

Abstract
Objectives
Pyruvate kinase deficiency (PK deficiency) is a rare disorder caused by compound heterozygosity or homozygosity for > 300 mutations in the PKLR gene. To understand PK deficiency prevalence, we conducted a systematic literature review.
Methods
We queried Embase and Medline for peer‐reviewed references reporting PK deficiency prevalence/incidence, PKLR mutant allele frequency (MAF) among the general population, or crude results from which these metrics could be derived.
Results
Of 1390 references screened, 1296 were excluded after title/abstract review; 60 were excluded after full‐text review. Four of the remaining 34 studies were considered high‐quality for estimating PK deficiency prevalence. Two high‐quality studies identified cases from source populations of known sizes, producing estimates of diagnosed PK deficiency prevalence of 3.2 and 8.5 per million. Another high‐quality study derived an estimate of diagnosed PK deficiency prevalence of 6.5 per million by screening jaundiced newborns. The final high‐quality study estimated total diagnosed and undiagnosed PK deficiency prevalence to be 51 per million through extrapolation from observed MAFs.
Conclusions
We conclude that prevalence of clinically diagnosed PK deficiency is likely between 3.2 and 8.5 per million in Western populations, while the prevalence of diagnosed and undiagnosed PK deficiency could possibly be as high as 51 per million.
Keywords: incidence, PK deficiency, PKD, prevalence, pyruvate kinase deficiency, systematic literature review
Novelty Statement
1. What is the NEW aspect of your work? (ONE sentence)
This is a comprehensive and contemporary systematic review of studies of PK deficiency epidemiology which includes potential reasons for the observed variation in prevalence estimates.
2. What is the CENTRAL finding of your work? (ONE sentence)
The prevalence of clinically diagnosed PK deficiency is likely between 3.2 and 8.5 per million in Western populations, while the prevalence of diagnosed and undiagnosed PK deficiency could possibly be as high as 51 per million.
3. What is (or could be) the SPECIFIC clinical relevance of your work? (ONE sentence)
This work could help hematologists fully appreciate the potential for underdiagnosis and misdiagnosis of PK deficiency when diagnosing causes of hemolytic anemia, jaundice, and more.
1. INTRODUCTION
Pyruvate kinase (PK) deficiency is a rare congenital disorder characterized by diminished activity of the PK enzyme in red blood cells (RBCs). 1 Low PK enzyme activity can lead to lifelong chronic hemolysis and associated symptoms and complications such as anemia, jaundice, gallstones, thrombosis, and iron overload. 2 PK deficiency is a recessive disorder caused by compound heterozygosity or homozygosity for one or more of >300 known mutations 3 to the PKLR gene. The large number of PKLR mutations, which may have different impacts on PK function, likely contributes to the notable clinical heterogeneity of this disorder. 2 , 4
The prevalence of PK deficiency is unknown, with commonly cited estimates differing by orders of magnitude. Factors contributing to the uncertain prevalence include the extreme rarity of the disorder, the phenotypic similarity to other RBC disorders (leading to misdiagnosis), its heterogeneous clinical presentation, ethnic and geographic variability, and the current lack of adequate data from PK deficiency registries.
We conducted a systematic literature review (SLR) to assess the prevalence of PK deficiency and to better understand factors contributing to the wide range of reported values. We focused on studies reporting PK deficiency incidence, prevalence, survival duration, and/or mortality and studies reporting mutant allele frequencies (MAFs) for PKLR mutations among general populations.
2. METHODS
A protocol containing the full details of study eligibility criteria, data to be extracted, and quality assessment process was created and maintained. We queried Embase and Medline via Ovid, searched the single conference year of the Deutsche Gesellschaft für Hämatologie und Medizinische Onkologie (DGHO) that was not indexed by Embase/Medline, and evaluated other relevant references encountered over the course of the SLR. Study eligibility was assessed by a review of titles and abstracts, followed by full‐text review for potentially relevant studies. Data were extracted from studies meeting eligibility criteria, and quality assessment was performed using a custom tool. Study screening, data extraction, and quality assessment were conducted in duplicate by two independent reviewers, with disagreements resolved through consensus or deferral to a third reviewer.
2.1. Eligibility criteria
Peer‐reviewed publications and conference abstracts were included if they were published (or in press) in English before January 23, 2019. Studies describing PK deficiency epidemiology and studies describing MAFs for PKLR mutations among general populations were included. We defined PK deficiency epidemiology studies as those reporting point/period prevalence, incidence rate/proportion, survival duration, life expectancy, and/or mortality rate among source populations selected without respect to PK deficiency symptoms. PKLR MAF studies were those that reported MAFs (number of alleles with a mutation out of total alleles) for mutations to the PKLR gene known to cause PK deficiency or predicted to significantly impair PK enzyme function. We also included studies reporting crude results from which PK deficiency epidemiology or PKLR MAFs could be derived.
We excluded studies that were not written in English, based on humans, subjected to peer‐review, or were not the primary report of the data. We also excluded studies of PK deficiency prevalence/incidence conducted within a source population of patients with symptoms of PK deficiency such as anemia or jaundice, as these populations would likely be enriched with patients with PK deficiency and not reflective of general populations. Similarly, PKLR MAF studies were excluded if they only reported MAFs among PK deficiency patients. We excluded studies reporting MAFs for only PKLR mutations unlikely to cause reduction of PK function as stated by the authors or inferred from amino acid substitution prediction models. Duplicate references were removed, though complementary studies with overlapping study populations were retained. Population, Intervention, Comparison, Outcomes and Study Design (PICOS) parameters were used to describe the eligibility criteria (Table S1).
2.2. Information sources
2.2.1. Search
PK deficiency and PKLR Emtree and Medical Subject Heading (MeSH) terms and keywords were combined with epidemiology and gene frequency Emtree/MeSH terms and keywords in Ovid queries (Tables S2 and S3). The queries included references from January 1, 1974, (Embase) or January 1, 1946, (Medline) to January 23, 2019 (exclusive).
Conferences believed a priori to contain publications of PK deficiency epidemiology were indexed by Ovid except for a single conference year (2018) of the annual DGHO conference. The 2018 DGHO abstract booklet was searched for the terms “pyruvate kinase,” “PKD,” “PK deficiency,” and “PKLR.” Abstracts which contained ≥ 1 of these terms proceeded to title/abstract screening.
Finally, references encountered over the course of the literature review (eg, in the background section of another article) which appeared to be relevant were submitted to title/abstract screening. Each time a potentially relevant reference was encountered outside of the Ovid queries, screening terms were reevaluated.
2.2.2. Study selection
Titles and abstracts of all references were screened, and those meeting inclusion/exclusion criteria were selected for full‐text review. Data extraction and quality assessment were performed on studies meeting all eligibility criteria upon full‐text review.
2.2.3. Data extraction
Extracted data from eligible studies included the following: reference information, type and values of statistics reported, demographic and clinical characteristics of the study population, location of the study population, and study objectives and methods.
The primary outcome measure was PK deficiency prevalence. Where necessary, prevalence units were standardized. MAF estimates were converted to prevalence estimates assuming Hardy‐Weinberg equilibrium (HWE) to facilitate comparison.
2.2.4. Quality assessment and evidence synthesis
A tailor‐made, qualitative, quality assessment tool was created based on the Joanna Briggs Institute critical appraisal tools 5 and the STrengthening the Reporting of OBservational studies in Epidemiology (STROBE) Statement. 6
The generalizability, quality, and consistency of all outcome statistics were considered collectively to evaluate the weight and associated uncertainty of the evidence of PK deficiency prevalence.
3. RESULTS
3.1. Study selection and characteristics
The Ovid queries identified 1386 references (Tables S2 and S3). Additional potentially relevant references were identified from the 2018 DGHO conference abstract booklet (n = 1) and during review of other references (n = 3), for a total of 1390 references (Figure 1). Most studies were excluded during title/abstract screening (n = 1296) and upon full‐text review (n = 60) due to irrelevance (Figure 1). To facilitate comparison, the remaining 34 studies meeting eligibility criteria were grouped as follows: population‐based prevalence studies (n = 2); molecular PKLR screening studies (n = 11); and non‐molecular PK deficiency screening studies (n = 21). No studies reporting on mortality rates or life expectancy in PK deficiency were found.
FIGURE 1.
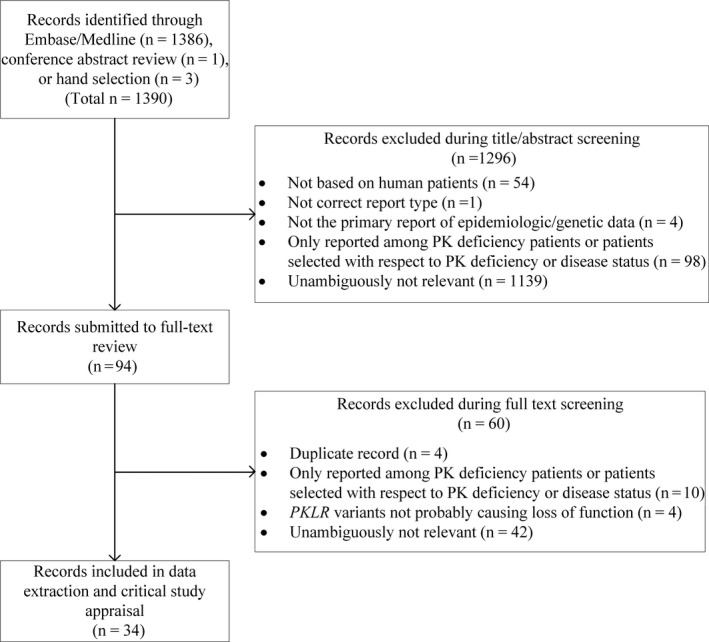
Flow diagram of studies meeting inclusion/exclusion criteria
Of the 34 studies meeting our inclusion criteria, four 7 , 8 , 9 , 10 were deemed to be high‐quality for purposes of estimating overall disease prevalence.
3.2. Results of population‐based prevalence studies
Two of the four studies considered to be high‐quality were population‐based prevalence studies reporting counts of diagnosed PK deficiency cases out of source populations of known sizes (Table 1). Carey et al 7 estimated PK deficiency prevalence to be approximately 3.2 per million (Table 2) based on 10 cases diagnosed from 1974 to 1999 in a PK deficiency registry in the Northern Health Region of the United Kingdom among a general population of 3.1 million, while de Medicis et al 8 estimated PK deficiency prevalence to be 8.5 per million based on 56 confirmed cases with hemolytic anemia recorded in a hematologic database out of a population of 6.5 million individuals in Quebec. We consider these studies to have minimal risk of bias for the purpose of estimating PK deficiency prevalence given the large source populations and the implicit requirement for a clinician to suspect PK deficiency, though they likely underestimate PK deficiency prevalence by not accounting for undiagnosed cases.
TABLE 1.
Summary table of studies selected for data extraction
| First author (year)Citation | Study purpose | Source population |
|---|---|---|
| Population‐based prevalence study | ||
| Carey (2000) 7 | To estimate the prevalence of PK deficiency in a general, white population | Former Northern Health Region of the United Kingdom |
| de Medicis (1992) 8 | To estimate the prevalence of PK deficiency associated with hemolytic anemia in Quebec | Quebec, Canada |
| Molecular PKLR screening study in a general population | ||
| Beutler (2000) 10 | To estimate the prevalence of PK deficiency in a general, predominately white population | 3785 anonymous individuals without PK deficiency in the USA, predominately white; 20 patients with PK deficiency, mostly white Americans |
| Chami (2016) 16 | To identify pleiotropic genetic mutations associated with RBC traits | Up to 130 273 individuals in 24 discovery cohorts and five ancestries: European, African American, Hispanic, East Asian, and South Asian ancestries |
| Figueroa (2018) 14 | To evaluate whether genetic mutations in kinases affect Tenofovir's efficacy in the treatment/prevention of HIV | 505 individuals in HIV prevention trials in Thailand, the USA, and South Africa |
| Manco (2001) 12 | To determine the frequency of allele 1456C‐T in a general population in Portugal | 616 unrelated Portuguese individuals |
| Svidnicki (2018) 13 | To evaluate PKLR mutations among Brazilian patients with PK deficiency and healthy individuals | 500 healthy Brazilian blood donors |
| Molecular PKLR screening study in areas with endemic malaria | ||
| Alves (2010) 18 | To evaluate the associations between malaria and sickle cell disease, G6PD, and PK deficiency | 257 unrelated samples (selected from 1056 total samples) from Santiago Island, Ecuador |
| Berghout (2012) 19 | To evaluate the frequency of PKLR mutations to address whether PKLR mutations are protective in malaria‐pressured areas | 387 unrelated individuals, primarily from African and Asian origin |
| Machado (2010) 17 | To evaluate possible associations between PKLR variants and malaria | 396 individuals, 316 from Africa (including malaria‐infected and uninfected children) and 80 from Portugal |
| Machado (2012) 20 | To determine PK deficiency prevalence in sub‐Saharan Africa, to evaluate PKLR variant frequency, and to evaluate the PK deficiency‐malaria association | 611 individuals in sub‐Saharan Africa for gene frequency analysis, 296 for enzyme assays |
| Non‐molecular PK deficiency screening study in a general population | ||
| Feng (1993) 32 | To establish a reference interval of PK activity and to determine PK deficiency prevalence in local Chinese population | 497 healthy men and 100 neonates in China |
| Fox (1969) 33 | To evaluate two different screening methods for erythrocyte enzyme deficiencies | 1000 blood samples from individuals in a general hospital in the USA |
| Fung (1969) 31 | To determine the incidence of PK deficiency and G6PD deficiency in the Chinese newborn in Hong Kong and the possible relation of PK deficiency with neonatal jaundice | 700 consecutive neonates in Hong Kong |
| Garcia (1979) 28 | To estimate prevalence of enzyme deficiencies including PK deficiency in a Spanish population | 1636 hematologically normal individuals in Spain |
| Mohrenweiser (1981) 34 | To evaluate the frequency of genetic variants resulting in total or near total loss of enzyme activity for nine human erythrocyte enzymes | 675 neonates and 200 of their parents in the USA (697 total assayed for PK deficiency) |
| Mohrenweiser (1987) 35 | To estimate the frequency of enzyme deficiency variants including PK deficiency | 2020 unrelated neonates in the USA |
| Tanaka (1971) 30 | To offer a thorough description of PK diagnosis and epidemiology | 146 normal individuals in the USA |
| Tsang (1993) 27 | To evaluate a new PK enzyme activity assay | 193 healthy adults in Hong Kong |
| Wu (1985) 29 | To estimate the prevalence of PK deficiency in a Chinese population of clinically normal neonates | 1159 random clinically normal infants from China |
| Non‐molecular PK deficiency screening study in areas with endemic malaria | ||
| El‐Hazmi (1986) 36 | To estimate PK deficiency and hexokinase deficiency in a population with a high frequency of G6PD deficiency | 3120 individuals in outpatient clinics in Saudi Arabia |
| Munyanganizi (2006) 37 | To determine the prevalence of hemoglobin variants including PK deficiency in Rwanda | 987 neonates in Rwanda |
| Upadhye (2018) 38 | To estimate the prevalence of hemoglobinopathies and enzyme deficiencies in India | 2400 neonates in India |
| Non‐molecular PK deficiency screening study in areas with high consanguinity | ||
| Abu‐Melha (1991) 44 | To explore the prevalence of PK deficiency in Saudis | 513 clinically normal neonates in Saudi Arabia |
| Akin (1997) 41 | To explore the prevalence of PK deficiency in a Turkish population | 1190 unrelated individuals from Turkey |
| Al‐Naama (1994) 42 | To explore erythrocyte activities including PK deficiency in Iraq | 1226 clinically normal neonates and 498 healthy students in Iraq (n = 1724 total) |
| Al‐Naama (1995) 43 | To estimate red blood cell enzyme deficiencies including PK deficiency in a population with high G6PD deficiency prevalence | 506 clinically normal neonates and 343 healthy adults (n = 849 total) |
| Christensen (2010) 9 | To report on neonates with PK deficiency, encourage clinicians to consider PK deficiency diagnosis, and discuss PK deficiency in a polygamous community | 153 830 neonates screened for jaundice in the USA |
| Yavarian (2008) 40 | To estimate PK deficiency prevalence in a south Iranian population | 4017 randomly selected individuals from Iran |
| Non‐molecular PK deficiency screening in other unique population | ||
| Mohrenweiser (1984) 45 | To evaluate the prevalence of rare electrophoretic mobility variants including PK deficiency among Native Americans | Approximately 10% of 629 Native Americans |
| Satoh (1983) 46 | To explore enzyme activity including PK in children of parents exposed to atomic bomb radiation in Japan | 3069 children of individuals exposed to atomic bomb radiation in Japan |
| Satoh (1985) 47 | To explore the frequency of inherited variations in enzyme thermostability including PK within a population of children of atomic bomb survivors | 974 children of individuals exposed to atomic bomb radiation in Japan |
| Molecular PKLR screening in other unique population | ||
| Lyon (2011) 22 | To explore the hypothesis that rare family specific genetics may account for the remaining missing heritability of ADHD | A family of 4 of whom 3 have ADHD |
| Robinson (2010) 21 | To explore the hypothesis that rare family specific genetics may account for the remaining missing heritability of ADHD | A family of 4 of whom 3 have ADHD |
Abbreviations: ADHD, attention‐deficit hyperactivity disorder; G6PD, glucose‐6‐phosphate dehydrogenase; HIV, human immunodeficiency virus; PK deficiency, pyruvate kinase deficiency; PK, pyruvate kinase; RBC, red blood cell; USA, United States of America.
TABLE 2.
Summary of results by study type
| First author (year)Citation | PKLR genes sequenced | Reported results | Prevalence estimates (per million) | Derivation |
|---|---|---|---|---|
| Population‐based prevalence study | ||||
| Carey (2000) 7 | N/A | Period prevalence: 3.2 per million | 3.2 | † |
| de Medicis (1992) 8 | N/A | Period prevalence: 1 per 117 206 | 8.5 | ‡ |
| Molecular PKLR screening study in a general population | ||||
| Beutler (2000) 10 | c.1456C > T, c.1468C > T, c.1484C > T, c.1529G > A | Estimated prevalence: 51 per million | 51 | † |
| Chami (2016) 16 | c.1456C > T | MAF for 1 PK deficiency‐causing allele: 0.30% | 9.0 | § |
| Figueroa (2018) 14 | Whole PKLR gene | MAF of 1.1% | 120 | § |
| Manco (2001) 12 | c.1456C > T | MAF of single allele: 0.325% (95% CI 0.001% to 0.649%) Estimated point prevalence: 10 per million | 10 | † |
| Svidnicki (2018) 13 | c.1456C > T, c.1529G > A | MAF for 2 common alleles: 0.10% | 1 | § |
| Molecular PKLR screening study in areas with endemic malaria | ||||
| Alves (2010) 18 | c.269T > A and c.1456C > T | MAF for two alleles: 0.0% | 0 | N/A |
| Berghout (2012) 19 | Whole PKLR gene | MAF: 1.5% (SD: 0.9%) Estimated point prevalence of PK deficiency: 1/4203 births assuming HWE | 240 | ‡ |
| Machado (2010) 17 | c.1456C > T and c.1614A > T | MAF for two alleles: 0.0% | 0 | N/A |
| Machado (2012) 20 | Whole PKLR gene |
MAF for 1 allele in children: 2.9% MAF for 1 allele in adults: 4.6% Low PK activity prevalence (Non‐molecular PK deficiency screening): 4.1% |
840 (children) § ; 2100 (adults) § ; 41 000 (Non‐molecular PK deficiency screening) ‡ | ‡ , § |
| Non‐molecular PK deficiency screening study in a general population | ||||
| Feng (1993) 32 | N/A | Point prevalence: 0.0% | 0 | N/A |
| Fox (1969) 33 | N/A | Point prevalence: 0.0% | 0 | N/A |
| Fung (1969) 31 | N/A |
Incidence/point prevalence in newborns (partial and total deficiency): 3.4% Incidence/ point prevalence in newborns (partial deficiency): 2.3% Incidence/point prevalence in newborns (total deficiency): 1.1% |
34 000 ‡ (partial and total deficiency) 23 000 ‡ (partial deficiency) 11 000 ‡ (total deficiency) |
‡ |
| Garcia (1979) 28 | N/A | Point prevalence: 0.24% | 2400 ‡ | ‡ |
| Mohrenweiser (1981) 34 | N/A | Incidence/point prevalence in newborns: 0.14% | 1400 ‡ | ‡ |
| Mohrenweiser (1987) 35 | N/A |
Incidence/ prevalence at birth using ≤ 65% normal activity threshold: 1.1% Incidence/prevalence at birth using < 50% normal activity threshold: 0.50% |
11 000 ‡ 5000 ‡ |
‡ |
| Tanaka (1971) 30 | N/A | Point prevalence: 1.4% | 14 000 ‡ | ‡ |
| Tsang (1993) 27 | N/A |
Point prevalence using modified technique: 0.0% Point prevalence using Beutler technique: 3.7% |
0 (modified technique) 37 000 (Beutler technique) |
‡ |
| Wu (1985) 29 | N/A |
Incidence/prevalence at birth (Beutler spot test): 2.2% Incidence/prevalence at birth (direct enzyme activity): 2.1% |
22 000 (Beutler spot test) 21 000 (enzyme activity screening) |
‡ |
| Non‐molecular PK deficiency screening study in areas with endemic malaria | ||||
| El‐Hazmi (1986) 36 | N/A |
Point prevalence of partial PK deficiency: 3.4% Prevalence of complete PK deficiency: 0.0% |
34 000 (partial) 0 (complete) |
¶ |
| Munyanganizi (2006) 37 | N/A | Point prevalence: 0.0% | 0 | N/A |
| Upadhye (2018) 38 | N/A | Point prevalence: 0.083% | 830 | †† |
| Non‐molecular PK deficiency screening study in areas with high consanguinity | ||||
| Abu‐Melha (1991) 44 | N/A |
Incidence/prevalence among neonates by fluorescence screening: 2.90% Incidence/prevalence among neonates by fluorescence screening and enzyme activity screening: 3.10% (95% CI: 1.5) |
29 000 (fluorescence screening) 31 000 (fluorescence screening and enzyme activity) |
‡ |
| Akin (1997) 41 | N/A |
Point prevalence of “heterozygous deficiency”: 1.10% Point prevalence of “homozygous deficiency”: 0.00% |
11 000 0 |
‡ |
| Al‐Naama (1994) 42 | N/A | Point prevalence of PK deficiency: 0.0% | 0 | N/A |
| Al‐Naama (1995) 43 | N/A | Point prevalence of partial PK deficiency among neonates: 1.4% | 14 000 (partial PK deficiency among neonates) | ‡ |
| Point prevalence of partial PK deficiency among adults: 0.0% | 0 (partial PK deficiency among adults) | |||
| Point prevalence of partial PK deficiency among all age groups: 0.82% | 8200 (partial PK deficiency among all age groups) | |||
| Point prevalence of PK deficiency among all age groups: 0.0% | 0 (complete PK deficiency) | |||
| Christensen (2010) 9 | N/A | Incidence proportion/prevalence at birth of hospital including polygamist community: 1/30 000 | 33 (total population) | ‡ |
| Incidence proportion/prevalence at birth of polygamist community: 1/250 | 4000 (polygamous community) | |||
| Implied incidence proportion/prevalence at birth of non‐polygamous population: 1 per 152 830 births | 6.5 (non‐polygamous population) | |||
| Yavarian (2008) 40 | N/A |
Point prevalence based on 60% PK activity: 1.9% Point prevalence of homozygous individuals: 0.27% |
19 000 2700 |
‡ |
| Non‐molecular PK deficiency screening in other unique population | ||||
| Mohrenweiser (1984) 45 | N/A | Point prevalence: 0.0% | 0 | N/A |
| Satoh (1983) 46 | N/A | Mutant allele frequency for all alleles: 1.4% | 14 000 | ‡ |
| Satoh (1985) 47 | N/A | Mutant allele frequency for all alleles: 0.0% | 0 | N/A |
| Molecular PKLR screening in other unique population | ||||
| Lyon (2011) 22 | Whole PKLR gene | Point prevalence: 1 PK deficiency patient out of 4 individuals studied = 25% | 250 000 | ‡‡ |
| Robinson (2010) 21 | Whole PKLR gene | Point prevalence: 1 PK deficiency patient out of 4 individuals studied = 25% | 250 000 | ‡‡ |
Abbreviations: HWE, Hardy‐Weinberg equilibrium; MAF, mutant allele frequency; PK deficiency, pyruvate kinase deficiency; PK, pyruvate kinase.
Estimate provided by study authors.
Units converted from authors’ estimate.
Estimate derived assuming HWE.
Average partial PK deficiency prevalence calculated from the individual estimates for each location.
Derived from two patients with PK deficiency among 2400 neonates.
Derived units from 1 PK deficiency case among 4 individuals sequenced.
3.3. Results of molecular PKLR screening studies
Eleven studies screened individuals for mutations to the PKLR gene and reported MAFs (Table 1). However, many of these studies were uninformative because they identified PKLR mutations of unknown clinical significance in a general population without PK deficiency.
3.3.1. Molecular PKLR screening studies in a general population
Five studies evaluated PKLR mutations in general populations (Table 1). The most informative of these for our purposes, by Beutler and Gelbart, 10 utilized PKLR mutation data to estimate overall disease prevalence. This study was among the four in the review that we considered to be high‐quality, in part because it addressed a potential source of bias present in other studies reporting on PKLR mutations. Namely, many molecular screening studies assumed that the penetrance of each mutation is 100%; that is, that each compound heterozygous or homozygous individual who carries a mutation will have symptoms of PK deficiency. This assumption may be incorrect, as the ability of a mutation to elicit clinical symptoms varies between individuals due to a variety of genetic and non‐genetic factors. 11 Beutler and Gelbart 10 attempted to account for variable penetrance levels by utilizing an “index mutation,” c.1529G > A, which they considered, based on clinical experience and its high frequency in a population with PK deficiency, to have 100% penetrance. They assumed the ratio of this index mutation to other PK deficiency‐causing mutations to be fixed and therefore to be equal between those with and without PK deficiency. Based on this assumption and MAFs from 20 PK deficiency cases and 3785 non‐cases, they derived an estimate of PK deficiency prevalence of 51 per million, albeit with a substantial amount of associated uncertainty (standard error = 32.5 per million). Notably, this estimate represents the prevalence of diagnosed and undiagnosed cases and so is distinct from the previously described population‐based studies which estimated the prevalence of diagnosed PK deficiency cases only. However, the accuracy of this estimate depends on the true distributions of PKLR mutations between those with and without PK deficiency, which are not fully known.
All of the other studies reporting on PKLR MAFs among general populations did not consider the full suite of known mutations to the PKLR gene 10 , 12 , 13 or included mutations that may not cause loss of PK function and, therefore, are less useful for our purposes. 14 All determined MAFs for the PKLR mutation c.1456C > T, one of the most common mutations in European patients with PK deficiency. 15 In the largest of these, 16 Chami and colleagues reported the MAF of c.1456C > T a to be 0.30% among up to 130 273 individuals. PKLR MAFs derived in the other four studies were generally consistent with this finding, reporting MAF ranges from 0.10% 13 to 0.33%. 12
In the only study to fully sequence the PKLR gene, Figueroa et al 14 reported a MAF for all PKLR mutations to be 1.09%, a high MAF that likely relates to the inclusion of mutations not known to cause loss of function.
3.3.2. Molecular PKLR screening studies in areas with endemic malaria
Four studies screened for PKLR variants in areas with endemic malaria to evaluate the hypothesis that malaria induces a selection pressure that increases PK deficiency prevalence which, if true, would undermine the generalizability of such studies (Table 1). Of the four studies, two did not identify any PKLR mutations in their study populations, 17 , 18 while two others found high MAFs between 1.50% 19 and 4.6%. 20 These studies are limited by the uncertainty around the clinical impact of the reported PKLR mutations.
3.3.3. Molecular PKLR screening studies in other unique populations
One study 21 , 22 used whole‐exome sequencing to investigate the heritability of attention‐deficit hyperactivity disorder. It was found incidentally that a child among four family members whose exomes were sequenced was a compound heterozygote for PKLR mutations. Though this study met our inclusion criteria, it is uninformative of PK deficiency prevalence.
3.4. Results of non‐molecular PK deficiency screening studies
Twenty‐one studies used enzymatic assays to identify individuals with low PK activity. These studies have a high risk of bias due to the poor accuracy of screening assays, which can be subjective, 23 incomparable prior to standardization, 24 , 25 not always reflective of in vivo PK function, 26 and sensitive to experimental procedures. 27 In particular, PK activity assays may overestimate PK deficiency prevalence because of incomplete lysis of RBC or underestimate PK deficiency prevalence due to incomplete white blood cell or platelet removal or blood donor contamination in a transfused case. PK activity assays may also fail to distinguish between heterozygotes and compound heterozygotes or homozygotes, leading to inflated estimates of PK deficiency prevalence.
3.4.1. Non‐molecular PK deficiency screening studies in a general population
Three studies screened for patients with PK deficiency among populations considered by the authors to be “normal” or “healthy,” observing PK deficiency prevalence values between 0.2% and 2.2%. 28 , 29 , 30 Given the extremely high prevalence values reported in these “healthy” populations, which do not comport with clinician experience of PK deficiency as a rare condition, it seems particularly likely that they are subjected to diagnostic limitations. Indeed, Tsang et al 27 argue that incomplete RBC lysis may produce artificially low PK activities and demonstrated in samples from 493 healthy adults that simply freezing RBC to promote lysis can reduce PK deficiency prevalence estimates from 3.7% to 0.0%.
Six enzyme screening studies that did not restrict to “normal” or “healthy” individuals may have contained similar diagnostic issues. In a study of 700 consecutive newborns in Hong Kong, the PK deficiency prevalence was observed to be 1.1% (total PK deficiency) and 3.4% (total and partial PK deficiency). 31 Notably, all eight newborns with total PK deficiency were jaundiced, lending credibility to the observed prevalence of PK deficiency. However, the extremely high prevalence merits increased scrutiny on the diagnostic methods, which were qualitative in nature and potentially unblinded to each individual's jaundice status.
One study by Feng and colleagues was coauthored by Tsang subsequent to her finding that incomplete RBC lysis may cause false‐positive results for PK deficiency in the enzyme assay. 32 This study, conducted among 497 healthy men and 100 neonates, found no cases of PK deficiency using a method that included freezing to promote lysis. 25 In another study, by Fox and colleagues, 33 no patients with PK deficiency were detected among 1000 individuals in the United States in a diverse population.
Mohrenweiser reported on a screening study of newborn infants at the University of Michigan in two manuscripts. In the first, 34 675 newborns and 200 adults were assayed for enzyme activity (697 total for PK activity) using a unique method which required presumptive cases to be confirmed in separate assays. With this method, one PK deficiency case out of 697 individuals was detected for a prevalence of 0.14%. This relatively high estimate of PK deficiency prevalence may reflect the true PK deficiency prevalence in the study population and/or random sampling with a small population: The lower bound of the 95% confidence interval (CI) is 37.5 cases per million, which is more consistent with other studies. In the other study on an overlapping population, 35 2013 unselected newborns were screened for PK deficiency. Using a threshold of <50% of normal activity to define PK deficiency, only one PK deficiency case was identified (0.50%). Again, small sample size may have inflated the impact of a single PK deficiency case (lower 95% CI: 12.6 cases per million).
3.4.2. Non‐molecular PK deficiency screening studies in areas with endemic malaria
We identified three studies in areas with endemic malaria that screened for PK deficiency with enzymatic techniques (Table 1). Given our concerns for enzyme assay accuracy and the poor generalizability of populations in malaria‐endemic regions, the risk of bias for these studies was considered to be high. These studies produced estimates of complete PK deficiency of 0.0% 36 , 37 to 0.083%. 38
3.4.3. Non‐molecular PK deficiency screening studies in areas with high consanguinity
Six studies investigated PK deficiency prevalence in areas with high consanguinity (Table 1). As with other studies based on enzyme assays and in consanguineous populations, these must be interpreted with caution for concerns related to assay accuracy and study population generalizability.
Despite these concerns, one study, by Christensen and colleagues, 9 was deemed to be of high‐quality and useful for the estimation of PK deficiency prevalence. The authors screened 153 830 live births in the USA for high bilirubin; those with neonatal jaundice were subsequently tested for PK deficiency. Five neonates were found to have PK deficiency (prevalence of approximately 1 case per 30 000 births); however, the authors speculated that a polygamous Amish community with a known founder effect 39 may be responsible for four of the five PK deficiency cases (prevalence of one case per 250 births in the polygamous community). The implied prevalence in the non‐polygamous community is one birth per 152 830 births or 6.5 per million. We considered this study design to have a low risk of bias because neonates required clinical suspicion of PK deficiency and confirmatory symptoms (ie, jaundice); however, the estimation of the number of births from the purportedly polygamous community may be subjected to error.
In another study by Yavarian et al, 40 4017 Iranian individuals were screened for low PK activity (<60% normal). The 74 individuals (1.9%) with low PK activity were then evaluated for PKLR mutations. Eleven individuals possessed two PKLR mutations (0.27% of the 4017 screened individuals). Because the authors used both molecular and enzymatic methods to identify PK deficiency cases, we considered the diagnoses of PK deficiency to have a low risk of bias. However, the study region has a high rate of consanguinity, limiting the generalizability of this study.
In a third study by Akin et al, 41 the authors found the heterozygous deficiency (<60%) prevalence to be 1.1% and the homozygous deficiency (<30% normal activity) prevalence to be 0.0%.
The three remaining non‐molecular PK deficiency screening studies in areas with high consanguinity reported populations described as “clinically normal” or “healthy,” further limiting their generalizability. The prevalence estimates in these studies ranged from 0.0% 42 , 43 to 3.1%. 44
3.4.4. Non‐molecular PK deficiency screening studies in other unique populations
Three studies used non‐molecular techniques to screen for PK deficiency in unique and potentially non‐generalizable populations. Mohrenweiser et al 45 screened approximately 10% of 629 Native American individuals who the study's authors suspected may have abnormally high frequencies of PK deficiency; however, none had PK deficiency. Two studies with overlapping study populations by Satoh et al 46 , 47 screened children of survivors of the atomic bombs in Japan and found prevalence values of 1.4% (threshold < 66% of normal) among 3069 children 46 and 0.0% (via a thermostability assay) among 974 children. 47
4. DISCUSSION
Identified studies varied widely in their reported or implied prevalence of PK deficiency. Much of this variation was attributable to known factors, such as the generalizability of source populations. Studies estimating prevalence in malaria‐endemic areas, among populations with high consanguinity or among “healthy,” asymptomatic patients, would not be expected to be applicable to a general population. PK activity assay methods likely played a large role in the heterogeneity of PK prevalence values as well, as these methods can be subjective (ie, visual examination), sensitive to chosen PK activity thresholds, and sensitive to experimental conditions. Similarly, studies of PKLR MAFs may not be indicative of disease prevalence due to the heterogeneous effects of different PKLR mutations. Finally, the definition of PK deficiency was a major source of heterogeneity between identified studies. Some studies reported only on clinically diagnosed cases; these studies would be expected to underestimate the true prevalence of PK deficiency by omitting undiagnosed cases. Others attempted to estimate the prevalence of diagnosed and undiagnosed PK deficiency.
We considered four studies to provide high‐quality estimates of PK deficiency prevalence. The population‐based studies reported prevalence values of 3.2 per million 7 and 8.5 per million. 8 Additionally, although the Christensen study 9 investigated an area with high consanguinity due to a polygamous population, the implied prevalence among non‐polygamous individuals was considered to be a high‐quality estimate (6.5 per million). As discussed previously, these studies likely underestimated PK deficiency prevalence because they considered only diagnosed and symptomatic clinical cases.
The Beutler and Gelbart study estimated diagnosed and undiagnosed PK deficiency prevalence to be 51 per million 10 and was deemed to have a low risk of bias. Most studies sequencing PKLR to estimate PK deficiency prevalence or MAFs contained two competing sources of bias: the measurement of only a subset of PKLR mutations (leading to prevalence underestimates) and the potential for some measured PKLR mutations to not cause PK deficiency or to not occur in PK deficiency patients at the same frequency as in the general population (leading to prevalence overestimates). The Beutler and Gelbart study is the only one that attempted to address both limitations. Notably, due to the small counts of patients with PK deficiency, the standard error of the reported prevalence estimate was high (32.5 per million), suggesting a large degree of uncertainty around the point estimate of 51 per million. An additional source of uncertainty in the Beutler and Gelbart estimate results from the unclear validity of the assumption that PK deficiency cases and non‐cases have the same distribution of PKLR mutations that impair PK enzyme function.
5. CONCLUSIONS
The prevalence of PK deficiency in general Western populations is likely greater than 3.2‐8.5 per million and may be as high as 51 per million. Future studies are needed to understand the clinical significance of various mutant alleles, which may inform more accurate, clinically relevant PK deficiency prevalence estimates, identify the degree of and reasons for underdiagnosis, and elucidate PK deficiency heterogeneity between populations. Some of this additional research is currently being conducted through existing patient registries.
CONFLICT OF INTEREST
CC and DC are employees of IQVIA. IQVIA received research funding from Agios Pharmaceuticals to conduct this study. KG, MS, LP, and ANB are employees of Agios pharmaceuticals. MHS has no potential conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
MHS drafted the manuscript. MS, CC, DC, KG, LP, and ANB provided critical revisions to the manuscript. MHS, MS, CC, DC, and ANB designed and executed the study. MS and ANB conceived of the study. All authors approved of the final manuscript version.
Supporting information
Table S1‐S3
ACKNOWLEDGMENTS
This study was supported by research funding from Agios Pharmaceuticals to IQVIA. The authors would like to thank Phil Prince, Sam Piteri, and John D'Alotto for their help in the conceptualization of this study.
Secrest MH, Storm M, Carrington C, et al. Prevalence of pyruvate kinase deficiency: A systematic literature review. Eur J Haematol. 2020;105:173–184. 10.1111/ejh.13424
ENDNOTE
In the study by Chami et al, this mutation was incorrectly referred to as c.1456T > G.
REFERENCES
- 1. Prchal JT.Pyruvate kinase deficiency. https://www.uptodate.com/contents/pyruvate‐kinase‐deficiency
- 2. Grace RF, Bianchi P, van Beers EJ, et al. Clinical spectrum of pyruvate kinase deficiency: Data from the Pyruvate Kinase Deficiency Natural History Study. Blood. 2018;131(20):2183‐2192. [DOI] [PubMed] [Google Scholar]
- 3. Bianchi P, Fermo E, Lezon‐Geyda K, et al. Genotype‐phenotype correlation and molecular heterogeneity in pyruvate kinase deficiency. Am J Hematol. 2020;95(5):472‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bianchi P, Fermo E, Glader B, et al. Addressing the diagnostic gaps in pyruvate kinase deficiency: Consensus recommendations on the diagnosis of pyruvate kinase deficiency. Am J Hematol. 2019;94(1):149‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Joanna Briggs Institute . Critical appraisal tools 2018. http://joannabriggs.org/research/critical‐appraisal‐tools.html
- 6. Von Elm E, Altman DG, Egger M, et al. The STrengthening the Reporting of OBservational Studies in Epidemiology (STROBE) statement: Guidelines for reporting observational studies. PLoS Med. 2007;4(10):e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carey PJ, Chandler J, Hendrick A, et al. Prevalence of pyruvate kinase deficiency in a northern European population in the north of England. Blood. 2000;96(12):4005‐4006. [PubMed] [Google Scholar]
- 8. de Medicis E, Ross P, Friedman R, et al. Hereditary nonspherocytic hemolytic anemia due to pyruvate kinase deficiency: A prevalence study in Quebec (Canada). Hum Hered. 1992;42(3):179‐183. [DOI] [PubMed] [Google Scholar]
- 9. Christensen R, Eggert L, Baer V, Smith K. Pyruvate kinase deficiency as a cause of extreme hyperbilirubinemia in neonates from a polygamist community. J Perinatol. 2010;30(3):233. [DOI] [PubMed] [Google Scholar]
- 10. Beutler E, Gelbart T. Estimating the prevalence of pyruvate kinase deficiency from the gene frequency in the general white population. Blood. 2000;95(11):3585‐3588. [PubMed] [Google Scholar]
- 11. Grace RF, Zanella A, Neufeld EJ, et al. Erythrocyte pyruvate kinase deficiency: 2015 status report. Am J Hematol. 2015;90(9):825‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Manco L, Abade A. Pyruvate kinase deficiency: Prevalence of the 1456C→T mutation in the Portuguese population. Clin Genet. 1456C;60(6):472‐473. [DOI] [PubMed] [Google Scholar]
- 13. Svidnicki MCCM, Santos A, Fernandez JAA, et al. Novel mutations associated with pyruvate kinase deficiency in Brazil. Hematol Transfus Cell Ther. 2018;40(1):5‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Figueroa DB, Tillotson J, Li M, et al. Discovery of genetic variants of the kinases that activate tenofovir among individuals in the United States, Thailand, and South Africa: HPTN067. PLoS ONE. 2018;13(4):e0195764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zanella A, Bianchi P, Baronciani L, et al. Molecular characterization of PK‐LR gene in pyruvate kinase–deficient Italian patients. Blood. 1997;89(10):3847‐3852. [PubMed] [Google Scholar]
- 16. Chami N, Chen M‐H, Slater AJ, et al. Exome genotyping identifies pleiotropic variants associated with red blood cell traits. Am J Human Genetic. 2016;99(1):8‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Machado P, Pereira R, Rocha AM, et al. Malaria: Looking for selection signatures in the human PKLR gene region. Br J Haematol. 2010;149(5):775‐784. [DOI] [PubMed] [Google Scholar]
- 18. Alves J, Machado P, Silva J, et al. Analysis of malaria associated genetic traits in Cabo Verde, a melting pot of European and sub Saharan settlers. Blood Cells Mol Dis. 2010;44(1):62‐68. [DOI] [PubMed] [Google Scholar]
- 19. Berghout J, Higgins S, Loucoubar C, Sakuntabhai A, Kain KC, Gros P. Genetic diversity in human erythrocyte pyruvate kinase. Genes Immun. 2012;13(1): 98‐102. [DOI] [PubMed] [Google Scholar]
- 20. Machado P, Manco L, Gomes C, et al. Pyruvate kinase deficiency in sub‐Saharan Africa: Identification of a highly frequent missense mutation (G829A; Glu277Lys) and association with malaria. PLoS ONE. 2012;7(10):e47071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robison R, Lyon GJ, Jiang T, et al. Full exome sequencing reveals the genetic basis of a case of idiopathic hemolytic anemia and suggests candidate rare variants for ADHD in a Utah pedigree. Neuropsychopharmacology. 2010;35(S1):S310. [Google Scholar]
- 22. Lyon GJ, Jiang T, Van Wijk R, et al. Exome sequencing and unrelated findings in the context of complex disease research: Ethical and clinical implications. Discovery Medicine. 2011;12(62):41. [PMC free article] [PubMed] [Google Scholar]
- 23. Beutler E, Halasz A. A series of new screening procedures for pyruvate kinase deficiency, glucose‐6‐phosphate dehydrogenase deficiency, and glutathione reductase deficiency. Blood. 1966;28(4):553‐562. [PubMed] [Google Scholar]
- 24. Miwa S. Recommended Methods for the Characterization of Red Cell Pyruvate Kinase Variants* International Committee for Standardization in Haematology. Br J Haematol. 1979;43(2):275‐286. [DOI] [PubMed] [Google Scholar]
- 25. Beutler E, Blume K, Kaplan J, Löhr G, Ramot B, Valentine W. International Committee for Standardization in Haematology: Recommended methods for red‐cell enzyme analysis. Br J Haematol. 1977;35(2):331‐340. [DOI] [PubMed] [Google Scholar]
- 26. Beutler E. Red cell enzyme defects as nondiseases and as diseases. Blood. 1979;54(1):1‐7. [PubMed] [Google Scholar]
- 27. Tsang SS, Feng C‐S. A modified screening procedure to detect pyruvate kinase deficiency. Am J Clin Pathol. 1993;99(2):128‐131. [DOI] [PubMed] [Google Scholar]
- 28. García C, Moragón C, López‐Fernández ME. Frequency of glutathione reductase, pyruvate kinase and glucose‐6‐phosphate dehydrogenase deficiency in a Spanish population. Hum Hered. 1979;29(5):310‐313. [DOI] [PubMed] [Google Scholar]
- 29. Wu ZL, Yu WD, Chen SC. Frequency of erythrocyte pyruvate kinase deficiency in Chinese infants. Am J Hematol. 1985;20(2):139‐144. [DOI] [PubMed] [Google Scholar]
- 30. Tanaka K, Paglia D. Pyruvate kinase deficiency. Sem Hematol. 1971;8:365‐396. [PubMed] [Google Scholar]
- 31. Fung R, Keung YK, Chung G. Screening of pyruvate kinase deficiency and G6PD deficiency in Chinese newborn in Hong Kong. Arch Dis Child. 1969;44(235):373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Feng CS, Tsang SS, Mak YT. Prevalence of pyruvate kinase deficiency among the Chinese: Determination by the quantitative assay. Am J Hematol. 1993;43(4):271‐273. [DOI] [PubMed] [Google Scholar]
- 33. Fox A, Menkes A. An evaluation of screening tests for erythrocyte enzyme deficiencies in a general hospital laboratory. J Am Osteopathic Assoc. 1969;69(3):255‐261. [PubMed] [Google Scholar]
- 34. Mohrenweiser HW. Frequency of enzyme deficiency variants in erythrocytes of newborn infants. Proc Natl Acad Sci. 1981;78(8):5046‐5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mohrenweiser HW. Functional hemizygosity in the human genome: Direct estimate from twelve erythrocyte enzyme loci. Hum Genet. 1987;77(3):241‐245. [DOI] [PubMed] [Google Scholar]
- 36. El‐Hazmi M, Al‐Swailem AR, Al‐Faleh FZ, Warsy A. Frequency of glucose‐6‐phosphate dehydrogenase, pyruvate kinase and hexokinase deficiency in the Saudi population. Hum Hered. 1986;36(1):45‐49. [DOI] [PubMed] [Google Scholar]
- 37. Munyanganizi R, Cotton F, Vertongen F, Gulbis B. Red blood cell disorders in Rwandese neonates: Screening for sickle cell disease and glucose‐6‐phosphate dehydrogenase deficiency. J Med Screen. 2006;13(3):129‐131. [DOI] [PubMed] [Google Scholar]
- 38. Upadhye D, Das RS, Ray J, et al. Newborn screening for hemoglobinopathies and red cell enzymopathies in Tripura State: A malaria‐endemic state in Northeast India. Hemoglobin. 2018;42(1):43‐46. [DOI] [PubMed] [Google Scholar]
- 39. Christensen RD, Yaish HM, Johnson CB, Bianchi P, Zanella A. Six children with pyruvate kinase deficiency from one small town: Molecular characterization of the PK‐LR gene. J Pediatrics. 2011;159(4):695‐697. [DOI] [PubMed] [Google Scholar]
- 40. Yavarian M, Karimi M, Shahriary M, Afrasiabi A. Prevalence of pyruvate kinase deficiency among the south Iranian population: Quantitative assay and molecular analysis. Blood Cells Mol Dis. 2008;40(3):308‐311. [DOI] [PubMed] [Google Scholar]
- 41. Akin H, Baykal‐Erkılıç A, Aksu A, Yücel G, Gümüşlü S. Prevalence of erythrocyte pyruvate kinase deficiency and normal values of enzyme in a Turkish population. Hum Hered. 1997;47(1):42‐46. [DOI] [PubMed] [Google Scholar]
- 42. Al‐Naama MM, Al‐Naama LM, Al‐Sadoon TA. Glucose‐6‐phosphate dehydrogenase, hexokinase and pyruvate kinase activities in erythrocytes of neonates and adults in Basrah. Ann Trop Paediatr. 1994;14(3):195‐200. [DOI] [PubMed] [Google Scholar]
- 43. Al‐Naama MM, Al‐Naama LM, Al‐Saadoon TA. Frequencies of glucose 6‐phosphate dehydrogenase pyruvate kinase and hexokinase deficiencies in the Basrah population of Iraq. Screening. 1995;4(1):27‐34. [Google Scholar]
- 44. Abu‐Melha A, Ahmed M, Knox‐Macaulay H, Al‐Sowayan S, El‐Yahia A. Erythrocyte pyruvate kinase deficiency in newborns of eastern Saudi Arabia. Acta Haematol. 1991;85(4):192‐194. [DOI] [PubMed] [Google Scholar]
- 45. Mohrenweiser H, Neel J. A "disproportion" between the frequency of rare electropmorphs and enzyme deficiency variants in Amerindians. Am J Hum Genet. 1984;36(3):655. [PMC free article] [PubMed] [Google Scholar]
- 46. Satoh C, Neel J, Yamashita A, Goriki K, Fujita M, Hamilton H. The frequency among Japanese of heterozygotes for deficiency variants of 11 enzymes. Am J Hum Genet. 1983;35(4):656. [PMC free article] [PubMed] [Google Scholar]
- 47. Satoh C, Neel JV, Miura A, et al. Inherited thermostability variants of seven enzymes in a Japanese population. Ann Hum Genet. 1985;49(1):11‐22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S3