

Abstract
Background and Aims
Genetically modified mice have been used extensively to study human disease. However, the data gained are not always translatable to humans because of major species differences. Liver‐humanized mice (LHM) are considered a promising model to study human hepatic and systemic metabolism. Therefore, we aimed to further explore their lipoprotein metabolism and to characterize key hepatic species‐related, physiological differences.
Approach and Results
Fah−/−, Rag2−/−, and Il2rg−/− knockout mice on the nonobese diabetic (FRGN) background were repopulated with primary human hepatocytes from different donors. Cholesterol lipoprotein profiles of LHM showed a human‐like pattern, characterized by a high ratio of low‐density lipoprotein to high‐density lipoprotein, and dependency on the human donor. This pattern was determined by a higher level of apolipoprotein B100 in circulation, as a result of lower hepatic mRNA editing and low‐density lipoprotein receptor expression, and higher levels of circulating proprotein convertase subtilisin/kexin type 9. As a consequence, LHM lipoproteins bind to human aortic proteoglycans in a pattern similar to human lipoproteins. Unexpectedly, cholesteryl ester transfer protein was not required to determine the human‐like cholesterol lipoprotein profile. Moreover, LHM treated with GW3965 mimicked the negative lipid outcomes of the first human trial of liver X receptor stimulation (i.e., a dramatic increase of cholesterol and triglycerides in circulation). Innovatively, LHM allowed the characterization of these effects at a molecular level.
Conclusions
LHM represent an interesting translatable model of human hepatic and lipoprotein metabolism. Because several metabolic parameters displayed donor dependency, LHM may also be used in studies for personalized medicine.
Abbreviations
- APO
apolipoprotein
- CETP
cholesteryl ester transfer protein
- DHM
double humanized mouse
- FRG
Fah −/−, Rag2 −/−, and Il2rg −/−
- FRGN
FRG NOD
- haPG
human aortic proteoglycan
- HDL
high‐density lipoprotein
- KO
knockout
- LDL
low‐density lipoprotein
- LDLR
LDL receptor
- LHM
liver‐humanized mice
- LMM
liver‐murinized mice
- Lp(a)
lipoprotein (a)
- LXR
liver X receptor
- NOD
nonobese diabetic
- PCSK9
proprotein convertase subtilisin/kexin type 9
- PLTP
phospholipid transfer protein
- VLDL
very low‐density lipoprotein
Genetically modified mice have been used extensively used as models of comparative physiology to study atherosclerosis and cardiometabolic diseases in general. However, the data gained from these studies are not always translatable to the clinic because of major species differences, particularly in hepatic metabolism. Therefore, the development of translatable models to study the human condition is an essential step for the future of basic and preclinical research. In addition, the development and integration of personalized medicine into health care requires access to, standardization, and validation of personalized and translatable models to test and refine innovative therapeutic protocols.
Liver‐humanized mice (LHM), in which the liver is repopulated with human hepatocytes, have been proposed as an improved model to study human liver and lipoprotein metabolism.1 One of the available mouse models is the FRG triple knockout (KO), characterized by the lack of fumarylacetoacetate hydrolase (Fah) and severe immunodeficiency (Rag2−/− and Il2rg−/−).2 After transplantation with human hepatocytes, liver cells bearing functional Fah/FAH are selected,2 resulting in liver chimerism of at least 80%. Moreover, backcrossing FRG‐KO mice with the nonobese diabetic (NOD) strain yields FRGN mice, which are more efficient for xenografts and show an improved humanization.3, 4 These chimeric models offer the unique opportunity to gain insights in the species‐related differences between human and mouse hepatic metabolism. For example, it is well known that cholesterol in low‐density lipoprotein (LDL) represents the major fraction of plasma cholesterol in humans, whereas mice are primarily a high‐density lipoprotein (HDL) species. However, the underlying reasons continue to be a matter of investigation. Differences are found not only in the distribution of lipoproteins, but also in their properties and in the proteins modifying them. Cholesteryl ester transfer protein (CETP), for instance, is expressed by humans but not in mice, and exchanging cholesteryl esters from HDL particles for triglycerides in very low‐density lipoprotein (VLDL) and LDL is considered the main reason for the species differences observed in cholesterol lipoprotein distribution.5, 6 FRG‐KO mice engrafted with human hepatocytes were reported to exhibit a human‐like lipoprotein profile1 and CETP.7 However, these latter results contradict other studies in humans showing Kupffer cells, rather than hepatocytes, as the main source for CETP.8 Therefore, it is crucial to assess thoroughly which metabolic features are present in humans, but not in mice, and are subsequently acquired by LHM.
An aspect of LHM that is intriguing in light of the future demands of personalized medicine is the ability to engraft them with human hepatocytes deriving from different donors. Because liver is a master regulator of metabolism, LHM may be useful to study individual disposition to disease, gender differences, and impact of risk or protective genetic variants.
Therefore, we aimed to validate LHM as a useful and translatable model of human lipoprotein metabolism by focusing on the liver‐related species differences responsible for the humanization of the lipoprotein profile, and the variability of response among different human donors. In addition, LHM are used extensively in drug discovery and development, demonstrating an improved predictive capacity over transgenic or wild‐type mice.9 Hence, we investigated whether LHM could also be used to predict the human response to novel therapeutic strategies, such as liver X receptor (LXR) agonism, which showed great potential when tested in classical mouse models.
Experimental Procedures
Study Design
The objective of this study was to corroborate LHM as a relevant model to study human lipoprotein metabolism by thorough characterization of the related physiological differences limiting the translatability to the human condition. The primary purpose was to assess which human features are acquired by this model and whether these are dependent on the human donor. Human and mouse reference samples were tested in all analyses, and FRGN mice transplanted with NOD mouse hepatocytes (liver‐murinized mice, LMM) served as negative control. To determine lipoprotein metabolism in LHM, we analyzed the lipid lipoprotein composition, CETP, and phospholipid transfer protein (PLTP/Pltp) expression at different levels, apolipoprotein (APO) B mRNA editing in liver, and apolipoproteins of lipoproteins isolated from serum. We also assessed LDL receptor (LDLR) in liver and circulating levels of human or mouse proprotein convertase subtilisin/kexin type 9 (PCSK9). Lipoprotein properties such as human aortic proteoglycan (haPG) binding or serum cholesterol efflux capacity were measured to understand the relevance of the changes in lipoprotein profile and metabolism in LHM. To investigate the use of LHM for elucidating the effects of LXR stimulation in humans, LHM (donors M1 and M3) were treated by gavage for 3 days (four treatments) with vehicle (methylcellulose 0.5% wt/vol) or 30 mg/kg/day of the LXR agonist GW3965. In LHM‐M1 animals, lipoprotein and intrahepatic metabolism were investigated, the latter by measurement of liver cholesterol precursors and composition, bile acid precursors and composition, and triglycerides. Moreover, gene expression was assessed with both RNA sequencing and real‐time quantitative PCR analyses to identify the effects of LXR stimulation in human hepatocytes. Liver histology was analyzed in the LHM‐M3 animals. Analyses were performed in a blinded fashion when possible. Within each group, outlier rejection was performed prospectively by the robust regression and outlier removal method (Q = 5%) for (HDL/LDL) cholesterol or VLDL cholesterol. Samples were analyzed in duplicate or triplicate, and each test was performed at least twice. More details are available in the Supporting Information.
Generation of the FRGN Strain and Hepatocyte Transplantation
FRGN mice were repopulated with hepatocytes as described previously.2, 3 Male mice were engrafted with primary hepatocytes from male NOD mouse (LMM, n = 7) and human donor A (LHM‐A, n = 4), donor B (LHM‐B, n = 3), female donor F1 (LHM‐F1, n = 11), female donor F2 (LHM‐F2, n = 12), male donor M1 (LHM‐M1, n = 15), male donor M2 (LHM‐M2, n = 5), and male donor M3 (LHM‐M3, n = 4). Hepatocytes and serum from human donors A and B were obtained from the Division of Surgery, Department of Clinical Science, Intervention and Technology, Karolinska Institute at Karolinska University Hospital Huddinge (Stockholm, Sweden), whereas human donor F1, F2, M1, M2 and M3 hepatocytes were obtained from commercially available sources. FRGN mice repopulated with primary hepatocytes were fed a Pico Lab High Energy Mouse Diet 5LJ5 (LabDiet; W.F. Fisher & Son, Somerville, NJ). Only mice with human hepatocyte repopulation greater than 70% (corresponding to circulating levels of human albumin > 3.5 mg/mL) were used in this study. The double humanized mouse (DHM) was engrafted with human hepatocytes from donor F2 and hematopoietic stem cells (DHM‐F2, n = 1), as described.10 Chimeric animals were generated at Yecuris Corp. (Tualatin, OR) or Karolinska Institute (Stockholm, Sweden). This study complies with the Declaration of Helsinki, and ethical approvals (2010/678‐31/3, S82‐13, and 2017/269‐31) were obtained from local authorities. All animal work was conducted according to approved Institutional Animal Care and Use Committee (Yecuris Corp.) protocol DN000024 and NIH OLAW assurance #A4664‐01. The protocols follow the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Quantification of Serum Lipoprotein Lipids
Lipoproteins were separated from 2.5 μL serum by size‐exclusion chromatography, and lipids were quantified with a real‐time detection method as previously described.11, 12
APOB/Apob mRNA Editing Assay
We developed in collaboration with TATAA Biocenter (Gothenburg, Sweden) the qPCR test of APOB/Apob probe SNP Assay (Cat# Apob500SNP; TATAA Biocenter). For details see the Supporting Information.
Isolation of Lipoproteins in Deuterium Oxide/Sucrose Buffer
Lipoproteins from 50 μL‐pooled serum samples were separated by sequential differential micro‐ultracentrifugation in D2O/sucrose.13 To keep the initial proportion of lipoprotein concentration, each fraction was limited to the same serum volume (50 μL).
Statistical Analysis
Due to different group sizes and a high prevalence of variables not normally distributed, we used a nonparametric analysis. Data are expressed as median and interquartile range, with the exception of the lipoprotein chromatograms and data from pooled samples (expressed as mean ± SEM). To avoid false‐positive results from the multiple testing, statistical significance was set at the level of α < 0.01 after Holm‐Bonferroni adjustment (α < 0.05 for post‐hoc analyses). In the LXR study, in which fewer comparisons were tested, the α‐value was kept to 0.05. Kruskal‐Wallis test followed by Dunn’s multiple comparison test was used for multigroup comparisons. Mann‐Whitney U Test was used for two‐group comparisons. Spearman rank correlation analysis was used to determine associations between continuous and ordinal parameters. Statistical analyses and graphs were performed using Statistica (TIBCO Software Inc., Palo Alto, CA) and GraphPad Prism (GraphPad Software, Inc., La Jolla, CA).
Results
Lipoprotein Distribution in FRGN Mice is Humanized After Engraftment With Human Hepatocytes
It is well known that humans transport plasma cholesterol primarily in LDL particles, whereas mice use HDL particles. Peripheral lipoprotein profiles were analyzed in LHM to understand how they resemble the human donor and to determine what contribution is made by the murine host. FRGN mice were transplanted with hepatocytes from human donor A (LHM‐A, n = 4), human donor B (LHM‐B, n = 3) and NOD mouse (LMM, n = 7), and cholesterol lipoprotein profiles were compared. As shown in Fig. 1A, lipoproteins from donor A and LHM‐A clearly displayed similar LDL profiles, with a LDL/HDL‐cholesterol ratio of 1.1 and 9.2, respectively. In contrast, LMM generated lipoprotein profiles that were HDL‐dominant (LDL/HDL cholesterol close to 0). As observed in donor A, the LDL fraction represented most of the cholesterol in LHM‐A, whereas the HDL fraction was proportionally lower. Nevertheless, the cholesterol lipoprotein profile from LHM‐A was much more similar to donor A than that of LMM. Similar results were observed for donor B and LHM‐B (Supporting Fig. S1).
Figure 1.

Cholesterol lipoprotein profile in LHM is humanized and depends on the human donor’s hepatocytes. Cholesterol lipoprotein profiles were assessed by size‐exclusion chromatography. (A) Cholesterol profile of the human donor A (n = 1), of the LHM engrafted with his primary hepatocytes (LHM‐A, n = 4), and of LMM (n = 7). Cholesterol profiles of LHM transplanted with F1 (n = 11) (B), F2 (n = 12) (C), M1 (n = 8) (D), or M2 (n = 5) (E) hepatocytes. The profiles represent the mean chromatograms, and the shaded area around them represents the SEM. See Supporting Fig. S1 for the cholesterol profiles of human donor B and LHM‐B, Supporting Fig. S2 for the correlation between liver repopulation and cholesterol phenotype, and Supporting Table S1 for genetic data of human donors.
Cholesterol Lipoprotein Profile in LHM Is Dependent on the Human Donor
To extend our investigation on donor specificity, 36 FRGN male mice were divided into four groups and repopulated with primary human hepatocytes from two female (F1 and F2) and two male donors (M1 and M2). These mice were highly humanized (>70%), and a summary of the genetic data of human donors is available in Supporting Table S1. As shown in Fig. 1B‐E, each group displayed a typical human lipoprotein profile with higher proportion of cholesterol in LDL than in HDL. Moreover, the profiles were highly consistent with small intragroup variability. For instance, LHM‐M2 showed a profile characteristic of a subject with a dyslipidemia (i.e., equal amount of cholesterol in VLDL and LDL). In addition, the differences originated from the human donors were statistically significant (P < 0.01) for all other main lipid lipoprotein components (Table 1). Among all of the groups, LHM‐F2 displayed the highest amount of serum total cholesterol (4.7 mmol/L), in contrast to LHM‐F1, which showed the lowest total cholesterol content (2.6 mmol/L). Importantly, the lipid levels observed were within the normal human physiological range (and higher compared with wild‐type mice). It is necessary to emphasize that the differences and precision of the lipoprotein composition were clearly dependent on the hepatocytes of the human donor, and not directly correlative of the level of humanization (Supporting Fig. S2).
Table 1.
Lipid Levels in Lipoproteins of LHM Receiving Hepatocytes From Different Donors
| LHM‐F1 (n = 11) | LHM‐F2 (n = 12) | LHM‐M1 (n = 8) | LHM‐M2 (n = 5) | Kruskal‐Wallis Test | |
|---|---|---|---|---|---|
| Total Cholesterol (mmol/L) | 2.6 (0.5) | 4.7 (1.0) | 3.1 (0.9) | 4.0 (0.6) | P < 0.01 |
| VLDL/remnants‐C (mmol/L) | 0.5 (0.2) | 0.4 (0.1) | 0.4 (0.3) | 1.5 (0.4) | P < 0.01 |
| LDL‐C (mmol/L) | 1.7 (0.4) | 2.9 (1.0) | 2.4 (0.6) | 2.4 (0.5) | P < 0.01 |
| HDL‐C (mmol/L) | 0.4 (0.2) | 1.4 (1.1) | 0.2 (0.2) | 0.3 (0.1) | P < 0.01 |
| (LDL/HDL)‐C | 4 (2) | 2 (2) | 17 (19) | 11 (5) | P < 0.01 |
| Total CE (mmol/L) | 2.1 (0.6) | 3.8 (0.8) | 2.4 (0.7) | 3.2 (0.2) | P < 0.01 |
| VLDL/remnants‐CE (mmol/L) | 0.4 (0.2) | 0.3 (0.1) | 0.3 (0.2) | 1.1 (0.3) | P < 0.01 |
| LDL‐CE (mmol/L) | 1.3 (0.4) | 2.3 (0.7) | 1.9 (0.5) | 1.9 (0.3) | P < 0.01 |
| HDL‐CE (mmol/L) | 0.3 (0.1) | 1.1 (0.9) | 0.1 (0.2) | 0.3 (0.1) | P < 0.01 |
| Total FC (mmol/L) | 0.5 (0.1) | 0.9 (0.2) | 0.7 (0.2) | 0.9 (0.4) | P < 0.01 |
| VLDL/remnants‐FC (mmol/L) | 0.1 (0.0) | 0.1 (0.0) | 0.1 (0.1) | 0.4 (0.1) | P < 0.01 |
| LDL‐FC (mmol/L) | 0.3 (0.1) | 0.6 (0.2) | 0.5 (0.1) | 0.5 (0.2) | P < 0.01 |
| HDL‐FC (mmol/L) | 0.1 (0.1) | 0.3 (0.2) | 0.1 (0.1) | 0.1 (0.0) | P < 0.01 |
| Total TG (mmol/L) | 0.8 (0.2) | 1.2 (0.4) | 0.8 (0.4) | 0.9 (0.1) | P < 0.01 |
| VLDL/remnants‐TG (mmol/L) | 0.3 (0.1) | 0.4 (0.2) | 0.3 (0.2) | 0.6 (0.0) | P < 0.01 |
| LDL‐TG (mmol/L) | 0.3 (0.1) | 0.7 (0.2) | 0.4 (0.2) | 0.3 (0.0) | P < 0.01 |
| HDL‐TG (mmol/L) | 0.1 (0.1) | 0.1 (0.1) | 0.1 (0.0) | 0.1 (0.0) | P = 0.24 |
| Total PL (mmol/L) | 1.8 (0.2) | 3.5 (0.6) | 1.9 (0.5) | 2.5 (0.8) | P < 0.01 |
| VLDL/remnants‐PL (mmol/L) | 0.2 (0.1) | 0.2 (0.1) | 0.2 (0.2) | 0.9 (0.2) | P < 0.01 |
| LDL‐PL (mmol/L) | 0.8 (0.2) | 1.1 (0.5) | 1.0 (0.3) | 1.3 (0.2) | P < 0.01 |
| HDL‐PL (mmol/L) | 0.8 (0.3) | 2.1 (0.7) | 0.6 (0.4) | 0.7 (0.2) | P < 0.01 |
Data are presented as the median and interquartile range. Kruskal‐Wallis test followed by Dunn’s multiple comparison test showed significant differences (P < 0.01) in all lipoprotein lipids (except for HDL triglycerides).
Abbreviations: C, cholesterol; CE, cholesteryl esters; FC, free (or unesterified) cholesterol; PL, phospholipids; TG, triglycerides.
Humanization of Lipoprotein Profile Is Independent of CETP
Plasma lipid transfer proteins, such as CETP and PLTP, are liver‐derived proteins that are important for lipoprotein metabolism. Because CETP is not expressed in mice, it is believed that its presence in humans is the main reason for the typical LDL distribution of cholesterol.5 To understand the drivers of humanization of lipoprotein profiles in LHM, we assessed the hepatic mRNA and protein expression of CETP, as well as circulating protein and activity. Surprisingly, we did not detect CETP mRNA in liver, nor protein expression in liver or in circulation (Supporting Table S2 and Fig. S3), even after using two different primer pairs for real‐time quantitative PCR, two different CETP antibodies, and two different enzyme‐linked immunosorbent assay kits. Moreover, CETP activity in circulation was present only in humans and not in LHM or LMM (Supporting Table S2). We had the possibility to analyze the serum from one FRGN mouse co‐transplanted with human hematopoietic stem cells and hepatocytes (DHM) from donor F2 (DHM‐F2), which presents human Kupffer cells in the chimeric liver.10 Therefore, as a proof of concept that CETP does not originate from hepatocytes, we analyzed CETP activity in DHM‐F2. As shown in Fig. 2, DHM‐F2 exhibited CETP activity in circulation, and its levels were about 50% of those observed in the human reference sample. To understand the impact of the appearance of CETP activity, we analyzed the cholesterol lipoprotein profile of DHM‐F2 and compared it with the LHM‐F2 group, whose mice were bearing only hepatocytes from the same donor. Consistently with the known function of CETP, DHM‐F2 exhibited higher levels of cholesterol in the APOB‐containing particles (i.e., VLDL and LDL) and lower levels of cholesterol in the HDL fraction (Fig. 2).
Figure 2.
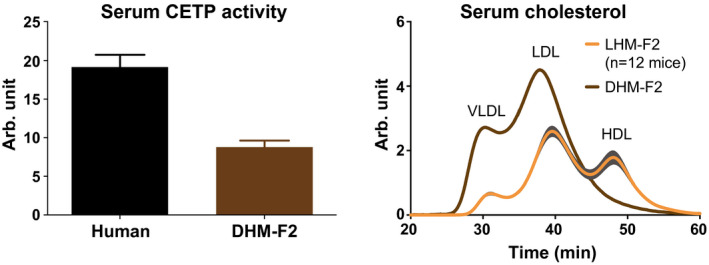
The chimeric mouse model of human liver with hepatocytes and Kupffer cells exhibits CETP activity and changes in the distribution of lipoprotein cholesterol. DHM was transplanted with human hepatocytes from donor F2 and hematopoietic stem cells (DHM‐F2, n = 1). CETP activity was analyzed with a fluorometric assay in kinetic measurement. Data are presented as mean and SEM from three different triplicates. Compared with LHM‐F2 (n = 12), DHM‐F2 also exhibited changes in cholesterol levels that were compatible with CETP activity in circulation. Cholesterol profiles represent the mean chromatograms, and the shaded area around them represents SEM. See Supporting Fig. S3 and Supporting Table S2 for CETP expression in LHM.
PLTP transfers phospholipids from triglyceride‐rich lipoproteins to HDL. LHM expressed both human PLTP and mouse Pltp mRNA in liver, although the human transcript was at low levels (Supporting Fig. S4). In LHM, PLTP activity in circulation was at comparable levels to those observed in the human reference sample, whereas it was lower compared to LMM (−57% ± 15%) and to the wild‐type mouse reference (−67% ± 12%) (Table 2).
Table 2.
PLTP Activity in Circulation of LHM
| PLTP Activity (Arb. Unit) | |
|---|---|
| Human | 131.3 ± 3.6 |
| LHM‐F1 | 115.5 ± 12.3 |
| LHM‐F2 | 286.2 ± 3.8 |
| LHM‐M1 | 126.9 ± 3.6 |
| LHM‐M2 | 108.4 ± 4.2 |
| LMM | 543.7 ± 48.7 |
| Mouse | 707.5 ± 30.4 |
Sera were pooled based on the group, and phospholipid transfer was analyzed with a fluorometric assay in kinetic measurement. Data are presented as mean ± SEM from three different triplicates. Human and mouse sera are reference samples. LMM served as negative control. See Supporting Fig. S4 for PLTP/Pltp expression in LHM.
LHM Express Higher Levels of Circulating APOB100 and PCSK9, and Lower Hepatic LDLR Compared With LMM
Because the humanization of LHM lipoprotein profile does not require CETP, we hypothesized that the distribution of cholesterol predominantly in LDL particles could result from a higher presence of APOB100 in circulation. Editing of APOB mRNA to generate APOB48 does not occur in human liver,14 and lipoproteins containing APOB100 instead of APOB48 have higher half‐life in circulation.15 We therefore hypothesized that hepatic APOB mRNA editing would not be present in LHM. Hence, a real‐time quantitative PCR–based assay to evaluate the mRNA editing of both APOB and Apob in tandem was specifically developed. As expected, we observed a markedly lower editing of liver APOB mRNA in LHM compared with LMM (P < 0.01) (Fig. 3A). Subsequently, we investigated the APO composition in lipoproteins isolated from serum, paying particular attention to APOB. As shown in Fig. 3B for LDL and HDL (and in Supporting Fig. S5 for VLDL), we isolated the major APOs specific for lipoprotein fractions. We could identify both APOB100 and, to a lesser extent, APOB48 in all LDL samples from LHM (band quantification in Supporting Fig. S6). However, in LHM the APOB100 levels were more similar to those of the human reference sample than to the mouse reference and LMM (Fig. 3B and Supporting Fig. S6). Moreover, the distribution of APOs was donor‐specific: APOA1 (band at 25‐30 kDa) was present in the HDL from all samples, but only present in the LDL from LHM‐F2 (Fig. 3B), suggesting the presence of large and buoyant HDL that float at the density of LDL particles (1.019‐1.063 g/mL) specifically for this human donor. In LHM, we could also identify lipoprotein (a) (Lp[a]) in circulation, which is expressed in humans but not in mice (Supporting Fig. S7).
Figure 3.
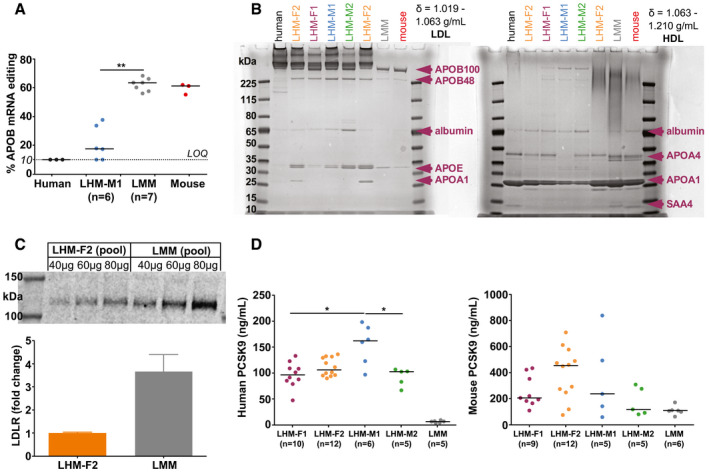
APOB phenotype in LHM. (A) Hepatic APOB mRNA editing quantified by real‐time quantitative PCR–based assay. Data are presented as the median (depicted by the line) of scattered dots. Mann‐Whitney U Test; **P < 0.01. Human and mouse served as reference samples. Samples below the limit of quantification (LOQ, 10%) were set equal to the LOQ in both statistical and graphical analyses. (B) Apolipoprotein composition of LDL and HDL (see Supporting Fig. S5 for VLDL and Supporting Fig. S6 for the quantification of APOB bands in LDL). Lipoproteins were isolated from pooled sera and underwent 4%‐12% Bis‐Tris gel electrophoresis and staining with Coomassie G‐250. Human and mouse sera are reference samples, and LMM served as negative control. Major apolipoproteins were identified by molecular weight. See Supporting Fig. S7 for Lp(a) in LHM. (C) Hepatic LDLR expression. Data are presented as mean and SEM from three different replicates. Quantification was done by western blot with sample titration using an antibody reactive to human and mouse LDLR. (D) Human or mouse circulating PCSK9 quantified by enzyme‐linked immunosorbent assay. Data are presented as the median (depicted by the line) of scattered dots. Kruskal‐Wallis test was followed by multiple comparisons; *P < 0.05. LMM served as specificity control in the human PCSK9 kit. Because of the lack of a common calibrator, concentrations from human or mouse PCSK9 kits could not be converted in molar units, annulling their direct comparison. Abbreviation: SAA, serum amyloid A.
Knowing the lower hepatic LDL clearance in humans compared with mice,16 we quantified the protein expression of human and mouse LDLR in the liver from LHM and LMM. The LHM‐F2 pool had approximately 75% lower levels of LDLR in liver compared with the LMM pool (Fig. 3C). Because PCSK9 regulates the protein expression of LDLR in both humans and mice, we assessed the circulating levels of human or mouse PCSK9 in LHM and LMM. In LHM samples, the levels of human PCSK9 were donor‐dependent. On the contrary, mouse PCSK9 levels were similar between LHM and LMM, with the loss of donor dependency (Fig. 3D). As result, LHM exhibited higher total levels of circulating PCSK9 than LMM, which contributes to the explanation of the lower expression of hepatic LDLR in LHM.
LHM Lipoproteins Bind Human Aortic Proteoglycans Similarly to Human Lipoproteins
To understand the relevance of the changes in lipoprotein profiles and to further characterize the lipoproteins of LHM, we assessed in vitro their binding to haPG. This property can predict the atherogenic potential of lipoproteins in circulation, as haPG binding in vivo is the first step in the response‐to‐retention hypothesis of atherogenesis.17, 18 LMM, which exhibited low levels of APOB100 in circulation, displayed almost no haPG binding, whereas LHM displayed haPG binding similar to that observed in humans (Fig. 4). Again, donor‐related differences were present and were modified after correction for serum total cholesterol (Fig. 4), suggesting the presence of a different number of lipoproteins with different characteristics depending on the human donor.
Figure 4.
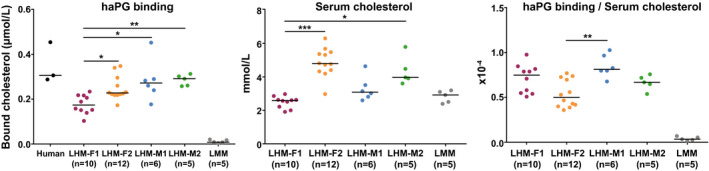
LHM lipoproteins bind haPG similarly to human lipoproteins. One microliter of serum was added to the immobilized proteoglycans, and after incubation the amount of bound cholesterol was determined. To correct for differences in serum cholesterol in each individual sample, the bound cholesterol was divided for the concentration of serum cholesterol. Data are presented as the median (depicted by the line) of scattered dots. Kruskal‐Wallis test was followed by multiple comparisons test; *P < 0.05, **P < 0.01, and ***P < 0.001. Human sera are reference samples; LMM served as negative control.
LXR Activation Determines an Unfavorable Phenotype of Circulating and Hepatic Lipids in LHM
The human trial with the LXR agonist BMS‐852927 showed negative outcomes regarding plasma lipoprotein and liver lipids.19 This result was likely attributable to species‐related differences between humans and mice. To assess the potential of LHM as a model in which to predict the pharmacodynamics of compounds aimed to target nuclear receptors in human liver, we treated LHM‐M1 mice with the LXR agonist GW3965. As shown in Fig. 5A, LXR activation in LHM‐M1 induced a substantial increase of serum cholesterol and triglycerides (P < 0.01), as reported in hamster and cynomolgus monkey.20 Interestingly, the highest proportional increase for cholesterol was not observed in the LDL fraction (+144%), but in the VLDL fraction (+309%). To investigate the reasons underlying the increase in serum cholesterol following LXR stimulation, we explored LDLR expression. LDLR mRNA in liver decreased 60% after treatment with GW3965 (Supporting Table S3), although its protein levels were unchanged (Supporting Fig. S8). PCSK9 is an LXR‐independent regulator of LDLR (and APOB) catabolism, and its hepatic mRNA or circulating protein levels did not change after treatment with GW3965 (Supporting Table S3 and Fig. S9). MYLIP (myosin regulatory light chain interacting protein, also known as IDOL, inducible degrader of LDLR) is an LXR‐target gene degrading LDLR21; however, no differences were found at the hepatic mRNA level in LHM‐M1 after LXR stimulation (Supporting Table S3). CETP is another LXR target gene,20 but we could not detect any mRNA (Supporting Table S3) or protein (Supporting Fig. S3) in the liver of LHM‐M1 treated with GW3965, eliminating the possibility of evaluating any effect on this gene. As shown in Fig. 5B, we assessed the haPG binding of lipoproteins, which was higher after LXR stimulation. Nonetheless, lipoproteins in LHM‐M1 treated with GW3965 maintained the same binding capacity after correction for serum cholesterol. We also analyzed the serum cholesterol efflux capacity (Fig. 5C), as LXR activation is known to affect this property in mice.22 GW3965 stimulation increased (P = 0.05) the adenosine triphosphate binding cassette subfamily A member 1 (ABCA1)‐mediated efflux to lipid‐poor APOA1 or nascent/preβ‐HDL. Aqueous diffusion, the concentration gradient‐driven cholesterol transport toward mature HDL, was significantly increased in LHM‐M1 treated with GW3965 (P < 0.01).
Figure 5.
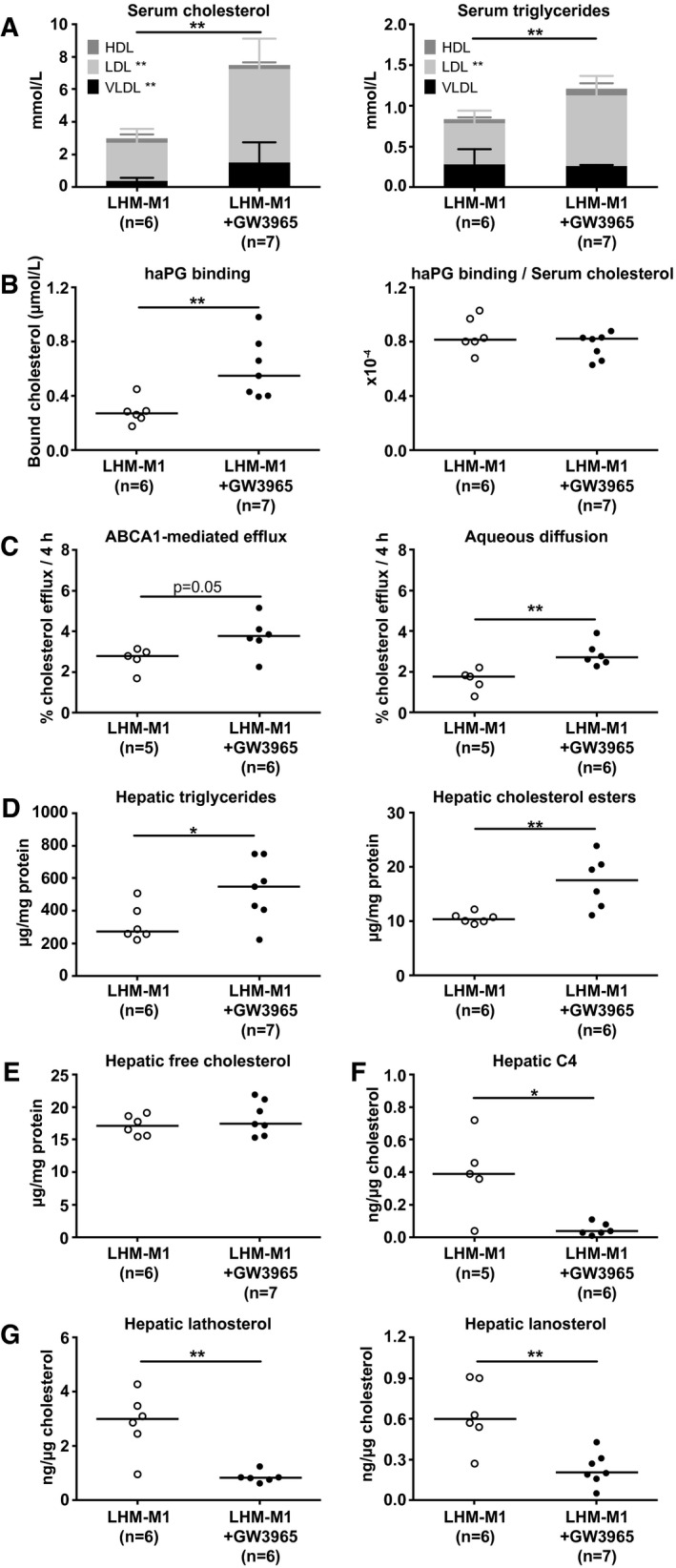
LXR stimulation by GW3965 determines an unfavorable phenotype of circulating and hepatic lipids in LHM‐M1. FRGN mice were repopulated with hepatocytes from the human donor M1 (LHM‐M1) and treated with either vehicle (n = 6) or GW3965 (n = 7). (A) Cholesterol and triglyceride lipoprotein distribution in serum. Bar data are presented as the median and interquartile range. (B) Lipoprotein binding to haPG. Data are presented as the median (depicted by the line) of scattered dots. (C) Serum cholesterol efflux capacity through adenosine triphosphate binding cassette subfamily A member 1 (ABCA1) or aqueous diffusion. Data are presented as the median (depicted by the line) of scattered dots. (D) Triglycerides and cholesteryl esters in liver. Data are presented as the median (depicted by the line) of scattered dots. (E) Free (or unesterified) cholesterol in the liver. Data are presented as the median (depicted by the line) of scattered dots. (F) 7alpha‐hydroxy‐4‐cholesten‐3‐one (C4) in liver. Data are presented as the median (depicted by the line) of scattered dots. (G) Cholesterol precursors lathosterol and lanosterol in the liver. Data are presented as the median (depicted by the line) of scattered dots. All data were analyzed by Mann‐Whitney U Test; *P < 0.05 and **P < 0.01. See also Supporting Fig. S8 (hepatic LDLR), Supporting Fig. S9 (circulating PCSK9), Supporting Fig. S10 (circulating transaminases), Supporting Fig. S11 (liver bile acids), Supporting Fig. S12 (graphical scheme of LXR stimulation in human liver), Supporting Fig. S13 (human and mouse liver transcriptome), Supporting Table S3 (hepatic expression of human genes), and Supporting Table S4 (small intestine expression of mouse genes).
Mirroring the lipoprotein phenotype observed in circulation, LXR activation by GW3965 in LHM‐M1 also resulted in intrahepatic accumulation of triglycerides and cholesteryl esters (Fig. 5D), without affecting the levels of free (or unesterified) cholesterol (Fig. 5E) or inducing liver damage (circulating transaminases in Supporting Fig. S10). LXR activation greatly reduced bile acid synthesis evaluated by the levels of 7alpha‐hydroxy‐4‐cholesten‐3‐one (C4) (Fig. 5F) and glycocholic acid (Supporting Fig. S11) in the liver. LHM‐M1 treated with GW3965 had lower cholesterol biosynthesis (Fig. 5G), mediated by down‐regulation of the sterol regulatory element binding transcription factor 2 (SREBF2) pathway (Supporting Table S3). Moreover, despite a 7‐fold induction of sterol regulatory element binding transcription factor 1 (SREBF1) variant c, its target genes involved in de novo lipogenesis were not up‐regulated (Supporting Table S3). The adverse changes in lipoprotein and liver metabolism were likely the result of the up‐regulation of lipid droplet–associated genes and the down‐regulation of genes involved in neutral lipid hydrolysis, together with the down‐regulation of cytochrome P450 family 7 subfamily A member 1 (CYP7A1), the rate‐limiting enzyme in neutral bile acid biosynthetic pathway (Supporting Table S3). A graphical representation of the effects of LXR stimulation on hepatic gene expression leading to the adverse changes in lipoprotein and lipid metabolism is presented in Supporting Fig. S12. Furthermore, a transcriptomic analysis of human and mouse genes in LHM‐M1 chimeric liver following LXR stimulation is available in Supporting Fig. S13.
To explore the potential development of steatosis and compensatory regeneration following LXR stimulation, we characterized the liver histology of another batch of animals (LHM‐M3) treated with GW3965. As shown in Fig. 6, LXR stimulation by GW3965 induced moderate macrovesicular steatosis and occasional ballooning. Changes in cell proliferation were not detected.
Figure 6.
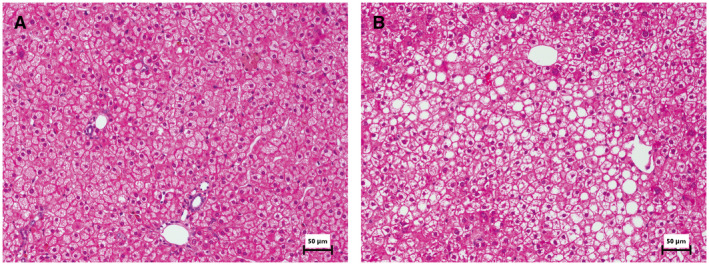
LXR stimulation by GW3965 results in moderate steatosis in LHM‐M3. FRGN mice were repopulated with hepatocytes from the human donor M3 (LHM‐M3) and treated with either vehicle (n = 2) or GW3965 (n = 2). LXR stimulation in LHM‐M3 determined higher levels of cholesterol and triglycerides both in serum (driven by the accumulation in VLDL and LDL) and in liver (Supporting Fig. S14). The liver left‐lateral lobe was paraffin‐embedded and slices were stained with hematoxylin and eosin. Representative images from one mouse per group are shown (scale bar = 50 µm). (A) Almost no signs of steatosis were observed in the vehicle group. (B) After LXR stimulation, predominantly moderate multifocal macrovesicular steatosis (sometimes hinting toward microvesicular) and rare ballooning could be appreciated. In both groups, no prominent/frequent mitotic figures were detected in human hepatocytes.
Gene‐expression analysis in LHM‐M1 small intestine revealed the up‐regulation of LXR target genes following treatment with GW3965, but no effect on FXR‐target genes (Supporting Table S4). This suggests neither alteration in the intestinal response of LXR activation, nor in the cross‐talk between the liver and the gut in LHM, apart from the previously reported unresponsiveness of human hepatocytes to the intestinal murine fibroblast growth factor (FGF) 15.1, 23
Discussion
The continuous growth in the prevalence of cardiometabolic disease, the development of new therapeutic approaches, and the need to implement personalized medicine in health care systems demand the development of advanced preclinical research models. In these models, it should be possible to elucidate the uniqueness of the individual and to obtain results that are translatable to the human patients. The use of chimeric models such as LHM is a clear step toward this direction. Donor‐dependent phenotypes and the unfavorable outcomes of the clinical trial using LXR agonist were observed and reproduced in LHM. Furthermore, the use of LHM models allowed a better understanding of human cholesterol lipoprotein metabolism and explained some of the liver‐related species differences between humans and mice.
CETP has been recognized as one of the main protein responsible for the difference in cholesterol distribution between humans (LDL animals) and mice (HDL—or rather non‐LDL—animals).5 By exchanging triglycerides in APOB‐containing lipoproteins for cholesteryl esters in HDL, CETP is supposed to increase the cholesterol content of VLDL and LDL.5, 6 For this reason, this protein is considered proatherogenic and pharmacologically targetable.5, 6 CETP is produced predominantly by the spleen, adipose tissue and liver in humans, but not in mice. Despite the attainment of a human‐like lipoprotein phenotype in mice repopulated with hepatocytes from four different human donors, we could not detect CETP protein or activity in the circulation. To confirm our findings, we also assayed the hepatic mRNA and protein expression of CETP and found none. Our results reflect what has been reported in humans, namely that CETP expression is not present in hepatocytes and is confined to Kupffer cells in the liver and to circulating macrophages.8 Moreover, when comparing the double chimeric mouse model of human liver and immune system (DHM‐F2) with chimeric mice of only human liver receiving hepatocytes from the same donor (LHM‐F2), an increase of cholesterol in the APOB‐containing lipoproteins accompanied by a decrease of cholesterol in HDL was observed. These results are in line with the phenotype of subjects bearing CETP homozygous loss‐of‐function mutations, which consists of low LDL and high HDL cholesterol levels.24, 25 Hence, our findings provide the ultimate proof that liver parenchymal hepatocytes do not contribute to CETP activity in circulation, which is only detected when human Kupffer cells are present.
In contrast to what was observed for CETP, we could detect in circulation the activity of PLTP (another plasma lipid transfer protein) to a comparable extent in both humans and LHM. The levels of PLTP activity were about one‐fifth of those observed in LMM or wild‐type mice, which are known to have higher PLTP activity than humans. Hepatic PLTP mRNA was extremely low in LHM, in contrast to Pltp mRNA, which was similar to LMM. Because the remaining mouse hepatocytes in our highly repopulated LHM represent less than 10%‐15% of parenchymal cells, their contribution to PLTP activity in circulation can be considered minor. Thus, our data suggest that hepatocytes are great contributors of PLTP activity in mouse, despite the much lower expression of Pltp in liver compared with other organs.26 As far as the effects of the reduction in PLTP activity in LHM, Pltp−/− mouse models show decreased levels of cholesterol in HDL, which accumulates in VLDL and LDL without an increase in plasma APOB levels.27 However, heterozygous Pltp+/‐ mice do not exhibit differences in the distribution of cholesterol in lipoproteins compared with wild type.27 Our results therefore suggest that PLTP does not greatly contribute to the generation of the human‐like cholesterol lipoprotein phenotype of LHM.
APOB lipoproteins and mRNA editing have been studied thoroughly in humans and other mammals.14, 28, 29 In both human and mice, APOB100 is the full‐size protein. APOB mRNA can be edited by APOB mRNA‐editing enzyme catalytic subunit 1 (APOBEC1), a process producing APOB48, which lacks the C‐terminus. In humans, APOB mRNA editing does not occur in the liver, but only in the intestine. In contrast, mice can edit Apob mRNA in both organs.14 Because APOB100 and APOB48 are known to confer different properties to lipoproteins,15 several models have been generated to mimic in mice the human physiology or pathology, such as Apobec1−/− and APOB100‐only mice, which both produce only APOB100.30, 31 However, both models acquire the typical LDL profile only when bred with Ldlr−/− mice, resulting in a dramatically nonphysiologic impaired uptake of APOB‐containing lipoproteins from the circulation.32, 33 Hence, the greatly reduced hepatic APOB mRNA editing and the lower LDLR protein expression in liver—the latter mediated by higher levels of circulating PCSK9—appear to be the principal drivers in humanizing the cholesterol lipoprotein phenotype of LHM. Moreover, the translatability to the human condition of the humanized lipoprotein profile in LHM is corroborated by the observation of a lipoprotein binding to haPG that is similar to the one observed in human reference samples.
LHM were also tested as a translatable human model of pharmacodynamics. The clinical trial testing the effect of an LXR agonist on lipid and lipoprotein metabolism was prematurely terminated principally because of the increase in plasma cholesterol and triglycerides.19 In parallel to what was observed in humans, treatment with the LXR agonist GW3965 resulted in severe hyperlipidemia in LHM. The strategy of targeting LXR to decrease atherosclerosis originated from different mouse models: C57BL/6 mice treated with GW3965 showed an increase of cholesterol in HDL,34 and in hypercholesterolemic Ldlr−/− or Apoe−/− mouse, LXR activation could inhibit atherosclerosis.35 Increase of cholesterol in LDL was only observed in species expressing CETP (i.e., hamster, cynomolgus monkey, and human).19, 36 Because CETP is an LXR‐target gene, it was suggested that its up‐regulation followed by pharmacological stimulation of LXR represented one of the main causes for the increase of cholesterol in LDL. Despite the lack of CETP expression, LHM displayed severe hypercholesterolemia after LXR activation, confirming the contribution of additional negative effects on cholesterol metabolism, as previously discussed.36
CYP7A1 up‐regulation by LXR stimulation is species‐dependent and is absent in primary human hepatocytes.37 In our LHM model, LXR stimulation decreased CYP7A1 mRNA levels, which translated into a decreased bile acid synthesis via the neutral pathway. This was the result of the up‐regulation of nuclear receptor subfamily 0 group B member 2 (NR0B2)/small heterodimer partner (SHP) and FGF19. Consequently, cholesterol accumulated in the liver and down‐regulated SREBF2 and its target genes (e.g., 3‐hydroxy‐3‐methylglutaryl‐CoA reductase [HMGCR] and LDLR). The triglyceride accumulation in the liver of LHM treated with GW3965 is likely independent on de novo lipogenesis despite an increase in mRNA levels of SREBF1 variant c, suggesting a lack of activation of this transcription factor. The increased hepatic triglycerides appear to be the result of a reduced hydrolysis of neutral lipids: PLIN2 (perilipin 2) and CIDEC (cell death‐inducing DFFA‐like effector c)—two lipid droplet–associated genes limiting the neutral lipids hydrolysis—were indeed up‐regulated in parallel to a down‐regulation of several carboxylesterases and lipases. Thus, we describe the unexpected molecular effects of LXR activation in human hepatocytes (Supporting Fig. S12), contributing to the explanation of the severe combined hyperlipidemia and triglyceride accumulation in the liver following LXR stimulation in humans.19
From our study, it is clear that FRGN mice receiving human hepatocytes display several parameters (including lipoprotein levels and properties, apolipoprotein distribution, circulating PCSK9, and Lp[a]) based on the human donor and independently of the degree of humanization. Therefore, LHM can be used in several preclinical settings to monitor the response based on the donor’s hepatocytes, allowing an efficient combination of translatable and personalized studies. Although examples of such applications are already available,7, 38 major limitations for the translatability of the model come in the form of residual activity of the recipient mouse liver, the interaction between donor–recipient tissues, and the peripheral metabolism of the recipient. The latter, for instance, is likely responsible for the differences observed in the cholesterol lipoprotein profile of donors A and B and the LHM transplanted with their hepatocytes (LHM‐A and LHM‐B). In addition, as shown here for CETP, not all liver‐related factors are acquired by LHM (without further humanization of the immune system). This aspect has to be considered also in the use of LHM to study cardiometabolic diseases with an inflammatory component such as atherosclerosis and nonalcoholic steatohepatitis, wherein the model immunodeficiency may prevent the achievement of a full disease spectrum. Even so, LHM represent an improvement in terms of human translatability compared with the use of ordinary wild‐type or genetically modified mouse models, paving the way for a preclinical approach for pharmacological and personalized research.
Author Contributions
M.E.M., M.P., L.‐L.V., K.Ö., S.L., E.M.W., K.R.S., and P.P. designed the hypotheses and the experiments. M.E.M., M.P., L.‐L.V., A.‐S.D., R.G.P.D., C.S., C.P., D.T., and K.M. performed the experiments and their analysis. M.E.M. performed the statistical analysis and wrote the first draft of the manuscript. All authors contributed to the scientific discussion of the data and of the manuscript.
Supporting information
Acknowledgment
We thank M. Olin, A. Lövgren‐Sandblom, L. Larsson, K. Littman, C. Hammarstedt, G. Bial, L. Foquet, G. Camejo, C. Pramfalk, and O. Ahmed for their expert technical support and helpful discussion throughout the work. For the help with the histology analysis, we thank Raoul V. Kuiper and the Unit for Morphological Phenotype Analysis (FENO, Clinical Research Center, Department of Laboratory Medicine, Karolinska Institutet). We also thank all of the consortium members of the European Union–funded research project HUMAN (Health and the Understanding of Metabolism, Aging and Nutrition), who have directly and indirectly contributed to the discussion of the results.
This work was supported by funding from HUMAN (Health and the Understanding of Metabolism, Aging and Nutrition) within the European Union’s Seventh Framework Program for research, technological development and demonstration (602757); the Swedish Research Council; the Swedish Heart‐Lung Foundation; Stockholm City Council; the Karolinska Institute; the Academy of Finland (315568); the NIHR GOSH BRC; and the kind donations from the Szeban Peto Foundation. The views expressed are those of the author(s) and not necessarily those of the funding agencies.
Potential conflict of interest: Dr. Grompe consults, owns stock, and received grants from Ambys Medicines and Logicbio Therapeutics. He founded, consults and owns stock in Yecuris Corporation. Dr. Strom owns stock in Yecuris Corporation. Mr. Bial and Ms. Wilson were employed by Yecuris Corporation. Dr. Parini owns stock in Galmed Pharmaceuticals.
References
Author names in bold designate shared co‐first authorship.
- 1. Ellis EC, Naugler WE, Parini P, Mork LM, Jorns C, Zemack H, et al. Mice with chimeric livers are an improved model for human lipoprotein metabolism. PLoS One 2013;8:e78550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Azuma H, Paulk N, Ranade A, Dorrell C, Al‐Dhalimy M, Ellis E, et al. Robust expansion of human hepatocytes in Fah−/−/Rag2−/−/Il2rg−/− mice. Nat Biotechnol 2007;25:903‐910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Foquet L, Wilson EM, Verhoye L, Grompe M, Leroux‐Roels G, Bial J, et al. Successful engraftment of human hepatocytes in uPA‐SCID and FRG((R)) KO mice. Methods Mol Biol 2017;1506:117‐130. [DOI] [PubMed] [Google Scholar]
- 4. Takenaka K, Prasolava TK, Wang JC, Mortin‐Toth SM, Khalouei S, Gan OI, et al. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat Immunol 2007;8:1313‐1323. [DOI] [PubMed] [Google Scholar]
- 5. de Grooth GJ, Klerkx AH, Stroes ES, Stalenhoef AF, Kastelein JJ, Kuivenhoven JA. A review of CETP and its relation to atherosclerosis. J Lipid Res 2004;45:1967‐1974. [DOI] [PubMed] [Google Scholar]
- 6. Blauw LL, Noordam R, Soidinsalo S, Blauw CA, Li‐Gao R, de Mutsert R, et al. Mendelian randomization reveals unexpected effects of CETP on the lipoprotein profile. Eur J Hum Genet 2019;27:422‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bissig‐Choisat B, Wang L, Legras X, Saha PK, Chen L, Bell P, et al. Development and rescue of human familial hypercholesterolaemia in a xenograft mouse model. Nat Commun 2015;6:7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Y, van der Tuin S, Tjeerdema N, van Dam AD, Rensen SS, Hendrikx T, et al. Plasma cholesteryl ester transfer protein is predominantly derived from Kupffer cells. Hepatology 2015;62:1710‐1722. [DOI] [PubMed] [Google Scholar]
- 9. Grompe M, Strom S. Mice with human livers. Gastroenterology 2013;145:1209‐1214. [DOI] [PubMed] [Google Scholar]
- 10. Wilson EM, Bial J, Tarlow B, Bial G, Jensen B, Greiner DL, et al. Extensive double humanization of both liver and hematopoiesis in FRGN mice. Stem Cell Res 2014;13:404‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parini P, Johansson L, Broijersen A, Angelin B, Rudling M. Lipoprotein profiles in plasma and interstitial fluid analyzed with an automated gel‐filtration system. Eur J Clin Invest 2006;36:98‐104. [DOI] [PubMed] [Google Scholar]
- 12. Pedrelli M, Davoodpour P, Degirolamo C, Gomaraschi M, Graham M, Ossoli A, et al. Hepatic ACAT2 knock down increases ABCA1 and modifies HDL metabolism in mice. PLoS One 2014;9:e93552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davidsson P, Hulthe J, Fagerberg B, Olsson BM, Hallberg C, Dahllof B, et al. A proteomic study of the apolipoproteins in LDL subclasses in patients with the metabolic syndrome and type 2 diabetes. J Lipid Res 2005;46:1999‐2006. [DOI] [PubMed] [Google Scholar]
- 14. Greeve J, Altkemper I, Dieterich JH, Greten H, Windler E. Apolipoprotein B mRNA editing in 12 different mammalian species: hepatic expression is reflected in low concentrations of apoB‐containing plasma lipoproteins. J Lipid Res 1993;34:1367‐1383. [PubMed] [Google Scholar]
- 15. Hussain MM, Kancha RK, Zhou Z, Luchoomun J, Zu H, Bakillah A. Chylomicron assembly and catabolism: role of apolipoproteins and receptors. Biochim Biophys Acta 1996;1300:151‐170. [DOI] [PubMed] [Google Scholar]
- 16. Dietschy JM, Turley SD. Control of cholesterol turnover in the mouse. J Biol Chem 2002;277:3801‐3804. [DOI] [PubMed] [Google Scholar]
- 17. Williams KJ, Tabas I. The response‐to‐retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol 1995;15:551‐561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Camejo G, Hurt‐Camejo E, Wiklund O, Bondjers G. Association of apo B lipoproteins with arterial proteoglycans: pathological significance and molecular basis. Atherosclerosis 1998;139:205‐222. [DOI] [PubMed] [Google Scholar]
- 19. Kirchgessner TG, Sleph P, Ostrowski J, Lupisella J, Ryan CS, Liu X, et al. Beneficial and adverse effects of an LXR agonist on human lipid and lipoprotein metabolism and circulating neutrophils. Cell Metab 2016;24:223‐233. [DOI] [PubMed] [Google Scholar]
- 20. Hong C, Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov 2014;13:433‐444. [DOI] [PubMed] [Google Scholar]
- 21. Hong C, Marshall SM, McDaniel AL, Graham M, Layne JD, Cai L, et al. The LXR‐Idol axis differentially regulates plasma LDL levels in primates and mice. Cell Metab 2014;20:910‐918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zanotti I, Poti F, Pedrelli M, Favari E, Moleri E, Franceschini G, et al. The LXR agonist T0901317 promotes the reverse cholesterol transport from macrophages by increasing plasma efflux potential. J Lipid Res 2008;49:954‐960. [DOI] [PubMed] [Google Scholar]
- 23. Naugler WE, Tarlow BD, Fedorov LM, Taylor M, Pelz C, Li B, et al. Fibroblast growth factor signaling controls liver size in mice with humanized livers. Gastroenterology 2015;149:728‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koizumi J, Mabuchi H, Yoshimura A, Michishita I, Takeda M, Itoh H, et al. Deficiency of serum cholesteryl‐ester transfer activity in patients with familial hyperalphalipoproteinaemia. Atherosclerosis 1985;58:175‐186. [DOI] [PubMed] [Google Scholar]
- 25. Niesor EJ, von der Mark E, Calabresi L, Averna M, Cefalu AB, Tarugi P, et al. Lipid and apoprotein composition of HDL in partial or complete CETP deficiency. Curr Vasc Pharmacol 2012;10:422‐431. [DOI] [PubMed] [Google Scholar]
- 26. Albers JJ, Wolfbauer G, Cheung MC, Day JR, Ching AF, Lok S, et al. Functional expression of human and mouse plasma phospholipid transfer protein: effect of recombinant and plasma PLTP on HDL subspecies. Biochim Biophys Acta 1995;1258:27‐34. [DOI] [PubMed] [Google Scholar]
- 27. Jiang XC, Bruce C, Mar J, Lin M, Ji Y, Francone OL, et al. Targeted mutation of plasma phospholipid transfer protein gene markedly reduces high‐density lipoprotein levels. J Clin Invest 1999;103:907‐914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Camus MC, Chapman MJ, Forgez P, Laplaud PM. Distribution and characterization of the serum lipoproteins and apoproteins in the mouse, Mus musculus. J Lipid Res 1983;24:1210‐1228. [PubMed] [Google Scholar]
- 29. Vezina CA, Milne RW, Weech PK, Marcel YL. Apolipoprotein distribution in human lipoproteins separated by polyacrylamide gradient gel electrophoresis. J Lipid Res 1988;29:573‐585. [PubMed] [Google Scholar]
- 30. Hirano K, Young SG, Farese RV, Jr. , Ng J, Sande E, Warburton C, et al. Targeted disruption of the mouse apobec‐1 gene abolishes apolipoprotein B mRNA editing and eliminates apolipoprotein B48. J Biol Chem 1996;271:9887‐9890. [DOI] [PubMed] [Google Scholar]
- 31. Farese RV, Jr. , Veniant MM, Cham CM, Flynn LM, Pierotti V, Loring JF, et al. Phenotypic analysis of mice expressing exclusively apolipoprotein B48 or apolipoprotein B100. Proc Natl Acad Sci U S A 1996;93:6393‐6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Powell‐Braxton L, Veniant M, Latvala RD, Hirano KI, Won WB, Ross J, et al. A mouse model of human familial hypercholesterolemia: markedly elevated low density lipoprotein cholesterol levels and severe atherosclerosis on a low‐fat chow diet. Nat Med 1998;4:934‐938. [DOI] [PubMed] [Google Scholar]
- 33. Veniant MM, Zlot CH, Walzem RL, Pierotti V, Driscoll R, Dichek D, et al. Lipoprotein clearance mechanisms in LDL receptor‐deficient “Apo‐B48‐only” and “Apo‐B100‐only” mice. J Clin Invest 1998;102:1559‐1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Collins JL, Fivush AM, Watson MA, Galardi CM, Lewis MC, Moore LB, et al. Identification of a nonsteroidal liver X receptor agonist through parallel array synthesis of tertiary amines. J Med Chem 2002;45:1963‐1966. [DOI] [PubMed] [Google Scholar]
- 35. Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci U S A 2002;99:7604‐7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Groot PH, Pearce NJ, Yates JW, Stocker C, Sauermelch C, Doe CP, et al. Synthetic LXR agonists increase LDL in CETP species. J Lipid Res 2005;46:2182‐2191. [DOI] [PubMed] [Google Scholar]
- 37. Menke JG, Macnaul KL, Hayes NS, Baffic J, Chao YS, Elbrecht A, et al. A novel liver X receptor agonist establishes species differences in the regulation of cholesterol 7alpha‐hydroxylase (CYP7a). Endocrinology 2002;143:2548‐2558. [DOI] [PubMed] [Google Scholar]
- 38. Cayo MA, Mallanna SK, Di Furio F, Jing R, Tolliver LB, Bures M, et al. A drug screen using human iPSC‐derived hepatocyte‐like cells reveals cardiac glycosides as a potential treatment for hypercholesterolemia. Cell Stem Cell 2017;20:478‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials