

Abstract
Cardiac sympathetic overactivity is a well‐established contributor to the progression of neurogenic hypertension and heart failure, yet the underlying pathophysiology remains unclear. Recent studies have highlighted the importance of acutely regulated cyclic nucleotides and their effectors in the control of intracellular calcium and exocytosis. Emerging evidence now suggests that a significant component of sympathetic overactivity and enhanced transmission may arise from impaired cyclic nucleotide signalling, resulting from compromised phosphodiesterase activity, as well as alterations in receptor‐coupled G‐protein activation. In this review, we address some of the key cellular and molecular pathways that contribute to sympathetic overactivity in hypertension and discuss their potential for therapeutic targeting.
Keywords: Autonomic Nervous System, Sympathetic Ganglion, Hypertension, Cardiovascular Disease, Cyclic Nucleotide, Protein Kinase, Sympathetic Nervous System, Intracellular Calcium
In healthy stellate neurons, Ca2+‐dependent exocytosis facilitates the release of noradrenaline (NA) onto cardiac myocytes, where post‐synaptic β1‐ARs and β2‐ARs are activated. Increases in extracellular NA acts on presynaptic α2‐ARs, and presynaptic β1‐ARs and β2‐ARs to a small degree. Acute regulation of cAMP and cGMP signalling is maintained by phosphodiesterases (PDEs). cAMP generation and PKA activity increases [Ca2+]i via phosphorylation of the N‐type Ca2+ Channel (ICaN; CaV2.2); endoplasmic reticulum (ER) store and mitochondrial Ca2+ release. In diseased sympathetic neurons, Ca2+‐dependent exocytosis facilitates the release of NA and adrenaline (Adr) onto cardiac myocytes, where post‐synaptic β2‐AR signalling is upregulated and preferentially activated. Chronic elevations in NA and Adr release also acts on presynaptic β‐ARs. Adr preferentially stimulates the β2‐AR isoform, thus augmenting cAMP generation and PKA activity in prehypertension, in a potentiating feed‐forward manner. Increased PKA activity raises [Ca2+]i via phosphorylation of the N‐type Ca2+ Channel (ICaN; CaV2.2), exacerbating the Ca2+ phenotype that may contribute to the initiation of hypertension.

Introduction
The autonomic nervous system, comprising the parasympathetic and sympathetic branches, provides a regulatory link between the central nervous system (CNS) and myocardium (Herring & Paterson, 2018). The notion of a mind–body connection has been proposed by many scientists throughout history, but it was perhaps first recorded in ad 30 by the Roman physician Celsus who wrote, ‘fear and anger and any other state of mind may often be apt to excite the pulse’ (Celsus & Spencer, 1935). Yet, the physiological mechanisms responsible for the relationship between the heart and the brain remained elusive until the 19th century, whereupon, it was discovered that heart rate could be accelerated or decelerated by stimulation of two antagonistic systems: sympathetic or parasympathetic nerve fibres (Gaskell, 1886; Langley, 1898; Woollard, 1926; Sheehan, 1936; Hoff, 1940). The ‘autonomic nervous system’, as coined by Langley in 1898 (Langley, 1898), is now known to play an integral role in cardiovascular homeostasis and cardiac responses to physical or emotional disturbances (Rozanski et al. 1999; Steptoe & Kivimaki, 2012; Tahsili‐Fahadan & Geocadin, 2017; Herring & Paterson, 2018).
The cervicothoracic sympathetic stellate ganglion located adjacently to T1–T4 preferentially innervates the heart (Gaskell, 1886; Korzina et al. 2011) and, as such, exerts the greatest control over heart rate acceleration, contractility and conduction velocity at the atrio‐ventricular node (Shivkumar et al. 2016). Chronic alteration in sympathetic/parasympathetic balance (dysautonomia) is a well‐established contributor to many cardiovascular diseases (CVDs) and is strongly linked to clinical outcome and prognosis (Brook & Julius, 2000; Palatini & Julius, 2004; Malpas, 2010; Parati & Esler, 2012; Mancia & Grassi, 2014). Increasing evidence suggests that essential hypertension is underpinned and maintained by sustained elevations in sympathetic nerve activity (SNA) and chronic end‐organ transmission (Iriuchijima, 1973; Judy et al. 1979; Esler et al. 1986; 1988; Grassi & Esler, 1999; Johansson et al. 1999; Guyenet, 2006; Wang et al. 2006; Malpas, 2010; Parati & Esler, 2012; Shanks et al. 2013 b; Esler, 2014; Oliveira‐Sales et al. 2014; Grassi et al. 2015; Oliveira‐Sales et al. 2016). Elevations in SNA are also frequently seen in normotensive progeny of hypertensive patients (Ferrara et al. 1988; Hausberg et al. 1998; Lopes et al. 2000; Piccirillo et al. 2000; Maver et al. 2004; Hamer, 2006; Pal et al. 2011; Johncy et al. 2015), suggesting a causative role and potential genetic basis (Judy et al. 1979; Horikoshi et al. 1985; Adams et al. 1989) for sympathetic overactivity in the aetiology of hypertension.
However, it is also well established that SNA is not uniformly altered within each ganglionic site (Grassi et al. 2015) and preclinical models have highlighted the critical role of elevated cardiac sympathetic nerve activity, specifically in the initiation and maintenance of hypertension (Souza et al. 2001; Petersson et al. 2002; Tan et al. 2010; Shanks et al. 2013 b; Larsen et al. 2016 a; Tromp et al. 2018), cardiac arrhythmia (Meredith et al. 1991) and heart failure (Kaye et al. 1995; Rundqvist et al. 1997; Watson et al. 2007; Ramchandra et al. 2009; Tu et al. 2014). Multiple levels of the neural axis comprising several integrated feedback loops are involved in the regulation of autonomic transmission, and may be disturbed in hypertension. These include cardio‐cardiac reflexes and intrinsic cardiac nerve activity that alter end‐organ transmission within the myocardium directly, intrathoracic reflexes and feedback mechanisms that modify sympathetic ganglionic efferent transmission, and spinal and lower brainstem regulation that modulate autonomic outflow (Shivkumar et al. 2016; Hanna et al. 2017). Sustained alterations in one or several of these feedback processes may directly contribute to an elevation in SNA, yet it is difficult to dissociate the primary causative events from secondary consequential factors. Nevertheless, the dominance of cardiac sympathetic neurons over myocyte function is observed. This is illustrated in Fig. 1, where co‐cultures of diseased stellate neurons and myocytes from rats predisposed to hypertension display enhanced myocyte cyclic adenosine monophosphate (cAMP) generation during neuronal stimulation compared to normal co‐cultures (Larsen et al. 2016 b). Moreover, cross‐culturing diseased stellate neurons provokes healthy myocytes into a prehypertensive state partially recapitulating the elevation in cAMP observed in diseased myocytes. Critically, however, healthy neurons cultured with diseased myocytes rescues the aberrant myocardial cAMP response restoring cAMP to levels seen in normal myocytes (Larsen et al. 2016 b). What are the mechanisms that underpin the sympathetic phenotype and lead to elevated cardiac sympathetic transmission?
Figure 1. Sympathetic neurons are a powerful driver of myocyte function in cardiovascular disease.
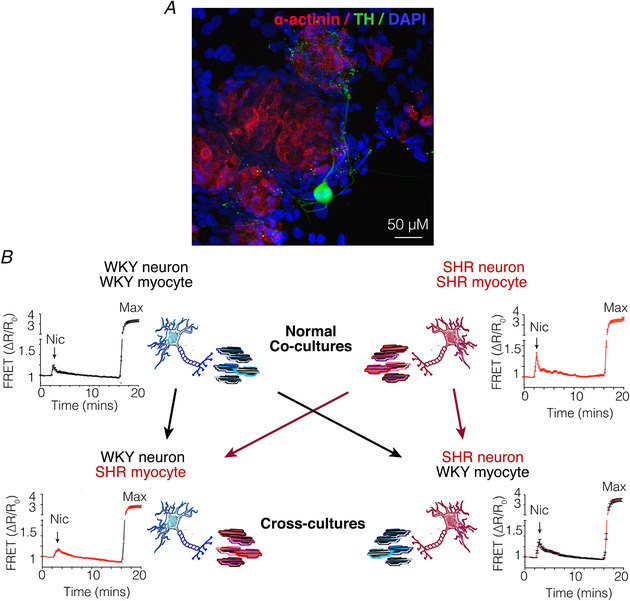
A, immunofluorescence depicting a co‐culture of sympathetic neurons and ventricular myocytes (reproduced from Larsen et al. 2016 b). Sympathetic neurons labelled with tyrosine hydroxylase (TH, green) densely innervate cultured cardiomyocytes labelled with sarcomeric α‐actinin (red). B, Wistar–Kyoto (WKY) or SHR sympathetic neurons were stimulated with nicotine (Nic) and the resulting myocyte cAMP was measured as a surrogate for sympathetic transmission, in mycoytes transduced with a cAMP Förster resonance energy transfer (FRET) sensor. FRET sensors were maximally stimulated (max) with an adenylyl cyclase (AC) activator forskolin (25 M) and a non‐specific phosphodiesterase (PDE) inhibitor 3‐isobutyl‐1‐methylxanthine (IBMX,100 M). In healthy co‐cultures (WKYn/WKYm), neuron‐evoked myocyte cAMP (17.05 ± 3.715, n = 29 cells) was significantly lower than cAMP measured in the diseased co‐culture myocytes (SHRn/SHRm; 44.02 ± 5.310, n = 36 cells; P < 0.0001). Cross‐cultures were established by plating diseased SHR neurons on top of healthy WKY myocytes (SHRn/WKYm) or healthy WKY neurons on top of diseased SHR myocytes (WKYn/SHRm). In the first cross‐culture (SHRn/WKYm), neuronal stimulation elevated myocyte cAMP (31.37 ± 5.194, n = 42 cells) to levels that were not significantly different from measured in the diseased (SHRn/SHRm) co‐cultures (P = 0.094), demonstrating that enhanced neuronal transmission elevates healthy‐myocyte cAMP to levels observed in disease. Moreover, in the second cross‐culture (WKYn/SHRm), stimulation of WKY neurons elevated SHR myocyte cAMP (15.67 ± 1.936, n = 24 cells) to levels that were not significantly different from that measured in healthy (WKYn/WKYm) co‐cultures (P = 0.76), demonstrating that healthy neurons attenuate the elevated myocyte cAMP response observed in SHR myocytes (modified from Larsen et al. 2016 b).
In models of neurogenic hypertension, several key sympathetic adaptations are reported, including increased neuronal firing rate and burst frequency (Iriuchijima, 1973; Briant et al. 2015), elevated and aberrant regulation of intracellular Ca2+ ([Ca2+]i) that facilitates exocytosis (Li et al. 2013; Larsen et al. 2016 a; Shanks et al. 2017; Tomek et al. 2017), decreased transmitter reuptake (Esler et al. 1981; Kimura et al. 1983; Esler et al. 1991; Rumantir et al. 2000 b; Shanks et al. 2013 a), and alterations in presynaptic feedback systems coupled to impaired intracellular signalling cascades (Wang et al. 2006; Shanks et al. 2013 b; Bardsley et al. 2018 b). In this brief review, we present the current evidence for the molecular and biochemical alterations that occur in stellate ganglia from rat and human patients that have a sympathetic phenotype and discuss their potential for therapeutic targeting.
Intrinsic excitability: control by cyclic nucleotides
The N‐type Ca2+ channel is the primary neuronal voltage‐gated Ca2+ channel (Catterall, 2003, 2011) and as such plays a critical role in determining the cytosolic Ca2+ concentration during an action potential in sympathetic neurons (Pruneau & Bélichard, 1992; Ino et al. 2001; Mori et al. 2002; Uhrenholt & Nedergaard, 2003; Tu et al. 2014; Larsen et al. 2016 a). Emerging evidence suggests that N‐type Ca2+ channel activity is elevated in cardiac sympathetic ganglia in the prehypertensive SHR (Fig. 2; Larsen et al. 2016 a) and in heart failure (Tu et al. 2014), indicating a synaptopathy that augments intracellular Ca2+ and raises the intrinsic excitability of these nerves (Briant et al. 2015). Voltage‐gated Ca2+ channel conductance is differentially regulated by kinase phosphorylation (Gray et al. 1998; Schroder, 2003; Mahapatra et al. 2012; Larsen et al. 2016 a) where processes that decrease cyclic guanosine monophosphate (cGMP)–protein kinase G (PKG) signalling, or elevate cAMP–protein kinase A (PKA) signalling result in a net increase in Ca2+ channel conductance (Brown & Birnbaumer, 1988; Leiser & Fleischer, 1996; Gray et al. 1998; D'Ascenzo et al. 2002; Schroder, 2003; Mahapatra et al. 2012; Zamponi et al. 2015; Sandoval et al. 2017). Thus, processes that selectively modulate the strength of cAMP or cGMP signals effectively regulate neuronal transmission (Pruneau & Bélichard, 1992; Leiser & Fleischer, 1996; Gray et al. 1998; Molderings et al. 2000; Ino et al. 2001; Mori et al. 2002; Tanaka et al. 2013; Yamada et al. 2014).
Figure 2. N‐type Ca2+ channel conductance is elevated in preSHR cardiac sympathetic neurons.
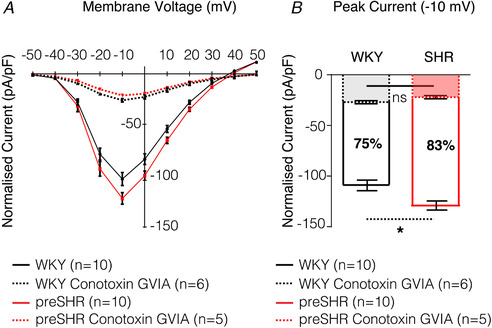
Whole cell voltage clamp was performed on cardiac sympathetic stellate neurons to investigate whole cell Ca2+ currents. A, the current–voltage relationship. Access to the cell was obtained in normal Tyrode's solution containing the following (in mM): 135 NaCl, 4.5 KCl, 11 glucose, 20 HEPES, 1MgCl2, 2 CaCl2, pH 7.4. To identify the Ca2+ current, normal Tyrode solution was replaced with a Ca2+‐isolating solution using Ba2+ as the charge carrier, containing the following (in mM): 135 TEACl, 10 HEPES, 4.5 KCl, 1 MgCl2, 4 glucose, 1 NaHCO3, 2 BaCl2, pH 7.40, either in the presence or absence of ω‐conotoxin GVIA (1 μM), which selectively blocks N‐Type Ca2+ channels (IC50 = 0.15 nM) (Sato et al. 1993). Ba2+ was used as the charge carrier to avoid Ca2+‐dependent current inactivation (Imredy & Yue, 1994). The internal solution contained the following (in mM): 140 CsCl, 10 HEPES, 0.1 CaCl2, 1 MgCl2, 4 MgATP, 1 EGTA, pH 7.30. All solutions had osmolarities of 300 mOsm L−1. B, the whole cell Ca2+ current is larger in preSHR sympathetic nerves (127.5 ± 5.94 pA pF−1, n = 10) compared to WKY cells (−108.0 ± 6.80 pA pF−1, n = 10, P = 0.045) where peak current was recorded at −10 mV. ω‐Conotoxin GVIA (1 μM), significantly reduced the N‐type Ca2+ channel current to similar levels in both strains. A 75% reduction was observed in cells cultured from WKY stellate ganglia (−26.88 ± 1.7 pA pF−1, n = 6) and an 83% reduction was measured in neurons cultured from preSHR ganglia (−22.04 ± 1.60 pA pF−1, n = 5, ns) where peak current remained at −10 mV. Solid lines represent the mean of the WKY (black) and preSHR (red) control data. Dashed lines represent the mean of WKY (black) and preSHR (red) in the presence of ω‐Conotoxin GVIA. Data are represented as mean ± SEM. (A and B modified from Larsen et al. 2016 a).
An increased cAMP–PKA/cGMP–PKG ratio exacerbates cardiac sympathetic activity
Nitric oxide (NO) is a significant neuronal modulator of sympatho‐vagal activity (Sears et al. 1998; Wang et al. 2007). In the SHR, impaired NO generation via neuronal nitric oxide synthase (nNOS; Wang et al. 2007; Danson et al. 2009; Lee et al. 2009; Li et al. 2013, 2015; Lu et al. 2015) and down‐regulation of soluble guanylyl cyclase (sGC; Li et al. 2013; Bardsley et al. 2018 a) lead to significant reductions in cGMP production and PKG activity (Li et al. 2013, 2015; Larsen et al. 2016 a). In the prehypertensive rat, deficits in cGMP–PKG signalling are directly linked to elevations in N‐type Ca2+ channel Ca2+ conductance (Larsen et al. 2016 a; Fig. 3) and may contribute to the increased firing rate and spike amplitude observed in models of disease (Briant et al. 2014; Tu et al. 2014). To understand the genetic basis for these observations, we carried out a comprehensive RNA sequencing study using ganglia from hypertensive and normotensive rats (Bardsley et al. 2018 a) and found that transcripts within the cGMP–PKG pathway were significantly under‐represented in the stellate ganglia of SHR with established hypertension. Notable transcripts included down‐regulation of protein kinase G II (Prkg2) and the α1‐sGC subunit (Gucy1a3). Genome wide association studies (GWAS) have also revealed a critical link between mutations in loci containing the gene Gucy1a3 and clinical hypertension (Ehret et al. 2011; Zheng et al. 2015; Wallace et al. 2016; Rippe et al. 2017; Seidel & Scholl, 2017), myocardial infarction (Erdmann et al. 2013; Wobst et al. 2015), atherosclerosis (Segura‐Puimedon et al. 2016; Wobst et al. 2016) and coronary artery disease (CARDIoGRAMplusC4D Consortium et al. 2013; Nikpay et al. 2015; Kessler et al. 2017).
Figure 3. Elevated Ca2+ conductance in preSHR stellate neurons is rescued with cGMP administration.
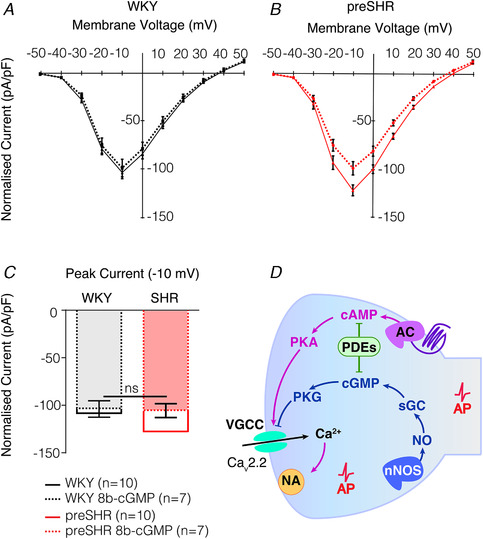
A–C, to ascertain whether cGMP signalling inhibits Ca2+ currents, whole cell voltage clamp was performed on sympathetic neurons young normotensive WKY (A) and young prehypertensive SHR (B) in the presence of a cGMP analogue, 8‐bromo‐cGMP (8b‐cGMP) (Larsen et al. 2016 a). Access to the cell was obtained in normal Tyrode solution containing the following (in mM): 135 NaCl, 4.5 KCl, 11 glucose, 20 HEPES, 1 MgCl2, 2 CaCl2, pH 7.4. To identify the Ca2+ current, the solution was replaced with a Ca2+‐isolating solution using Ba2+ as the charge carrier, containing the following (in mM): 135 TEACl, 10 Hepes, 4.5 KCl, 1 MgCl2, 4 glucose, 1 NaHCO3, 2 BaCl2, pH 7.40, in either the presence or the absence of 8b‐cGMP (100 μM). Ba2+ was used as the charge carrier to avoid Ca2+‐dependent current inactivation (Imredy & Yue, 1994). The internal solution contained the following (in mM): 140 CsCl, 10 Hepes, 0.1 CaCl2, 1 MgCl2, 4 MgATP, 1 EGTA, pH 7.30. All solutions had osmolarities of 300 mOsm L−1. 8b‐cGMP significantly reduced the elevated preSHR Ca2+ currents (−127.5 ± 5.94 pA pF−1, n = 10 to −105.2 ± 7.79 pA pF−1, n = 7) to levels that were no longer greater than WKY Ca2+ currents (−108.0 ± 6.80 pA pF−1, n = 10). Moreover, 8b‐cGMP had no significant effect on the WKY Ca2+ current, where peak currents were measured at −10 mV. Continuous lines represent the mean of the WKY (black) and preSHR (red) control data. Dashed lines represent the mean of WKY (black) and preSHR (red) in the presence of 8b‐cGMP. Data are represented as mean ± SEM. (A‐C are reproduced from Larsen et al. 2016 a). D, model diagram representing N‐type Ca2+ channel control by PKA and PKG, where PKA augments and PKG inhibits channel conductance. Pathways that are decreased (blue) or increased (pink) in disease are represented. AP, action potential; NA, noradrenaline; VGCC, voltage‐gated calcium channel; AC, adenylyl cyclase.
Reductions in cGMP–PKG or increases in cAMP–PKA augment Ca2+ conductance (Fig. 3) via site‐specific phosphorylation of the N‐type Ca2+ channel, where a shift towards cAMP–PKA signalling in hypertension facilitates exocytosis (Leiser & Fleischer, 1996; Gray et al. 1998; D'Ascenzo et al. 2002; Tanaka et al. 2013; Larsen et al. 2016 a). In support of the evidence for elevated cAMP–PKA activity in hypertension, we identified a significant down‐regulation in the gene encoding the type Iα regulatory subunit of PKA (Prkar1a) in our RNA sequencing dataset. This subunit plays a dominant role as an endogenous inhibitor of kinase activity (Bardsley et al. 2018 a) where loss‐of‐function mutations in Prkar1a are associated with a twofold greater responsiveness to cAMP and an excess of PKA type II activity (Stratakis et al. 2001). Knock‐out mouse models of Prkar1a display impaired axonal sorting, myelination and proliferation (Guo et al. 2013). In humans, Prkar1a mutations are characterised by endocrine overactivity, neural dysfunction and cardiac complications, which result in dysregulation of arterial blood pressure homeostasis, arrhythmia and cardiomyopathies (Stratakis, 2002; Horvath et al. 2010), highlighting the importance of cAMP–PKA signalling in neuronal and cardiovascular regulation. Consequently, it appears that in cardiac sympathetic nerves from prehypertensive rats, several processes that favour excitatory cAMP–PKA signalling are up‐regulated, whereas pathways coupled to NO–cGMP are critically impaired early in disease, thus exacerbating or underpinning the observed Ca2+ phenotype (Li et al. 2013, 2015; Larsen et al. 2016 a; Fig. 3 D).
Phosphodiesterase enzymes: the centre of balance for cyclic nucleotides
Phosphodiesterase enzymes (PDEs) regulate ion channel activity through selective termination of cAMP and/or cGMP signalling (Tanaka et al. 2013; Zhao et al. 2016); therefore, the acute spatial and temporal regulation of cyclic nucleotide (cN) levels by PDEs is critical for maintaining a fine balance between PKA‐ and/or PKG‐mediated effects (Zaccolo & Movsesian, 2007; Stangherlin & Zaccolo, 2012). The cN signal is acutely maintained by the PDE superfamily, comprising 11 isoforms (Stangherlin & Zaccolo, 2012), which confine individual and unique cAMP/cGMP signals to distinct subcellular compartments, enabling the regulation of multiple effector responses at any given time (Lefkimmiatis & Zaccolo, 2014). Indeed, cAMP is localised in close proximity to its effectors and regulators, where PKA, PDEs and phosphatases are tethered to A‐kinase anchoring proteins forming signalosomes that restrict the duration and magnitude of the cAMP–PKA signal within specific subcellular domains (Musheshe et al. 2018). Moreover, PDE isoforms are also subject to feedback inhibition and/or potentiation where specific isoforms are sensitive to cNs themselves (Zaccolo & Movsesian, 2007; Zhao et al. 2016), kinase activity (Zaccolo & Movsesian, 2007; Francis et al. 2011) and/or intracellular Ca2+/calmodulin‐dependent protein kinase signalling (Maurice, 2003; Bender, 2006; Francis et al. 2011). Sustained elevations in cAMP generation or alterations in PDE activity underpin several cardiovascular pathologies including cardiac hypertrophy (Zaccolo & Movsesian, 2007; Sprenger et al. 2015; Zoccarato et al. 2015) and sympathetic overactivity in hypertension (Larsen et al. 2016 a; Liu et al. 2018) where cAMP signals saturate the available PDEs and diffuse into neighbouring compartments leading to aberrant effector activity (Larsen et al. 2016 a; Zhao et al. 2017).
Phosphodiesterases in the cardiac sympathetic ganglia
We have previously reported that the activity of specific PDEs involved in the cross‐talk between cAMP and cGMP pathways (PDE2a, PDE3) are impaired in cardiac sympathetic nerves in prehypertension (Li et al. 2015; Bardsley et al. 2016; Larsen et al. 2016 a), and that cGMP pathways are preferentially diminished (Larsen et al. 2016 a). However, a distinct contrast has also been identified in the hydrolysing activity of the wider PDE family within the sympathetic ganglia between normotensive and prehypertensive strains (Fig. 4 A). To understand the genetic basis for these observations, we carried out a gene ontology analysis from our RNA sequencing dataset and found that the genetic family representing ‘phosphoric ester hydrolase activity’ was significantly over‐represented in established hypertension (Davis et al. 2018; Bardsley et al. 2018 a), supporting preclinical reports and several clinical studies (Katz et al. 2000; Bender, 2006; Nagendran et al. 2007; Zaccolo & Movsesian, 2007; Lee et al. 2015; Maass et al. 2015; Zoccarato et al. 2015; Boda et al. 2016; Vettel et al. 2017; Assenza et al. 2018; Baliga et al. 2018; Bardsley et al. 2018 a). It was observed that over 30 genes linked to the PDE superfamily are differentially expressed in the SHR stellate ganglia and that many of these mapped to regulators of PDE activity (Bardsley et al. 2018 a; Fig. 4 B), adding a further layer of complexity to the systems involved in cN control. Moreover, changes in transcripts do not necessarily lead to changes in protein levels. For example, RNA sequencing data revealed a decrease in Pde2a expression (Bardsley et al. 2018 a), whereas PDE2A activity and protein levels are reportedly raised in SHR and human stellates (Li et al. 2015; Liu et al. 2018). Furthermore, over‐expression of PDE2A in neuronal stellates recapitulates the Ca2+ phenotype and enhanced sympathetic response seen in disease (Li et al. 2015), illustrating complex interactions that may be related to microdomain signalling of various isoforms of PDE2A (Zhao et al. 2016).
Figure 4. Phosphodiesterase (PDE) activity is impaired in preSHR neurons and has a genetic component.
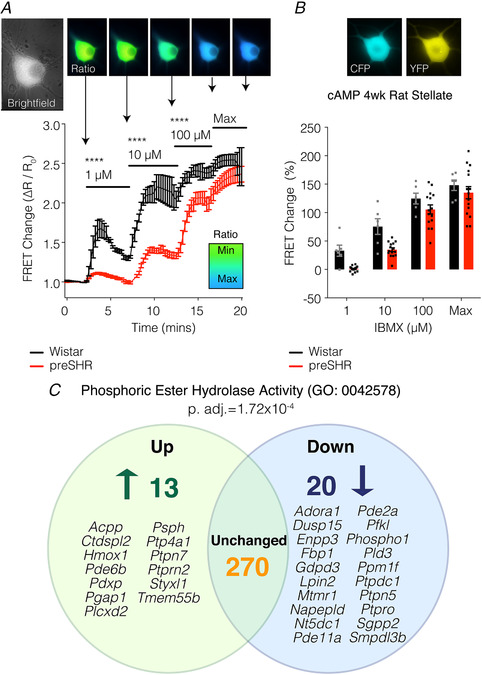
A and B, to investigate whether cytosolic PDE signalling is impaired in preSHR sympathetic neurons, a non‐specific PDE inhibitor, 3‐isobutyl‐1‐methylxanthine (IBMX; inhibits PDEs 1–7, 10–11), was administered to sympathetic stellate neurons (1–100 μM). The resulting intracellular cAMP was measured using real‐time Förster resonance energy transfer (FRET) in cells transduced with the adenovirus encoding the Epac‐SH187 biosensor (Klarenbeek et al. 2015). A, there was significantly greater IBMX‐stimulated cAMP in Wistar vs. preSHR neurons at all concentrations measured (two‐way repeated measures ANOVA; P < 0.05) supporting the evidence that there is a differential PDE profile in preSHR vs. control stellate neurons. At 100 μM IBMX, FRET responses were close to sensor saturation. B, peak FRET changes are depicted. Data are expressed as mean ± SEM. C, we investigated whether transcriptomic changes could be identified in SHR stellate ganglia with established hypertension. Using RNA sequencing, it was observed that the molecular function gene ontology (GO) group encoding ‘phosphoric ester hydrolase activity’ (GO:0042578) was significantly over‐represented in the SHR ganglia at 16 weeks. Thirty‐three genes were found to be differentially expressed and many of these mapped to regulators of PDE and kinase activity (figure reproduced from Bardsley et al. 2018 a).
Phosphodiesterases in the myocardium
Within the cardiac sympathetic axis, intrinsic electrical pacemaker activity arising from the sinoatrial node (SAN) dictates resting heart rate, which is increased by sympathetic noradrenaline and activation of myocardial Gαs‐coupled β‐adrenergic receptors (βARs). Elevation in myocardial cAMP–PKA activity regulates a large number of phospho‐sensitive processes (Yaniv et al. 2015; Behar et al. 2016) and in particular plays a key role in elevating intracellular Ca2+ via phosphorylation of the L‐type Ca2+ channel (Cav1.2, Cav1.3) (Zhao et al. 2016, 2017; Hua et al. 2012) as well as phospholamban, which increases Ca2+ reuptake by the sarcoplasmic reticulum (SR), facilitating rapid repolarisation (Simmerman & Jones, 1998; Mattiazzi & Kranias, 2014; Akaike et al. 2017). Conversely, mediators that elevate cGMP–PKG, such as NO coupled to sGCs or activation of membrane‐bound particulate guanylyl cyclase (pGC) receptors (e.g. ANP, BNP), oppose the actions of cAMP–PKA, thus limiting intracellular Ca2+. Sustained elevations in cAMP–PKA activity (Sprenger et al. 2015) and/or reductions in cardiac NO–cGMP signalling (Heaton et al. 2006; Dawson et al. 2008; Baliga et al. 2018) that elevate [Ca2+]i (Leiser & Fleischer, 1996; Mattiazzi & Kranias, 2014; Zhao et al. 2016, 2017) are involved in cardiac remodelling and hypertrophy (Sprenger et al. 2015; Zoccarato et al. 2015), arrhythmia (Kalla et al. 2016) and heart failure (Kaye et al. 1995; Mehel et al. 2013; Florea & Cohn, 2014). In the SHR model, atrial myocytes display a greater cAMP response to βAR stimulation (Heaton et al. 2006), and lower basal levels of NO–cGMP (Heaton et al. 2006). Gene transfer approaches targeted to the SAN to up‐regulate neuronal nitric oxide synthase (nNOS) or its anchoring protein CAPON (Lu et al. 2015) successfully reduce the surface density and activity of L‐type Ca2+ currents (Danson et al. 2005) and decrease intracellular concentrations of cAMP via the proposed activation of PDE2a (Danson et al. 2005), highlighting a novel therapeutic potential for targeting cNs and their effectors within the myocardium directly. The intricacy of cN regulation, the inability to target specific PDE isoforms that reside in precise intracellular compartments, and the high‐level of functional redundancy observed in the PDE superfamily perhaps help to explain the lack of clinical efficacy achieved by selective PDE inhibitors. Computational protein design, protein engineering and the application of targeted vector systems may provide innovative solutions to these problems.
Neurohormonal and endocrine signalling: effects on presynaptic sympathetic nerves
Impaired neurohormonal regulation plays a critical role in the pathogenesis and progression of cardiovascular diseases (Malpas, 2010). Plasma and tissue levels of noradrenaline (NA), adrenaline (Adr), angiotensin II (AngII), aldosterone and other mediators are significantly altered in hypertension and heart failure and correlate with the severity of disease (Catt et al. 1971; Dang et al. 1999; Grassi & Esler, 1999; Romero & Reckelhoff, 1999; Rupp & Jäger, 2001; Schiffer et al. 2009; Riet et al. 2015; Shinohara et al. 2015; Najafi et al. 2016). Therapeutics aimed at opposing elevated adrenergic and/or antagonising renin–angiotensin–aldosterone signalling are gold‐standard treatment strategies for blood pressure maintenance (van den Meiracker et al. 1995; Hansson et al. 1999; White et al. 2003; Flack et al. 2007; Ram, 2010; Nussberger & Bohlender, 2013; Williams et al. 2015; Frishman, 2016; Ghazi & Drawz, 2017; Rubattu et al. 2018; Wiysonge et al. 2018). Nevertheless, their precise mechanisms of action still remain unclear (Nussberger et al. 1986; van den Meiracker et al. 1995; Nussberger & Bohlender, 2013; Riet et al. 2015; Watanabe et al. 2017).
NA transmission plays a dominant role in vascular constriction and cardiac output (Herring & Paterson, 2018), whereas sustained elevations are involved in hypertension (Shanks et al. 2013 b), arrhythmia (Meredith et al. 1991) and heart failure (Kaye et al. 1995; Florea & Cohn, 2014). In the 1980s, it was demonstrated that the activation of presynaptic β‐ARs facilitates transmission within several peripheral ganglia (Lokhandwala & Eikenburg, 1983; Majewski, 1983; Misu & Kubo, 1986; Nedergaard & Abrahamsen, 1990; Apparsundaram & Eikenburg, 1995), yet little is known about the physiological or pathophysiological relevance of these receptors in hypertension. Recently, we demonstrated that activation of sympathetic stellate presynaptic β‐AR receptors leads to cAMP–PKA activation that is significantly elevated in the prehypertensive SHR and is predominantly β2‐AR mediated (Bardsley et al. 2018 b) (Fig. 5). This increase in cAMP–PKA signalling augments high K+‐evoked Ca2+ liberation in neurons from prehypertensive rats, reflecting ion channel involvement (Bardsley et al. 2018 b). These findings suggest a feed‐forward potentiating mechanism exists for catecholaminergic regulation of cardiac sympathetic transmission, which exacerbates the cAMP/cGMP imbalance in disease (Fig. 6). To give these observations contextual relevance, we also confirmed the presence of β‐ARs in human stellate ganglia, highlighting an alternative site of action for the efficacy achieved with sustained clinical β‐blocker therapy (Ram, 2010; Frishman, 2016; Wiysonge et al. 2018).
Figure 5. β‐AR signalling is elevated in preSHR neurons.
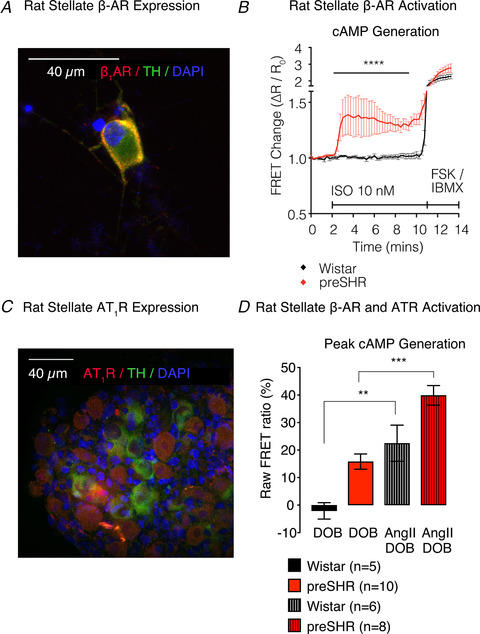
A, we identified the presence of β‐adrenergic receptors (β‐ARs) on tyrosine hydroxylase (TH) positive cardiac sympathetic neurons. B, activation of presynaptic β‐ARs with isoprenaline (10 nM) led to a significantly larger cAMP generation in preSHR (56%; n = 12) vs. Wistar neurons (7%; n = 12; 2‐way ANOVA; P < 0.001), which was measured using real‐time cAMP in cells expressing the Epac‐SH187 biosensor (A and B are reproduced from Bardsley et al. 2018 b). C, we also identified the presence of AT1Rs on TH positive neurons. D, we investigated whether AT1R could elevate B1‐AR‐evoked cAMP. Dobutamine (DOB) alone elevated cAMP in preSHR neurons (reproduced from Bardsley et al. 2018 b). Moreover, AngII augments DOB‐evoked cAMP generation in Wistar neurons (n = 5, 6; P = 0.0073) and SHR neurons (n = 10, 8; P = 0.0005). We also measured a strain‐dependent effect following administration of DOB only (P = 0.0015) and in the presence of DOB with AngII (P = 0.0283). Data are represented as mean ± SEM.
Figure 6. Adrenaline is released from preSHR neurons.
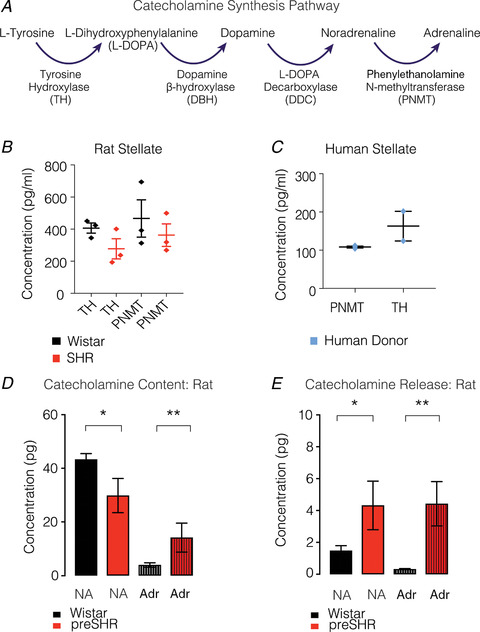
A, the catecholamine synthesis pathway, highlights the role of Phenylethanolamine‐N‐methyltransferase (PNMT) in the conversion from noradrenaline (NA) to Adrenaline (Adr). B and C, tyrosine hydroxylase (TH) and PNMT were measured in adult rat (B) and human (C) stellate ganglia (reproduced from Bardsley et al. 2018 b). D, using high pressure liquid chromatography with electrochemical detection (HPLC‐EC), we measured significantly higher total NA in Wistar (43.3 ± 2.173 pg; n = 8) compared with preSHR neurons (29.82 ± 6.366 pg; n = 4; P = 0.0294). In the same samples, we also measured a significantly greater total content of Adr in preSHR (14.14 ± 5.399 pg) compared with that measured in Wistar ganglia (3.937 ± 0.820 pg, P = 0.0019). E, electrical field stimulation of whole rat stellate ganglia led to the release of NA that was significantly higher in samples obtained from preSHR (4.32 ± 1.523 pg) vs. Wistar ganglia (1.477 ± 0.316 pg; P = 0.0396). The concentrations of neurally mediated Adr release were also significantly higher in preSHR (4.424 ± 1.391 pg, n = 4) compared with Wistar stellates (0.3201 ± 0.0325 pg; n = 8; P = 0.0028) (figure reproduced from Bardsley et al. 2018 b).
The renin–angiotensin system (RAS) is critically involved in blood pressure regulation and fluid volume homeostasis (Hall, 1986; Herring & Paterson, 2018) and alterations in RAS signalling are strongly associated with the aetiology of cardiovascular disease (Dang et al. 1999; Weir & Dzau, 1999; Rupp & Jäger, 2001; Crowley et al. 2006; Riet et al. 2015). AngII is a bioactive product of RAS that is synthesised through sequential cleavage of angiotensinogen and angiotensin I by the enzymes renin and angiotensin converting enzyme (ACE) (Weir & Dzau, 1999). Classically, AngII synthesis was thought to predominantly result from the activity of renal‐derived renin, but emerging evidence has highlighted a critical role for ‘intracrine’ or intracellular RAS synthesis (Re & Bryan, 1984; Re, 2003) within several organ and tissue sites including the brain, heart and vasculature (Phillips et al. 1993). AngII and RAS peptide reactivity within the brain is primarily observed in areas involved in sympathetic outflow and blood pressure control, including the paraventricular nucleus of the hypothalamus (Li et al. 2012; Biancardi et al. 2014) nucleus tractus solitarius (Li et al. 2012; Shan et al. 2013; Biancardi et al. 2014), rostroventral lateral medulla (Li et al. 2012; Biancardi et al. 2014) and subfornical organ (Hendel & Collister, 2005; Cao et al. 2012; Li et al. 2012), where the effects of AngII are primarily transduced via activation its cognate Gq‐coupled receptor AT1R (Sakai et al. 2004; Tan et al. 2004; Zhu et al. 2004; Sakai & Sigmund, 2005; Wang et al. 2012; Shan et al. 2013; Biancardi et al. 2014; Young & Davisson, 2015).
Evidence suggests that AngII signalling is enhanced in the CNS in hypertension (Chai et al. 1993; Gironacci et al. 2004; Schiffer et al. 2009; Young & Davisson, 2015; Santos et al. 2018), heart failure (Wang et al. 2012) and post‐myocardial infarction (Tan et al. 2004). AngII also has a direct stimulatory effect on peripheral sympathetic neurons themselves (Cox et al. 2000; DiBona, 2000; Ma et al. 2001; Fernandez et al. 2003; Talaia et al. 2006; Wang et al. 2012; Berg, 2013). Critically, mice lacking the AngII receptor AT1R within catecholaminergic neurons develop fewer pathological effects following chronic AngII infusions. This includes attenuated sympathetic activation, reduced hypertensive responses and amelioration of ventricular hypertrophy (Jancovski et al. 2013). Collectively, this demonstrates the potential importance of neuronal AngII–AT1R activation in the aetiology of sympathetic overactivity and neurogenic hypertension.
The close relationship between elevated AngII and sympathetic overactivity in cardiovascular disease is intriguing (Hilgers et al. 1993; Cox et al. 2000; Goldsmith, 2004; Berg, 2013) and has raised questions surrounding membrane level receptor–receptor interactions and cross‐talk between AngII and adrenergic signalling cascades (Grant & McGrath, 1988; Barki‐Harrington et al. 2003; Tilley, 2011; Saulière et al. 2012; Bellot et al. 2015; Liu et al. 2017; Tóth et al. 2018). Specifically, AT1R‐α2c adrenergic receptor (AT1R‐α2c‐AR) heterodimers have been observed, where activation by NA promotes atypical enhanced cAMP–PKA signalling by converting an α2c‐AR autoinhibitory signal to excitatory positive feedback signalling (Bellot et al. 2015). Moreover, activation of the AT1Rα2c‐AR heterodimer facilitates NA hypersecretion and sympathetic overactivity in sympathetic neurons in vivo (Bellot et al. 2015). Heterodimer formation has also been found to occur between AT1R‐β2‐AR (Barki‐Harrington et al. 2003; Tóth et al. 2017), which enhances the membrane stability of β2‐AR and prolongs cAMP signalling. These results support our observations that AngII augments presynaptic β‐AR‐evoked cAMP (Fig. 5) and suggests a potential synergistic role for NA–AngII‐mediated effects in provoking sympathetic overactivity in hypertension and cardiovascular pathophysiology (Barki‐Harrington et al. 2003; Lourdes González‐Hernández et al. 2010; Christensen et al. 2011; Berg, 2013; Bellot et al. 2015; Liu et al. 2017; Tóth et al. 2018).
Alterations in cardiac sympathetic transmitter release
Two simultaneous observations led to the concept of Adr as a pathological entity in the progression of hypertension. First, it was observed that Adr infusions underpin sustained increases in blood pressure post‐infusion (Majewski et al. 1981; Brown & Macquin, 1982; Brown & Dollery, 1984); and secondly, that plasma Adr is elevated in hypertensive patients (Franco‐Morselli et al. 1977; Brown & Macquin, 1981). Brown & Macquin (1981) proposed the ‘adrenaline hypothesis’ of essential hypertension (Brown & Dollery, 1984), which highlights a dominant role for Adr in facilitating NA release through actions at presynaptic β‐ARs (Abboud et al. 1964; Floras et al. 1988, 1990). The source of Adr, however, was not fully resolved with reports suggesting chronic neuronal uptake and enhanced release of circulating Adr derived from the adrenals as the primary site (Brown & Macquin, 1981; Majewski, 1983; Horikoshi et al. 1985; Blankestijn et al. 1988; Misu et al. 1988; Floras, 1992; Gudmundsdottir et al. 2008). Evidence has pointed to the possible synthesis of Adr in sympathetic nerves in patients with hypertension and stress disorder (Esler et al. 2008), but the in situ synthesis of Adr and a role for cardiac sympathetic Adr in the aetiology of hypertension are far from well‐established.
Our RNA sequencing dataset provided a comprehensive profile of neurotransmitters and their respective synthesising enzymes in rat stellate ganglia (Bardsley et al. 2018 a). Alongside the presence of classical transmitters and sympathetic markers, we also observed the transcript encoding phenylethanolamine N‐methyltransferase (PNMT), the enzyme involved in the conversion of NA to Adr (Bardsley et al. 2018 a). Protein concentrations of PNMT were detectable in rat and human stellate ganglia. To ascertain whether the presence of PNMT results in physiological concentrations and release of Adr, we electrically stimulated stellate ganglia from normotensive and hypertensive rats. Levels of both NA and Adr were elevated in the perfusate collected from prehypertensive SHR ganglia, whereas only NA could be detected in perfusate from healthy rat ganglia, and Adr was not observed (Fig. 6; Bardsley et al. 2018 b). In support of this observation, a 20‐year follow‐up of the Oslo study on normotensive, prehypertensive and male patients with established hypertension has identified arterial Adr as an independent predictor of blood pressure elevation (Gudmundsdottir et al. 2008), re‐raising the question of the importance of Adr in the pathophysiology of hypertension (Rumantir et al. 2000 a). It is now evident that Adr synthesis occurs directly within cardiac sympathetic nerves in diseases associated with sympathetic overactivity (Esler et al. 2008), and that this neurotransmitter switching takes place before elevations in arterial blood pressure are observed (Bardsley et al. 2018 b). In addition to the observed elevation in β‐AR‐mediated cAMP–PKA–Ca2+ signalling in prehypertensive rat stellate ganglia, these data support the notion of a causal role for Adr in the pathophysiology of neurogenic hypertension.
Targeting sympathetic overactivity: where are we now?
Hypertension is central in determining cardiovascular risk and is a strong predictive indicator of morbidity and mortality; however, there still remains an unmet clinical need for disease‐modifying and prophylactic interventions. Cardiac sympathetic hyperactivity is a key feature of human hypertension that is also seen in animal models of cardiovascular disease (Esler, 2010; Larsen et al. 2016 a), yet interventions that target this sympathetic phenotype are problematic to develop, due to the anatomical location of the cardiac sympathetic ganglia (Kwon et al. 2018) and the challenge in unravelling the underlying pathophysiological mechanisms. Surgical techniques such as sympathectomy per se, provide symptomatic relief and lead to fewer cardiovascular co‐morbidities in hypertension (Morrissey et al. 1953) and reduce the incidence of ventricular arrhythmia (Ajijola et al. 2014; Irie et al. 2017), yet these techniques are not without risk (Ajijola et al. 2014). Current pharmacological approaches including β‐blockers and AngII inhibitors are mainstay therapeutic strategies for early hypertension and many other cardiovascular diseases associated with dysautonomia (Wiysonge et al. 2017). However, their efficacy may also be explained via reductions in peripheral sympatho‐transmission. Approaches that aim to modulate sympathetic overactivity may have both a therapeutic and a physiological advantage over surgical techniques. Optimal neuromodulation of sympathetic tone will counteract hypertension‐induced cardiovascular damage whilst retaining a level of sympathetic reserve that will still enable cardiac performance during physical exertion. Gene transfer therapies that modulate cyclic nucleotide activity have had some success in improving neuronal activity, and a new era of genetic and protein modification techniques might be predicted to underpin the primary areas of advancement in this field. Moreover, the application of bioinformatics and the integration of machine‐learning techniques with primary research may provide novel approaches for assisting diagnoses and prediction (LaFreniere et al. 2016; Kublanov et al. 2017; Savage, 2017; Poplin et al. 2018) as well as providing clarity regarding the complex interactions between pathways and their associated cellular and molecular processes (Cunningham, 2017; Wang et al. 2017; Xie et al. 2017; Cholley et al. 2018; Costello & Martin, 2018; Pavillon et al. 2018), as a way to facilitate precise therapeutic targeting.
Conclusion
Sympathetic overactivity is a well‐established contributor to hypertension and CVD. Increased intracellular Ca2+ augments neurotransmission early in disease before increases in blood pressure develop. This Ca2+ phenotype is underpinned by an impaired cAMP/cGMP balance that is weighted in favour of cAMP–PKA‐dependent activity. Evidence suggests that this alteration in cN signalling results from changes in presynaptic receptor expression and signalling pathways, as well as critical changes in PDE activity. Pharmacological, surgical and genetic techniques aimed at reducing sympathetic tone or raising vagal transmission have had reasonable levels of success reducing hypertension and improving cardiac function (Morrissey et al. 1953; Heaton et al. 2007; Sabbah et al. 2011; Rathi et al. 2013; Ajijola et al. 2014; Sverrisdottir et al. 2014; Shivkumar et al. 2016; Irie et al. 2017); nevertheless, no prophylactic strategies have yet successfully entered the clinical arena, emphasising a critical need for translational advancements in this field.
Additional information
Competing interests
None of the authors have any conflicts of interests.
Author contributions
Both authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding information
This work was funded by the Wellcome Trust OXION (105409/Z/14/Z) and British Heart Foundation (RG/17/14/33085).
Biographies
David J. Paterson is Professor of Physiology and Head of the Department of Physiology, Anatomy & Genetics at the University of Oxford. He graduated from the Universities of Otago (NZ), Western Australia and Oxford, gaining his DPhil from Oxford and DSc from the University of Western Australia. He is a group leader in the British Heart Foundation Centre of Research Excellence at Oxford, and is Honorary Director of the Burdon Sanderson Cardiac Science Centre. As a cardiac neurobiologist, his research focuses on the neural control of the cardiorespiratory system in normal and diseased states. In 2014 he was made an Honorary Fellow of The Royal Society of New Zealand, and in 2018 was awarded the Carl Ludwig Distinguished Lectureship from the American Physiological Society and made President‐elect of The Physiological Society.

Emma N. Bardsley is a final year Wellcome trust OXION DPhil student in the Department of Physiology, Anatomy & Genetics at the University of Oxford. She graduated from King's College London in 2014 and moved to Oxford to pursue her DPhil research in David Paterson's laboratory. Her main research interests are in using cellular and molecular approaches with transcriptomics to investigate how second messengers impact on neuronal and cardiac function in health and disease.
Edited by: Ole Petersen & Alicia D'Souza
This review was presented at the symposium ‘2018 Gordon Research Conference on Cardiac Regulatory Mechanisms’, which took place at Colby‐Sawyer College, New London, NH, USA, 3–8 June 2018.
This is an Editor's Choice article from the 15 July 2020 issue.
Contributor Information
E. N. Bardsley, Email: emma.bardsley@dpag.ox.ac.uk.
D. J. Paterson, Email: david.paterson@dpag.ox.ac.uk.
References
- Abboud FM, Eckstein JW & Zimmerman BG (1964). Effect of dichloroisoproterenol on vascular responses to catecholamines. J Clin Invest 43, 316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MA, Bobik A & Korner PI (1989). Differential development of vascular and cardiac hypertrophy in genetic hypertension. Relation to sympathetic function. Hypertension 14, 191–202. [DOI] [PubMed] [Google Scholar]
- Ajijola OA, Vaseghi M, Mahajan A & Shivkumar K (2014). Bilateral cardiac sympathetic denervation: why, who and when? Expert Rev Cardiovasc Ther 10, 947–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike T, Du N, Lu G, Minamisawa S, Wang Y & Ruan H (2017). A sarcoplasmic reticulum localized protein phosphatase regulates phospholamban phosphorylation and promotes ischemia reperfusion injury in the heart. JACC Basic Transl Sci 2, 160–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apparsundaram S & Eikenburg DC (1995). Role of prejunctional beta adrenoceptors in rat cardiac sympathetic neurotransmission. J Pharmacol Exp Ther 272, 519–526. [PubMed] [Google Scholar]
- Assenza MR, Barbagallo F, Barrios F, Cornacchione M, Campolo F, Vivarelli E, Gianfrilli D, Auletta L, Soricelli A, Isidori AM, Lenzi A, Pellegrini M & Naro F (2018). Critical role of phosphodiesterase 2A in mouse congenital heart defects. Cardiovasc Res 114, 830–845. [DOI] [PubMed] [Google Scholar]
- Baliga RS, Preedy MEJ, Dukinfield MS, Chu SM, Aubdool AA, Bubb KJ, Moyes AJ, Tones MA & Hobbs AJ (2018). Phosphodiesterase 2 inhibition preferentially promotes NO/guanylyl cyclase/cGMP signaling to reverse the development of heart failure. Proc Natl Acad Sci U S A 115, E7428–E7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardsley EN, Davis H, Ajijola OA, Buckler KJ, Ardell JL, Shivkumar K & Paterson DJ (2018. a). RNA sequencing reveals novel transcripts from sympathetic stellate ganglia during cardiac sympathetic hyperactivity. Sci Rep 8, 8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardsley EN, Davis H, Buckler KJ & Paterson DJ (2018. b). Neurotransmitter switching coupled to β‐adrenergic signaling in sympathetic neurons in prehypertensive states. Hypertension 71, 1226–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardsley EN, Larsen HE & Paterson DJ (2016). Impaired cAMP‐cGMP cross‐talk during cardiac sympathetic dysautonomia. Channels (Austin) 11, 178–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barki‐Harrington L, Luttrell LM & Rockman HA (2003). Dual inhibition of beta‐adrenergic and angiotensin II receptors by a single antagonist: a functional role for receptor‐receptor interaction in vivo. Circulation 108, 1611–1618. [DOI] [PubMed] [Google Scholar]
- Behar J, Ganesan A, Zhang J & Yaniv Y (2016). The autonomic nervous system regulates the heart rate through cAMP‐PKA dependent and independent coupled‐clock pacemaker cell mechanisms. Front Physiol 7, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellot M, Galandrin S, Boularan C, Matthies HJ, Despas F, Denis C, Javitch J, Mazères S, Sanni SJ, Pons V, Seguelas MH, Hansen JL, Pathak A, Galli A, Sénard JM & Galés C (2015). Dual agonist occupancy of AT1‐R‐α2C‐AR heterodimers results in atypical Gs‐PKA signaling. Nat Chem Biol 11, 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender AT (2006). Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58, 488–520. [DOI] [PubMed] [Google Scholar]
- Berg T (2013). Angiotensin AT1‐α2C‐adrenoceptor interaction disturbs α2A‐auto‐inhibition of catecholamine release in hypertensive rats. Front Neurol 4, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancardi VC, Son SJ, Ahmadi S, Filosa JA & Stern JE (2014). Circulating angiotensin II gains access to the hypothalamus and brain stem during hypertension via breakdown of the blood‐brain barrier. Hypertension 63, 572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankestijn PJ, Man in't Veld AJ, Tulen J, van den Meiracker AH, Boomsma F, Moleman P, Ritsema van Eck HJ, Derkx FH, Mulder P & Lamberts SJ (1988). Support for adrenaline‐hypertension hypothesis: 18 hour pressor effect after 6 hours adrenaline infusion. Lancet 2, 1386–1389. [DOI] [PubMed] [Google Scholar]
- Boda H, Uchida H, Takaiso N, Ouchi Y, Fujita N, Kuno A, Hata T, Nagatani A, Funamoto Y, Miyata M, Yoshikawa T, Kurahashi H & Inagaki H (2016). A PDE3A mutation in familial hypertension and brachydactyly syndrome. J Hum Genet 61, 701–703. [DOI] [PubMed] [Google Scholar]
- Briant LJB, Paton JFR, Pickering AE & Champneys AR (2015). Modelling the vascular response to sympathetic postganglionic nerve activity. J Theor Biol 371, 102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briant LJB, Stalbovskiy AO, Nolan MF, Champneys AR & Pickering AE (2014). Increased intrinsic excitability of muscle vasoconstrictor preganglionic neurons may contribute to the elevated sympathetic activity in hypertensive rats. J Neurophysiol 112, 2756–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook RD & Julius S (2000). Autonomic imbalance, hypertension, and cardiovascular risk. Am J Hypertens 13, 112S–122S. [DOI] [PubMed] [Google Scholar]
- Brown AM & Birnbaumer L (1988). Direct G protein gating of ion channels. Am J Physiol 254, H401–H410. [DOI] [PubMed] [Google Scholar]
- Brown MJ & Dollery CT (1984). Adrenaline and hypertension. Clin Exp Hypertens A6, 539–549. [DOI] [PubMed] [Google Scholar]
- Brown MJ & Macquin I (1981). Is adrenaline the cause of essential hypertension? Lancet 2, 1079–1082. [DOI] [PubMed] [Google Scholar]
- Brown MJ & Macquin I (1982). Catecholamine neurotransmitters and the heart. Acta Medica Scandinavica 211, 34–39. [DOI] [PubMed] [Google Scholar]
- Cao X, Peterson JR, Wang G, Anrather J, Young CN, Guruju MR, Burmeister MA, Iadecola C & Davisson RL (2012). Angiotensin II‐dependent hypertension requires cyclooxygenase 1‐derived prostaglandin E2 and EP1 receptor signaling in the subfornical organ of the brain. Hypertension 59, 869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARDIoGRAMplusC4D Consortium et al (2013). Large‐scale association analysis identifies new risk loci for coronary artery disease. Nat Methods 45, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catt KJ, Zimmet PZ, Cain MD, Cran E, Best JB & Coghlan JP (1971). Angiotensin II blood‐levels in human hypertension. Lancet 297, 459–464. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2003). International union of pharmacology. XL. Compendium of voltage‐gated ion channels: calcium channels. Pharmacol Rev 55, 579–581. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2011). Voltage‐gated calcium channels. Cold Spring Harb Perspect Biol 3, a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celsus AC & Spencer WG (1935). De Medicina (On Medicine). Loeb Classical Library. Harvard University Press, Cambridge, MA, USA: /W. Heinemann, Ltd, London. [Google Scholar]
- Chai CY, Hellmann W, Tseng CJ & Luft FC (1993). Angiotensinogen mRNA and pressor reactions to angiotensin in brain stem areas of spontaneously hypertensive rats. Clin Exp Hypertens 15, 709–725. [DOI] [PubMed] [Google Scholar]
- Cholley P‐E, Moehlin J, Rohmer A, Zilliox V, Nicaise S, Gronemeyer H & Mendoza‐Parra MA (2018). Modeling gene‐regulatory networks to describe cell fate transitions and predict master regulators. NPJ Syst Biol Appl 4, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen GL, Knudsen S, Schneider M, Aplin M, Gammeltoft S, Sheikh SP & Hansen JL (2011). AT1 receptor Gαq protein‐independent signalling transcriptionally activates only a few genes directly, but robustly potentiates gene regulation from the β2‐adrenergic receptor. Mol Cell Endocrinol 331, 49–56. [DOI] [PubMed] [Google Scholar]
- Costello Z & Martin HG (2018). A machine learning approach to predict metabolic pathway dynamics from time‐series multiomics data. NPJ Syst Biol Appl 4, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox SL, Schelb V, Trendelenburg AU & Starke K (2000). Enhancement of noradrenaline release by angiotensin II and bradykinin in mouse atria: Evidence for cross‐talk between Gq/11 protein‐ and Gi/o protein‐coupled receptors. Br J Pharmacol 129, 1095–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim H‐S, Smithies O, Le TH & Coffman TM (2006). Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A 103, 17985–17990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham RJ (2017). The application of deep convolutional neural networks to ultrasound for modelling of dynamic states within human skeletal muscle. bioRxiv, doi: 10.1101/157479. [DOI] [Google Scholar]
- Dang AM, Zheng DY, Wang B, Zhang YQ, Zhang PH, Yu MF, Liu GZ & Liu LS (1999). The role of the renin‐angiotensin and cardiac sympathetic nervous systems in the development of hypertension and left ventricular hypertrophy in spontaneously hypertensive rats. Hypertens Res 22, 217–221. [DOI] [PubMed] [Google Scholar]
- D'Ascenzo M, Martinotti G, Azzena GB & Grassi C (2002). cGMP/protein kinase G‐dependent inhibition of N‐type Ca2+ channels induced by nitric oxide in human neuroblastoma IMR32 cells. J Neurosci 22, 7485–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danson E, Choate J & Paterson DJ (2005). Cardiac nitric oxide: Emerging role for nNOS in regulating physiological function. Pharmacol Ther 106, 57–74. [DOI] [PubMed] [Google Scholar]
- Danson EJ, Li D, Wang L, Dawson TA & Paterson DJ (2009). Targeting cardiac sympatho‐vagal imbalance using gene transfer of nitric oxide synthase. J Mol Cell Cardiol 46, 482–489. [DOI] [PubMed] [Google Scholar]
- Davis H, Bardsley EN & Paterson DJ (2018). Data descriptor: Transcriptional profiling of stellate ganglia from normotensive and spontaneously hypertensive rat strains. Sci Data, 10.1038/sdata.2018.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TA, Li D, Woodward T, Barber Z, Wang L & Paterson DJ (2008). Cardiac cholinergic NO‐cGMP signaling following acute myocardial infarction and nNOS gene transfer. Am J Physiol Heart Circ Physiol 295, H990–H998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiBona GF (2000). Nervous kidney: Interaction between renal sympathetic nerves and the renin‐angiotensin system in the control of renal function. Hypertension 36, 1083–1088. [DOI] [PubMed] [Google Scholar]
- Ehret GB et al (2011). Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 478, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdmann J, Stark K, Esslinger UB, Rumpf PM, Koesling D, de Wit C, Kaiser FJ, Braunholz D, Medack A, Fischer M, Zimmermann ME, Tennstedt S, Graf E, Eck S, Aherrahrou Z, Nahrstaedt J, Willenborg C, Bruse P, Brænne I, Nöthen MM, Hofmann P, Braund PS, Mergia E, Reinhard W, Burgdorf C, Schreiber S, Balmforth AJ, Hall AS, Bertram L, Steinhagen‐Thiessen E, Li SC, März W, Reilly M, Kathiresan S, McPherson R, Walter U; CARDIoGRAM , Ott J, Samani NJ, Strom TM, Meitinger T, Hengstenberg C & Schunkert H (2013). Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature 504, 432–436. [DOI] [PubMed] [Google Scholar]
- Esler M (2010). Sympathetic nervous activation in essential hypertension: commonly neglected as a therapeutic target, usually ignored as a drug side effect. Hypertension 55, 1090–1091. [DOI] [PubMed] [Google Scholar]
- Esler M (2014). Sympathetic nervous system moves toward center stage in cardiovascular medicine from Thomas Willis to resistant hypertension. Hypertension 63, e25–e32. [DOI] [PubMed] [Google Scholar]
- Esler M, Eikelis N, Schlaich M, Lambert G, Alvarenga M, Kaye D, El‐Osta A, Guo L, Barton D, Pier C, Brenchley C, Dawood T, Jennings G & Lambert E (2008). Human sympathetic nerve biology: parallel influences of stress and epigenetics in essential hypertension and panic disorder. Ann N Y Acad Sci 1148, 338–348. [DOI] [PubMed] [Google Scholar]
- Esler M, Jackman G, Bobik A, Leonard P, Kelleher D, Skews H, Jennings G & Korner P (1981). Norepinephrine kinetics in essential hypertension. Defective neuronal uptake of norepinephrine in some patients. Hypertension 3, 149–156. [DOI] [PubMed] [Google Scholar]
- Esler M, Jennings G, Biviano B, Lambert G & Hasking G (1986). Mechanism of elevated plasma noradrenaline in the course of essential hypertension. J Cardiovasc Pharmacol 8, S39–S43. [DOI] [PubMed] [Google Scholar]
- Esler M, Jennings G, Korner P, Willett I, Dudley F, Hasking G, Anderson W & Lambert G (1988). Assessment of human sympathetic nervous‐system activity from measurements of norepinephrine turnover. Hypertension 11, 3–20. [DOI] [PubMed] [Google Scholar]
- Esler MD, Wallin G, Dorward PK, Eisenhofer G, Westerman R, Meredith I, Lambert G, Cox HS & Jennings G (1991). Effects of desipramine on sympathetic nerve firing and norepinephrine spillover to plasma in humans. Am J Physiol 260, R817–R823. [DOI] [PubMed] [Google Scholar]
- Fernandez SF, Huang M‐H, Davidson BA, Knight PR & Izzo JL (2003). Modulation of angiotensin II responses in sympathetic neurons by cytosolic calcium. Hypertension 41, 56–63. [DOI] [PubMed] [Google Scholar]
- Ferrara LA, Moscato TS, Pisanti N, Marotta T, Krogh V, Capone D & Mancini M (1988). Is the sympathetic nervous system altered in children with familial history of arterial hypertension? Cardiology 75, 200–205. [DOI] [PubMed] [Google Scholar]
- Flack JM, Atlas SA, Pool JL & White WB (2007). Renin‐angiotensin aldosterone system and hypertension: current approaches and future directions. J Manag Care Spec Pharm 13, 1–39. [Google Scholar]
- Floras JS (1992). Epinephrine and the genesis of hypertension. Hypertension 19, 1–18. [DOI] [PubMed] [Google Scholar]
- Floras JS, Aylward PE, Mark AL & Abboud FM (1990). Adrenaline facilitates neurogenic vasoconstriction in borderline hypertensive subjects. J Hypertens 8, 443–448. [DOI] [PubMed] [Google Scholar]
- Floras JS, Aylward PE, Victor RG, Mark AL & Abboud FM (1988). Epinephrine facilitates neurogenic vasoconstriction in humans. J Clin Invest 81, 1265–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florea VG & Cohn JN (2014). The autonomic nervous system and heart failure. Circ Res 114, 1815–1826. [DOI] [PubMed] [Google Scholar]
- Francis SH, Blount MA & Corbin JD (2011). Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev 91, 651–690. [DOI] [PubMed] [Google Scholar]
- Franco‐Morselli R, Elghozi JL, Joly E, Di Giuilio S & Meyer P (1977). Increased plasma adrenaline concentrations in benign essential hypertension. Br Med J 2, 1251–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frishman WH (2016). Beta‐adrenergic receptor blockers in hypertension: alive and well. Prog Cardiovasc Dis 59, 247–252. [DOI] [PubMed] [Google Scholar]
- Gaskell WH (1886). On the structure, distribution and function of the nerves which innervate the visceral and vascular systems. J Physiol 7, 1–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazi L & Drawz P (2017). Advances in understanding the renin‐angiotensin‐aldosterone system (RAAS) in blood pressure control and recent pivotal trials of RAAS blockade in heart failure and diabetic nephropathy. F1000Res 6, F1000 Faculty Rev‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gironacci MM, Valera MS, Yujnovsky I & Peña C (2004). Angiotensin‐(1‐7) inhibitory mechanism of norepinephrine release in hypertensive rats. Hypertension 44, 783–787. [DOI] [PubMed] [Google Scholar]
- Goldsmith SR (2004). Interactions between the sympathetic nervous system and the RAAS in heart failure. Curr Heart Fail Rep 1, 45–50. [DOI] [PubMed] [Google Scholar]
- Grant TL & McGrath JC (1988). Interactions between angiotensin‐II and alpha‐adrenoceptor agonists mediating pressor‐responses in the pithed rat. Br J Pharmacol 95, 1229–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi G & Esler M (1999). How to assess sympathetic activity in humans. J Hypertens 17, 719–734. [DOI] [PubMed] [Google Scholar]
- Grassi G, Mark A & Esler M (2015). The sympathetic nervous system alterations in human hypertension. Circ Res 116, 976–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray PC, Scott JD & Catterall WA (1998). Regulation of ion channels by cAMP‐dependent protein kinase and A‐kinase anchoring proteins. Curr Opin Neurobiol 8, 330–334. [DOI] [PubMed] [Google Scholar]
- Gudmundsdottir H, Strand AH, Høieggen A, Reims HM, Westheim AS, Eide IK, Kjeldsen SE & Os I (2008). Do screening blood pressure and plasma catecholamines predict development of hypertension? Twenty‐year follow‐up of middle‐aged men. Blood Press 17, 94–103. [DOI] [PubMed] [Google Scholar]
- Guo L, Lee AA, Rizvi TA, Ratner N & Kirschner LS (2013). The protein kinase A regulatory subunit R1A (Prkar1a) plays critical roles in peripheral nerve development. J Neurosci 33, 17967–17975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG (2006). The sympathetic control of blood pressure. Nat Rev Neurosci 7, 335–346. [DOI] [PubMed] [Google Scholar]
- Hall JE (1986). Control of sodium‐excretion by angiotensin‐II – intrarenal mechanisms and blood‐pressure regulation. Am J Physiol 250, R960–R972. [DOI] [PubMed] [Google Scholar]
- Hamer M (2006). The effects of exercise on haemodynamic function in relation to the familial hypertension risk model. J Hum Hypertens 20, 313–319. [DOI] [PubMed] [Google Scholar]
- Hanna P, Rajendran PS, Ajijola OA, Vaseghi M, Armour JA, Ardell JL & Shivkumar K (2017). Cardiac neuroanatomy – Imaging nerves to define functional control. Auton Neurosci 207, 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson L, Lindholm LH, Niskanen L, Lanke J, Hedner T, Niklason A, Luomanmäki K, Dahlöf B, de Faire U, Mörlin C, Karlberg BE, Wester PO, Björck J‐E; the Captopril Prevention Project CAPPP study group (1999). Effect of angiotensin‐converting‐enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: The Captopril Prevention Project (CAPPP) randomised trial. Lancet 353, 611–616. [DOI] [PubMed] [Google Scholar]
- Hausberg M, Sinkey CA, Mark AL, Hoffman RP & Anderson EA (1998). Sympathetic nerve activity and insulin sensitivity in normotensive offspring of hypertensive parents. Am J Hypertens 11, 1312–1320. [DOI] [PubMed] [Google Scholar]
- Heaton DA, Lei M, Li D, Golding S, Dawson TA, Mohan RM & Paterson DJ (2006). Remodeling of the cardiac pacemaker L‐type calcium current and its beta‐adrenergic responsiveness in hypertension after neuronal NO synthase gene transfer. Hypertension 48, 443–452. [DOI] [PubMed] [Google Scholar]
- Heaton DA, Li D, Almond SC, Dawson TA, Wang L, Channon KM & Paterson DJ (2007). Gene transfer of neuronal nitric oxide synthase into intracardiac ganglia reverses vagal impairment in hypertensive rats. Hypertension 49, 380–388. [DOI] [PubMed] [Google Scholar]
- Hendel MD & Collister JP (2005). Contribution of the subfornical organ to angiotensin II‐induced hypertension. Am J Physiol Heart Circ Physiol 288, H680–H685. [DOI] [PubMed] [Google Scholar]
- Herring N & Paterson DJ (2018). Levick's Introduction to Cardiovascular Physiology, 6th edn. CRC Press, Boca Raton. [Google Scholar]
- Hilgers KF, Veelken R, Rupprecht G, Reeh PW, Luft FC & Mann JF (1993). Angiotensin II facilitates sympathetic transmission in rat hind limb circulation. Hypertension 21, 322–328. [DOI] [PubMed] [Google Scholar]
- Hoff HE (1940). The history of vagal inhibition. Bull Hist Med 8, 461–496. [Google Scholar]
- Horikoshi Y, Tajima I, Igarashi H, Inui M, Kasahara K & Noguchi T (1985). The adreno‐sympathetic system, the genetic predisposition to hypertension, and stress. Am J Med Sci 289, 186–191. [DOI] [PubMed] [Google Scholar]
- Horvath A, Bertherat J, Groussin L, Guillaud‐Bataille M, Tsang K, Cazabat L, Libé R, Remmers E, René‐Corail F, Faucz FR, Clauser E, Calender A, Bertagna X, Carney JA & Stratakis CA (2010). Mutations and polymorphisms in the gene encoding regulatory subunit type 1‐alpha of protein kinase A (PRKAR1A): an update. Hum Mutat 31, 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua R, Adamczyk A, Robbins C, Ray G & Rose RA (2012). Distinct patterns of constitutive phosphodiesterase activity in mouse sinoatrial node and atrial myocardium. PLoS One 7, e47652–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imredy JP & Yue DT (1994). Mechanism of Ca2+‐sensitive inactivation of L‐type Ca2+channels. Neuron 12, 1301–1318. [DOI] [PubMed] [Google Scholar]
- Ino M, Yoshinaga T, Wakamori M, Miyamoto N, Takahashi E, Sonoda J, Kagaya T, Oki T, Nagasu T, Nishizawa Y, Tanaka I, Imoto K, Aizawa S, Koch S, Schwartz A, Niidome T, Sawada K & Mori Y (2001). Functional disorders of the sympathetic nervous system in mice lacking the α1B subunit (Cav2.2) of N‐type calcium channels. Proc Natl Acad Sci U S A 98, 5323–5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie T, Yamakawa K, Hamon D, Nakamura K, Shivkumar K & Vaseghi M (2017). Cardiac sympathetic innervation via middle cervical and stellate ganglia and antiarrhythmic mechanism of bilateral stellectomy. Am J Physiol Heart Circ Physiol 312, H392–H405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iriuchijima J (1973). Sympathetic discharge rate in spontaneously hypertensive rats. Jpn Heart J 14, 350–356. [DOI] [PubMed] [Google Scholar]
- Jancovski N, Bassi JK, Carter DA, Choong Y‐T, Connelly A, Nguyen T‐P, Chen D, Lukoshkova EV, Menuet C, Head GA & Allen AM (2013). Stimulation of angiotensin type 1A receptors on catecholaminergic cells contributes to angiotensin‐dependent hypertension. Hypertension 62, 866–871. [DOI] [PubMed] [Google Scholar]
- Johansson M, Elam M, Rundqvist B, Eisenhofer G, Herlitz H, Lambert G & Friberg P (1999). Increased sympathetic nerve activity in renovascular hypertension. Circulation 99, 2537–2542. [DOI] [PubMed] [Google Scholar]
- Johncy SS, Karthik CS, Bondade SY & Jayalakshmi MK (2015). Altered cardiovascular autonomic function in young normotensive offspring of hypertensive parents—Is obesity an additional risk factor? J Basic Clin Physiol Pharmacol 26, 531–537. [DOI] [PubMed] [Google Scholar]
- Judy WV, Watanabe AM, Murphy WR, Aprison BS & Yu PL (1979). Sympathetic nerve activity and blood pressure in normotensive backcross rats genetically related to the spontaneously hypertensive rat. Hypertension 1, 598–604. [DOI] [PubMed] [Google Scholar]
- Kalla M, Chotalia M, Coughlan C, Hao G, Crabtree MJ, Tomek J, Bub G, Paterson DJ & Herring N (2016). Protection against ventricular fibrillation via cholinergic receptor stimulation and the generation of nitric oxide. J Physiol 594, 3981–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz SD, Balidemaj K, Homma S, Wu H, Wang J & Maybaum S (2000). Acute type 5 phosphodiesterase inhibition with sildenafil enhances flow‐mediated vasodilation in patients with chronic heart failure. J Am Coll Cardiol 36, 845–851. [DOI] [PubMed] [Google Scholar]
- Kaye DM, Lefkovits J, Jennings GL, Bergin P, Broughton A & Esler MD (1995). Adverse consequences of high sympathetic nervous activity in the failing human heart. J Am Coll Cardiol 26, 1257–1263. [DOI] [PubMed] [Google Scholar]
- Kessler T, Wobst J, Wolf B, Eckhold J, Vilne B, Hollstein R, von Ameln S, Dang TA, Sager HB, Moritz Rumpf P, Aherrahrou R, Kastrati A, Björkegren JLM, Erdmann J, Lusis AJ, Civelek M, Kaiser FJ & Schunkert H (2017). Functional characterization of the GUCY1A3 coronary artery disease risk locus. Circulation 136, 476–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Miura Y, Adachi M, Nezu M, Toriyabe S, Sugawara T, Ishizuka Y, Noshiro T & Takahashi M (1983). The effect of sodium depletion on plasma norepinephrine kinetics in patients with essential hypertension. Jpn Circ J 47, 1232–1241. [DOI] [PubMed] [Google Scholar]
- Klarenbeek J, Goedhart J, van Batenburg A, Groenewald D & Jalink K (2015). Fourth‐generation Epac‐based FRET sensors for cAMP feature exceptional brightness, photostability and dynamic range: characterization of dedicated sensors for FLIM, for ratiometry and with high affinity. PLoS One 10, e0122513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzina MB, Korobkin AA, Vasilieva OA & Maslyukov PM (2011). Morphological characteristics of the stellate ganglion in white rats. Neurosci Behav Physiol 41, 436–439. [Google Scholar]
- Kublanov VS, Dolganov AY, Belo D & Gamboa H (2017). Comparison of machine learning methods for the arterial hypertension diagnostics. Appl Bionics Biomech 2017, 5985479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon OJ, Pendekanti S, Fox JN, Yanagawa J, Fishbein MC, Shivkumar K, Lambert HW & Ajijola OA (2018). Morphological spectra of adult human stellate ganglia: implications for thoracic sympathetic denervation. Anat Rec (Hoboken) 301, 1244–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFreniere D, Zulkernine F, Barber D & Martin K (2016). Using machine learning to predict hypertension from a clinical dataset. 2016 IEEE Symposium Series on Computational Intelligence (SSCI) IEEE. https://ieeexplore.ieee.org/document/7849886/.
- Langley JN (1898). On the union of cranial autonomic (visceral) fibres with the nerve cells of the superior cervical ganglion. J Physiol 23, 240–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen HE, Bardsley EN, Lefkimmiatis K & Paterson DJ (2016. a). Dysregulation of neuronal Ca2+ channel linked to heightened sympathetic phenotype in prohypertensive states. J Neurosci 36, 8562–8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen HE, Lefkimmiatis K & Paterson DJ (2016. b). Sympathetic neurons are a powerful driver of myocyte function in cardiovascular disease. Sci Rep 6, 38898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C‐W, Li D, Channon KM & Paterson DJ (2009). L‐arginine supplementation reduces cardiac noradrenergic neurotransmission in spontaneously hypertensive rats. J Mol Cell Cardiol 47, 149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, Jo SH, Danner T, Zhang M, Rainer PP, Bedja D, Kirk JA, Ranek MJ, Dostmann WR, Kwon C, Margulies KB, Van Eyk JE, Paulus WJ, Takimoto E & Kass DA (2015). Phosphodiesterase 9A controls nitric‐oxide‐ independent cGMP and hypertrophic heart disease. Nature 519, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkimmiatis K & Zaccolo M (2014). cAMP signaling in subcellular compartments. Pharmacol Ther 143, 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiser M & Fleischer N (1996). cAMP‐dependent regulation of cardiac L‐type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Diabetes 45, 1412–1418. [DOI] [PubMed] [Google Scholar]
- Li D, Lu C‐J, Hao G, Wright H, Woodward L, Liu K, Vergari E, Surdo NC, Herring N, Zaccolo M & Paterson DJ (2015). Efficacy of B‐type natriuretic peptide is coupled to phosphodiesterase 2a in cardiac sympathetic neurons. Hypertension 66, 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Nikiforova N, Lu C‐J, Wannop K, McMenamin M, Lee C‐W, Buckler KJ & Paterson DJ (2013). Targeted neuronal nitric oxide synthase transgene delivery into stellate neurons reverses impaired intracellular calcium transients in prehypertensive rats. Hypertension 61, 202–207. [DOI] [PubMed] [Google Scholar]
- Li W, Peng H, Cao T, Sato R, McDaniels SJ, Kobori H, Navar LG & Feng Y (2012). Brain‐targeted (pro)renin receptor knockdown attenuates angiotensin II‐dependent hypertension. Hypertension 59, 1188–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C‐H, Gong Z, Liang ZL, Liu ZX, Yang F, Sun YJ, Ma ML, Wang YJ, Ji CR, Wang YH, Wang MJ, Cui FA, Lin A, Zheng WS, He DF, Qu CX, Xiao P, Liu CY, Thomsen AR, Joseph Cahill T 3rd, Kahsai AW, Yi F, Xiao KH, Xue T, Zhou Z, Yu X & Sun JP (2017). Arrestin‐biased AT1R agonism induces acute catecholamine secretion through TRPC3 coupling. Nat Commun 8, 14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Li D, Hao G, McCaffary D, Neely O, Woodward L, Ioannides D, Lu C‐J, Brescia M, Zaccolo M, Tandri H, Ajijola OA, Ardell JL, Shivkumar K & Paterson DJ (2018). Phosphodiesterase 2A as a therapeutic target to restore cardiac neurotransmission during sympathetic hyperactivity. JCI Insight 3, 98694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokhandwala MF & Eikenburg DC (1983). Presynaptic receptors and alterations in norepinephrine release in spontaneously hypertensive rats. Life Sci 33, 1527–1542. [DOI] [PubMed] [Google Scholar]
- Lopes HF, Silva HB, Consolim‐Colombo FM, Barreto Filho JA, Riccio GM, Giorgi DM & Krieger EM (2000). Autonomic abnormalities demonstrable in young normotensive subjects who are children of hypertensive parents. Braz J Med Biol Res 33, 51–54. [DOI] [PubMed] [Google Scholar]
- Lourdes González‐Hernández Mde, Godínez‐Hernández D, Bobadilla‐Lugo RA & López‐Sánchez P (2010). Angiotensin‐II type 1 receptor (AT1R) and alpha‐1D adrenoceptor form a heterodimer during pregnancy‐induced hypertension. Auton Autacoid Pharmacol 30, 167–172. [DOI] [PubMed] [Google Scholar]
- Lu C‐J, Hao G, Nikiforova N, Larsen HE, Liu K, Crabtree MJ, Li D, Herring N & Paterson DJ (2015). CAPON modulates neuronal calcium handling and cardiac sympathetic neurotransmission during dysautonomia in hypertension. Hypertension 65, 1288–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XY, Chapleau MW, Whiteis CA, Abboud FM & Bielefeldt K (2001). Angiotensin selectively activates a subpopulation of postganglionic sympathetic neurons in mice. Circ Res 88, 787–793. [DOI] [PubMed] [Google Scholar]
- Maass PG, Aydin A, Luft FC, Schächterle C, Weise A, Stricker S, Lindschau C, Vaegler M, Qadri F, Toka HR, Schulz H, Krawitz PM, Parkhomchuk D, Hecht J, Hollfinger I, Wefeld‐Neuenfeld Y, Bartels‐Klein E, Mühl A, Kann M, Schuster H, Chitayat D, Bialer MG, Wienker TF, Ott J, Rittscher K, Liehr T, Jordan J, Plessis G, Tank J, Mai K, Naraghi R, Hodge R, Hopp M, Hattenbach LO, Busjahn A, Rauch A, Vandeput F, Gong M, Rüschendorf F, Hübner N, Haller H, Mundlos S, Bilginturan N, Movsesian MA, Klussmann E, Toka O & Bähring S (2015). PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat Methods 47, 647–653. [DOI] [PubMed] [Google Scholar]
- Mahapatra S, Marcantoni A, Zuccotti A, Carabelli V & Carbone E (2012). Equal sensitivity of Cav1.2 and Cav1.3 channels to the opposing modulations of PKA and PKG in mouse chromaffin cells. J Physiol 590, 5053–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski H (1983). Modulation of noradrenaline release through activation of presynaptic β‐adrenoreceptors. J Auton Pharmacol 3, 47–60. [DOI] [PubMed] [Google Scholar]
- Majewski H, Tung LH & Rand MJ (1981). Adrenaline‐induced hypertension in rats. J Cardiovasc Pharmacol 3, 179–185. [DOI] [PubMed] [Google Scholar]
- Malpas SC (2010). Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol Rev 90, 513–557. [DOI] [PubMed] [Google Scholar]
- Mancia G & Grassi G (2014). The autonomic nervous system and hypertension. Circ Res 114, 1804–1814. [DOI] [PubMed] [Google Scholar]
- Mattiazzi A & Kranias EG (2014). The role of CaMKII regulation of phospholamban activity in heart disease. Front Pharmacol 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice DH (2003). Cyclic nucleotide phosphodiesterase activity, expression, and targeting in cells of the cardiovascular system. Mol Pharmacol 64, 533–546. [DOI] [PubMed] [Google Scholar]
- Maver J, Štrucl M & Accetto R (2004). Autonomic nervous system activity in normotensive subjects with a family history of hypertension. Clin Auton Res 14, 369–375. [DOI] [PubMed] [Google Scholar]
- Mehel H, Emons J, Vettel C, Wittköpper K, Seppelt D, Dewenter M, Lutz S, Sossalla S, Maier LS, Lechêne P, Leroy J, Lefebvre F, Varin A, Eschenhagen T, Nattel S, Dobrev D, Zimmermann WH, Nikolaev VO, Vandecasteele G, Fischmeister R & El‐Armouche A (2013). Phosphodiesterase‐2 is up‐regulated in human failing hearts and blunts β‐adrenergic responses in cardiomyocytes. J Am Coll Cardiol 62, 1596–1606. [DOI] [PubMed] [Google Scholar]
- Meredith IT, Broughton A, Jennings GL & Esler MD (1991). Evidence of a selective increase in cardiac sympathetic activity in patients with sustained ventricular arrhythmias. N Engl J Med 325, 618–624. [DOI] [PubMed] [Google Scholar]
- Misu Y & Kubo T (1986). Presynaptic β‐adrenoceptors. Med Res Rev 6, 197–225. [DOI] [PubMed] [Google Scholar]
- Misu Y, Kuwahara M, Amano H & Kubo T (1988). Adrenaline as an endogenous agonist for presynaptic beta‐adrenoceptors and their relevance to the development of hypertension in spontaneously hypertensive rats. J Hypertens Suppl 6, S572–S574. [DOI] [PubMed] [Google Scholar]
- Molderings GJ, Likungu J & Göthert M (2000). N‐Type calcium channels control sympathetic neurotransmission in human heart atrium. Circulation 101, 403–407. [DOI] [PubMed] [Google Scholar]
- Mori Y, Nishida M, Shimizu S, Ishii M, Ino M, Sawada K & Niidome T (2002). Ca2+ channel α1B subunit (Cav2.2) knockout mouse reveals a predominant role of N‐type channels in the sympathetic regulation of the circulatory system. Trends Cardiovasc Med 12, 270–275. [DOI] [PubMed] [Google Scholar]
- Morrissey DM, Brookes VS & Cooke WT (1953). Sympathectomy in the treatment of hypertension – review of 122 cases. Lancet 264, 403–412. [DOI] [PubMed] [Google Scholar]
- Musheshe N, Schmidt M & Zaccolo M (2018). cAMP: from long‐range second messenger to nanodomain signalling. Trends Pharmacol Sci 39, 209–222. [DOI] [PubMed] [Google Scholar]
- Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, St Aubin C, Webster L, Rebeyka IM, Ross DB, Light PE, Dyck JRB & Michelakis ED (2007). Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation 116, 238–248. [DOI] [PubMed] [Google Scholar]
- Najafi A, Sequeira V, Kuster DWD & van der Velden J (2016). β‐Adrenergic receptor signalling and its functional consequences in the diseased heart. Eur J Clin Invest 46, 362–374. [DOI] [PubMed] [Google Scholar]
- Nedergaard OA & Abrahamsen J (1990). Modulation of noradrenaline release by activation of presynaptic beta‐adrenoceptors in the cardiovascular system. Ann N Y Acad Sci 604, 528–544. [DOI] [PubMed] [Google Scholar]
- Nikpay M et al (2015). A comprehensive 1,000 Genomes‐based genome‐wide association meta‐analysis of coronary artery disease. Nat Methods 47, 1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussberger J & Bohlender J (2013). Pharmacotherapy: optimal blockade of the renin–angiotensin–aldosterone system. Nat Methods 10, 183–184. [DOI] [PubMed] [Google Scholar]
- Nussberger J, Brunner DB, Waeber B & Brunner HR (1986). Specific measurement of angiotensin metabolites and in vitro generated angiotensin II in plasma. Hypertension 8, 476–482. [DOI] [PubMed] [Google Scholar]
- Oliveira‐Sales EB, Colombari E, Abdala AP, Campos RR & Paton JFR (2016). Sympathetic overactivity occurs before hypertension in the two‐kidney, one‐clip model. Exp Physiol 101, 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira‐Sales EB, Toward MA, Campos RR & Paton JFR (2014). Revealing the role of the autonomic nervous system in the development and maintenance of Goldblatt hypertension in rats. Auton Neurosci 183, 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal GK, Pal P, Nanda N, Lalitha V, Dutta TK & Adithan C (2011). Sympathovagal imbalance in prehypertensive offspring of two parents versus one parent hypertensive. Int J Hypertens 2011, 263170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palatini P & Julius S (2004). Elevated heart rate: a major risk factor for cardiovascular disease. Clin Exp Hypertens 26, 637–644. [DOI] [PubMed] [Google Scholar]
- Parati G & Esler M (2012). The human sympathetic nervous system: Its relevance in hypertension and heart failure. Eur Heart J 33, 1058–1066. [DOI] [PubMed] [Google Scholar]
- Pavillon N, Hobro AJ, Akira S & Smith NI (2018). Noninvasive detection of macrophage activation with single‐cell resolution through machine learning. Proc Natl Acad Sci U S A 115, E2676–E2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersson MJ, Rundqvist B, Johansson M, Eisenhofer G, Lambert G, Herlitz H, Jensen G & Friberg P (2002). Increased cardiac sympathetic drive in renovascular hypertension. J Hypertens 20, 1181–1187. [DOI] [PubMed] [Google Scholar]
- Phillips MI, Speakman EA & Kimura B (1993). Levels of angiotensin and molecular biology of the tissue renin angiotensin systems. Regul Pept 43, 1–20. [DOI] [PubMed] [Google Scholar]
- Piccirillo G, Viola E, Nocco M, Durante M, Tarantini S & Marigliano V (2000). Autonomic modulation of heart rate and blood pressure in normotensive offspring of hypertensive subjects. J Lab Clin Med 135, 145–152. [DOI] [PubMed] [Google Scholar]
- Poplin R, Varadarajan AV, Blumer K, Liu Y, McConnell MV, Corrado GS, Peng L & Webster DR (2018). Prediction of cardiovascular risk factors from retinal fundus photographs via deep learning. Nat Biomed Eng 2, 158–164. [DOI] [PubMed] [Google Scholar]
- Pruneau D & Bélichard P (1992). Haemodynamic and humoral effects of ω‐conotoxin GVIA in normotensive and spontaneously hypertensive rats. Eur J Pharmacol 211, 329–335. [DOI] [PubMed] [Google Scholar]
- Ram CVS (2010). Beta‐blockers in hypertension. Asian‐Australas J Anim Sci 106, 1819–1825. [DOI] [PubMed] [Google Scholar]
- Ramchandra R, Hood SG, Denton DA, Woods RL, McKinley MJ, McAllen RM & May CN (2009). Basis for the preferential activation of cardiac sympathetic nerve activity in heart failure. Proc Natl Acad Sci U S A 106, 924–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathi P, Agarwal V & Kumar A (2013). Sympathetic hyperactivity in children of hypertensive parents. Ann Neurosci 20, 4–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Re R & Bryan SE (1984). Functional intracellular renin‐angiotensin systems may exist in multiple tissues. Clin Exp Hypertens 6, 1739–1742. [DOI] [PubMed] [Google Scholar]
- Re RN (2003). The intracrine hypothesis and intracellular peptide hormone action. Bioessays 25, 401–409. [DOI] [PubMed] [Google Scholar]
- Riet Te L, Van Esch JHM, Roks AJM, Van Den Meiracker AH & Danser AHJ (2015). Hypertension: renin‐angiotensin‐aldosterone system alterations. Circ Res 116, 960–975. [DOI] [PubMed] [Google Scholar]
- Rippe C, Zhu B, Krawczyk KK, Van Bavel E, Albinsson S, lund JSX, Bakker ENTP & Rd KSX (2017). Hypertension reduces soluble guanylyl cyclase expression in the mouse aorta via the Notch signaling pathway. Sci Rep, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero JC & Reckelhoff JF (1999). Role of angiotensin and oxidative stress in essential hypertension. Hypertension 34, 943–949. [DOI] [PubMed] [Google Scholar]
- Rozanski A, Blumenthal JA & Kaplan J (1999). Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation 99, 2192–2217. [DOI] [PubMed] [Google Scholar]
- Rubattu S, Cotugno M, Forte M, Stanzione R, Bianchi F, Madonna M, Marchitti S & Volpe M (2018). Effects of dual angiotensin type 1 receptor/neprilysin inhibition vs. angiotensin type 1 receptor inhibition on target organ injury in the stroke‐prone spontaneously hypertensive rat. J Hypertens 36, 1902–1914. [DOI] [PubMed] [Google Scholar]
- Rumantir MS, Jennings GL, Lambert GW, Kaye DM, Seals DR & Esler MD (2000. a). The “adrenaline hypothesis” of hypertension revisited: evidence for adrenaline release from the heart of patients with essential hypertension. J Hypertens 18, 717–723. [DOI] [PubMed] [Google Scholar]
- Rumantir MS, Kaye DM, Jennings GL, Vaz M, Hastings JA & Esler MD (2000. b). Phenotypic evidence of faulty neuronal norepinephrine reuptake in essential hypertension. Hypertension 36, 824–829. [DOI] [PubMed] [Google Scholar]
- Rundqvist B, Elam M, Bergmann‐Sverrisdottir YB, Eisenhofer G & Friberg P (1997). Increased cardiac adrenergic drive precedes generalized sympathetic activation in human heart failure. Circulation 95, 169–175. [DOI] [PubMed] [Google Scholar]
- Rupp H & Jäger B (2001). The renin‐angiotensin system and the sympathetic nervous system in hypertension and congestive heart failure: Implications for therapeutic interventions. J Clin Basic Cardiol 4, 47–51. [Google Scholar]
- Sabbah HN, Ilsar I, Zaretsky A, Rastogi S, Wang M & Gupta RC (2011). Vagus nerve stimulation in experimental heart failure. Heart Fail Rev 16, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K, Chapleau MW, Morimoto S, Cassell MD & Sigmund CD (2004). Differential modulation of baroreflex control of heart rate by neuron‐ vs. glia‐derived angiotensin II. Physiol Genomics 20, 66–72. [DOI] [PubMed] [Google Scholar]
- Sakai K & Sigmund CD (2005). Molecular evidence of tissue renin‐angiotensin systems: A focus on the brain. Curr Hypertens Rep 7, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval A, Duran P, Gandini MA, Andrade A, Almanza A, Kaja S & Felix R (2017). Regulation of L‐type Cav1.3 channel activity and insulin secretion by the cGMP‐PKG signaling pathway. Cell Calcium 66, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos RAS, Sampaio WO, Alzamora AC, Motta‐Santos D, Alenina N, Bader M & Campagnole‐Santos MJ (2018). The ACE2/angiotensin‐(1–7)/mas axis of the renin‐angiotensin system: focus on angiotensin‐(1–7). Physiol Rev 98, 505–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Park NG, Kohno T, Maeda T, Kim JI, Kato R & Takahashi M (1993). Role of basic residues for the binding of omega‐conotoxin GVIA to N‐type calcium channels. Biochem Biophys Res Commun 194, 1292–1296. [DOI] [PubMed] [Google Scholar]
- Saulière A, Bellot M, Paris H, Denis C, Finana F, Hansen JT, Altié M‐F, Seguelas MH, Pathak A, Hansen JL, Sénard JM & Galés C (2012). Deciphering biased‐agonism complexity reveals a new active AT1 receptor entity. Nat Chem Biol 8, 622–630. [DOI] [PubMed] [Google Scholar]
- Savage N (2017). Machine learning: Calculating disease. Nature 550, S115–S117. [DOI] [PubMed] [Google Scholar]
- Schiffer S, Pummer S, Witte K & Lemmer B (2009). Cardiovascular regulation in TGR(mREN2)27 rats: 24h variation in plasma catecholamines, angiotensin peptides, and telemetric heart rate variability. Chronobiol Int 18, 461–474. [DOI] [PubMed] [Google Scholar]
- Schroder F (2003). Single L‐type Ca2+ channel regulation by cGMP‐dependent protein kinase type I in adult cardiomyocytes from PKG I transgenic mice. Cardiovasc Res 60, 268–277. [DOI] [PubMed] [Google Scholar]
- Sears CE, Choate JK & Paterson DJ (1998). Effect of nitric oxide synthase inhibition on the sympatho‐vagal control of heart rate. J Auton Nerv Syst 73, 63–73. [DOI] [PubMed] [Google Scholar]
- Segura‐Puimedon M, Mergia E, Al‐Hasani J, Aherrahrou R, Stoelting S, Kremer F, Freyer J, Koesling D, Erdmann J, Schunkert H, de Wit C & Aherrahrou Z (2016). Proatherosclerotic effect of the α1‐subunit of soluble guanylyl cyclase by promoting smooth muscle phenotypic switching. Am J Pathol 186, 2220–2231. [DOI] [PubMed] [Google Scholar]
- Seidel E & Scholl UI (2017). Genetic mechanisms of human hypertension and their implications for blood pressure physiology. Physiol Genomics 49, 630–652. [DOI] [PubMed] [Google Scholar]
- Shan Z, Zubcevic J, Shi P, Jun JY, Dong Y, Murça TM, Lamont GJ, Cuadra A, Yuan W, Qi Y, Li Q, Paton JFR, Katovich MJ, Sumners C & Raizada MK (2013). Chronic knockdown of the nucleus of the solitary tract AT1 receptors increases blood inflammatory‐endothelial progenitor cell ratio and exacerbates hypertension in the spontaneously hypertensive rat. Hypertension 61, 1328–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks J, Herring N, Johnson E, Liu K, Li D & Paterson DJ (2017). Overexpression of sarcoendoplasmic reticulum calcium ATPase 2a promotes cardiac sympathetic neurotransmission via abnormal endoplasmic reticulum and mitochondria Ca2+ regulation. Hypertension 69, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks J, Mane S, Ryan R & Paterson DJ (2013. a). Ganglion‐specific impairment of the norepinephrine transporter in the hypertensive rat. Hypertension 61, 197–193. [DOI] [PubMed] [Google Scholar]
- Shanks J, Manou‐Stathopoulou S, Lu C‐J, Li D, Paterson DJ & Herring N (2013. b). Cardiac sympathetic dysfunction in the prehypertensive spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol 305, H980–H986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan D (1936). Discovery of the autonomic nervous system. Arch Neurol Psychiatry 35, 1081–1115. [Google Scholar]
- Shinohara K, Kishi T, Hirooka Y & Sunagawa K (2015). Circulating angiotensin II deteriorates left ventricular function with sympathoexcitation via brain angiotensin II receptor. Physiol Rep 3, e12514–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivkumar K, Ajijola OA, Anand I, Armour JA, Chen PS, Esler M, De Ferrari GM, Fishbein MC, Goldberger JJ, Harper RM, Joyner MJ, Khalsa SS, Kumar R, Lane R, Mahajan A, Po S, Schwartz PJ, Somers VK, Valderrabano M, Vaseghi M & Zipes DP (2016). Clinical neurocardiology defining the value of neuroscience‐based cardiovascular therapeutics. J Physiol 594, 3911–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmerman HK & Jones LR (1998). Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol Rev 78, 921–947. [DOI] [PubMed] [Google Scholar]
- Souza HC, Ballejo G, Salgado MC, Da Silva VJ & Salgado HC (2001). Cardiac sympathetic overactivity and decreased baroreflex sensitivity in L‐NAME hypertensive rats. Am J Physiol Heart Circ Physiol 280, H844–H850. [DOI] [PubMed] [Google Scholar]
- Sprenger JU, Perera RK, Steinbrecher JH, Lehnart SE, Maier LS, Hasenfuss G & Nikolaev VO (2015). In vivo model with targeted cAMP biosensor reveals changes in receptor‐microdomain communication in cardiac disease. Nat Commun 6, 6965. [DOI] [PubMed] [Google Scholar]
- Stangherlin A & Zaccolo M (2012). Phosphodiesterases and subcellular compartmentalized cAMP signaling in the cardiovascular system. Am J Physiol Heart Circ Physiol 302, H379–H390. [DOI] [PubMed] [Google Scholar]
- Steptoe A & Kivimaki M (2012). Stress and cardiovascular disease. Nat Rev Cardiol 9, 360–370. [DOI] [PubMed] [Google Scholar]
- Stratakis CA (2002). Mutations of the gene encoding the protein kinase A type I‐alpha regulatory subunit (PRKAR1A) in patients with the “complex of spotty skin pigmentation, myxomas, endocrine overactivity, and schwannomas” (Carney complex). Ann N Y Acad Sci 968, 3–21. [DOI] [PubMed] [Google Scholar]
- Stratakis CA, Kirschner LS & Carney JA (2001). Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab 86, 4041–4046. [DOI] [PubMed] [Google Scholar]
- Sverrisdottir YB, Green AL, Aziz TZ, Bahuri NFA, Hyam J, Basnayake SD & Paterson DJ (2014). Differentiated baroreflex modulation of sympathetic nerve activity during deep brain stimulation in humans. Hypertension 63, 1000–1010. [DOI] [PubMed] [Google Scholar]
- Tahsili‐Fahadan P & Geocadin RG (2017). Heart‐brain axis: effects of neurologic injury on cardiovascular function. Circ Res 120, 559–572. [DOI] [PubMed] [Google Scholar]
- Talaia C, Queiroz G, Pinheiro H, Moura D & Gonçalves J (2006). Involvement of G‐protein βγ subunits on the influence of inhibitory α2‐autoreceptors on the angiotensin AT1‐receptor modulation of noradrenaline release in the rat vas deferens. Neurochem Int 49, 698–707. [DOI] [PubMed] [Google Scholar]
- Tan J, Wang H & Leenen FHH (2004). Increases in brain and cardiac AT1 receptor and ACE densities after myocardial infarct in rats. Am J Physiol Heart Circ Physiol 286, H1665–H1671. [DOI] [PubMed] [Google Scholar]
- Tan Z‐Y, Lu Y, Whiteis CA, Simms AE, Paton JFR, Chapleau MW & Abboud FM (2010). Chemoreceptor hypersensitivity, sympathetic excitation, and overexpression of ASIC and TASK channels before the onset of hypertension in SHR. Circ Res 106, 536–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Tanaka R, Harada S, Kohda Y, Matsumura H, Shimamoto C, Sawabe Y, Marunaka Y, Kuwabara H, Takahashi Y, Ito S & Nakahari T (2013). A PKG inhibitor increases Ca2+‐regulated exocytosis in guinea pig antral mucous cells: cAMP accumulation via PDE2A inhibition. Am J Physiol Gastrointest Liver Physiol 304, G773–G780. [DOI] [PubMed] [Google Scholar]
- Tilley DG (2011). Functional relevance of biased signaling at the angiotensin II type 1 receptor. Endocr Metab Immune Disord Drug Targets 11, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomek J, Rodriguez B, Bub G & Heijman J (2017). β‐Adrenergic receptor stimulation inhibits proarrhythmic alternans in postinfarction border zone cardiomyocytes: A computational analysis. Am J Physiol Heart Circ Physiol 313, H338–H353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tóth AD, Gyombolai P, Szalai B, Várnai P, Turu G & Hunyady L (2017). Angiotensin type 1A receptor regulates β‐arrestin binding of the β2‐adrenergic receptor via heterodimerization. Mol Cell Endocrinol 442, 113–124. [DOI] [PubMed] [Google Scholar]
- Tóth AD, Turu G, Hunyady L & Balla A (2018). Novel mechanisms of G‐protein‐coupled receptors functions: AT1 angiotensin receptor acts as a signaling hub and focal point of receptor cross‐talk. Best Pract Res Clin Endocrinol Metab 32, 69–82. [DOI] [PubMed] [Google Scholar]
- Tromp TR, Mahesh D, Joles JA & Ramchandra R (2018). Direct recording of cardiac and renal sympathetic nerve activity shows differential control in renovascular hypertension. Hypertension 71, 1108–1116. [DOI] [PubMed] [Google Scholar]
- Tu H, Liu J, Zhang D, Zheng H, Patel KP, Cornish KG, Wang W‐Z, Muelleman RL & Li Y‐L (2014). Heart failure‐induced changes of voltage‐gated Ca2+ channels and cell excitability in rat cardiac postganglionic neurons. Am J Physiol Cell Physiol 306, C132–C142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhrenholt TR & Nedergaard OA (2003). Calcium channels involved in noradrenaline release from sympathetic neurones in rabbit carotid artery. Pharmacol Toxicol 92, 226–233. [DOI] [PubMed] [Google Scholar]
- van den Meiracker AH, Admiraal PJJ, Janssen JA, Kroodsma JM, de Ronde WAM, Boomsma F, Sissmann J, Blankestijn PJ, Mulder PGM, Man in t Veld AJ & Schalekamp MADH (1995). Hemodynamic and biochemical effects of the AT1 receptor antagonist irbesartan in hypertension. Hypertension 25, 22–29. [DOI] [PubMed] [Google Scholar]
- Vettel C, Lindner M, Dewenter M, Lorenz K, Schanbacher C, Riedel M, Lämmle S, Meinecke S, Mason FE, Sossalla S, Geerts A, Hoffmann M, Wunder F, Brunner FJ, Wieland T, Mehel H, Karam S, Lechêne P, Leroy J, Vandecasteele G, Wagner M, Fischmeister R & El‐Armouche A (2017). Phosphodiesterase 2 protects against catecholamine‐induced arrhythmia and preserves contractile function after myocardial infarction. Circ Res 120, 120–132. [DOI] [PubMed] [Google Scholar]
- Wallace S, Guo DC, Regalado E, Mellor‐Crummey L, Bamshad M, Nickerson DA, Dauser R, Hanchard N, Marom R, Martin E, Berka V, Sharina I, Ganesan V, Saunders D, Morris SA & Milewicz DM (2016). Disrupted nitric oxide signaling due to GUCY1A3 mutations increases risk for moyamoya disease, achalasia and hypertension. Clin Genet 90, 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Henrich M, Buckler KJ, McMenamin M, Mee CJ, Sattelle DB & Paterson DJ (2007). Neuronal nitric oxide synthase gene transfer decreases [Ca2+]i in cardiac sympathetic neurons. J Mol Cell Cardiol 43, 717–725. [DOI] [PubMed] [Google Scholar]
- Wang L, Li D, Plested CP, Dawson T, Teschemacher AG & Paterson DJ (2006). Noradrenergic neuron‐specific overexpression of nNOS in cardiac sympathetic nerves decreases neurotransmission. J Mol Cell Cardiol 41, 364–370. [DOI] [PubMed] [Google Scholar]
- Wang W, Yang Y, Yin J & Gong X (2017). Different protein‐protein interface patterns predicted by different machine learning methods. Sci Rep 7, 16023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Seto S‐W & Golledge J (2012). Angiotensin II, sympathetic nerve activity and chronic heart failure. Heart Fail Rev 19, 187–198. [DOI] [PubMed] [Google Scholar]
- Watanabe R, Suzuki J‐I, Wakayama K, Maejima Y, Shimamura M, Koriyama H, Nakagami H, Kumagai H, Ikeda Y, Akazawa H, Morishita R, Komuro I & Isobe M (2017). A peptide vaccine targeting angiotensin II attenuates the cardiac dysfunction induced by myocardial infarction. Sci Rep 7, 43920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson AMD, Hood SG, Ramchandra R, McAllen RM & May CN (2007). Increased cardiac sympathetic nerve activity in heart failure is not due to desensitization of the arterial baroreflex. Am J Physiol Heart Circ Physiol 293, H798–H804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir MR & Dzau VJ (1999). The renin‐angiotensin‐aldosterone system: A specific target for hypertension management. Am J Hypertens 12, 205S–213S. [DOI] [PubMed] [Google Scholar]
- White WB, Carr AA, Krause S, Jordan R, Roniker B & Oigman W (2003). Assessment of the novel selective aldosterone blocker eplerenone using ambulatory and clinical blood pressure in patients with systemic hypertension. Am J Cardiol 92, 38–42. [DOI] [PubMed] [Google Scholar]
- Williams B, MacDonald TM, Morant S, Webb DJ, Sever P, McInnes G, Ford I, Cruickshank JK, Caulfield MJ, Salsbury J, Mackenzie I, Padmanabhan S, Brown MJ; The British Hypertension Society's PATHWAY Studies Group (2015). Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug‐resistant hypertension (PATHWAY‐2): a randomised, double‐blind, crossover trial. Lancet 386, 2059–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiysonge CS, Bradley HA, Volmink J & Mayosi BM (2018). Cochrane corner: beta‐blockers for hypertension. Heart 104, 282–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiysonge CS, Bradley HA, Volmink J, Mayosi BM & Opie LH (2017). Beta‐blockers for hypertension. Cochrane Database Syst Rev 285, 2719–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobst J, Ameln S, Wolf B, Wierer M, Dang TA, Sager HB, Tennstedt S, Hengstenberg C, Koesling D, Friebe A, Braun SL, Erdmann J, Schunkert H & Kessler T (2016). Stimulators of the soluble guanylyl cyclase: promising functional insights from rare coding atherosclerosis‐related GUCY1A3 variants. Basic Res Cardiol 111, 51. [DOI] [PubMed] [Google Scholar]
- Wobst J, Rumpf PM, Dang TA, Segura‐Puimedon M, Erdmann J & Schunkert H (2015). Molecular variants of soluble guanylyl cyclase affecting cardiovascular risk. Circ J 79, 463–469. [DOI] [PubMed] [Google Scholar]
- Woollard HH (1926). The innervation of the heart. J Anat 60, 345–373. [PMC free article] [PubMed] [Google Scholar]
- Xie Z‐R, Chen J & Wu Y (2017). Predicting protein‐protein association rates using coarse‐grained simulation and machine learning. Sci Rep 7, 46622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y, Kinoshita H, Kuwahara K, Nakagawa Y, Kuwabara Y, Minami T, Yamada C, Shibata J, Nakao K, Cho K, Arai Y, Yasuno S, Nishikimi T, Ueshima K, Kamakura S, Nishida M, Kiyonaka S, Mori Y, Kimura T, Kangawa K & Nakao K (2014). Inhibition of N‐type Ca2+ channels ameliorates an imbalance in cardiac autonomic nerve activity and prevents lethal arrhythmias in mice with heart failure. Cardiovasc Res 104, 183–193. [DOI] [PubMed] [Google Scholar]
- Yaniv Y, Ganesan A, Yang D, Ziman BD, Lyashkov AE, Levchenko A, Zhang J & Lakatta EG (2015). Real‐time relationship between PKA biochemical signal network dynamics and increased action potential firing rate in heart pacemaker cells. Kinetics of PKA activation in heart pacemaker cells. J Mol Cell Cardiol 86, 168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CN & Davisson RL (2015). Angiotensin‐II, the brain, and hypertension: An update. Hypertension 66, 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccolo M & Movsesian MA (2007). cAMP and cGMP signaling cross‐talk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ Res 100, 1569–1578. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Striessnig J, Koschak A & Dolphin AC (2015). The physiology, pathology, and pharmacology of voltage‐gated calcium channels and their future therapeutic potential. Pharmacol Rev 67, 821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao CY, Greenstein JL & Winslow RL (2016). Roles of phosphodiesterases in the regulation of the cardiac cyclic nucleotide cross‐talk signaling network. J Mol Cell Cardiol 91, 215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao CY, Greenstein JL & Winslow RL (2017). Mechanisms of the cyclic nucleotide cross‐talk signaling network in cardiac L‐type calcium channel regulation. J Mol Cell Cardiol 106, 29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Rao DC & Shi G (2015). An update on genome‐wide association studies of hypertension. Appl Inform 2, 10. [Google Scholar]
- Zhu G‐Q, Gao L, Li Y, Patel KP, Zucker IH & Wang W (2004). AT1 receptor mRNA antisense normalizes enhanced cardiac sympathetic afferent reflex in rats with chronic heart failure. Am J Physiol Heart Circ Physiol 287, H1828–H1835. [DOI] [PubMed] [Google Scholar]
- Zoccarato A, Surdo NC, Aronsen JM, Fields LA, Mancuso L, Dodoni G, Stangherlin A, Livie C, Jiang H, Sin YY, Gesellchen F, Terrin A, Baillie GS, Nicklin SA, Graham D, Szabo‐Fresnais N, Krall J, Vandeput F, Movsesian M, Furlan L, Corsetti V, Hamilton G, Lefkimmiatis K, Sjaastad I & Zaccolo M (2015). Cardiac hypertrophy is inhibited by a local pool of cAMP regulated by phosphodiesterase 2. Circ Res 117, 707–719. [DOI] [PubMed] [Google Scholar]