

Abstract
The crosstalk between endothelial cells and cardiomyocytes has emerged as a requisite for normal cardiac development, but also a key pathogenic player during the onset and progression of cardiac disease. Endothelial cells and cardiomyocytes are in close proximity and communicate through the secretion of paracrine signals, as well as through direct cell‐to‐cell contact. Here, we provide an overview of the endothelial cell–cardiomyocyte interactions controlling heart development and the main processes affecting the heart in normal and pathological conditions, including ischaemia, remodelling and metabolic dysfunction. We also discuss the possible role of these interactions in cardiac regeneration and encourage the further improvement of in vitro models able to reproduce the complex environment of the cardiac tissue, in order to better define the mechanisms by which endothelial cells and cardiomyocytes interact with a final aim of developing novel therapeutic opportunities.
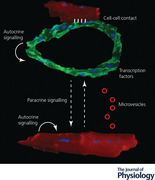
Keywords: Endothelial cell, cardiomyocyte, intercellular cross‐talk
Cardiomyocytes (labelled in red by anti‐α‐actinin antibodies) and endothelial cells (labelled in green by anti‐CD31 antibodies) crosstalk in multiple ways, including paracrine communication (dashed arrows) through either secreted molecules or vesicles (red circles), direct cell–cell contact (hinges) and autocrine signalling (curved arrows). This crosstalk plays an important role during embryonic development, normal post‐natal life and several pathological conditions, thus representing a novel target for the treatment of cardiovascular disorders.
Endothelial cell–cardiomyocyte crosstalk during development
During early embryonic development, oxygen efficiently diffuses from the blood into the cardiac lumen to the thin ventricular walls. As the ventricular mass increases, oxygen diffusion is no longer sufficient to meet the requirement of the developing myocardium, and coronary vessels start to emerge and infiltrate the whole heart (∼E10.5). Recent evidence has identified three possible origins for coronary endothelial cells (ECs), the proepicardium, the endocardium and the sinus venosus, although the relative contribution of each source still remains a matter of debate (Red‐Horse et al. 2010; Wu et al. 2012; Chen et al. 2014). Whatever their origin, the ongoing molecular crosstalk between ECs and cardiomyocytes (CMs) in the developing myocardium is exciting in light of its possible relevance for cardiac regeneration, as the ultimate goal of cardiac regeneration is to rebuild a myocardial architecture with a fully integrated vascular network. Indeed, ECs contribute to the physiological and structural maturation of CMs, as shown by studies in engineered cardiac tissue that recapitulate heart development (described later). In this section, we will discuss the communications between CMs and ECs during embryonic development that might also be relevant for heart regeneration.
Cell‐specific gene inactivation strategies have revealed multiple bidirectional signals occurring in the developing heart (Fig. 1).
Figure 1.
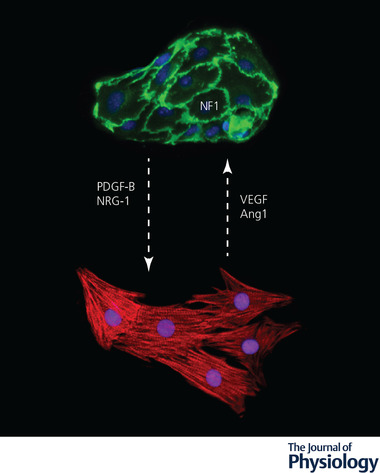
Endothelial cell–cardiomyocyte crosstalk during development
Secreted molecules and transcription factors mediate a bidirectional signalling between developing endothelial cells (labelled by anti‐CD31 antibodies in green) and cardiomyocytes (labelled by anti‐α‐actinin antibodies in red). The dashed arrows indicate the direction of the signals. Nuclei are stained in blue by 4′,6‐diamidino‐2‐phenylindole (DAPI). Ang1, angiopoietin‐1; NF1, neurofibromin‐1; NRG‐1, neuregulin‐1; PDGF, platelet‐derived growth factor; VEGF, vascular endothelial growth factor.
On the one hand, CMs secrete a few growth factors able to modulate the growth of blood vessels. Vascular endothelial growth factor (VEGF)‐A, herein referred to as VEGF, is a key controller of angiogenesis. The deletion of the gene encoding this factor, specifically in CMs, determines reduced number of coronary microvessels, thinned ventricular walls, depressed basal contractile function, induction of hypoxia‐responsive genes involved in energy metabolism and abnormal response to β‐adrenergic stimulation (Giordano et al. 2001). Despite the fact that CMs constitute less than one‐third of the total number of cells in the heart, the CM‐specific deletion of the VEGF gene decreases total mRNA levels by more than 85%, pointing to cardiac myocytes as the major source of this growth factor (Giordano et al. 2001). Of note, the structure of the coronary arteries is not affected in this condition, indicating that different mechanisms support the development of both the cardiac microvasculature and the major epicardial coronary arteries. Beside stimulating angiogenesis, CM‐derived VEGF also controls endocardial‐to‐mesenchymal transformation during the formation of cardiac cushions (i.e. the endocardium‐derived structures that build the heart septa and valves). This requires endocardial calcineurin/nuclear factor of activated T cells (NFAT) signalling to repress VEGF expression in the myocardium underlying the site of prospective valve formation (∼E9). This repression of VEGF is essential for endocardial cells to transform into mesenchymal cells (Chang et al. 2004). Soon after the onset of endocardial cushion formation, VEGF is specifically upregulated in the atrioventricular (AV) field of the heart tube, where it negatively regulates endocardial‐to‐mesenchymal transformation and terminates the formation of endocardial cushions (Dor et al. 2001). CM‐derived Angiopoetin‐1 (Ang1) also plays a major role in cardiac vessel development. Transgenic mice expressing Ang1 in CMs in an inducible manner die at ∼E12.5 because of major cardiac malformations, including dilated atria, significant thinning of the myocardial wall, outflow tract collapse and absence of coronary arteries (Ward et al. 2004).
On the other hand, developing ECs send multiple signals that contribute to myocardial development. For instance, EC‐specific deletion of the platelet‐derived growth factor (PDGF)‐B gene causes major cardiac abnormalities, including myocardial wall thinning, chamber dilation, septal defects and hypertrabeculation (Bjarnegard et al. 2004). Similar developmental defects are observed in mice with endothelium‐restricted deletion of the tumour‐suppressor gene neurofibromin (NF1), which results in dilated endocardial cushions, septal abnormalities and double‐outlet right ventricle (Gitler et al. 2003). Of note, these defects also occur in NF1 knock‐out mice, but not in mice missing NF1 in CMs, pointing to ECs as the most relevant source of NF1 for proper cardiac development (Gitler et al. 2003). Yet, the pathway linking NF1 expression in ECs to proper CM organization and cardiac tissue formation during development is largely unknown.
Finally, perhaps the most characterized pathway of EC–CM crosstalk during development is neuregulin (NRG) signalling. ECs in the developing endocardium secrete abundant levels of NRG‐1, which binds the ErbB2 and ErbB4 receptors expressed by CMs, essentially contributing to the formation of both endocardial cushions and myocardial trabeculae (Meyer & Birchmeier, 1995). This essential role of NRG‐1 in promoting trabeculation is consistent with its capacity to induce the proliferation, survival and hypertrophy of neonatal cardiomyocytes (Zhao et al. 1998).
Endothelial cell–cardiomyocyte crosstalk during normal post‐natal life
In the adult heart, capillaries are densely distributed throughout the myocardium, with the number of ECs being approximately 3 times higher than that of CMs. Yet, the large CM mass exceeds that of the smaller ECs by 25 times (Brutsaert, 2003). While ECs within the coronary arteries functions as in any other arterial vascular system in the body, essentially providing blood supply to the myocardium, capillary and endocardial ECs are in close proximity to CMs and provide multiple autocrine and paracrine signals controlling cardiac function (Fig. 2).
Figure 2.
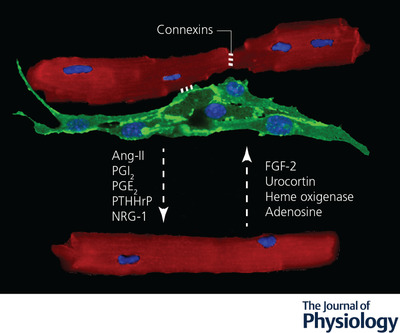
Endothelial cell–cardiomyocyte crosstalk during post‐natal life
Secreted molecules and gap junctions mediate a bidirectional signalling between adult endothelial cells (labelled by anti‐CD31 antibodies in green) and cardiomyocytes (labelled by anti‐α‐actinin antibodies in red). Nuclei are stained in blue by DAPI. The dashed arrows indicate the direction of the signals. Molecules secreted by endothelial cells are indicated on the left, those secreted by cardiomyocytes on the right, while those secreted by both cell types are in the middle. Ang‐II, angiotensin‐II; ET‐1, endothelin‐1; FGF2, fibroblast growth factor 2; NO, nitric oxide; NRG‐1, neuregulin‐1; PGE2, prostaglandin E2; PGI2, prostacyclin; PTHrP, parathyroid hormone‐related peptide.
In the normal adult heart, ECs release various factors controlling CM contractility. A first player is nitric oxide (NO), which is synthetized by the NO synthase class of enzymes. Three isoforms of NO synthase exist, including the neuronal (NOS1), the inducible (NOS2) and the endothelial (NOS3) isoforms. NOS1 and NOS3 are constitutively active and produce NO in response to calcium, whereas NOS2 is inducible and increases its production of NO upon adequate stimulation (i.e. cytokines) and independently from calcium activation. In the adult heart, NO is mainly produced by ECs and, consistent with its activity on vascular smooth muscle cells, controls the onset of ventricular relaxation (positive lusitropy), thus optimizing pump function at every cardiac contraction (Hsieh et al. 2006b). This effect has been attributed to cGMP‐ and protein kinase G (PKG)‐mediated phosphorylation of troponin I, leading to myofilament calcium desensitization, relaxation hastening and improved distensibility (Layland et al. 2002). In contrast, the inotropic effect exerted by low concentrations of NO relies on mechanisms independent of cGMP and possibly involving the activation of calcium and potassium channels through either G protein‐mediated response (Layland et al. 2002), formation of peroxynitrite (Malan et al. 2003) or direct channel S‐nitrosylation by NO (Kohr et al. 2011). Generation of peroxynitrite is maximal in conditions of oxidative stress, when high levels of superoxide anion react with NO. At the same time, ECs are able to protect CM from oxidative stress by modulating several buffering systems at different levels, such as ATP‐binding cassette transporter subfamily G member 2 (ABCG2) for the secretion of glutathione (Higashikuni et al. 2012), NRG‐1 (Kuramochi et al. 2004) (see also below), tetrahydrobiopterin (BH4) (Leucker et al. 2013) and thioredoxin (Trx) (Subramani et al. 2016). While most of these buffering systems have been discovered in conditions of cardiac stress (i.e. ischaemia–reperfusion injury and pressure overload), it is plausible that they play a major role in maintaining cardiac homeostasis.
Besides ECs, CMs also express the different endothelial NO synthases, although at lower levels compared to ECs. This supports a possible autocrine NO signalling in CMs, warranting further studies using transgenic animals with EC‐ and CM‐specific deletion of NO signalling.
Cardiac ECs also produce abundant levels of endothelin‐1 (ET‐1), a potent vasoactive peptide. In the adult heart, ET‐1 can bind either ETB receptors expressed by ECs or ETA receptors expressed by smooth muscle cells and CMs. The activation of ETB receptors stimulates the release of nitric oxide and prostanoids, also preventing apoptosis (Niwa et al. 2000). In contrast, stimulated ETA receptors induce cell contraction by increasing intracellular calcium in both smooth muscle cells and CMs (Rich & McLaughlin, 2003). Yet, CM‐specific ETA receptor knockout mice have normal cardiac function and physiologically respond to either angiotensin II (Ang‐II) or isoproterenol, indicating a minor role of this pathway in the homeostasis of the adult heart. While the ETB receptor is traditionally considered to be mainly expressed by ECs and its mRNA was not detected in human CMs (Modesti et al. 1999), recent evidence indicates that CM‐specific ETB receptor knockout mice are particularly resistant to extreme hypoxia, consistent with previous reports showing the presence of ETB on CM nuclear membrane and its role controlling nuclear calcium signalling in an intracrine manner (Merlen et al. 2013; Stobdan et al. 2018).
EC Ang‐II, prostacyclin (PGI2) and prostaglandin (PG)E2 additionally contribute to cardiac contraction control in physiological conditions, with different responses observed dependent on dose, species and experimental condition (in vivo versus isolated hearts/CMs) (Brutsaert, 2003).
Beside producing small molecules and peptides, ECs also secrete numerous proteins, including growth factors and matricellular proteins, able to modulate CM structure and function (Frangogiannis, 2012). For instance, cardiac ECs express and secrete parathyroid hormone‐related peptide (PTHrP), which exerts positive inotropic, chronotropic and lusitropic effects in adult ventricular CMs (Jansen et al. 2003). Over the last decades, numerous publications have described the cardioprotective role of a series of factors endogenously produced in the heart, including periostin, tenascin‐C, thrombospondins, frizzled‐related protein 3, insulin‐like growth factor 1, connective tissue growth factor, Dickkopf‐3, bone morphogenetic protein‐2 and ‐4, interleukin‐1β and ‐6, placental growth factor (PLGF), leukemia inhibitory factor, Wnt1‐induced secreted protein, midkine and adrenomedullin, without investigating their cellular source. Considering the abundance and location of cardiac ECs, it is reasonable that these cells represent an important source of most of these cardioprotective proteins, able to modulate cardiac contractility and CM remodelling under stress conditions (Segers et al. 2018).
In addition to its major role during development, NRG‐1 is also essential for normal function of the adult heart, where it continues to be expressed by microvascular ECs, but not by those laying large vessels. In accordance, both the ErbB2 and ErbB4 receptors are expressed by adult CMs. The first evidence for a role of the NRG‐1 signalling pathway in cardiac homeostasis came from clinical studies in patients with metastatic mammary carcinoma treated with an anthracycline in combination with trastuzumab (Herceptin), a monoclonal antibody against ErbB2. This treatment is associated with a particularly high risk of cardiotoxic cardiomyopathy and heart failure (Slamon et al. 2001), consistent with the experimental observation that postnatal disruption of NRG‐1/ErbB signalling by gene targeting in mice leads to dilated cardiomyopathy (Crone et al. 2002).
Much less is known about the signals produced by CMs and able to regulate EC biology. Besides NO, additional CM‐secreted molecules, including ET1, fibroblast growth factor (FGF)‐2, urocortin, haem oxygenase and adenosine, are known to control the vascular tone, although the mechanism of their action and in particular whether they mainly target ECs, smooth muscle cells or both, remain not fully understood (Tirziu et al. 2010). Recent data indicate that the release of pro‐angiogenic molecules by CMs in the adult heart is constitutively inhibited by a mechanism requiring the presence of the transcription factor RBPJ (recombination signal binding protein for immunoglobulin κ J region) and inhibition of hypoxia‐inducible factors (HIFs), but independent from Notch1. Deletion of rbpj in cardiomyocytes results in VEGF up‐regulation, increased capillary density and cardioprotection from hypoxia (Diaz‐Trelles et al. 2016). Thus, CMs can produce both pro‐angiogenic and angiostatic factors, the activity of which has to be finely tuned in normal and stress conditions.
In addition to soluble factors, intercellular communications can occur via extracellular vesicles (Colombo et al. 2014). These vesicles shuttle a wide range of functional lipids, proteins, mRNAs, microRNAs and long non‐coding RNAs. In particular, the relevance of exosomal transfer in the communication between CMs and ECs has so far been studied in vitro and in response to pathological stimuli, such as diabetes or ischaemia, and is therefore discussed in the next sections.
Although largely unexplored, direct cell–cell contact might occur between ECs and CMs. Both cell types express connexins, proteins that form gap junctions in the plasma membrane, allowing the movement of ions, second messengers and metabolites. The most abundant connexins (Cx) in the heart are Cx43, Cx40 and Cx45, which are variably expressed by CMs throughout the different cardiac regions and are responsible for their coupling during normal cardiac rhythm. ECs also express various connexins, including Cx37, Cx40 and Cx43. The first evidence for the existence of CM–EC gap junctions was provided in 2004 by an in vitro study showing that Cx43 junctions were associated to improved CM survival and structural organization (Narmoneva et al. 2004). Whilst the functional relevance of this heterocellular gap junction has never been documented in vivo, impaired EC–CM coupling has been proposed as one of the mechanisms leading to arrhythmia and heart failure in patients with hyperhomocysteinaemia. In these patients, the elevated levels of homocysteine determine Cx43 degradation and disruption of the gap junctions, together with peri‐vascular fibrosis, both of which could reasonably contribute to the high risk for sudden cardiac death that characterizes this condition (Givvimani et al. 2011).
Endothelial cell–cardiomyocyte crosstalk in diabetes and metabolic dysfunction
The adult heart is composed of cells that are markedly heterogeneous in their metabolism. CMs are the most energy and oxygen demanding cells in the heart due to their continuous contraction. In healthy conditions, CMs produce most of their ATP through oxidative phosphorylation (OxPhos), using mainly fatty acids (70%), but also other substrates (glucose, ketones and amino acids) as carbon sources (Kolwicz et al. 2013). ECs instead produce 85% of their ATP anaerobically through glycolysis, limiting fatty acid oxidation (FAO) and OxPhos to favour the biosynthesis of nucleotides and other metabolites required for replication and angiogenesis (Potente & Carmeliet, 2017).
Alterations in cardiac metabolism during metabolic syndrome and diabetes are driving forces leading to heart failure, as underscored by the 50–80% of deaths for cardiovascular diseases in patients with diabetes (http://www.who.int/healthinfo/global_burden_disease/estimates/en/). Although most of these cases are due to atherosclerosis, ‘diabetic cardiomyopathy’, i.e. myocardial dysfunction in the absence of general cardiovascular risk factors, is emerging as a significant cause of heart failure in diabetic patients.
In type 2 diabetes, which often involves not only hyperglycaemia but also increased levels of circulating triglycerides, both CMs and ECs undergo massive changes in their metabolism. In particular CMs reduce the expression of GLUT4, resulting in decreased glucose consumption, together with increased fatty acid uptake and oxidation (An & Rodrigues, 2006) through the activation of either AMP‐activated protein kinase (acute diabetes) or peroxisome proliferator‐activated receptor α (chronic diabetes) (Finck et al. 2002; Kewalramani et al. 2007). This leads to excessive production of ATP, with consequent down‐regulation of FAO‐associated genes and accumulation of lipids, leading to cardiac ‘lipotoxicity’ and diastolic dysfunction. Moreover, the low level of glucose oxidation increases the metabolic flux toward accessory pathways, such as the polyol and hexosamine biosynthetic pathway, leading to accumulation of advanced end glycation products (AGE) and increased protein O‐GlcNAcylation (Fulop et al. 2007).
On the other hand, ECs exposed to hyperglycaemia experience massive oxidative stress, due to their incapacity to metabolize the excess of glucose in the blood (Potente & Carmeliet, 2017). In response to oxidative stress, polyADP‐ribose polymerase 1 inactivates glyceraldehyde phosphate dehydrogenase, diverting the glucose flux from glycolysis to accessory pathways and leading to AGE accumulation, similar to what is observed in CMs, in addition to increased superoxide levels (Du et al. 2003). The increased reactive oxygen species (ROS) levels induce eNOS uncoupling and generation of peroxynitrite, leading to decreased NO availability and vasoconstriction (Sasaki et al. 2008). These metabolic shifts lead to an overall increase in oxygen consumption, vascular dysfunction and toxic lipid accumulation sustained by oxygen shortfall and consequent FAO inhibition.
Clarifying the intercellular crosstalk between CMs and ECs in modulating their respective metabolism could suggest innovative approaches to prevent heart failure in diabetic patients (Fig. 3). Most of the signals from CMs to ECs modulate the ‘gatekeeper’ function of the endothelium in controlling the uptake of fatty acids from the bloodstream. Fatty acids circulate inside lipoproteins and have to be hydrolysed to free fatty acids by lipoprotein lipase (LPL), anchored to the luminal side of the EC (Olivecrona & Olivecrona, 2010). In the heart, CMs are the major source of LPL, which is secreted and anchored to the heparan sulfate proteoglycans associated with the myocyte membrane. ECs secrete heparanase to release LPLs, which are internalized through glycosylphosphatidylinositol‐anchored high density lipoprotein binding protein 1 (GPIHBP1) and eventually translocated into the luminal side (Camps et al. 1990). High glucose levels induce the re‐localization of endothelial heparanase from the perinuclear compartment to the plasma membrane and its secretion by coronary ECs, consistent with the increased LPL translocation into the vessel lumen observed in mouse models of diabetes (Wang et al. 2009).
Figure 3.
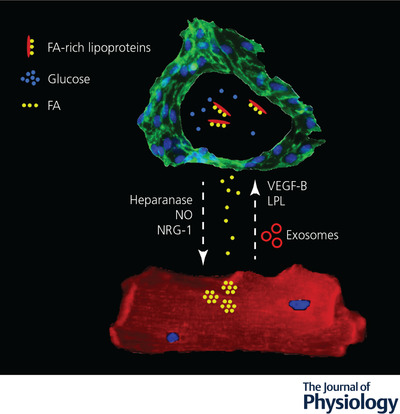
Endothelial cell–cardiomyocyte crosstalk in diabetes and metabolic dysfunction
Secreted molecules and exosomes (red circles) mediate a bidirectional signalling between endothelial cells (labelled by anti‐CD31 antibodies in green) and cardiomyocytes (labelled by anti‐α‐actinin antibodies in red) upon exposure to high levels of glucose (blue dots) and fatty acids (yellow dots). FA, fatty acids; LPL, lipoprotein lipase; NO, nitric oxide; NRG‐1, neuregulin‐1; VEGF‐B, vascular endothelial growth factor‐B.
VEGF‐B is emerging as one of the key regulator of FA uptake in the diseased heart. This factor is abundantly expressed by adult CMs and binds the VEGFR‐1 and neuropilin‐1 (NRP‐1) receptors, expressed by ECs. VEGF‐B−/− mice show decreased expression of fatty acid transporter (FATP)‐3 and ‐4 in ECs and reduced lipid droplets in CMs. Consistently, the exogenous administration of VEGF‐B enhances FA uptake in cultured ECs by biding to VEGFR‐1 and NRP‐1. When fed a high fat diet or crossed with diabetic mouse lines, VEGF‐B−/− mice maintain an increased insulin sensitivity, with improved blood lipid and glucose profiles (Hagberg et al. 2012). However, the CM‐specific overexpression of VEGF‐B does not alter FA uptake and lipid accumulation, but rather leads to a metabolic shift from FAO to glucose oxidation, as well as to an expanded coronary tree through the activation of VEGFR‐2 in cardiac ECs (Kivela et al. 2014). Remarkably, VEGF‐B expression is decreased in different human cardiomyopathies, consistent with its major role in controlling both CM and EC metabolism in both healthy and diseased heart (Bry et al. 2014).
In addition to secreted proteins, exosomes produced by CMs have been shown to regulate the metabolism of ECs, particularly in response to altered glucose levels. Exosomes derived from glucose‐starved CMs increase the proliferation and tube formation capacity of human umbilical vein ECs (HUVECs) and stimulate both glucose uptake and glycolysis in cardiac microvascular ECs in vitro (Garcia et al. 2016). Furthermore, CMs isolated from the Goto–Kakizaki rat, a model of type 2 diabetes, inhibit the angiogenic properties of cardiac ECs through the exosomal transfer of miR‐320 (Wang et al. 2014).
Do ECs regulate CM metabolism? Although very little is known in this respect, one possible player is NRG‐1, which is normally secreted by ECs and upregulated upon ischaemia, as mentioned earlier. This up‐regulation appears inhibited in type 1 diabetes, as diabetic rats fail to overexpress NRG‐1 and exhibit impaired membrane localization of ErbB2 and ErbB4 receptors in CMs after myocardial infarction. Of note, NRG‐1 increases glucose uptake in cultured CMs via ErbB2–ErbB4 heterodimers, phosphoinositide 3‐kinase α and Akt.
Another likely player in the process is NO, which is abundantly secreted by ECs and stimulates glucose uptake by CMs via a cGMP‐dependent mechanism (Tada et al. 2000). Whether this crosstalk is relevant in disease conditions characterized by poor NO availability, including diabetes and hypercholesterolaemia, is still unknown.
Endothelial cell–cardiomyocyte crosstalk during cardiac remodelling
Despite its highly structured architecture, the heart is an organ capable of extensive remodelling. Immediately after birth, the heart faces a pressure overload, which physiologically induces CM hypertrophy, resulting in the gross enlargement of the organ. In adult life, the same happens to the mother's heart during pregnancy as well as to the hearts of athletes as a result of extreme and/or repetitive exercise. Physiological hypertrophy is reversible and considered an adaptive process aimed at decreasing the ventricular wall stress by increasing its thickness. Morphologically, the increase in CM area is accompanied by enhanced angiogenesis in the absence of fibrosis. In contrast, pathological hypertrophy is irreversible and is typically associated with loss of myocytes, fibrotic replacement, decreased capillary density, cardiac dysfunction, and increased risk of heart failure and sudden death. It usually represents the evolution of a first phase of compensatory hypertrophy, sustained by a prolonged wall stress and caused by various disorders, including systemic or pulmonary hypertension, ischaemic, genetic or metabolic cardiomyopathy.
As physiological hypertrophy normally occurs during the first weeks after birth supporting the post‐natal growth of the heart, it is not surprising that the same molecules mediating EC–CM crosstalk during development also play a major role in this type of cardiac remodelling (Fig. 4). On the one hand, CMs undergoing a first phase of compensatory hypertrophy produce pro‐angiogenic factors (i.e. VEGF) (Zheng et al. 2001; Zentilin et al. 2010) able to stimulate blood vessel growth to supply enough nutrients to the growing myocardial mass. On the other hand, enhanced coronary angiogenesis per se, as can be achieved by cardiac‐specific overexpression of angiogenic factors, leads to cardiac hypertrophy in the absence of any additional pro‐hypertrophic stimulation (Tirziu et al. 2007). The precise mechanism by which increased angiogenesis induces CM hypertrophy still needs to be fully elucidated, but it reasonably involves the secretion of EC‐derived pro‐hypertrophic factors.
Figure 4.
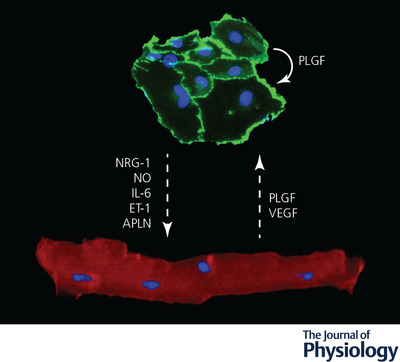
Endothelial cell–cardiomyocyte crosstalk during cardiac remodelling
Secreted molecules mediate a bidirectional signalling between adult endothelial cells (labelled by anti‐CD31 antibodies in green) and cardiomyocytes (labelled by anti‐α‐actinin antibodies in red) during pathological remodelling. Nuclei are stained in blue by DAPI. The dashed arrows indicate the direction of the signals. Molecules acting in autocrine manner are indicated by curved arrows. APLN, apelin; ET‐1, endothelin‐1; IL‐6, interleukin‐6; NO, nitric oxide; NRG‐1, neuregulin‐1; PLGF, placental growth factor; VEGF, vascular endothelial growth factor.
In pathological conditions, ECs control and promote cardiac remodelling by the production of secreted factors with both paracrine and autocrine effects. To limit the degree of complexity, here we keep the focus on signals secreted by cardiac ECs exerting a direct effect on CMs (Fig. 4).
Besides its role in controlling cardiac homeostasis, as discussed above, NO blunts the hypertrophic response of cultured CMs exposed to potent pro‐hypertrophic agents, such as noradrenaline and Ang‐II. Accordingly, NOS3−/− mice spontaneously develop cardiac hypertrophy and exhibit exaggerated hypertrophy after both myocardial infarction and chronic pressure overload compared to wild‐type mice (Scherrer‐Crosbie et al. 2001).
As mentioned before, NO stimulates the production of cGMP by activating soluble guanylyl cyclase. Increased levels of cGMP activate PKG, which is a negative regulator of the pro‐hypertrophic transcriptional regulator NFAT (Fiedler et al. 2002). This axis is emerging as a key player in the development of HF with preserved ejection fraction (HFpEF), a syndromic condition characterized by the impairment of proper left ventricular filling during diastole due to increased wall stiffness. In the context of HFpEF, various comorbidities, especially obesity, elicit a systemic pro‐inflammatory state, which is sensed by ECs that respond by producing ROS, which in turn limit NO availability, as mentioned before. Limited NO availability results in low cGMP levels and impaired PKG activity, leading to hypo‐phosphorylation of titin (Fiedler et al. 2002) and subsequent increase in CM stiffness.
Release of inflammatory cytokines, including tumour necrosis factor‐α, interleukin (IL)‐1, IL‐6 and IL‐11, represent an interesting example of multicellular crosstalk during pathological cardiac remodelling. These molecules can be produced by immune cells but also by other cell types including ECs. Besides the aforementioned effect of inflammatory mediators on NO availability, some of these cytokines play a more prominent role during pathological cardiac remodelling. For instance, IL‐6 is emerging as a pro‐hypertrophic and pro‐fibrotic cytokine, as IL‐6 knockout mice undergo attenuated cardiac remodelling in response to both pressure overload (Zhao et al. 2016) and administration of Ang‐II, a potent mediator of CM hypertrophy (Gonzalez et al. 2015). While the relevant cellular source of IL‐6 exerting these effects is still undefined, interesting insights into the molecular mechanisms involved have been shed by studying the Noonan syndrome, which is determined by mutations in the Ras–extracellular signal‐regulated kinase (ERK) pathway. Patients in which the disease is caused by kinase‐activating RAF1 alleles typically develop pathological left ventricular hypertrophy, which is well reproduced in knock‐in mice harbouring the Raf1(L613V/+) mutation. Interestingly, the EC‐specific expression of the mutant protein is sufficient to drive cardiac hypertrophy and the secretion of IL‐6 by Raf1(L613V/+)‐expressing ECs is essential in promoting CM hypertrophy, clearly pointing to this interleukin as a key mediator of the EC–CM crosstalk during cardiac remodelling (Yin et al. 2017).
As mentioned earlier, ET‐1 is a potent vasoconstrictive peptide secreted by ECs, acting on ECs and CMs, which both express ET receptors. Evidence that it may act as a paracrine regulator of cardiac hypertrophy first came from studies on cultured neonatal rat ventricular CMs, which responded to ET‐1 by increasing protein synthesis and cell size, as well as by reactivating the fetal gene programme (Ito et al. 1991). In final stages of pressure overload‐induced cardiac hypertrophy, ET‐1 is upregulated and is likely responsible for the enhanced vasoconstriction and compromised endothelial NO function, which induces oxidative stress and eventually leads to both CM loss and vascular degeneration (Tsai et al. 2017). Yet, the EC‐specific deletion of ET‐1 worsens the degree of hypertrophy and cardiac function in the same animal model, indicating that either excessive or insufficient ET‐1 signalling is detrimental in this context and that adequate levels of ET‐1 are required for a proper cardiac adaptation of the heart to the increased pressure (Heiden et al. 2014). Furthermore, ET‐1 is up‐regulated by ECs in response to Ang‐II through a mechanism involving the receptor‐mediated mobilization of intracellular calcium and activation of protein kinase C (Emori et al. 1991). Finally, ECs increase the production of ET‐1 in response to mechanical stretch, which invariably occurs during pressure overload (Hasdai et al. 1997). Collectively, these data indicate that ECs sense various signals associated to increased blood pressure, i.e. Ang‐II during renovascular hypertension, or more generally mechanical stretch, and respond by releasing ET‐1, which is able to induce CM hypertrophy (van Wamel et al. 2001). Despite these multiple indications of the involvement of the ET‐1 system in the pathogenesis of cardiac dysfunction, results with ET receptor antagonists in congestive heart failure have been disappointing. While some earlier studies have suggested benefit (Luscher et al. 2002), larger studies have been neutral, with indication of possible worsening of the patient's conditions (O'Connor et al. 2003), similar to what has been more recently observed in patients with HFpEF and pulmonary hypertension (Koller et al. 2017).
NRG‐1 secreted by ECs also induces CM hypertrophic growth, consistent with the observation that mice with cardiac specific deletion of the ErbB2 receptor develop dilated cardiomyopathy. Inhibition of the NRG‐1/ErbB signalling during pregnancy results in left ventricle dilation and decreased cardiac output, associated with decreased survival to pregnancy (Lemmens et al. 2011). Thus, EC–CM crosstalk via NRG‐1/ErbB signalling also plays a crucial role during cardiac adaptation to stress. During both pregnancy and pressure overload, levels of PLGF are increased upon production by both CMs and ECs. This induces capillary growth and fibroblast proliferation, which secondarily supports cardiac hypertrophy through intermediate paracrine growth factors, including IL‐6 (Yao et al. 2005; Accornero et al. 2011).
Finally, a major player in the EC–CM crosstalk during cardiac remodelling is Apelin (APLN), a secreted protein binding to the G‐protein coupled receptor APJ. In the heart, APLN is mainly produced by endocardial cells and ECs, while its APJ receptor is expressed by CMs, ECs and smooth muscle cells (Kleinz et al. 2005). Consistent with the broad expression of the receptor, the acute administration of APLN reduces the vasomotor tone with positive chronotropic and inotropic effect, as well as a trend towards an increase in lusitropy (Ashley et al. 2005). While initial biochemical studies suggested a beneficial effect of APLN during cardiac remodelling based on the ability of APJ to activate Gαi, thus activating cardio‐protective pathways (Rockman et al. 2002), genetic models have recently revealed a more complex scenario. While APJ knockout mice are partially protected from pressure‐overload cardiac hypertrophy and transition to heart failure, APLN knockout mice develop a normal hypertrophic response. This apparent discrepancy suggests that the APJ receptor may be activated also by APLN‐independent mechanisms. In fact, the APJ receptor can also act as a mechanosensor, able to transduce pro‐hypertrophic stimuli in a G‐protein‐independent, but β‐arrestin‐dependent manner, upon cell membrane stretch (Scimia et al. 2012). Recently, the generation of both CM‐ and EC‐specific APJ knockout animals has revealed that its role as ligand‐independent stretch sensor controlling cardiac hypertrophy is essentially exerted on CMs, with minimal, if not null, role of ECs in this process (Parikh et al. 2018).
Endothelial cell–cardiomyocyte crosstalk during ischaemia
Cardiac ischaemia is a clinical condition in which the importance of CM‐derived factors influencing the vasculature is crucial. Increasing evidence indicates that a major, if not the only, mechanism by which cell therapy can be beneficial after myocardial infarction is by replenishing cytokines and growth factors that can no longer be produced by the dead CMs and that act on endothelial cells inducing angiogenesis. At the same time, CMs also convey anti‐angiogenic signals to ECs. As mentioned before, RBPJ in CMs antagonizes HIFs and inhibits the secretion of pro‐angiogenic molecules, thereby desensitizing the angiogenic response to hypoxia (Diaz‐Trelles et al. 2016).
Whether ischaemic ECs convey protective or toxic signals (or both) to CMs is controversial. On the one hand, multiple evidence supports the idea that ECs can improve CM survival after ischaemia. The generation of ROS, including H2O2, in response to ischaemia, stimulates the release of NRG‐1 from ECs and the activation of ErbB4 signalling in CMs, with their protection from apoptotic cell death induced by ischaemia–reperfusion (Kuramochi et al. 2004). Accordingly, NRG‐1 heterozygous knockout mice are hypersensitive to doxorubicin‐induced cardiotoxicity (Liu et al. 2005), which is known to be sustained by the generation of ROS, similar to ischaemia–reperfusion injury. On the other hand, under hypoxic conditions cardiac ECs release transforming growth factor β, which in turn promotes endothelial to mesenchymal transition (EndoMT) in an autocrine loop, but also induces apoptosis in CMs (Sniegon et al. 2017). While several studies have reported that EndoMT contributes to the development of cardiac fibrosis after ischaemia or in other pathological settings (Xu et al. 2016), recent progresses in genetic lineage tracing has challenged its role in vivo after cardiac injury (Li et al. 2018), warranting further studies to confirm the extent and function of EndoMT after cardiac ischaemia.
Multiple secreted factors have been implicated in the response to cardiac ischaemia, often exerting pleiotropic functions, including maintenance and expansion of the coronary vasculature, remodelling of the extracellular matrix and protection of the CMs that have survived in the peri‐infarct region. One example is follistatin‐like 1 (Fstl1), which is highly expressed by ischaemic CMs and acts in an autocrine manner to promote myocyte survival by binding to the disco‐interacting protein 2 homolog A (DIP2A) receptor. This receptor was shown to be expressed and induce Akt activation in ECs (Ouchi et al. 2010). However, its actual expression by cardiac ECs and whether it could mediate post‐infarction coronary angiogenesis still remain open questions.
An additional, well documented, cardioprotective factor is Ang1. Besides its role in cardiac development, which was mentioned before, Ang1 promotes CM survival via integrin‐β1‐mediated ERK activation and preserves cardiac function upon ischaemia–reperfusion in mice (Lee et al. 2011). At the same time, Ang1 also acts on cardiac ECs by regulating VE‐cadherin phosphorylation and preventing vascular leakage in different models of cardiac ischaemia (Takahashi et al. 2003).
Finally, PDGF is an important signal mediating EC–CM crosstalk in ischaemic conditions. PDGF is a dimeric glycoprotein that can be composed of two A subunits (PDGF‐AA), two B subunits (PDGF‐BB), or one of each (PDGF‐AB). Primary cardiac microvascular ECs constitutively express PDGF‐A, but not PDGF‐B. Their co‐culture with cardiac myocytes induces the expression of PDGF‐B, resulting in the secretion of PDGF‐AB. PDGF‐AB in turn acts in an autocrine manner to up‐regulate the expression of von Willebrand factor, VEGF and VEGFR‐2, thus enhancing the haemostatic and angiogenic properties of ECs (Edelberg et al. 1998). While the mechanism by which ECs sense the presence of CMs and start producing PDGF‐B are completely unknown, it is interesting to note that aged ECs lose this response. Consistently, treatment of the heart with PDGF‐AB restored the angiogenic capacity of the aged myocardium in a rat model of cardiac ischaemia, limiting the extent of myocardial infarction (Edelberg et al. 2002). PDGF‐BB also activates Akt phosphorylation in CMs in a time‐ and dose‐dependent manner, thus preventing apoptosis after myocardial infarction (Hsieh et al. 2006a).
As already introduced before, intercellular communications can also occur via extracellular vesicles, including exosomes. Over the last few years, several studies have reported that exosomes modulate angiogenesis in various organs, including the heart, and that, in particular, exosomes produced by ischaemic CMs can stimulate cardiac angiogenesis, at least in part through the delivery of pro‐angiogenic miRNAs (i.e. miR‐222 and miR‐143) (Ribeiro‐Rodrigues et al. 2017). An overall view of the EC–CM crosstalk in the ischaemic heart is provided in Fig. 5.
Figure 5.
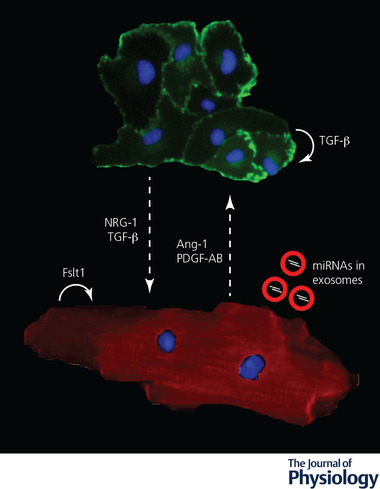
Endothelial cell‐cardiomyocyte crosstalk during ischaemia
Secreted molecules mediate a bidirectional signalling between adult endothelial cells (labelled by anti‐CD31 antibodies in green) and cardiomyocytes (labelled by anti‐α‐actinin antibodies in red) upon ischaemia. Nuclei are stained in blue by DAPI. The dashed arrows indicate the direction of the signals. Molecules secreted by endothelial cells are indicated on the left, while those secreted by both cell types are in the middle. Molecules acting in autocrine manner are indicated by curved arrows. Ang1, angiopoietin‐1; Fstl1, follistatin‐like 1; NRG‐1, neuregulin‐1; PDGF, platelet‐derived growth factor; TGF‐β, transforming growth factor β.
Endothelial cell–cardiomyocyte crosstalk in cardiac regeneration
Different from the adult mammalian heart, which has a very limited regenerative potential and heals any injury through scarring, the heart of some lower vertebrates, such as fish and amphibians, is able to regenerate new myocardium after damage. In both zebrafish and axolotls the regenerative process starts with the formation of a blood clot, followed by CM cell cycle re‐entry and proliferation (Flink, 2002; Poss, 2007). Neonatal rodents can also regenerate their heart, producing new CMs after either the resection of the cardiac apex or the induction of a myocardial infarction, while they lose this regenerative capacity after the first week of life (Porrello et al. 2011; Sadek et al. 2014). Furthermore, anecdotal evidence indicates that the human heart might also possess some regenerative potential during early post‐natal life (Haubner et al. 2016).
Several studies have shown that the epicardium, once properly activated, can give rise to fibroblasts, smooth muscle cells, endothelial cells and, to a lesser extent, also cardiomyocytes, able to colonize the damaged myocardium promoting cardiac repair. The role of the epicardium in cardiac regeneration has been extensively discussed elsewhere (Cao & Poss, 2018; Simões & Riley, 2018) and does not fall into the focus of this review.
While the major focus in cardiac regeneration has been on the formation of new CMs, it is obvious that the regenerated area has to be properly vascularized. This occurs through the proliferation and migration of ECs residing in the vessels of the surrounding cardiac tissue (Marin‐Juez et al. 2016). Less obvious is the observation that in both zebrafish and neonatal mice, ECs invade the damaged area well before CMs start proliferating, indicating that angiogenesis invariably precedes heart regeneration (Marin‐Juez et al. 2016). This also suggests that the new vessels not only provide oxygen and nutrients to proliferating CMs, but likely regulate the regenerative process, similar to what occurs in the liver, in the lung and in the pancreas (Ding et al. 2011, 2014; Kao et al. 2015).
The identification of the molecular mechanisms by which ECs and CMs communicate in these models stands as a crucial step toward the development of innovative and effective strategies promoting cardiac regeneration.
In vitro models to study the crosstalk between endothelial cells and cardiomyocytes
Most of the current knowledge on the molecular mechanisms controlling heart development and disease has been obtained using in vivo models. Yet, animal models are expensive, have a low‐throughput power and do not allow one to dissect the intercellular crosstalk in detail. In vitro models instead can be manipulated to control different variables, are cheaper and suitable for automation for big data analysis. Although the complex architecture of the cardiac muscle and its mechanical function are very difficult to reproduce ‘in a dish’, artificial cardiac tissue has been produced over the years using a variety of approaches, which have been very useful in investigating the EC–CM crosstalk.
Adult CMs isolated from the murine heart maintain a structured sarcomeric organization, mature electrical properties and beat. Unfortunately, these cells are difficult to isolate and maintain in culture, rapidly lose their mature phenotype in the presence of serum and do not easily integrate into three‐dimensional in vitro models. Neonatal murine ventricular CMs, together with CMs derived from either induced pluripotent stem cells (iPSC) or human embryonic stem cells (hESC) are the most common sources of myocytes for in vitro studies, due to the possibility of obtaining large numbers of cells and also of introducing genetic modifications to mimic pathological conditions. Yet, these cells have an immature phenotype, characterized by small size, immature cytoskeletal and sarcomeric organization and abnormal electrical properties.
ECs are easier to isolate, but their organ source and the age of donor are highly variable in the various models used so far. Several studies have relied on the use of cells derived from extra‐cardiac tissues, such as HUVECs or murine ECs derived from the aorta. Alternatively, commonly employed ECs are human or murine cardiac microvasculature ECs, human coronary ECs, iPSC‐ and hESC‐derived ECs.
The EC–CM in vitro crosstalk has been analysed using either classical co‐culture systems or more sophisticated three‐dimensional engineered tissues. The easiest set‐up is the two‐dimensional co‐culture of CMs and ECs. This is an easy and inexpensive approach, only requiring a medium that allows preservation of both cell types and is the only platform suitable to study the crosstalk between adult CMs and other cells. More specifically, the existence of soluble signals acting in a paracrine manner between ECs and CMs has been defined using transwell systems, using both neonatal (Zhang et al. 2015) and adult (Chiu et al. 2016) cardiac cells.
Since two‐dimensional cultures fail to recapitulate many features of the native tissue, including multiple cell–cell interaction, cell shape and the stiffness of the substrate used for cell adhesion (Baker & Chen, 2012), multiple approaches of three‐dimensional tissue engineering have been developed in an attempt to create functional cardiac tissues or organoids.
Different scaffolds have been considered, composed of a variety of bio‐materials, including chitosan (Hussain et al. 2013), hydrogels (Narmoneva et al. 2004) and porous sponges of poly‐l‐lactic and polylactic‐glycolic acid (Caspi et al. 2007), on which CMs and ECs can be seeded or encapsulated at different ratios. Alternatively, CMs can be seeded on a pre‐formed vascular network (Narmoneva et al. 2004). Two‐ and three‐dimensional miniaturized chips have also been developed for cell micro‐patterning and anisotropic cultures to achieve controlled cell–cell interaction and proper myofibril orientation. However, these systems are still poorly exploited for the study of EC–CM crosstalk (Kofron & Mende, 2017). A promising approach stems from the cell sheet engineering technology, which allows generation of dense and multi‐stratified tissue patches suitable for both drug screening and tissue transplant studies. ECs co‐cultured in CM sheets are able to spontaneously create a microvasculature network within the cardiac tissue and to connect with the capillaries of the host myocardium upon transplantation (Sekine et al. 2008; Stevens et al. 2009).
Beside scaffold‐based cultures or controlled cell layering, many three‐dimensional systems exploit the capacity of cells to self‐aggregate forming cardiac organoids. For instance, embryoid bodies seeded on an EC feeder layer show enhanced differentiation towards the CM phenotype, with faster and synchronized beating (Pasquier et al. 2017). Stem cell‐derived CMs self‐organize into a spherical cardiac microtissue when cultured in either hanging drops or spherical‐bottom low adhesion plates (Fennema et al. 2013). These CM organoids display enhanced contractility and functional maturation when primary coronary ECs are incorporated into the microtissue. Interestingly, the same effect is not achieved if ECs have extra‐cardiac origin, suggesting that organotypic signals are required to promote hESC‐derived CM maturation (Ravenscroft et al. 2016). Instead of mixing the two cell types, hESCs could be induced to differentiate toward a cardiac mesodermal fate, in order to generate simultaneously both ECs and CMs (Giacomelli et al. 2017). Overall, these models support a major role for cardiac ECs in promoting CM structural and functional maturation.
To further reproduce cardiac microenvironment cues, several studies have introduced cyclic stretch or electrical stimuli on both two‐ and three‐dimensional co‐culture systems to mimic the rhythmic beating of the heart. Mechanical stretch induces several changes in in vitro cardiac tissues, such as hypertrophy, metabolic shift, increased contractility and stem cell differentiation (Tulloch et al. 2011). However, a standardized method for cyclic stretch conditioning of engineered cardiac tissue has not been achieved yet, due to the variability of technological devices used to apply mechanical stretch and the heterogeneity of culture systems.
Despite the recent optimization of media and biomaterials used to recreate the complex cardiac three‐dimensional architecture in a dish, the models currently available are very heterogeneous, difficult to compare and do not provide a standard for the effective identification and validation of the mechanisms and molecules involved in the EC–CM crosstalk.
Conclusions
While revising and discussing the mechanisms by which ECs and CMs communicate during heart development and disease, it emerged that a substantial amount of information is available on the signals generated by ECs and able to influence CM structure and function. Yet, the way CMs can modulate EC biology in the heart still remains poorly investigated. While it is known that pro‐hypertrophic stimuli increase myocardial vascularization through the secretion of angiogenic factors by CMs, the exact mechanisms coordinating the workload with the sprouting of new vessels in the heart still remain elusive. The generation of CM‐specific knockout animals has provided conflicting results, indicating that these cells are able to send both stimulatory and inhibitory signals to ECs and that these signals are probably integrated within a complex scenario. Multiple cell types, hormones, metabolites and growth factors contribute to the regulation of survival, function and regeneration of cardiac vessels in both normal and pathological conditions. This complexity could explain, at least in part, the reasons why most of the clinical trials aimed at inducing therapeutic angiogenesis in the heart have failed so far, since in all cases these trials have been based on the delivery of a single factor (van der Laan et al. 2009; Mitsos et al. 2012).
In addition, scanty information is available on the relevance of the EC–CM crosstalk during a series of pathological conditions that often affect the heart, including the onset and perpetuation of arrhythmias, genetic cardiomyopathies, valve disease and cardiac transplantation.
Most of our current knowledge concerns the role of proteins secreted by either ECs or CMs and capable of acting on either or both cell types in a paracrine or autocrine fashion. However, it is becoming clear that ECs and CMs can communicate through alternative mechanisms, including the establishment of gap junctions and direct cell‐to‐cell contact, as well as the secretion of vesicles. In addition, ECs act as mechanosensors, coupling myocardial contraction to haemodynamic changes. For instance, TRPC channels, which are diffusely present in both atrial and ventricular CMs (Seo et al. 2014), are also expressed by endocardial ECs and increase the amplitude of calcium transients in normal CMs. Notably, this positive inotropic effect is lost in response to acute stretch. Whether these types of inter‐cellular connections play a major role during normal heart development and/or disease progression still remains largely unexplored.
Furthermore, for many molecules able to impact on either EC or CM function, the exact cellular source and the stimuli leading to their production are not yet defined. While this review specifically focuses on the direct crosstalk between ECs and CMs, many of these paracrine signals also exert pleiotropic actions on other cell types residing in the heart, such as fibroblasts, endocardial and pericardial cells, resident macrophages, and circulating immune cells. At the same time, other cells in the heart, as well as other organs, produce the same molecules, and their levels are importantly modified by several physiological and pathological conditions. For instance, ageing is known to be associated with both EC and CM dysfunction, resulting in heart hypertrophy and stiffness. During ageing, expression of both APLN and its APJ receptor are decreased and APLN infusion in 15‐month‐old mice reduces cardiac hypertrophy (Rai et al. 2017). On the other hand, ET‐1 increases in aged hearts, although its pathogenic role in determining cardiac stiffness seems to be related to its effect on cardiac fibroblasts rather than CMs (Wang et al. 2015). Additional complexity stems from the fact the heart is at the centre of a complex organism, in which multiple tissues actively produce large amounts of soluble molecules that flows through the cardiac pump and thus can further modulate cardiac EC and CM function, possibly influencing their crosstalk. For example, the thyroid hormones l‐thyroxine (T4) and 3,5,3′‐triiodo‐l‐thyronine (T3) are well known inducers of CM hypertrophy and, at the same time, exert a pro‐angiogenic function through direct binding to integrin αvβ3 on the EC surface, also modulating VEGF and FGF signalling (Luidens et al. 2010). Furthermore, LPL activity on the EC luminal surface is modulated by angiopoietin‐like proteins that can be produced by the heart but also by the liver (Kersten, 2014). Finally, both Ang‐II and ET‐1 are produced through a complex multi‐organ cascade, relying on angiotensinogen secretion by the liver, its conversion to Ang‐I by renin produced by the kidney and finally conversion of Ang‐I into Ang‐II by angiotensin converting enzyme, mainly produced by lung ECs (Boehm & Nabel, 2002). Further investigation is required to understand to what extent these signals are interconnected and whether they result in either synergic or antagonistic effects.
The establishment of more complex in vitro models, integrating multiple cell types in a three‐dimensional structure, and their miniaturization to render them suitable to screening approaches, rather than investigating the effect of a single molecule on a single cell type, will likely help shed light on several of these outstanding questions.
Finally, how this information could be exploited to interfere with the onset and pathogenesis of cardiac disease is a matter of active research, which might lead to innovative therapeutic approaches in the near future.
Additional information
Competing interests
The authors have no competing interests to declare.
Author contributions
All authors have contributed to the conception, drafting and revising of the work. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship and all those who qualify for authorship are listed.
Funding
This work was supported by a grant AIRC IG 2016 19032 to S.Z.; European Research Council (ERC) Advanced Grant 787971, Leducq Foundation Transatlantic Network of Excellence Grant 14CVD04 and Swiss National Science Foundation Grant ‘Sinergia’ to M.G.
Biography
Serena Zacchigna and Mauro Giacca serve as Professors of Molecular Biology at the University of Trieste, Italy and Principle Investigators of the Cardiovascular Biology and Molecular Medicine laboratories of the International Centre for Genetic Engineering and Biotechnology (ICGEB) in Trieste, Italy, respectively. Mauro Giacca also serves as the Director‐General of the ICGEB. Andrea Colliva and Luca Braga are post‐docs in the same laboratories. The main research interests of Serena Zacchigna focus around the mechanisms leading to new blood vessel formation in ischaemic muscles and hearts, whereas Mauro Giacca is a leader in the field of cardiac regeneration, having particular interest in understanding why mammalian cardiomyocytes permanently exit from the cell cycle at birth and how this can be overcome therapeutically. The two laboratories have been extensively collaborating over the last few years to explore the intercellular crosstalk between cardiomyocytes and cardiac endothelial cells.

Edited by: Don Bers & Bjorn Knollmann
A. Colliva and L. Braga contributed equally to this article.
This review was presented at the symposium ‘2018 Gordon Research Conference on Cardiac Regulatory Mechanisms’, which took place at Colby‐Sawyer College, New London, NH, USA, 3–8 June 2018.
This is an Editor's Choice article from the 15 July 2020 issue.
References
- Accornero F, van Berlo JH, Benard MJ, Lorenz JN, Carmeliet P & Molkentin JD (2011). Placental growth factor regulates cardiac adaptation and hypertrophy through a paracrine mechanism. Circ Res 109, 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An D & Rodrigues B (2006). Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol 291, H1489–H1506. [DOI] [PubMed] [Google Scholar]
- Ashley EA, Powers J, Chen M, Kundu R, Finsterbach T, Caffarelli A, Deng A, Eichhorn J, Mahajan R, Agrawal R, Greve J, Robbins R, Patterson AJ, Bernstein D & Quertermous T (2005). The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc Res 65, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker BM & Chen CS (2012). Deconstructing the third dimension: how 3D culture microenvironments alter cellular cues. J Cell Sci 125, 3015–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjarnegard M, Enge M, Norlin J, Gustafsdottir S, Fredriksson S, Abramsson A, Takemoto M, Gustafsson E, Fassler R & Betsholtz C (2004). Endothelium‐specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development 131, 1847–1857. [DOI] [PubMed] [Google Scholar]
- Boehm M & Nabel EG (2002). Angiotensin‐converting enzyme 2—a new cardiac regulator. N Engl J Med 347, 1795–1797. [DOI] [PubMed] [Google Scholar]
- Brutsaert DL (2003). Cardiac endothelial‐myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol Rev 83, 59–115. [DOI] [PubMed] [Google Scholar]
- Bry M, Kivela R, Leppanen VM & Alitalo K (2014). Vascular endothelial growth factor‐B in physiology and disease. Physiol Rev 94, 779–794. [DOI] [PubMed] [Google Scholar]
- Camps L, Reina M, Llobera M, Vilaro S & Olivecrona T (1990). Lipoprotein lipase: cellular origin and functional distribution. Am J Physiol 258, C673–C681. [DOI] [PubMed] [Google Scholar]
- Cao J & Poss KD (2018). The epicardium as a hub for heart regeneration. Nat Rev Cardiol 15, 631–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi O, Lesman A, Basevitch Y, Gepstein A, Arbel G, Habib IH, Gepstein L & Levenberg S (2007). Tissue engineering of vascularized cardiac muscle from human embryonic stem cells. Circ Res 100, 263–272. [DOI] [PubMed] [Google Scholar]
- Chang CP, Neilson JR, Bayle JH, Gestwicki JE, Kuo A, Stankunas K, Graef IA & Crabtree GR (2004). A field of myocardial‐endocardial NFAT signaling underlies heart valve morphogenesis. Cell 118, 649–663. [DOI] [PubMed] [Google Scholar]
- Chen HI, Sharma B, Akerberg BN, Numi HJ, Kivela R, Saharinen P, Aghajanian H, McKay AS, Bogard PE, Chang AH, Jacobs AH, Epstein JA, Stankunas K, Alitalo K & Red‐Horse K (2014). The sinus venosus contributes to coronary vasculature through VEGFC‐stimulated angiogenesis. Development 141, 4500–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu AP, Wan A, Lal N, Zhang D, Wang F, Vlodavsky I, Hussein B & Rodrigues B (2016). Cardiomyocyte VEGF regulates endothelial cell GPIHBP1 to relocate lipoprotein lipase to the coronary lumen during diabetes mellitus. Arterioscler Thromb Vasc Biol 36, 145–155. [DOI] [PubMed] [Google Scholar]
- Colombo M, Raposo G & Thery C (2014). Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30, 255–289. [DOI] [PubMed] [Google Scholar]
- Crone SA, Zhao YY, Fan L, Gu Y, Minamisawa S, Liu Y, Peterson KL, Chen J, Kahn R, Condorelli G, Ross J Jr, Chien KR & Lee KF (2002). ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat Med 8, 459–465. [DOI] [PubMed] [Google Scholar]
- Diaz‐Trelles R, Scimia MC, Bushway P, Tran D, Monosov A, Monosov E, Peterson K, Rentschler S, Cabrales P, Ruiz‐Lozano P & Mercola M (2016). Notch‐independent RBPJ controls angiogenesis in the adult heart. Nat Commun 7, 12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M, Penfold ME, Shido K, Rabbany SY & Rafii S (2014). Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature 505, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding BS, Nolan DJ, Guo P, Babazadeh AO, Cao Z, Rosenwaks Z, Crystal RG, Simons M, Sato TN, Worgall S, Shido K, Rabbany SY & Rafii S (2011). Endothelial‐derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell 147, 539–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor Y, Camenisch TD, Itin A, Fishman GI, McDonald JA, Carmeliet P & Keshet E (2001). A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development 128, 1531–1538. [DOI] [PubMed] [Google Scholar]
- Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C & Brownlee M (2003). Inhibition of GAPDH activity by poly(ADP‐ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest 112, 1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelberg JM, Aird WC, Wu W, Rayburn H, Mamuya WS, Mercola M & Rosenberg RD (1998). PDGF mediates cardiac microvascular communication. J Clin Invest 102, 837–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelberg JM, Lee SH, Kaur M, Tang L, Feirt NM, McCabe S, Bramwell O, Wong SC & Hong MK (2002). Platelet‐derived growth factor‐AB limits the extent of myocardial infarction in a rat model: feasibility of restoring impaired angiogenic capacity in the aging heart. Circulation 105, 608–613. [DOI] [PubMed] [Google Scholar]
- Emori T, Hirata Y, Ohta K, Kanno K, Eguchi S, Imai T, Shichiri M & Marumo F (1991). Cellular mechanism of endothelin‐1 release by angiotensin and vasopressin. Hypertension 18, 165–170. [DOI] [PubMed] [Google Scholar]
- Fennema E, Rivron N, Rouwkema J, van Blitterswijk C & de Boer J (2013). Spheroid culture as a tool for creating 3D complex tissues. Trends Biotechnol 31, 108–115. [DOI] [PubMed] [Google Scholar]
- Fiedler B, Lohmann SM, Smolenski A, Linnemuller S, Pieske B, Schroder F, Molkentin JD, Drexler H & Wollert KC (2002). Inhibition of calcineurin‐NFAT hypertrophy signaling by cGMP‐dependent protein kinase type I in cardiac myocytes. Proc Natl Acad Sci U S A 99, 11363–11368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD & Kelly DP (2002). The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J Clin Invest 109, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flink IL ( 2002). Cell cycle reentry of ventricular and atrial cardiomyocytes and cells within the epicardium following amputation of the ventricular apex in the axolotl, Amblystoma mexicanum: confocal microscopic immunofluorescent image analysis of bromodeoxyuridine‐labeled nuclei. Anat Embryol (Berl) 205, 235–244. [DOI] [PubMed] [Google Scholar]
- Frangogiannis NG (2012). Matricellular proteins in cardiac adaptation and disease. Physiol Rev 92, 635–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop N, Mason MM, Dutta K, Wang P, Davidoff AJ, Marchase RB & Chatham JC (2007). Impact of Type 2 diabetes and aging on cardiomyocyte function and O‐linked N‐acetylglucosamine levels in the heart. Am J Physiol Cell Physiol 292, C1370‐1378. [DOI] [PubMed] [Google Scholar]
- Garcia NA, Moncayo‐Arlandi J, Sepulveda P & Diez‐Juan A (2016). Cardiomyocyte exosomes regulate glycolytic flux in endothelium by direct transfer of GLUT transporters and glycolytic enzymes. Cardiovasc Res 109, 397–408. [DOI] [PubMed] [Google Scholar]
- Giacomelli E, Bellin M, Orlova VV & Mummery CL (2017). Co‐differentiation of human pluripotent stem cells‐derived cardiomyocytes and endothelial cells from cardiac mesoderm provides a three‐dimensional model of cardiac microtissue. Curr Protoc Hum Genet 95, 21.29.1–21.29.22. [DOI] [PubMed] [Google Scholar]
- Giordano FJ, Gerber HP, Williams SP, VanBruggen N, Bunting S, Ruiz‐Lozano P, Gu Y, Nath AK, Huang Y, Hickey R, Dalton N, Peterson KL, Ross J Jr, Chien KR & Ferrara N (2001). A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci U S A 98, 5780–5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD, Zhu Y, Ismat FA, Lu MM, Yamauchi Y, Parada LF & Epstein JA (2003). Nf1 has an essential role in endothelial cells. Nat Genet 33, 75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givvimani S, Qipshidze N, Tyagi N, Mishra PK, Sen U & Tyagi SC (2011). Synergism between arrhythmia and hyperhomo‐cysteinemia in structural heart disease. Int J Physiol Pathophysiol Pharmacol 3, 107–119. [PMC free article] [PubMed] [Google Scholar]
- Gonzalez GE, Rhaleb NE, D'Ambrosio MA, Nakagawa P, Liu Y, Leung P, Dai X, Yang XP, Peterson EL & Carretero OA (2015). Deletion of interleukin‐6 prevents cardiac inflammation, fibrosis and dysfunction without affecting blood pressure in angiotensin II‐high salt‐induced hypertension. J Hypertens 33, 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg CE, Mehlem A, Falkevall A, Muhl L, Fam BC, Ortsater H, Scotney P, Nyqvist D, Samen E, Lu L, Stone‐Elander S, Proietto J, Andrikopoulos S, Sjoholm A, Nash A & Eriksson U (2012). Targeting VEGF‐B as a novel treatment for insulin resistance and type 2 diabetes. Nature 490, 426–430. [DOI] [PubMed] [Google Scholar]
- Hasdai D, Holmes DR Jr, Garratt KN, Edwards WD & Lerman A (1997). Mechanical pressure and stretch release endothelin‐1 from human atherosclerotic coronary arteries in vivo. Circulation 95, 357–362. [DOI] [PubMed] [Google Scholar]
- Haubner BJ, Schneider J, Schweigmann U, Schuetz T, Dichtl W, Velik‐Salchner C, Stein JI & Penninger JM (2016). Functional recovery of a human neonatal heart after severe myocardial infarction. Circ Res 118, 216–221. [DOI] [PubMed] [Google Scholar]
- Heiden S, Vignon‐Zellweger N, Masuda S, Yagi K, Nakayama K, Yanagisawa M & Emoto N (2014). Vascular endothelium derived endothelin‐1 is required for normal heart function after chronic pressure overload in mice. PLoS One 9, e88730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashikuni Y, Sainz J, Nakamura K, Takaoka M, Enomoto S, Iwata H, Tanaka K, Sahara M, Hirata Y, Nagai R & Sata M (2012). The ATP‐binding cassette transporter ABCG2 protects against pressure overload‐induced cardiac hypertrophy and heart failure by promoting angiogenesis and antioxidant response. Arterioscler Thromb Vasc Biol 32, 654–661. [DOI] [PubMed] [Google Scholar]
- Hsieh PC, Davis ME, Gannon J, MacGillivray C & Lee RT (2006a). Controlled delivery of PDGF‐BB for myocardial protection using injectable self‐assembling peptide nanofibers. J Clin Invest 116, 237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh PC, Davis ME, Lisowski LK & Lee RT (2006b). Endothelial‐cardiomyocyte interactions in cardiac development and repair. Annu Rev Physiol 68, 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain A, Collins G, Yip D & Cho CH (2013). Functional 3‐D cardiac co‐culture model using bioactive chitosan nanofiber scaffolds. Biotechnol Bioeng 110, 637–647. [DOI] [PubMed] [Google Scholar]
- Ito H, Hirata Y, Hiroe M, Tsujino M, Adachi S, Takamoto T, Nitta M, Taniguchi K & Marumo F (1991). Endothelin‐1 induces hypertrophy with enhanced expression of muscle‐specific genes in cultured neonatal rat cardiomyocytes. Circ Res 69, 209–215. [DOI] [PubMed] [Google Scholar]
- Jansen J, Gres P, Umschlag C, Heinzel FR, Degenhardt H, Schluter KD, Heusch G & Schulz R (2003). Parathyroid hormone‐related peptide improves contractile function of stunned myocardium in rats and pigs. Am J Physiol Heart Circ Physiol 284, H49–H55. [DOI] [PubMed] [Google Scholar]
- Kao DI, Lacko LA, Ding BS, Huang C, Phung K, Gu G, Rafii S, Stuhlmann H & Chen S (2015). Endothelial cells control pancreatic cell fate at defined stages through EGFL7 signaling. Stem Cell Rep 4, 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S (2014). Physiological regulation of lipoprotein lipase. Biochim Biophys Acta 1841, 919–933. [DOI] [PubMed] [Google Scholar]
- Kewalramani G, An D, Kim MS, Ghosh S, Qi D, Abrahani A, Pulinilkunnil T, Sharma V, Wambolt RB, Allard MF, Innis SM & Rodrigues B (2007). AMPK control of myocardial fatty acid metabolism fluctuates with the intensity of insulin‐deficient diabetes. J Mol Cell Cardiol 42, 333–342. [DOI] [PubMed] [Google Scholar]
- Kivela R, Bry M, Robciuc MR, Rasanen M, Taavitsainen M, Silvola JM, Saraste A, Hulmi JJ, Anisimov A, Mayranpaa MI, Lindeman JH, Eklund L, Hellberg S, Hlushchuk R, Zhuang ZW, Simons M, Djonov V, Knuuti J, Mervaala E & Alitalo K (2014). VEGF‐B‐induced vascular growth leads to metabolic reprogramming and ischemia resistance in the heart. EMBO Mol Med 6, 307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinz MJ, Skepper JN & Davenport AP (2005). Immunocytochemical localisation of the apelin receptor, APJ, to human cardiomyocytes, vascular smooth muscle and endothelial cells. Regul Pept 126, 233–240. [DOI] [PubMed] [Google Scholar]
- Kofron CM & Mende U (2017). In vitro models of the cardiac microenvironment to study myocyte and non‐myocyte crosstalk: bioinspired approaches beyond the polystyrene dish. J Physiol 595, 3891–3905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohr MJ, Aponte AM, Sun J, Wang G, Murphy E, Gucek M & Steenbergen C (2011). Characterization of potential S‐nitrosylation sites in the myocardium. Am J Physiol Heart Circ Physiol 300, H1327–H1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller B, Steringer‐Mascherbauer R, Ebner CH, Weber T, Ammer M, Eichinger J, Pretsch I, Herold M, Schwaiger J, Ulmer H & Grander W (2017). Pilot study of endothelin receptor blockade in heart failure with diastolic dysfunction and pulmonary hypertension (BADDHY‐Trial). Heart Lung Circ 26, 433–441. [DOI] [PubMed] [Google Scholar]
- Kolwicz SC Jr, Purohit S & Tian R (2013). Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res 113, 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuramochi Y, Cote GM, Guo X, Lebrasseur NK, Cui L, Liao R & Sawyer DB (2004). Cardiac endothelial cells regulate reactive oxygen species‐induced cardiomyocyte apoptosis through neuregulin‐1β/erbB4 signaling. J Biol Chem 279, 51141–51147. [DOI] [PubMed] [Google Scholar]
- Layland J, Li JM & Shah AM (2002). Role of cyclic GMP‐dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol 540, 457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SW, Won JY, Lee HY, Lee HJ, Youn SW, Lee JY, Cho CH, Cho HJ, Oh S, Chae IH & Kim HS (2011). Angiopoietin‐1 protects heart against ischemia/reperfusion injury through VE‐cadherin dephosphorylation and myocardiac integrin‐β1/ERK/caspase‐9 phosphorylation cascade. Mol Med 17, 1095–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmens K, Doggen K & De Keulenaer GW (2011). Activation of the neuregulin/ErbB system during physiological ventricular remodeling in pregnancy. Am J Physiol Heart Circ Physiol 300, H931–H942. [DOI] [PubMed] [Google Scholar]
- Leucker TM, Ge ZD, Procknow J, Liu Y, Shi Y, Bienengraeber M, Warltier DC & Kersten JR (2013). Impairment of endothelial‐myocardial interaction increases the susceptibility of cardiomyocytes to ischemia/reperfusion injury. PLoS One 8, e70088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lui KO & Zhou B (2018). Reassessing endothelial‐to‐mesenchymal transition in cardiovascular diseases. Nat Rev Cardiol 15, 445–456. [DOI] [PubMed] [Google Scholar]
- Liu FF, Stone JR, Schuldt AJ, Okoshi K, Okoshi MP, Nakayama M, Ho KK, Manning WJ, Marchionni MA, Lorell BH, Morgan JP & Yan X (2005). Heterozygous knockout of neuregulin‐1 gene in mice exacerbates doxorubicin‐induced heart failure. Am J Physiol Heart Circ Physiol 289, H660–H666. [DOI] [PubMed] [Google Scholar]
- Luidens MK, Mousa SA, Davis FB, Lin HY & Davis PJ (2010). Thyroid hormone and angiogenesis. Vascul Pharmacol 52, 142–145. [DOI] [PubMed] [Google Scholar]
- Luscher TF, Enseleit F, Pacher R, Mitrovic V, Schulze MR, Willenbrock R, Dietz R, Rousson V, Hurlimann D, Philipp S, Notter T, Noll G, Ruschitzka F & Heart Failure ETRBT (2002). Hemodynamic and neurohumoral effects of selective endothelin A (ETA) receptor blockade in chronic heart failure: the Heart Failure ETA Receptor Blockade Trial (HEAT). Circulation 106, 2666–2672. [DOI] [PubMed] [Google Scholar]
- Malan D, Levi RC, Alloatti G, Marcantoni A, Bedendi I & Gallo MP (2003). Cyclic AMP and cyclic GMP independent stimulation of ventricular calcium current by peroxynitrite donors in guinea pig myocytes. J Cell Physiol 197, 284–296. [DOI] [PubMed] [Google Scholar]
- Marin‐Juez R, Marass M, Gauvrit S, Rossi A, Lai SL, Materna SC, Black BL & Stainier DY (2016). Fast revascularization of the injured area is essential to support zebrafish heart regeneration. Proc Natl Acad Sci U S A 113, 11237–11242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlen C, Farhat N, Luo X, Chatenet D, Tadevosyan A, Villeneuve LR, Gillis MA, Nattel S, Thorin E, Fournier A & Allen BG (2013). Intracrine endothelin signaling evokes IP3‐dependent increases in nucleoplasmic Ca2+ in adult cardiac myocytes. J Mol Cell Cardiol 62, 189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D & Birchmeier C (1995). Multiple essential functions of neuregulin in development. Nature 378, 386–390. [DOI] [PubMed] [Google Scholar]
- Mitsos S, Katsanos K, Koletsis E, Kagadis GC, Anastasiou N, Diamantopoulos A, Karnabatidis D & Dougenis D (2012). Therapeutic angiogenesis for myocardial ischemia revisited: basic biological concepts and focus on latest clinical trials. Angiogenesis 15, 1–22. [DOI] [PubMed] [Google Scholar]
- Modesti PA, Vanni S, Paniccia R, Bandinelli B, Bertolozzi I, Polidori G, Sani G & Neri Serneri GG (1999). Characterization of endothelin‐1 receptor subtypes in isolated human cardiomyocytes. J Cardiovasc Pharmacol 34, 333–339. [DOI] [PubMed] [Google Scholar]
- Narmoneva DA, Vukmirovic R, Davis ME, Kamm RD & Lee RT (2004). Endothelial cells promote cardiac myocyte survival and spatial reorganization: implications for cardiac regeneration. Circulation 110, 962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa Y, Nagata N, Oka M, Toyoshima T, Akiyoshi H, Wada T & Nakaya Y (2000). Production of nitric oxide from endothelial cells by 31‐amino‐acid‐length endothelin‐1, a novel vasoconstrictive product by human chymase. Life Sci 67, 1103–1109. [DOI] [PubMed] [Google Scholar]
- O'Connor CM, Gattis WA, Adams KF Jr, Hasselblad V, Chandler B, Frey A, Kobrin I, Rainisio M, Shah MR, Teerlink J, Gheorghiade M; Randomized Intravenous TeZosentan Study‐4 Investigators (2003). Tezosentan in patients with acute heart failure and acute coronary syndromes: results of the Randomized Intravenous TeZosentan Study (RITZ‐4). J Am Coll Cardiol 41, 1452–1457. [DOI] [PubMed] [Google Scholar]
- Olivecrona G & Olivecrona T (2010). Triglyceride lipases and atherosclerosis. Curr Opin Lipidol 21, 409–415. [DOI] [PubMed] [Google Scholar]
- Ouchi N, Asaumi Y, Ohashi K, Higuchi A, Sono‐Romanelli S, Oshima Y & Walsh K (2010). DIP2A functions as a FSTL1 receptor. J Biol Chem 285, 7127–7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh VN, Liu J, Shang C, Woods C, Chang AC, Zhao M, Charo DN, Grunwald Z, Huang Y, Seo K, Tsao PS, Bernstein D, Ruiz‐Lozano P, Quertermous T & Ashley EA (2018). Apelin and APJ orchestrate complex tissue‐specific control of cardiomyocyte hypertrophy and contractility in the hypertrophy‐heart failure transition. Am J Physiol Heart Circ Physiol 315, H348–H356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier J, Gupta R, Rioult D, Hoarau‐Vechot J, Courjaret R, Machaca K, Al Suwaidi J, Stanley EG, Rafii S, Elliott DA, Abi Khalil C & Rafii A (2017). Coculturing with endothelial cells promotes in vitro maturation and electrical coupling of human embryonic stem cell‐derived cardiomyocytes. J Heart Lung Transplant 36, 684–693. [DOI] [PubMed] [Google Scholar]
- Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN & Sadek HA (2011). Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss KD (2007). Getting to the heart of regeneration in zebrafish. Semin Cell Dev Biol 18, 36–45. [DOI] [PubMed] [Google Scholar]
- Potente M & Carmeliet P (2017). The link between angiogenesis and endothelial metabolism. Annu Rev Physiol 79, 43–66. [DOI] [PubMed] [Google Scholar]
- Rai R, Ghosh AK, Eren M, Mackie AR, Levine DC, Kim SY, Cedernaes J, Ramirez V, Procissi D, Smith LH, Woodruff TK, Bass J & Vaughan DE (2017). Downregulation of the apelinergic axis accelerates aging, whereas its systemic restoration improves the mammalian healthspan. Cell Rep 21, 1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenscroft SM, Pointon A, Williams AW, Cross MJ & Sidaway JE (2016). Cardiac non‐myocyte cells show enhanced pharmacological function suggestive of contractile maturity in stem cell derived cardiomyocyte microtissues. Toxicol Sci 152, 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Red‐Horse K, Ueno H, Weissman IL & Krasnow MA (2010). Coronary arteries form by developmental reprogramming of venous cells. Nature 464, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro‐Rodrigues TM, Laundos TL, Pereira‐Carvalho R, Batista‐Almeida D, Pereira R, Coelho‐Santos V, Silva AP, Fernandes R, Zuzarte M, Enguita FJ, Costa MC, Pinto‐do OP, Pinto MT, Gouveia P, Ferreira L, Mason JC, Pereira P, Kwak BR, Nascimento DS & Girao H (2017). Exosomes secreted by cardiomyocytes subjected to ischaemia promote cardiac angiogenesis. Cardiovasc Res 113, 1338–1350. [DOI] [PubMed] [Google Scholar]
- Rich S & McLaughlin VV (2003). Endothelin receptor blockers in cardiovascular disease. Circulation 108, 2184–2190. [DOI] [PubMed] [Google Scholar]
- Rockman HA, Koch WJ & Lefkowitz RJ (2002). Seven‐transmembrane‐spanning receptors and heart function. Nature 415, 206–212. [DOI] [PubMed] [Google Scholar]
- Sadek HA, Martin JF, Takeuchi JK, Leor J, Nie Y, Giacca M & Lee RT (2014). Multi‐investigator letter on reproducibility of neonatal heart regeneration following apical resection. Stem Cell Rep 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki N, Yamashita T, Takaya T, Shinohara M, Shiraki R, Takeda M, Emoto N, Fukatsu A, Hayashi T, Ikemoto K, Nomura T, Yokoyama M, Hirata K & Kawashima S (2008). Augmentation of vascular remodeling by uncoupled endothelial nitric oxide synthase in a mouse model of diabetes mellitus. Arterioscler Thromb Vasc Biol 28, 1068–1076. [DOI] [PubMed] [Google Scholar]
- Scherrer‐Crosbie M, Ullrich R, Bloch KD, Nakajima H, Nasseri B, Aretz HT, Lindsey ML, Vancon AC, Huang PL, Lee RT, Zapol WM & Picard MH (2001). Endothelial nitric oxide synthase limits left ventricular remodeling after myocardial infarction in mice. Circulation 104, 1286–1291. [DOI] [PubMed] [Google Scholar]
- Scimia MC, Hurtado C, Ray S, Metzler S, Wei K, Wang J, Woods CE, Purcell NH, Catalucci D, Akasaka T, Bueno OF, Vlasuk GP, Kaliman P, Bodmer R, Smith LH, Ashley E, Mercola M, Brown JH & Ruiz‐Lozano P (2012). APJ acts as a dual receptor in cardiac hypertrophy. Nature 488, 394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segers VFM, Brutsaert DL & De Keulenaer GW (2018). Cardiac remodeling: Endothelial cells have more to say than just NO. Front Physiol 9, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine H, Shimizu T, Hobo K, Sekiya S, Yang J, Yamato M, Kurosawa H, Kobayashi E & Okano T (2008). Endothelial cell coculture within tissue‐engineered cardiomyocyte sheets enhances neovascularization and improves cardiac function of ischemic hearts. Circulation 118, S145–S152. [DOI] [PubMed] [Google Scholar]
- Seo K, Rainer PP, Shalkey Hahn V, Lee DI, Jo SH, Andersen A, Liu T, Xu X, Willette RN, Lepore JJ, Marino JP Jr, Birnbaumer L, Schnackenberg CG & Kass DA (2014). Combined TRPC3 and TRPC6 blockade by selective small‐molecule or genetic deletion inhibits pathological cardiac hypertrophy. Proc Natl Acad Sci U S A 111, 1551–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões FC & Riley PR (2018). The ontogeny, activation and function of the epicardium during heart development and regeneration. Development 145, dev155994. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Leyland‐Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J & Norton L (2001). Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344, 783–792. [DOI] [PubMed] [Google Scholar]
- Sniegon I, Priess M, Heger J, Schulz R & Euler G (2017). Endothelial mesenchymal transition in hypoxic microvascular endothelial cells and paracrine induction of cardiomyocyte apoptosis are mediated via TGFβ1/SMAD signaling. Int J Mol Sci 18, E2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens KR, Kreutziger KL, Dupras SK, Korte FS, Regnier M, Muskheli V, Nourse MB, Bendixen K, Reinecke H & Murry CE (2009). Physiological function and transplantation of scaffold‐free and vascularized human cardiac muscle tissue. Proc Natl Acad Sci U S A 106, 16568–16573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stobdan T, Zhou D, Williams AT, Cabrales P & Haddad GG (2018). Cardiac‐specific knockout and pharmacological inhibition of endothelin receptor type B lead to cardiac resistance to extreme hypoxia. J Mol Med (Berl) 96, 975–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramani J, Kundumani‐Sridharan V, Hilgers RH, Owens C & Das KC (2016). Thioredoxin uses a GSH‐independent route to deglutathionylate endothelial nitric‐oxide synthase and protect against myocardial infarction. J Biol Chem 291, 23374–23389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada H, Thompson CI, Recchia FA, Loke KE, Ochoa M, Smith CJ, Shesely EG, Kaley G & Hintze TH (2000). Myocardial glucose uptake is regulated by nitric oxide via endothelial nitric oxide synthase in Langendorff mouse heart. Circ Res 86, 270–274. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Ito Y, Morikawa M, Kobune M, Huang J, Tsukamoto M, Sasaki K, Nakamura K, Dehari H, Ikeda K, Uchida H, Hirai S, Abe T & Hamada H (2003). Adenoviral‐delivered angiopoietin‐1 reduces the infarction and attenuates the progression of cardiac dysfunction in the rat model of acute myocardial infarction. Mol Ther 8, 584–592. [DOI] [PubMed] [Google Scholar]
- Tirziu D, Chorianopoulos E, Moodie KL, Palac RT, Zhuang ZW, Tjwa M, Roncal C, Eriksson U, Fu Q, Elfenbein A, Hall AE, Carmeliet P, Moons L & Simons M (2007). Myocardial hypertrophy in the absence of external stimuli is induced by angiogenesis in mice. J Clin Invest 117, 3188–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirziu D, Giordano FJ & Simons M (2010). Cell communications in the heart. Circulation 122, 928–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SH, Lu G, Xu X, Ren Y, Hein TW & Kuo L (2017). Enhanced endothelin‐1/Rho‐kinase signalling and coronary microvascular dysfunction in hypertensive myocardial hypertrophy. Cardiovasc Res 113, 1329–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulloch NL, Muskheli V, Razumova MV, Korte FS, Regnier M, Hauch KD, Pabon L, Reinecke H & Murry CE (2011). Growth of engineered human myocardium with mechanical loading and vascular coculture. Circ Res 109, 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Laan AM, Piek JJ & van Royen N (2009). Targeting angiogenesis to restore the microcirculation after reperfused MI. Nat Rev Cardiol 6, 515–523. [DOI] [PubMed] [Google Scholar]
- van Wamel AJ, Ruwhof C, van der Valk‐Kokshoom LE, Schrier PI & van der Laarse A (2001). The role of angiotensin II, endothelin‐1 and transforming growth factor‐β as autocrine/paracrine mediators of stretch‐induced cardiomyocyte hypertrophy. Mol Cell Biochem 218, 113–124. [DOI] [PubMed] [Google Scholar]
- Wang F, Kim MS, Puthanveetil P, Kewalramani G, Deppe S, Ghosh S, Abrahani A & Rodrigues B (2009). Endothelial heparanase secretion after acute hypoinsulinemia is regulated by glucose and fatty acid. Am J Physiol Heart Circ Physiol 296, H1108–H1116. [DOI] [PubMed] [Google Scholar]
- Wang X, Guo Z, Ding Z, Khaidakov M, Lin J, Xu Z, Sharma SG, Jiwani S & Mehta JL (2015). Endothelin‐1 upregulation mediates aging‐related cardiac fibrosis. J Mol Cell Cardiol 80, 101–109. [DOI] [PubMed] [Google Scholar]
- Wang X, Huang W, Liu G, Cai W, Millard RW, Wang Y, Chang J, Peng T & Fan GC (2014). Cardiomyocytes mediate anti‐angiogenesis in type 2 diabetic rats through the exosomal transfer of miR‐320 into endothelial cells. J Mol Cell Cardiol 74, 139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward NL, Van Slyke P, Sturk C, Cruz M & Dumont DJ (2004). Angiopoietin 1 expression levels in the myocardium direct coronary vessel development. Dev Dyn 229, 500–509. [DOI] [PubMed] [Google Scholar]
- Wu B, Zhang Z, Lui W, Chen X, Wang Y, Chamberlain AA, Moreno‐Rodriguez RA, Markwald RR, O'Rourke BP, Sharp DJ, Zheng D, Lenz J, Baldwin HS, Chang CP & Zhou B (2012). Endocardial cells form the coronary arteries by angiogenesis through myocardial‐endocardial VEGF signaling. Cell 151, 1083–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Tan X, Hulshoff MS, Wilhelmi T, Zeisberg M & Zeisberg EM (2016). Hypoxia‐induced endothelial‐mesenchymal transition is associated with RASAL1 promoter hypermethylation in human coronary endothelial cells. FEBS Lett 590, 1222–1233. [DOI] [PubMed] [Google Scholar]
- Yao YG, Yang HS, Cao Z, Danielsson J & Duh EJ (2005). Upregulation of placental growth factor by vascular endothelial growth factor via a post‐transcriptional mechanism. FEBS Lett 579, 1227–1234. [DOI] [PubMed] [Google Scholar]
- Yin JC, Platt MJ, Tian X, Wu X, Backx PH, Simpson JA, Araki T & Neel BG (2017). Cellular interplay via cytokine hierarchy causes pathological cardiac hypertrophy in RAF1‐mutant Noonan syndrome. Nat Commun 8, 15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentilin L, Puligadda U, Lionetti V, Zacchigna S, Collesi C, Pattarini L, Ruozi G, Camporesi S, Sinagra G, Pepe M, Recchia FA & Giacca M (2010). Cardiomyocyte VEGFR‐1 activation by VEGF‐B induces compensatory hypertrophy and preserves cardiac function after myocardial infarction. FASEB J 24, 1467–1478. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Li H, Wei R, Ma J, Zhao Y, Lian Z & Liu Z (2015). Endothelial cells regulate cardiac myocyte reorganisation through β1‐integrin signalling. Cell Physiol Biochem 35, 1808–1820. [DOI] [PubMed] [Google Scholar]
- Zhao L, Cheng G, Jin R, Afzal MR, Samanta A, Xuan YT, Girgis M, Elias HK, Zhu Y, Davani A, Yang Y, Chen X, Ye S, Wang OL, Chen L, Hauptman J, Vincent RJ & Dawn B (2016). Deletion of interleukin‐6 attenuates pressure overload‐induced left ventricular hypertrophy and dysfunction. Circ Res 118, 1918–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YY, Sawyer DR, Baliga RR, Opel DJ, Han X, Marchionni MA & Kelly RA (1998). Neuregulins promote survival and growth of cardiac myocytes. Persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J Biol Chem 273, 10261–10269. [DOI] [PubMed] [Google Scholar]
- Zheng W, Seftor EA, Meininger CJ, Hendrix MJ & Tomanek RJ (2001). Mechanisms of coronary angiogenesis in response to stretch: role of VEGF and TGF‐β. Am J Physiol Heart Circ Physiol 280, H909–H917. [DOI] [PubMed] [Google Scholar]