

Abstract
Itacitinib is a novel, selective, Janus kinase 1 inhibitor in development for treatment of graft‐versus‐host disease. The objective of this study was to assess pharmacokinetics and safety of 300‐mg itacitinib dosed in participants with normal renal function (n = 10), severe renal impairment (n = 8), and end‐stage renal disease (ESRD) on hemodialysis (n = 8). Serial plasma and urine samples (urine from normal and severe groups only) were collected before dosing until 72 hours after dosing. In the ESRD group, itacitinib was evaluated in 2 periods, when dosed before (period 1) and after (period 2) a hemodialysis session. Geometric mean ratios (90% confidence interval) in participants with severe renal impairment, ESRD period 1 and ESRD period 2 relative to participants with normal renal function were 1.65 (1.13‐2.39), 0.71 (0.49‐1.03), and 0.83 (0.57‐1.20) for maximum plasma drug concentration and 2.23 (1.56‐3.18), 0.81 (0.57‐1.16), and 0.95 (0.66‐1.35) for area under the plasma concentration–time curve from time zero to infinity. Itacitinib was well tolerated, and 3 grade 1 treatment‐emergent adverse events were reported over the course of the study. Given the magnitude of exposure changes in participants with severe renal impairment or ESRD and the historic risk‐benefit profile, no dose adjustment is recommended for itacitinib in patients with impaired renal function, although the final dosage recommendation will be based on cumulative pharmacokinetics and safety from this study and from the pivotal graft‐versus‐host disease trial. Additionally, itacitinib may be administered to patients undergoing dialysis regardless of the time of dialysis.
Keywords: dialysis, itacitinib, graft‐versus‐host disease, Janus kinase inhibitor, pharmacokinetics, renal impairment
Allogeneic hematopoietic stem cell transplants (HSCTs) may be a potentially curative treatment option for patients with hematologic malignancies. 1 However, allogeneic HSCT may lead to serious and even fatal complications such as graft‐versus‐host disease (GVHD), where alloreactive T cells attack healthy tissue rather than tumor cells. 2 Chronic GVHD occurs in 30% to 70% of patients, 2 and there is an urgent unmet clinical need for new therapies for chronic GVHD. 3 Itacitinib (INCB039110) adipate is currently in a late‐phase trial for the treatment of patients with chronic GVHD. 4 It is a novel and potent inhibitor of the Janus kinase (JAK) family of protein tyrosine kinases with preferential selectivity for JAK1 that is implicated in the pathophysiology of GVHD. 5 Itacitinib is administered in the form of a sustained‐release (SR) tablet. This formulation allows for once‐daily dosing with a longer half‐life and a reduced peak‐to‐trough ratio compared to the immediate‐release formulation 6 and can be administered without regard to food.
The absorption, distribution, metabolism, and excretion properties of itacitinib have been characterized in healthy volunteers following administration of 14C‐itacitinib oral solution (∼4.9 mg) with a 300‐mg dose of itacitinib delivered as SR tablets and demonstrated that elimination of itacitinib occurs primarily by oxidative metabolism. 7 In vitro data indicate that metabolism is mediated by cytochrome P450 (CYP) 3A (data on file, Incyte Corporation). The coadministration of the strong CYP3A inhibitor itraconazole with itacitinib showed an increase in the maximum plasma drug concentration (Cmax) and area under the plasma concentration–time curve from time zero to infinity (AUC0‐∞) of approximately 3‐fold and 5‐fold, respectively, whereas the coadministration of the strong CYP3A4 inducer rifampin showed a decrease in the Cmax and AUC0‐∞ of approximately 80%. 8 The renal elimination of itacitinib is minimal, with approximately 8.4% of the dose of SR itacitinib eliminated in the urine as unchanged drug. 7
Even for drugs with minimal renal clearance, a pharmacokinetic study should be performed in individuals with impaired renal function when the drug is likely to be used in patients with renal impairment. Dosage adjustments may be required 9 in these patients because renal failure can alter hepatic drug metabolism 10 , 11 and drug transporter systems. 12 Renal impairment is commonly seen in patients after HSCT 13 , 14 ; therefore, it is important to characterize the effect of renal impairment on itacitinib pharmacokinetics to identify if dosage adjustments may be required in patients with renal impairment.
The objective of this clinical study was to evaluate the effect of renal impairment on the plasma exposure of itacitinib. Participants with end‐stage renal disease (ESRD) were included to determine how itacitinib should be dosed for patients on dialysis. This study also evaluated plasma protein binding of itacitinib in participants with severe renal impairment and ESRD compared with participants with normal renal function, determined the renal clearance and dialysate clearance of itacitinib, and determined the fraction of itacitinib that is excreted in urine and dialysate.
Methods
The study was conducted in accordance with the International Conference on Harmonization Good Clinical Practice guidelines, applicable regulations, guidelines governing clinical study conduct, and by the principles in the Declaration of Helsinki. The study protocol was approved by the institutional review boards of the study sites (Orlando Clinical Research Center, Orlando, Florida; Riverside Clinical Research Center, Edgewater, Florida; and Orange County Research Center, Tustin, California), and all participants gave written informed consent before participation in the study.
Study Design and Participants
This study was a single‐dose, open‐label, parallel‐group study of itacitinib in participants with severe renal impairment or ESRD and participants with normal renal function (controls) matched by demographic characteristics including age (±10 years), sex, and body mass index (±20%). Participants were assigned to renal function groups according to the estimated glomerular filtration rate (eGFR) as calculated by the Modification of Diet in Renal Disease equation, and use of hemodialysis. Participants with normal renal function (eGFR ≥90 mL/min/1.73 m2; n = 10), severe renal impairment (eGFR <30 mL/min/1.73 m2, not on dialysis; n = 8), and ESRD (eGFR <30 mL/min/1.73 m2, on dialysis; n = 8) were enrolled. The enrollment of participants with mild and moderate renal impairment was optional based on the results from the severe cohort.
In the normal renal function and severe renal impairment groups (Figure 1A), all participants received a single 300‐mg dose (3 × 100 mg SR tablets) of itacitinib after a medium‐fat meal. In the ESRD group (Figure 1B), participants received a single 300‐mg dose (3 × 100 mg SR tablets) after a medium‐fat meal in 2 treatment periods: 4 hours before the start of a hemodialysis session in the first treatment period and 1 hour after the end of a hemodialysis session in the second treatment period. Participants were confined to the study site 1 day before dosing and remained at the site until study procedures were completed on day 4. The medium‐fat meal was defined as a meal of around 500 calories, providing approximately 35% calories from fat. The composition of the meal was similar in all study participants.
Figure 1.
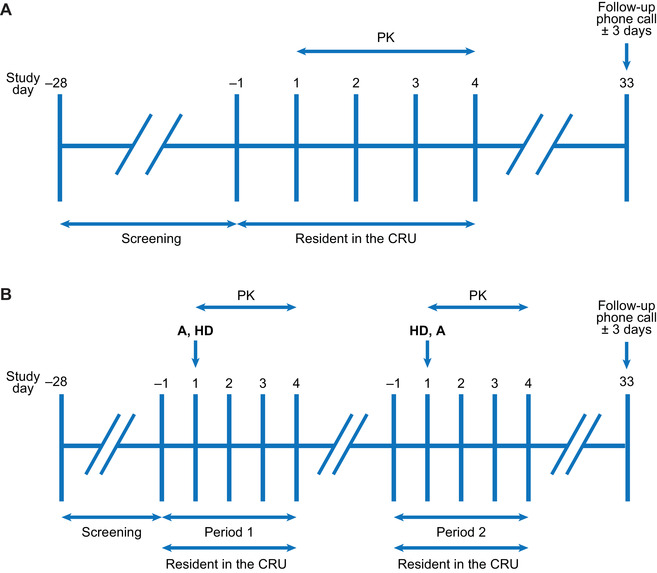
Clinical study design for normal renal function and severe renal impairment groups (A) and end‐stage renal disease groups (B). A, 3 × 100‐mg itacitinib dose; CRU, clinical research unit; HD, hemodialysis; PK, pharmacokinetics.
Pharmacokinetic Sampling and Bioanalysis
The blood samples for the assay of itacitinib in the plasma were collected into lavender‐top dipotassium ethylenediaminetetraacetic acid–containing collection tubes before dosing (0 hours) and at 0.5, 1, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, and 72 hours after dosing. Urine samples were collected in containers without preservatives over the following intervals: −12 to 0, 0 to 8, 8 to 24, 24 to 48, and 48 to 72 hours after dosing. In the ESRD group (period 1 only), dialysate samples were collected during the hemodialysis session at 4 (start of the hemodialysis session), 5, 6, 7, and 8 (end of the hemodialysis session) hours after dosing. The plasma samples were assayed by a validated liquid chromatography–tandem mass spectrometry (LC‐MS/MS) method with a linear range of 5 to 5000 nmol/L. Detailed methods on the itacitinib plasma assay have been described elsewhere. 8 Three plasma analytical quality control (QC) samples were included in each plasma analysis run: a low QC sample (15 nmol/L), a middle QC sample (250 nmol/L) and a high QC sample (4000 nmol/L). Interassay precision for this assay ranged from 1.7% to 3.5% (CV%), and interassay accuracy ranged from –3.3% to 1.6%. The analysis of the urine samples used a validated LC‐MS/MS method with a linear range of 0.025 to 25 μmol/L. In this method, 25 μL of the urine sample was placed in individual wells of a 96‐well plate. An aliquot of 500 μL of internal standard (dissolved in 50:50 methanol:water) was added, following which the plate was capped, vortexed, and centrifuged. The plate was then placed in the autosampler tray and injected into an LC‐MS/MS system for analysis, using the same LC‐MS/MS conditions as those described elsewhere. 8 Three urine analytical QC samples were included in each urine analysis run: low QC sample (0.075 μmol/L), middle QC sample (1 μmol/L), and high QC sample (20 μmol/L). Interassay precision of this assay ranged from 1.6% to 4.6% (CV%), and interassay accuracy ranged from –0.9% to –0.4%. For the dialysate, the samples were diluted 1:9 (dialysate:plasma, v:v) and analyzed using a similar validated plasma method, albeit with a lower linear range of 0.3 to 300 nmol/L. This matrix matching of the dialysate samples to plasma for analysis using this plasma method was partially validated before sample analysis. Three plasma analytical QC samples were included in the single dialysate sample analysis run: low QC sample (0.9 nmol/L), middle QC sample (20 nmol/L), and high QC sample (240 nmol/L). Interassay precision was not calculated since QC samples were analyzed in duplicate for the run. The interassay accuracy for this assay ranged from –0.8% to 0.5%.
Plasma protein binding was analyzed by a non–Good Laboratory Practices assay. Unbound fraction of itacitinib was determined by equilibrium dialysis using venous plasma samples collected 4 hours after dosing. Briefly, a semipermeable membrane with a molecule weight cutoff of 10 000 daltons separated the plasma‐containing compartment and plasma‐free compartment containing phosphate buffer. The system was allowed to equilibrate at 37°C for 2 hours, then samples were collected from each compartment for determination of itacitinib concentrations by LC‐MS/MS. Samples from the plasma‐containing compartment were normalized with an equal volume of phosphate buffer, and samples from the plasma‐free compartment were normalized with an equal volume of plasma prior to analysis using the plasma method with the range of 5 to 5000 nmol/L. A partial validation of this 1:1 matrix‐matching prior to sample analysis assured data integrity. Three plasma analytical QC samples were included in each protein binding sample analysis run: a low QC sample (15 nmol/L), a middle QC sample (250 nmol/L), and a high QC sample (4000 nmol/L). Interassay precision for this analysis ranged from 0.9% to 4.0% (CV%), and interassay accuracy ranged from –0.5% to 3.3%. The resulting protein binding sample concentrations were used to calculate a fraction unbound for individual participants.
Pharmacokinetics and Statistical Analyses
Standard noncompartmental pharmacokinetic methods were used to analyze itacitinib plasma concentrations using Phoenix WinNonlin version 8.0 (Certara, Princeton, New Jersey). The Cmax and time to maximum plasma drug concentration (tmax) values were taken directly from the observed plasma concentration data. The terminal phase disposition rate constant (λz) was estimated using a log‐linear regression of the concentration data in the terminal elimination phase, and the terminal elimination half‐life was estimated as ln(2)/λz. Total AUC0‐∞ was calculated as AUC0‐t + Ct/λz, where Ct was the last measurable concentration. Apparent clearance was calculated as dose/AUC0‐∞ with the apparent volume of distribution during the terminal phase calculated as dose/(λz × AUC0‐∞). All estimated pharmacokinetic parameters were summarized by descriptive statistics.
The percentage of the itacitinib dose eliminated in urine was calculated by the amount of itacitinib recovered in urine divided by the administered dose and multiplied by 100. Renal clearance was determined by the cumulative amount of itacitinib in urine divided by the plasma AUC over 72 hours. The clearance of itacitinib by the dialysate was determined by the cumulative amount of itacitinib in dialysate divided by the plasma AUC during dialysis (from 4 to 8 hours). The Cmax, AUC0‐t, and AUC0‐∞ pharmacokinetic parameters were log‐transformed and compared between renal function groups with the normal renal function group as reference using 3 separate 1‐factor analyses of variance (severe vs normal renal function groups, ESRD period 1 vs normal renal function groups, and ESRD period 2 vs normal renal function groups) using SAS Enterprise Guide version 7.1 (SAS Institute, Cary, North Carolina). The geometric mean ratios and 90% confidence intervals were determined for the comparison of itacitinib exposures in each renal impairment group vs the normal renal function group.
Safety Assessments
Each adverse event (AE) was coded using the Medical Dictionary for Regulatory Activities System Organ Class and Preferred Term (version 21.0). All AEs were listed and summarized using descriptive methodology. AEs for each renal function group were presented by association with the study drug and severity. The severity of AEs was described and graded using grades 1 to 4 from the Toxicity Grading Scale for Healthy Adult and Adolescent Volunteers Enrolled in Preventive Vaccine Clinical Trials. 15 Descriptive statistics were calculated for clinical laboratory test data, 12‐lead electrocardiograms, and vital signs by renal function group at each assessment time.
Results
Participant Disposition
Twenty‐six participants (16 men and 10 women) were enrolled and completed the clinical study. The demographic characteristics across all 3 renal function groups are shown in Table 1. In the healthy participants, demographic characteristics (ie, age, sex, and body mass index) were matched to both severe renal impairment and ESRD groups.
Table 1.
Study Demographic Information Across Renal Function Groups in the Clinical Study
| Demographic Characteristic | Normal Renal Function (n = 10) | Severe Renal Impairment (n = 8) | ESRD (n = 8) |
|---|---|---|---|
| Age, y, median (range) | 62.5 (44.0‐76.0) | 73.0 (56.0‐80.0) | 55.0 (41.0‐62.0) |
| Height, cm, median (range) | 173 (163‐185) | 170 (156‐188) | 174 (161‐188) |
| Weight, kg, median (range) | 84.7 (72.7‐106.5) | 89.5 (66.1‐99.7) | 96.7 (75.0‐124.7) |
| BMI, kg/m2, median (range) | 29.60 (23.6‐37.0) | 29.55 (25.8‐36.8) | 30.45 (26.3‐37.7) |
| Sex, n (%) | |||
| Male | 6 (60.0) | 5 (62.5) | 5 (62.5) |
| Female | 4 (40.0) | 3 (37.5) | 3 (37.5) |
| Race, n (%) | |||
| White | 4 (40.0) | 7 (87.5) | 1 (12.5) |
| African American | 5 (50.0) | 1 (12.5) | 7 (87.5) |
| Asian | 0 | 0 | 0 |
| Other | 1 (10.0) | 0 | 0 |
| eGFR at check‐in, mL/min/1.73 m2, median (range) | 96.5 (88‐125) | 28 (13‐32)a | 7.0 (5.0‐7.0) |
BMI, body mass index; eGFR, estimated glomerular filtration rate; ESRD, end‐stage renal disease.
Classification of participants was based on values at screening. One subject in the severe group had an eGFR of 27 mL/min/1.73 m2 at screening, which shifted to 32 mL/min/1.73 m2 at check‐in.
Pharmacokinetic Results
The mean itacitinib plasma concentrations versus time profiles by renal function group are presented in Figure 2. The Cmax and AUC data stratified by renal impairment group, with overlay of the individual level data are presented in Figures S1 and S2. A summary of the pharmacokinetic parameters of itacitinib after administration in each of the renal function groups is shown in Table 2. In participants with normal renal function, the geometric mean itacitinib Cmax and AUC0‐∞ were 700 nmol/L and 3050 nmol•hr/L, respectively, with a median tmax of 3 hours. Participants with severe renal impairment showed an increased Cmax and AUC0‐∞ of 1150 nmol/L and 6780 nmol•hr/L, with a median tmax of 4 hours. For participants with ESRD, the geometric mean Cmax and AUC0‐∞ were 495 nmol/L and 2480 nmol•hr/L, respectively, with a median tmax of 3 hours when itacitinib was dosed before the start of a hemodialysis session (treatment period 1). When itacitinib was dosed after a hemodialysis session (treatment period 2), the geometric mean Cmax and AUC0‐∞ were 580 nmol/L and 2890 nmol•hr/L, respectively, with a median tmax of 5 hours. When pharmacokinetics in the renal impairment groups were compared with the normal renal function group, the geometric mean ratio (90% confidence interval) was 1.65 (1.13‐2.39), 0.71 (0.49‐1.03), and 0.83 (0.57‐1.20) for Cmax and 2.23 (1.56‐3.18), 0.81 (0.57‐1.16), and 0.95 (0.66‐1.36) for AUC0‐∞ for the severe renal impairment, ESRD treatment period 1, and ESRD treatment period 2 groups, respectively (Table 2 and Figure 3).
Figure 2.
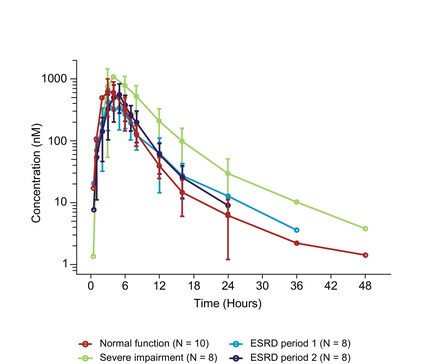
Itacitinib plasma concentrations (mean ± standard deviation) vs time after dosing stratified by renal function groups. Plasma concentrations are shown at time points when the majority of data points were measurable. ESRD, end‐stage renal disease.
Table 2.
Summary of Itacitinib Pharmacokinetic Parameters by Renal Function Group
| Group/Comparison | Cmax, nmol/L | tmax, h | AUC0‐t, nmol•hr/L | AUC0‐∞, nmol•hr/L | Half‐life, h | CL/F, L/h | Vz/F, L |
|---|---|---|---|---|---|---|---|
| Normal renal function (n = 10) | 821 ± 588 | 3.0 (2.0‐6.0) | 3290 ± 1630 | 3360 ± 1640 | 6.18 ± 6.88 | 194 ± 78.7 | 1400 ± 1150 |
| Severe renal impairment (n = 8) | 1250 ± 521 | 4.0 (3.0‐8.0) | 7250 ± 2660 | 7350 ± 2690 | 7.96 ± 7.28 | 89.5 ± 54.1 | 812± 507 |
| ESRD period 1 (n = 8) | 522 ± 172 | 3.0 (3.0‐6.0) | 2620 ± 1190 | 2670 ± 1210 | 5.27 ± 1.92 | 234 ± 86.7 | 1690 ± 800 |
| ESRD period 2 (n = 8) | 623 ± 253 | 5.0 (3.0, 6.0) | 3020 ± 1260 | 3080 ±1260 | 4.32 ± 1.43 | 199 ± 67.3 | 1280 ± 724 |
| Geometric mean ratios and 90%CIs | |||||||
| Severe renal impairment vs normal renal function | 1.65 (1.13‐2.39) | … | 2.24 (1.57‐3.20) | 2.23 (1.56‐3.18) | … | … | … |
| ESRD period 1 vs normal renal function | 0.71 (0.49‐1.03) | ‐ | 0.81 (0.57‐1.16) | 0.81 (0.57‐1.16) | … | … | … |
| ESRD period 2 vs normal renal function | 0.83 (0.57‐1.20) | … | 0.95 (0.66‐1.35) | 0.95 (0.66‐1.36) | … | … | … |
AUC, area under the plasma concentration curve; CI, confidence interval; CL/F, apparent oral clearance; Cmax, maximum plasma drug concentration; ESRD, end‐stage renal disease; SD, standard deviation; tmax, time to reach maximum plasma drug concentration; Vz/F, apparent volume of distribution during the terminal phase.
Pharmacokinetic parameters are shown as mean ± standard deviation; tmax is shown as median (range).
Figure 3.
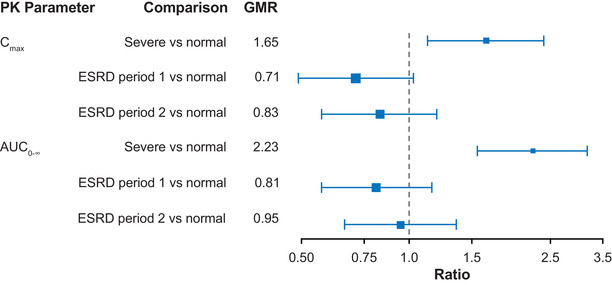
GMR and 90% confidence intervals for Cmax and AUC0‐∞ comparison for severe renal impairment and ESRD vs normal renal function. AUC0‐∞, area under the plasma concentration time curve from time zero to infinity; Cmax, maximum plasma drug concentration; ESRD, end‐stage renal disease; GMR, geometric mean ratio; PK, pharmacokinetic.
Plasma Protein Binding
The protein binding of itacitinib was independent of renal function and similar across all evaluated renal function groups with an average fraction unbound of 30.4%, 37.1%, and 39.6% in participants with normal renal function, severe renal impairment, and ESRD, respectively.
Renal Clearance and Dialysate Clearance of Itacitinib in Participants With Varying Degrees of Renal Function
In participants with normal renal function, the average renal clearance of itacitinib was 11.8 L/h, whereas itacitinib renal clearance was 3.03 L/h in the severe renal impairment group. The renal clearance was approximately 6% and 3% of total itacitinib clearance in the normal renal function and severe renal impairment groups, respectively. The fraction of the dose eliminated in the urine as itacitinib was 7.2% and 3.7% in the normal renal function and severe renal impairment groups, respectively. Itacitinib dialysate clearance was 2.01 L/h, which was approximately 1% of the total clearance of itacitinib in this group. The average fraction of the dose excreted in dialysate as itacitinib was 0.9%.
Safety Results
The single 300‐mg oral dose of itacitinib was well tolerated when administered to participants with normal renal function and participants with severe renal impairment and ESRD. Over the course of the study, 3 participants with severe renal impairment reported a total of 3 treatment emergent AEs. The treatment‐emergent AEs reported in this study were pruritus, constipation, and hyperkalemia. These were mild in severity (grade 1), resolved over the course of the study, and were considered to be not related to the study drug. There were no clinically significant findings in the clinical laboratory test results, vital signs, or 12‐lead electrocardiograms.
Discussion
This clinical study was performed to evaluate the impact of renal impairment and hemodialysis on the pharmacokinetics of a single oral dose of itacitinib. Patients with severe renal impairment showed an approximately 2‐fold increase in itacitinib exposure compared with patients with normal renal function.
From a pilot study in patients with acute GVHD receiving itacitinib with corticosteroids, in patients concomitantly taking potent CYP3A4 inhibitors (mainly posaconazole), there was an approximate 2‐fold increase in exposure of itacitinib relative to the exposure in patients not taking a strong CYP3A4 inhibitor. 16 Despite this increased exposure, itacitinib was well tolerated with the incidence and severity of AEs as expected in this population, and there was no exacerbation of corticosteroid relative AEs. This formed the basis of the therapeutic window supporting a 2‐fold increase in exposure may not be clinically relevant. Consequently, this clinical study did not enroll patients with mild or moderate renal impairment. Additional dosage adjustment of itacitinib is not currently recommended for participants with severe renal impairment, although final dosage recommendations will be based on the totality of data from this study and late‐phase patient studies in GVHD. Consequently, this clinical study did not enroll patients with mild and moderate renal impairment.
The fraction of the itacitinib dose eliminated as unchanged drug in the urine was 7.2% in the normal renal function group and 3.7% in the severe renal impairment group. These results support previously generated data 7 that renal clearance plays a minor role in the elimination of itacitinib. Although it is not clear why there was a 2‐fold increase in plasma exposure of itacitinib in the severe renal impairment group compared with the normal renal function group, potential explanations include itacitinib pharmacokinetic variability and altered bioavailability or nonrenal clearance mechanisms 17 , 18 in patients with renal failure. For the participants with ESRD, hemodialysis may have resulted in improvement of hepatic metabolic activity as shown by Nolin et al, 19 resulting in plasma exposures that were more similar to that seen in the healthy matched participants. When comparing exposures in the severe renal impairment group vs the ESRD group, it should be noted that there are differences in racial composition in the 2 groups. Seven of 8 participants in the ESRD group were African American vs only 1 of 8 participants in the severe renal impairment group. This may affect the pharmacokinetics of itacitinib due to differences in metabolism across racial groups. For example, a greater proportion of African Americans express the intermediate or extensive metabolizer phenotype of CYP3A5 while whites predominantly express the poor CYP3A5 metabolizer phenotype. 20 Therefore, there is a possibility that such difference may have contributed to the higher itacitinib exposures in the severe renal impairment group. The contribution of race to differences in pharmacokinetics will be determined in a population pharmacokinetics analysis. Finally, the protein‐binding data showed that itacitinib protein binding was independent of renal function and was similar to prior in vitro plasma protein‐binding data where the unbound fraction was approximately 35% (data on file, Incyte Corporation).
Conclusions
In summary, itacitinib was well tolerated across all participants with normal renal function and renal impairment and demonstrated a consistent safety profile. Although itacitinib exposure in the severe renal impairment group showed 1.6‐fold increase in Cmax and 2.2‐fold increase in AUC0‐∞ relative to healthy matched participants with normal renal function, this increase in itacitinib exposure may not be clinically relevant. This is based on prior characterization of the risk‐benefit profile in patients with acute GVHD who demonstrated a similar magnitude of increase in itacitinib exposure due to coadministration of a potent CYP3A inhibitor, mainly posaconazole. No dose adjustment is currently recommended for itacitinib in patients with severe renal impairment. However, final recommendations will be based on the totality of data from this study and the therapeutic window defined using exposure/response analyses from the late‐phase studies in patients with GVHD. Given that there was little effect on Cmax and AUC in the ESRD group compared with the normal renal function group, no dosage adjustment is recommended for itacitinib in patients with ESRD on hemodialysis and itacitinib can be administered without regard to time of dialysis.
Conflicts of Interest
Incyte Corporation contributed to the study design, research, and interpretation of the data and the writing, review, and approval of the manuscript. N.S., A.M.B., N.E., G.Z., S.P., Z.X., B.Y., X.C., S.Y., and N.P. are current or former employees of Incyte Corporation and may hold Incyte corporation stock or stock options. T.M. is an employee and equity owner of Orlando Clinical Research Center and declares no other conflicts of interest.
Funding
This research was supported by Incyte Corporation.
Data Sharing
Access to individual subject‐level data is not available for this study.
Supporting information
Supporting Information
Acknowledgments
Editorial assistance was provided by Envision Pharma Group, Inc. (Philadelphia, Pennsylvania), and funded by Incyte Corporation.
References
- 1. Magenau J, Couriel DR. Hematopoietic stem cell transplantation for acute myeloid leukemia: to whom, when, and how. Curr Oncol Rep. 2013;15(5):436‐444. [DOI] [PubMed] [Google Scholar]
- 2. Lee SJ, Flowers ME. Recognizing and managing chronic graft‐versus‐host disease. Hematology Am Soc Hematol Educ Program. 2008;2008:134‐141. [DOI] [PubMed] [Google Scholar]
- 3. Martin PJ, Lee SJ, Przepiorka D, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft‐versus‐Host Disease: VI. The 2014 Clinical Trial Design Working Group Report. Biol Blood Marrow Transplant. 2015;21(8):1343‐1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Incyte Corporation . GRAVITAS‐309: Itacitinib or placebo in combination with corticosteroids as initial treatment for chronic graft‐versus‐host disease [ClinicalTrials.gov Identifier: NCT03584516]. https://clinicaltrials.gov/ct2/show/NCT03584516. Accessed August 29, 2019.
- 5. Schroeder MA, Choi J, Staser K, DiPersio JF. The role of Janus kinase signaling in graft‐versus‐host disease and graft versus leukemia. Biol Blood Marrow Transplant. 2018;24(6):1125‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Srinivas N, Barbour AM, Modi D, et al. Pharmacokinetic considerations in the development and selection of the phase 3 formulation of itacitinib for the treatment of graft‐versus‐host disease. Presented at: American College of Clinical Pharmacology Annual Meeting, September 20‐22, 2019; Chicago, IL. [Google Scholar]
- 7. Boer J, Barbour A, Kennedy K, et al. Human absorption, metabolism and elimination of itacitinib in healthy male adult volunteers. Presented at: American College of Clinical Pharmacology Annual Meeting, September 23‐25, 2018; Bethesda, MD. [Google Scholar]
- 8. Barbour AM, Punwani N, Epstein N, et al. Effect of itraconazole or rifampin on itacitinib pharmacokinetics when administered orally in healthy subjects. J Clin Pharmacol. 2019;59(12):1641‐1647. [DOI] [PubMed] [Google Scholar]
- 9.Renal Impairment Guidance Working Group, Center for Drug Evaluation and Research. Draft guidance for industry: pharmacokinetics in patients with impaired renal function—study design, data analysis, and impact on dosing and labelling. Rockville, MD: US Food and Drug Administration; 2010. [Google Scholar]
- 10. Dreisbach AW. The influence of chronic renal failure on drug metabolism and transport. Clin Pharmacol Ther. 2009;86(5):553‐556. [DOI] [PubMed] [Google Scholar]
- 11. Nolin TD, Naud J, Leblond FA, Pichette V. Emerging evidence of the impact of kidney disease on drug metabolism and transport. Clin Pharmacol Ther. 2008;83(6):898‐903. [DOI] [PubMed] [Google Scholar]
- 12. Sun H, Frassetto L, Benet LZ. Effects of renal failure on drug transport and metabolism. Pharmacol Ther. 2006;109(1‐2):1‐11. [DOI] [PubMed] [Google Scholar]
- 13. Cohen EP, Pais P, Moulder JE. Chronic kidney disease after hematopoietic stem cell transplantation. Semin Nephrol. 2010;30(6):627‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hazar V, Gungor O, Guven AG, et al. Renal function after hematopoietic stem cell transplantation in children. Pediatr Blood Cancer. 2009;53(2):197‐202. [DOI] [PubMed] [Google Scholar]
- 15. US Food and Drug Administration . Toxicity Grading scale for healthy adult and adolescent volunteers enrolled in preventive vaccine clinical trials. Rockville, MD: US Food and Drug Administration; 2007. [Google Scholar]
- 16. Srinivas N, Barbour AM, Xun E, Chen X, Yeleswaram S. Pharmacokinetics of itacitinib from a pilot study in patients with acute graft‐versus‐host disease in the presence or absence of organ involvement. Presented at: American Society of Clinical Pharmacology and Therapeutics Annual Meeting, March 13‐16, 2019; Washington, DC. [Google Scholar]
- 17. Dreisbach AW, Lertora JJ. The effect of chronic renal failure on hepatic drug metabolism and drug disposition. Semin Dial. 2003;16(1):45‐50. [DOI] [PubMed] [Google Scholar]
- 18. Elston AC, Bayliss MK, Park GR. Effect of renal failure on drug metabolism by the liver. Br J Anaesth. 1993;71(2):282‐290. [DOI] [PubMed] [Google Scholar]
- 19. Nolin TD, Appiah K, Kendrick SA, Le P, McMonagle E, Himmelfarb J. Hemodialysis acutely improves hepatic CYP3A4 metabolic activity. J Am Soc Nephrol. 2006;17(9):2363‐2367. [DOI] [PubMed] [Google Scholar]
- 20. Campagne O, Mager DE, Tornatore KM. Population pharmacokinetics of tacrolimus in transplant recipients: what did we learn about sources of interindividual variabilities? J Clin Pharmacol. 2019;59(3):309‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information