al. Serum C‐reactive protein as a prognostic biomarker in amyotrophic lateral sclerosis. JAMA Neurol. 2017;74:660‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Aricha R, Kaspi H, Cudkowicz M, et al. Modulation of CSF miRNAs in ALS phase 2 study participants treated with MSC‐NTF (NurOwn). Presented at: 70th Annual Meeting of the American Academy of Neurology; April 21‐27, 2018; Los Angeles. California.. [Google Scholar]
- 48. Kovalchuk MO, Heuberger J, Sleutjes B, et al. Acute effects of riluzole and retigabine on axonal excitability in patients with amyotrophic lateral sclerosis: a randomized, double‐blind, placebo‐controlled, crossover trial. Clin Pharmacol Ther. 2018;104:1136‐1145. [DOI] [PubMed] [Google Scholar]
- 49. Berry JD, Cudkowicz ME, Windebank AJ, et al. NurOwn phase 2 randomized clinical trial in ALS: safety, clinical and biomarker results. Neurology. 2019;93(24):e2294‐e2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhao W, Beers DR, Bell S, et al. TDP‐43 activates microglia through NF‐kappaB and NLRP3 inflammasome. Exp Neurol. 2015;273:24‐35. [DOI] [PubMed] [Google Scholar]
- 51. Rooney J, Burke T, Vajda A, Heverin M, Hardiman O. What does the ALSFRS‐R really measure? A longitudinal and survival analysis of functional dimension subscores in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2017;88:381‐385. [DOI] [PubMed] [Google Scholar]
- 52. Cedarbaum JM, Stambler N, Malta E, et al. The ALSFRS‐R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. 1999;169:13‐21. [DOI] [PubMed] [Google Scholar]
- 53. Kaufmann P, Levy G, Thompson JL, et al. The ALSFRSr predicts survival time in an ALS clinic population. Neurology. 2005;64:38‐43. [DOI] [PubMed] [Google Scholar]
- 54. Kimura F, Fujimura C, Ishida S, et al. Progression rate of ALSFRS‐R at time of diagnosis predicts survival time in ALS. Neurology. 2006;66:265‐267. [DOI] [PubMed] [Google Scholar]
- 55. Castrillo‐Viguera C, Grasso DL, Simpson E, Shefner J, Cudkowicz ME. Clinical significance in the change of decline in ALSFRS‐R. Amyotroph Lateral Scler. 2010;11:178‐180. [DOI] [PubMed] [Google Scholar]
- 56. Bedlack RS, Vaughan T, Wicks P, et al. How common are ALS plateaus and reversals? Neurology. 2016;86:808‐812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Czaplinski A, Yen AA, Appel SH. Forced vital capacity (FVC) as an indicator of survival and disease progression in an ALS clinic population. J Neurol Neurosurg Psychiatry. 2006;77:390‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Andrews JA, Meng L, Kulke SF, et al. Association between decline in slow vital capacity and respiratory insufficiency, use of assisted ventilation, tracheostomy, or death in patients with amyotrophic lateral sclerosis. JAMA Neurol. 2018;75:58‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fortis S, Corazalla EO, Wang Q, Kim HJ. The difference between slow and forced vital capacity increases with increasing body mass index: a paradoxical difference in low and normal body mass indices. Respir Care. 2015;60:113‐118. [DOI] [PubMed] [Google Scholar]
- 60. Shefner JM, Liu D, Leitner ML, et al. Quantitative strength testing in ALS clinical trials. Neurology. 2016;87:617‐624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Andres PL, Skerry LM, Munsat TL, et al. Validation of a new strength measurement device for amyotrophic lateral sclerosis clinical trials. Muscle Nerve. 2012;45:81‐85. [DOI] [PubMed] [Google Scholar]
- 62. Rutkove SB, Caress JB, Cartwright MS, et al. Electrical impedance myography as a biomarker to assess ALS progression. Amyotroph Lateral Scler. 2012;13:439‐445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain. 2008;131:1540‐1550. [DOI] [PubMed] [Google Scholar]
- 64. Vucic S, Kiernan MC. Transcranial magnetic stimulation for the assessment of neurodegenerative disease. Neurotherapeutics. 2017;14:91‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang J, Kothalkar PV, Kim M, et al. Automatic prediction of intelligible speaking rate for individuals with ALS from speech acoustic and articulatory samples. Int J Speech Lang Pathol. 2018;20:669‐679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Grolez G, Moreau C, Danel‐Brunaud V, et al. The value of magnetic resonance imaging as a biomarker for amyotrophic lateral sclerosis: a systematic review. BMC Neurol. 2016;16:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Antonescu F, Adam M, Popa C, Tuta S. A review of cervical spine MRI in ALS patients. J Med Life. 2018;11:123‐127. [PMC free article] [PubMed] [Google Scholar]
- 68. Bozik ME, Mitsumoto H, Brooks BR, et al. A post hoc analysis of subgroup outcomes and creatinine in the phase III clinical trial (EMPOWER) of dexpramipexole in ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:406‐413. [DOI] [PubMed] [Google Scholar]
- 69. O'Reilly EJ, Liu D, Johns DR, et al. Serum urate at trial entry and ALS progression in EMPOWER. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:120‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shepheard SR, Chataway T, Schultz DW, Rush RA, Rogers ML. The extracellular domain of neurotrophin receptor p75 as a candidate biomarker for amyotrophic lateral sclerosis. PLoS One. 2014;9:e87398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shepheard SR, Wuu J, Cardoso M, et al. Urinary p75(ECD): a prognostic, disease progression, and pharmacodynamic biomarker in ALS. Neurology. 2017;88:1137‐1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lu CH, Macdonald‐Wallis C, Gray E, et al. Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis. Neurology. 2015;84:2247‐2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lu CH, Petzold A, Topping J, et al. Plasma neurofilament heavy chain levels and disease progression in amyotrophic lateral sclerosis: insights from a longitudinal study. J Neurol Neurosurg Psychiatry. 2015;86:565‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Steinacker P, Huss A, Mayer B, et al. Diagnostic and prognostic significance of neurofilament light chain NF‐L, but not progranulin and S100B, in the course of amyotrophic lateral sclerosis: data from the German MND‐net. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:112‐119. [DOI] [PubMed] [Google Scholar]
- 75. Steinacker P, Verde F, Fang L, et al. Chitotriosidase (CHIT1) is increased in microglia and macrophages in spinal cord of amyotrophic lateral sclerosis and cerebrospinal fluid levels correlate with disease severity and progression. J Neurol Neurosurg Psychiatry. 2018;89:239‐247. [DOI] [PubMed] [Google Scholar]
- 76. Henkel JS, Beers DR, Wen S, et al. Regulatory T‐lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med. 2013;5:64‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cooper‐Knock J, Green C, Altschuler G, et al. A data‐driven approach links microglia to pathology and prognosis in amyotrophic lateral sclerosis. Acta Neuropathol Commun. 2017;5:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Peters OM, Ghasemi M, Brown RH Jr. Emerging mechanisms of molecular pathology in ALS. J Clin Invest. 2015;125:1767‐1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bali T, Self W, Liu J, et al. Defining SOD1 ALS natural history to guide therapeutic clinical trial design. J Neurol Neurosurg Psychiatry. 2017;88:99‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. El Fatimy R, Li S, Chen Z, et al. MicroRNA‐132 provides neuroprotection for tauopathies via multiple signaling pathways. Acta Neuropathol. 2018;136:537‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Freischmidt A, Muller K, Ludolph AC, Weishaupt JH. Systemic dysregulation of TDP‐43 binding microRNAs in amyotrophic lateral sclerosis. Acta Neuropathol Commun. 2013;1:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Iyer A, Zurolo E, Prabowo A, et al. MicroRNA‐146a: a key regulator of astrocyte‐mediated inflammatory response. PLoS One. 2012;7:e44789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lu LF, Boldin MP, Chaudhry A, et al. Function of miR‐146a in controlling Treg cell‐mediated regulation of Th1 responses. Cell. 2010;142:914‐929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Saba R, Sorensen DL, Booth SA. MicroRNA‐146a: a dominant, negative regulator of the innate immune response. Front Immunol. 2014;5:578. [DOI] [PMC free article] [PubMed] [Google Scholar]