

Abstract
The severe cutaneous adverse reaction epidermal necrolysis (EN) which includes toxic epidermal necrolysis and the milder Stevens‐Johnson syndrome is characterized by epidermal loss due to massive keratinocyte apoptosis and/or necroptosis. EN is often caused by a drug mediating a specific TCR‐HLA interaction via the (pro)hapten, pharmacological interaction or altered peptide loading mechanism involving a self‐peptide presented by keratinocytes. (Memory) CD8 + T cells are activated and exhibit cytotoxicity against keratinocytes via the perforin/granzyme B and granulysin pathway and Fas/FasL interaction. Alternatively drug‐induced annexin release by CD14 + monocytes can induce formyl peptide receptor 1 death of keratinocytes by necroptosis. Subsequent keratinocyte death stimulates local inflammation, activating other immune cells producing pro‐inflammatory molecules and downregulating regulatory T cells. Widespread epidermal necrolysis and inflammation can induce life‐threatening systemic effects, leading to high mortality rates. Research into genetic susceptibility aims to identify risk factors for eventual prevention of EN. Specific HLA class I alleles show the strongest association with EN, but risk variants have also been identified in genes involved in drug metabolism, cellular drug uptake, peptide presentation and function of CD8 + T cells and other immune cells involved in cytotoxic responses. After the acute phase of EN, long‐term symptoms can remain or arise mainly affecting the skin and eyes. Mucosal sequelae are characterized by occlusions and strictures due to adherence of denuded surfaces and fibrosis following mucosal inflammation. In addition, systemic pathology can cause acute and chronic hepatic and renal symptoms. EN has a large psychological impact and strongly affects health‐related quality of life among EN survivors.
Search strategy PubMed was searched until Feb 20 2019 for articles about EN using following search term: "Stevens‐Johnson Syndrome" OR "SJS"[tiab] OR "Stevens‐Johnson Syndrome"[tiab] OR "Lyell's syndrome"[tiab] OR "Stevens Johnson Syndrome"[tiab] OR "Lyell syndrome"[tiab] OR "SJS/TEN"[tiab]. To find information regarding the specific topics, terms were added such as "pathogenesis", "sequelae", "long‐term", "susceptibility" and "HLA". Relevant references from the articles found were used as well.
Introduction
The severe cutaneous adverse reaction epidermal necrolysis (EN) includes toxic epidermal necrolysis (TEN) and the milder Stevens‐Johnson syndrome (SJS) and is characterized by epidermal loss due to massive keratinocyte cell death through apoptosis/necroptosis. Frequently, the eyes and mucous membranes are affected as well. 1 , 2 TEN and SJS are distinguished based on the extent of skin detachment. 2 By definition, SJS involves < 10% of the body surface area (BSA), whereas the overlap syndrome SJS/TEN shows detachment of 10%–30% of BSA and TEN > 30%. 3 Epidermal necrolysis (EN) has been proposed as unifying term for SJS and TEN. 4
EN is often drug‐induced but has also been related to infections or other causes. The adverse reaction is rare with an incidence estimated at 2–7 cases per million persons per year. Of these, 0.4–1.9 cases per million persons per year are diagnosed with TEN. 2 , 5 There is no consensus about adequate, specific treatment strategies, and mortality rates are high (23% at 6 weeks, 34% at 1 year). 6 Despite several decades of ongoing research, pathogenesis is still unclear. 7 However, advances in genetics have revealed interactions of specific HLA alleles with EN‐associated drugs, giving more insight in EN pathogenesis. 8 , 9 As survivors of EN suffer from a variety of sequelae affecting for instance the eyes and respiration, a disregarded chronic phase of the illness has recently been highlighted. 10 , 11
With EN being a rare life‐threatening disease, global research efforts are essential for better understanding of the disease process and susceptibility. Increasing knowledge on disease pathology might improve treatment strategies and elucidate risk factors for both short and long‐term consequences of EN. Moreover, enhancing common knowledge among medical practitioners and patients would alleviate uncertainty, which is a large psychological burden. Hence, this review on EN will shortly describe the disease and then give an overview on current knowledge of pathogenesis, genetic susceptibility and sequelae.
Epidermal necrolysis in short
EN starts with a prodromal phase of typically 48–72 h, presenting with specific systemic symptoms such as fever and cough. 5 Subsequently, erythematous macules and atypical target lesions develop and spread rapidly within a few days. Blisters appear and the epidermis detaches progressively up to one week. Mucosal lesions occur in almost all patients. Most frequently, the oropharynx, eyes, genitals and anus are affected, but also the nose, oesophagus, trachea and bronchi can be involved, which can lead to respiratory problems. The severity of mucosal involvement is not correlated with the area of skin affected. 5 Re‐epithelialization usually takes 1–3 weeks. 5 However, the acute phase is often followed by sequelae as described later on. 11
The massive keratinocyte death leads to systemic effects as the barrier function of the skin is lost, leading to thermal dysregulation and fluid loss affecting electrolyte and perfusion homeostasis. Furthermore, the local inflammatory response can induce systemic inflammation. 5 Body systems such as the kidneys and cardiovascular system can also be affected by complications. 8 The major cause of death in EN is sepsis, but gastrointestinal bleeding, pulmonary embolism, oedema and/or acute respiratory distress syndrome (ARDS), and myocardial infarction can be fatal as well. 12 To predict acute phase morbidity in patients, the Severity of Illness Score for Toxic Epidermal Necrolysis (SCORTEN) has been developed by Bastuji‐Garin and colleagues (Table 1) and validated in several studies. 13 , 14 , 15 , 16 , 17 , 18 Although mortality in patients with respiratory involvement might be underestimated, SCORTEN is the golden standard in predicting the prognosis of EN. 19 However, the use of SCORTEN is being questioned as supportive care has been improved since its development and several factors such as age and percentage of body surface area affected are not included. 20 Recently, the ABCD10 score has been proposed by Noe et al., which uses age, serum bicarbonate level, active cancer, dialysis and the extent of epidermal detachment to estimate mortality. The ABCD10 score highlights the negative prognostic significance of renal insufficiency, although it does not predict outcome significantly differently from SCORTEN and requires further validation and investigation into its clinical utility. 21 , 22
Table 1.
SCORTEN Severity of Illness Score for Toxic Epidermal Necrolysis Assessment score developed for prediction of acute phase morbidity in EN
| Criteria: 1 point per condition | Total score | Mortality rate (%) |
|---|---|---|
|
0–1 | 3.2 |
|
2 | 12.2 |
|
3 | 35.5 |
|
4 | 58.3 |
|
5 or more | 90.0 |
|
||
|
About 75% of EN cases are drug‐induced. 23 A slightly higher proportion for TEN specifically, with 80%–95% drug‐induced cases, 24 might be explained by misclassification of SJS, which resembles the infection‐induced erythema multiforme major. Main drug groups associated with EN include anticonvulsants, antibiotics and nonsteroidal anti‐inflammatory drugs (NSAIDs) (Table 2). 25 , 26 , 27 Research into causal drugs in an European and Israeli population indicated allopurinol as most frequent drug‐related cause. 28 Recently, novel targeted cancer drugs have been implicated in EN. 29 , 30 , 31 , 32 , 33
Table 2.
Drugs most commonly reported to induce epidermal necrolysis sorted according to drug groups
| Group | Drugs |
|---|---|
| Antibacterials | Sulphonamides (e.g. sulphamethoxazole, sulphasalazine), penicillins (e.g. amoxicillin), quinolones (e.g. ciprofloxacin) |
| Anticonvulsants | Phenytoin, carbamazepine, lamotrigine, phenobarbitone |
| NSAIDs | Oxicam‐NSAIDs (e.g. piroxicam), diclofenac, phenylbutazone |
| Antiretrovirals | Nevirapine, abacavir |
| Antituberculous | Isoniazid, ethambutol |
| Antigout | Allopurinol |
As the mean time to onset of EN usually ranges from 6 to 14 days after intake of the culprit drug, and many patients use multiple drugs simultaneously, determining the causal drug can be challenging. 24 To improve the assessment of drug causality, the algorithm of drug causality for epidermal necrolysis (ALDEN) has been developed. 34 ALDEN assigns a score based on the presence of the drug prior and during disease progression, drug notoriety, previous adverse reactions and presence of other aetiological causes.
Known non‐drug‐related causes of EN are infection with Mycoplasma pneumonia, viral infections and connective tissue diseases such as systemic lupus erythematosus. 35 , 36 , 37 However, a recent cohort study including 189 patients showed that only 5 of 17 non‐drug‐related cases could be shown to be caused by infection and connective tissue disease, leaving 12 cases unexplained. 36 The absence of a clear cause can potentially be explained by unintended drug intake, for instance via meat from treated farm animals, or by an unexpectedly long delay between drug intake and EN.
At the moment, the cornerstones of medical management mainly consist of direct discontinuation of the causal drug and supportive care, preferably in burn centres. As the main concern is sepsis, patients have to be barrier‐nursed, signs of systemic infection must be carefully monitored and cultures of affected skin must be performed regularly. Antibiotic prophylaxis should be avoided, and antibiotics should only be applied if signs of infections occur. Common causes of sepsis in EN are Staphylococcus aureus and Pseudomonas aeruginosa. 8 , 38
To date, no treatment has truly demonstrated superiority over supportive care. 39 Currently however, various systemic adjuvant therapies are used in different centres: corticosteroids, intravenous immunoglobulins (IVIGs), tumour necrosis factor (TNF) inhibitors and cyclosporine. IVIGs are thought to inhibit cellular apoptosis, whereas the other therapies target the evoked immune reactions. Some studies investigating the use of corticosteroids have shown increased rates of infection and complications, but evidence for the harm or benefit of current treatments are inconclusive. 38 , 40 As EN is rare, most evidence is based on case reports and case series. Only two randomized controlled trials (RCTs) have been performed. An RCT investigating the use of thalidomide, a drug showing among others immunosuppressive activity, demonstrated increased mortality compared with a placebo, whereupon the study was terminated. 41 In contrast, the TNF inhibitor etanercept showed faster skin healing and less gastrointestinal haemorrhages compared to treatment with corticosteroids in an RCT. 42 The efficacy of cyclosporine is still under discussion. Although this immunosuppressant showed improved survival in a recent meta‐analysis, an epidemiological study containing 174 patients did not show a beneficial effect. 43 , 44 A recent review advised to consider the use of IVIG, etanercept or cyclosporine as systemic therapy options alongside standard supportive care. 40
Pathogenesis
Several mechanisms involving different cell types and inflammatory mediators have been proposed as explanations for the pathogenesis of EN. Often, studies address a single mechanism, but it is likely that the currently proposed mechanisms should be considered as complementary (Fig. 1). 9
Figure 1.
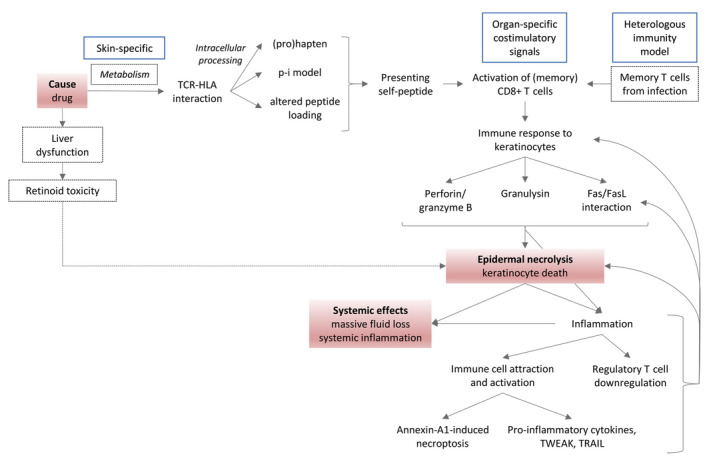
Integrated model of EN pathogenesis based on existing literature. The causal drug is thought to mediate a specific TCR‐HLA interaction involving a keratinocyte self‐peptide via the (pro)hapten, pharmacological interaction (p‐i) or altered peptide loading mechanism. (Memory) CD8 + T cells are activated and exhibit cytotoxicity against keratinocytes via the perforin/granzyme B and granulysin pathway and Fas/FasL interaction. In parallel, keratinocyte cytotoxicity can be induced by monocyte‐derived annexin A1 binding to the keratinocyte formyl peptide receptor 1. The subsequent keratinocyte death enhances local inflammation by attraction and activation of other immune cells producing pro‐inflammatory cytokines and death receptor ligands (TWEAK, TRAIL) and by downregulation of regulatory T cells. Local inflammation stimulates FasL expression on keratinocytes and perpetuation of keratinocyte cell death. Widespread epidermal necrolysis and inflammation can induce systemic effects. Skin‐specific drug metabolism, organ‐specific co‐stimulatory signals and local memory T cells from previous infections are thought to contribute to the skin‐specificity of this adverse reaction. An alternative to date less validated mechanism by which drugs could induce keratinocyte death is via liver dysfunction and retinoid toxicity. Red boxes: key events in pathogenesis; blue boxes: hypotheses on the localization of the adverse reaction in skin; dashed boxes/lines: hypotheses not (yet) generally accepted by EN research community.
EN is generally considered as type IV hypersensitivity reaction, characterized by antigen recognition by T cells which subsequently induce an immune response. 8 Studying the cell types in blister fluid and skin biopsies indeed showed a predominant fraction of CD8 + T cells. 45 , 46 , 47 , 48 , 49 Furthermore, NK cells and a subset of cytotoxic T cells exhibiting NK‐cell characteristics (NKT cells) have been found in blister fluid. 47 , 48 , 49 Indeed, in vitro incubation of blister fluid from acute stage EN with keratinocytes has shown cytotoxicity whereas control blister fluid from burn injuries did not. 49 Macrophages and dendritic cells have also been detected in skin biopsies and are involved in enhancing the immune response. 50 , 51
The cytotoxic T‐cell reaction in EN is thought to be induced by the interaction of a T‐cell receptor (TCR) and keratinocyte human leucocyte antigen (HLA) molecule. In the majority of cases, this TCR‐HLA interaction is drug‐induced, for which three models have been proposed (Fig. 2). 52 , 53 First, the (pro)hapten model involves intracellular processing of (a metabolite derived from) the drug, whereupon a drug‐derived peptide is covalently conjugated to an endogenous peptide. This novel (pro)hapten is presented by a HLA class I molecule and recognized by a specific TCR on the cytotoxic T‐cell membrane. Examples of drugs implemented in this model are penicillin and sulphamethoxazole. 54 , 55 Second, the model of pharmacological interaction with immune receptors (p‐i model) describes non‐covalent binding of the drug to either the TCR or HLA molecule to initiate the T‐cell response in a peptide‐independent way. 56 The third model is characterized by an altered peptide repertoire presented by the HLA molecule, due to the drug occupying and changing the HLA peptide‐binding groove. The peptides presented are thus no longer recognized as self‐peptides and hence elicit a T‐cell response. Abacavir is an example of this mechanism, as experiments have shown that the peptide repertoire presented by abacavir‐treated antigen‐presenting cells (APCs) differed markedly from untreated cells. 57 , 58 , 59
Figure 2.
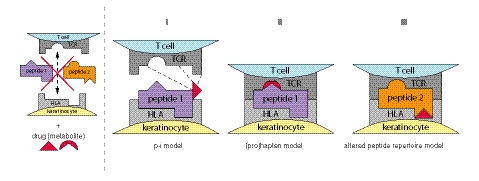
Proposed models of drug‐mediated TCR‐HLA interaction in EN. HLA molecules expressed on the membrane of antigen‐presenting cells (APCs – here keratinocyte) show specificity for peptides they can present. Subsequently, the HLA‐peptide complex is recognized by a specific T‐cell receptor (TCR) on the T‐cell membrane. Peptides 1 and 2 cannot bind the TCR and HLA depicted constituting a TCR‐HLA interaction, unless a drug (metabolite) is present and an interaction is made following one of these three models: I) The (pro)hapten model involves covalent binding of the drug or its metabolite during intracellular peptide processing, forming a novel HLA‐peptide complex that is recognized by a TCR. II) In the pharmacological interaction (p‐i) model, the drug binds non‐covalently to either the TCR or HLA molecule, enabling the formation of an unusual TCR‐HLA complex. III) The altered peptide repertoire model consists of the drug binding to the HLA peptide‐binding groove, changing the range of peptides the HLA molecule can present and thus altering the recognition by TCRs.
All three aforementioned models share breaching of T‐cell tolerance as unusual self‐peptides are presented, evoking an immune response. The skin‐specific reaction induced by EN‐related drugs could be explained by the involvement of a keratinocyte‐specific peptide or by skin‐specific metabolism of the drug involved. 60 , 61 It has also been suggested that skin‐specific co‐stimulatory signals might constitute the environment required for the aberrant immune response to occur. 9 The so‐called heterologous immunology model could also offer an explanation for the skin‐specific reaction. This model is based on the assumption that one TCR can cross‐react, recognizing different peptides, for instance by molecular mimicry of the peptide‐HLA complex. 62 White and colleagues proposed that a preceding skin infection with a pathogen could predispose someone to EN by generating a pathogen‐derived peptide‐HLA complex and evoking a cytotoxic T‐cell response that later cross‐reacts with drug‐induced peptide‐HLA complexes. 53 Memory T cells generated during the pathogen infection become reactivated upon drug exposure, constituting the adverse drug reaction. As memory T cells persist at the site of antigen encounter, the pathogen infection site determines the location of the adverse reaction. Although the reactivation of the T‐cell response might start at the previous infection site, the extension of the second immune response depends on the distribution of the antigen involved in this TCR‐HLA interaction. This might explain the expansive tissue involvement seen in EN. 53 Direct evidence for the heterologous model is still lacking.
Multiple players have been identified in the induction of keratinocyte apoptosis with a focus on the perforin/granzyme pathway, Fas/Fas Ligand (FasL) pathway and release of granulysin. 7 , 8 These three mechanisms are all observed in (NK)T cells and NK cells. The perforin/granzyme and Fas/FasL pathway are dependent on cell–cell contact of the killing cells and their target cell. 63 In the first case, NK/T cells bind their target cell and secrete granules containing perforin and granzyme B. Perforin creates channels in the target cell membrane, allowing granzyme B to enter and activate the intrinsic apoptotic pathway. 8 Levels of perforin and granzyme B in peripheral blood and blister fluid of EN patients were shown to correlate with disease severity. 64 Inhibition of the perforin/granzyme pathway decreased lymphocyte cytotoxicity to target cells in vitro. 65
Secondly, Fas is a death receptor triggering the intrinsic apoptotic pathway when binding to FasL displayed on the surface of another cell. 8 FasL can also be released as soluble FasL (sFasL), but sFasL is much less potent in inducing apoptosis. 66 , 67 Conflicting data exist on the role of the Fas/FasL pathway in EN pathogenesis. 68 Fas is constitutively expressed on keratinocytes and increased levels of sFasL have been detected in serum and blister fluid from EN patients. 69 , 70 , 71 However, given the inability of sFasL to induce apoptosis, cytotoxicity is unlikely mediated by sFasL, hence pointing towards membrane‐bound FasL binding the death receptor. 65 , 66 , 67 Although Abe et al. did not detect FasL on keratinocytes in 3 EN skin biopsies, others did show FasL expression in control and TEN keratinocytes. 69 , 71 , 72 It is thought that FasL is retained intracellularly in keratinocytes in physiological conditions, preventing its cytotoxic function. In EN, the enhanced expression of FasL leads to localization to the cell membrane mediating cytotoxicity. 71 , 72 FasL expression in keratinocytes is thought to be upregulated by cytokines released by T cells in a nitric oxide (NO)‐dependent manner. TNFα and IFNγ were shown to induce iNOS expression in keratinocytes, enhancing NO levels which subsequently stimulated FasL expression. 67 This way, initial T‐cell cytotoxicity based on cell–cell contact can generate a signalling cascade following which keratinocytes express FasL and induce death of neighbouring cells. Nonetheless, questions remain whether keratinocyte FasL expression could also be protective by targeting T cells instead of neighbouring keratinocytes, as well as whether enhanced production of sFasL can counteract Fas/FasL‐cytotoxicity. 66 , 71 , 73
Thirdly, NK and T cells produce granulysin, a pro‐inflammatory molecule inducing cell death by disruption of the target cell membrane. Being independent of cell–cell interaction or receptor binding, granulysin can induce widespread cell damage. 63 Granulysin has been detected in EN skin biopsies and in blister fluid and serum of EN patients, where its levels correlated with disease severity. 49 Expression of granulysin by (NK)T and NK cells is thought to be enhanced by IL‐15 secreted by keratinocytes. 7 , 74 Chung and colleagues showed in vitro keratinocyte cytotoxicity of granulysin, whereas antibody‐mediated depletion of granulysin prevented in vitro cytotoxic effects of blister fluid. Moreover, injection of granulysin in immunocompromised mice induced blister formation. 49
Thus, (NK)T cells and NK cells are thought to use the aforementioned pathways to induce keratinocyte apoptosis in an expansive manner, starting by cell–cell contact‐dependent perforin/granzyme‐mediated apoptosis and further activating FasL on keratinocytes and producing the soluble mediator granulysin. As inflammation is induced, other cell types and cytokines participate in the massive keratinocyte loss. Alarmins are released from damaged keratinocytes, attracting innate immune cells. 75 Monocytes infiltrate and boost the T cells by enhancing their proliferation and cytotoxicity. 76 In addition, monocytes and macrophages produce TNFα. Besides being involved in upregulation of FasL in keratinocytes, TNFα participates in enhancing HLA class I expression on keratinocytes, making them more susceptible to T cell‐mediated cytotoxicity. 8 , 71 Furthermore, TNFα can function as death receptor ligand, inducing apoptosis via the death receptor TNF‐R1. Also death receptor ligands TWEAK and TRAIL produced by monocytes and macrophages can be involved in keratinocyte death. 63
Moreover, activated monocytes are thought to be involved in inducing keratinocyte necroptosis, which is proposed as an additional cytotoxic mechanism in EN besides apoptosis. 77 During necroptosis, a form of programmed cell death is induced showing necrotic features such as mitochondrial swelling and blebbing of the cellular membrane. Saito and colleagues demonstrated that monocyte‐derived annexin A1 binds to the formyl peptide receptor 1 (FPR1) on keratinocytes, thereby inducing necroptosis. Treatment of an EN mouse model with a blocker of necroptosis prevented the development of EN‐like symptoms.
In contrast to the activation of pro‐inflammatory cells, regulatory T cells (Tregs) are suppressed. Although Tregs do not show altered frequencies in the skin, their inhibitory function is decreased. 78 This contributes to the escalation of the immune reaction against keratinocytes.
Mawson et al. have hypothesized that EN‐related drugs indirectly cause elevated plasma levels of retinoids leading to toxicity against keratinocytes, as EN symptoms resemble hypervitaminosis A and the drugs implicated in EN could interact with retinoid metabolism. However, evidence for this hypothesis is limited in contrast to the TCR‐HLA models explaining the induced keratinocyte apoptosis by various drugs. 79
Genetic susceptibility
HLA risk alleles
The strongest genetic associations found for EN are specific HLA class I alleles, emphasizing the role of CD8 + T cells in EN pathogenesis. 27 , 80 , 81 , 82 , 83 , 84 , 85 , 86 , 87 , 88 , 89 , 90 , 91 , 92 , 93 , 94 , 95 , 96 , 97 , 98 , 99 , 100 , 101 , 102 , 103 , 104 , 105 , 106 These risk factors are often drug‐specific. For instance, HLA‐B*15:2 is a risk factor for carbamazepine‐related EN, whereas HLA‐B*58:1 predisposes for EN induced by allopurinol. Moreover, the association between HLA alleles and drugs can be related to a specific severe cutaneous drug reaction. HLA‐B*15:2 is related to carbamazepine‐induced EN, but not to other carbamazepine‐induced reactions such as drug reaction with eosinophilia and systemic symptoms (DRESS). In contrast, HLA‐B*58:1 is associated to both DRESS and EN caused by allopurinol. 27 HLA‐A*31:1 is mainly related to carbamazepine‐induced DRESS and maculopapular exanthema instead of EN. 81 , 107 , 108 The HLA risk alleles identified for EN also seem to differ among populations. For example, the association between carbamazepine and HLA‐B*15:2 was detected in Han Chinese, but not in Europeans, where HLA‐B*57:1 appeared to be a risk allele for carbamazepine‐induced EN. 27 , 109 These population‐specific genetic determinants suggest a role of other (genetic) factors in EN development, differing among populations. However, the 'findability' of an HLA risk allele depends on its population prevalence and regional difference in drug prescription, which might also influence risk allele identification. The different associations with HLA alleles identified led to population‐specific guidelines for genetic testing before drug intake in order to reach cost‐effectiveness. 39 , 110
The HLA risk alleles appear to be necessary but not sufficient for developing EN after drug intake, as illustrated by the predicting values of genetic testing. The negative predictive value (NPV) is 100% for both HLA‐B*15:2 and HLA‐B*58:1 testing in Southeast Asians for carbamazepine‐ and allopurinol‐induced EN, respectively, whereas the positive predicting values (PPV) are respectively only 2%–8% for carbamazepine‐ and 2%–3% for allopurinol‐related EN. 39 , 111
The requirement of an HLA risk allele for EN development is in line with the central role of the TCR‐HLA interaction in EN pathogenesis: a drug must be able to constitute a TCR‐HLA interaction, which can only occur for HLA molecules to which the drug can (non)covalently bind. Several studies have elucidated the interaction of drugs with HLA risk alleles. For carbamazepine, the adverse reaction is likely established via the p‐i hypothesis, where the drug binds non‐covalently to the HLA molecule or TCR to activate T cells. A direct interaction between HLA‐B*15:2 and carbamazepine was shown by Wei et al. Carbamazepine‐specific cytotoxic T cells only exhibited cytotoxicity towards cells expressing HLA‐B*15:2 or closely related HLA molecules. Intracellular processing was not required for cytotoxicity. 112 Computational modelling confirmed binding of carbamazepine in the HLA binding groove and interaction with the TCR to establish the TCR‐HLA complex. 112 , 113 , 114 Carbamazepine did not alter the peptide repertoire of HLA‐B*15:2, excluding the altered peptide model of EN pathogenesis. 115
Contrary to carbamazepine, the prohapten model has been proposed for sulphamethoxazole. This drug is rapidly metabolized and autoxidized into nitro sulphamethoxazole (SMX‐NO), which is chemically reactive and able to bind intracellular proteins. 116 HLA‐mediated presentation of SMX‐NO haptenated proteins is required for T‐cell activation. 117
Novel HLA‐drug interactions could possibly be predicted based on structural features shared by these risk alleles. A shared binding pocket motif was identified in different HLA‐C risk alleles for nevirapine‐induced hypersensitivity. 118 Likewise, carbamazepine appeared to bind to several HLA‐B alleles sharing a conserved binding pocket. 52
Specific genetic susceptibility for severe mucosal complications has been studied extensively. Genetic associations of HLA‐A*02:6 and HLA‐B*44:3 were detected in EN patients with severe mucosal involvement only. 101 This could emphasize the role of HLA‐presented peptides as determinants of the localization of the adverse reaction.
Other risk factors
Still, the low PPVs of HLA risk alleles indicate that other factors must be involved in developing EN. Apart from the HLA molecules, a specific TCR and peptide presented are required to establish the TCR‐HLA interaction. 119 , 120 , 121 Recently, a preferential TCR clonotype was identified, binding to carbamazepine which can subsequently constitute the interaction between this TCR and HLA‐B*15:2. 114 Regarding the sequence and quantity of the peptide presented by the HLA, variants in genes related to proteasomal function or peptide generation can contribute to the risk for EN. 120 , 122 , 123 The function of metabolic genes and drug transporters influence the availability of the drug (metabolite) involved in the TCR‐HLA interaction. For instance, genetic variants in CYP2B6 and GSTM1 have been associated with nevirapine‐caused EN and CYP2C9 with EN due to phenytoin. 120 , 124 , 125 , 126 , 127 , 128 , 129 , 130 , 131 Furthermore, several players in the processes following the TCR‐HLA interaction, such as the induction of apoptosis or the immune response involved, have been suggested to facilitate the occurrence of disease (Fig. 3 and Table 3).
Figure 3.
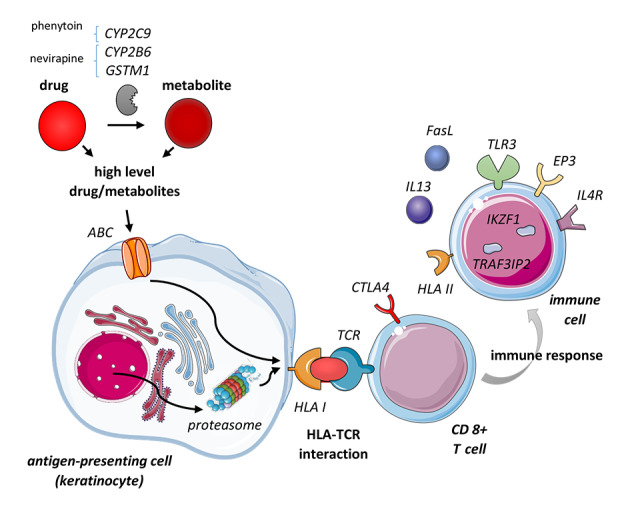
Risk variants identified in various genes involved in EN pathogenesis. Genetic associations have been shown in genes involved in drug metabolism influencing the levels of the drug or its metabolite involved (CYP2B6, CYP2C9, GSTM1); in cellular uptake and peptide presentation by antigen‐presenting cells (ABC transporters, proteasome); in the HLA‐TCR interaction (HLA I, TCR repertoire); in factors influencing CD8 + T‐cell function (CTLA4); in other immune cells shaping the subsequent immune response (HLA II, IL4R, IL13, EP3, TLR3, IKZF1, TRAF3IP2, FasL).
Table 3.
Risk components identified in development of EN
| Component/process within pathogenesis | Risk components/pathways | Reference | |
|---|---|---|---|
| HLA‐TCR interaction | HLA class I | Various HLA class I risk alleles | 27, 80, 81 |
| TCR clonotype: presence of a TCR able to interact with drug/HLA complex | Random TCR recombination | 119, 120 | |
| Immunologic history | |||
| Thymic TCR selection influenced by proteasomal activity | |||
| Availability of drug/drug metabolite | Metabolic genes: e.g. CYP2B6 and GSTM1 (nevirapine), CYP2C9 (phenytoin) | 124, 125, 126 | |
| Drug transporters, e.g. ABC transport pathway | 120, 130 | ||
| Peptide sequence and quantity | Peptide generation: e.g. ERAP2, proteasomal function | 120, 122, 123 | |
| Immune response | Lymphocyte proliferation | Apoptosis | 120 |
| Proteasomal pathway | 120, 137 | ||
| Lymphocyte activation | Co‐inhibitory pathways PD‐1 or CTLA4 | 135 | |
| CD4 + T cells | Th17‐cells producing IL‐17 | 136 | |
| Regulation of cytokine production | Proteasomal pathway | 121 | |
| Act1 signalling | 133 | ||
| Innate immune response | TLR3, IL4R, IL‐13, PTGER3 | 138, 139 | |
| Other | PSORS1C1, HCP5 | 95, 132, 134 |
Although numerous studies have investigated genetic risk factors for EN, studies are often underpowered due to small sample sizes. Another challenge in the interpretation of EN‐related SNPs is the possibility of genetic linkage of closely located genes, meaning that a suspected variant could be inherited with another variant actually influencing disease development. Many findings on genetic associations still require validation in other cohorts and functional studies to confirm its influence on disease susceptibility. Furthermore, epigenetic and environmental factors have been described, but are out of the scope of this review. 132
Disease sequelae
Following the acute phase of EN, many survivors experience chronic complications of the disease. These sequelae affect the skin and eyes, but can also be otorhinolaryngeal, pulmonary, urogenital, gastrointestinal or hepatic or related to the kidneys (Table 4). Moreover, patients frequently suffer from psychological sequelae. 11 , 133 , 134 As information is scarcely available, sequelae are often underrecognized and insufficiently treated. A cohort study of 17 patients showed a discrepancy between medical follow‐up and presence of complications, as for instance only 6% of patients were followed by an ophthalmologist, while 67% suffered from ophthalmological complications. 135 Therefore, it is important to raise awareness towards the chronic phase of EN and to unravel the pathogenic mechanisms involved to achieve adequate management or even prevention of sequelae. Collaboration between medical centres is essential to reach these goals. An example is the International Registry for Toxic Epidermal Necrolysis (IRTEN; www.irten.org), a large prospective registry cohort of TEN patients recently established, which is more extensively described later in this review. 136
Table 4.
Overview of sequelae per organ, based on Lee et al. Br J Dermatol. 2017; 177(4): 924–935 11 ; Saeed et al. Burns. 2016; 42: 20–27 146 and Dodiuk‐Gad et al. Br J Dermatol. 2016; 175(2): 422–424 147
| Organ | Sequelae |
|---|---|
| Skin | Dyspigmentation |
| Abnormal scarring | |
| Eruptive nevi | |
| Nail changes | |
| Telogen effluvium | |
| Chronic pruritus | |
| Hyperhidrosis | |
| Photosensitivity | |
| Heterotopic ossification | |
| Ectopic sebaceous glands | |
| Eyes | Corneal complications: |
| |
| Conjunctival complications: | |
| |
| Eyelid complications: | |
| |
| Mouth | Synechiae formation |
| Oral ulcers | |
| Depapillation of tongue | |
| Dental growth abnormalities | |
| Ear, nose, throat | Hypopharyngeal stenosis |
| Nasal septal synechiae | |
| External auditory canal stenosis | |
| Synechiae between ear pinna and scalp | |
| Pulmonary | Interstitial lung disease |
| Respiratory tract obstruction | |
| Bronchiectasis | |
| Bronchitis | |
| Bronchiolitis obliterans | |
| Urogenital/gynaecological | Vulvar and vaginal adenosis |
| Vaginal stenosis | |
| Fusion of labia minora and majora | |
| Gastrointestinal | Oesophageal strictures |
| Intestinal ulceration | |
| Hepatic | Vanishing bile duct syndrome |
| Renal | Chronic renal insufficiency |
| Glomerulonephritis | |
| Psychological | Post‐traumatic stress disorder |
| Anxiety | |
| Depression | |
| Psychological distress |
Dermatological sequelae
In the majority of patients, re‐epithelialized skin shows dyspigmentation (hyper‐ or hypopigmentation, Table 4). Re‐epithelization, which can take up to 3–6 months, usually occurs without scarring as the dermis is only slightly affected. 2 , 137 However, delayed onset of re‐epithelialization, in case of delayed withdrawal of the culprit drug, unrelieved skin pressure or secondary infections, can increase the risk of hypertrophic scarring. 11 , 137 , 138 It is thought that the extension of re‐epithelialization allows pro‐inflammatory cytokines, T cells and macrophages to accumulate in the skin, which subsequently influences scar formation. 137 Application of skin grafts or surgical interventions can also induce abnormal scarring. 11
The altered cutaneous micro‐environment during regeneration is also thought to be involved in the abnormal eruption of nevi and conversion of pre‐existing nevi into atypical nevi by inducing melanocyte proliferation. 139 , 140 When the local environment stabilizes again after resolution of EN, the nevi stabilize as well and remain benign. 139 , 140
Nail changes and nail loss are other complications of EN. Complete arrest of nail matrix production during acute EN is proposed to lead to nail shedding. 11 Involvement of nail changes correlates with disease severity, as more overlap/TEN patients than SJS patients present with this symptom. 141
Although rarely seen, heterotopic ossification has been reported as complication of EN. In EN, abnormal bone formation is thought to be due to hypoxia resulting from massive local tissue death. 142 Prolonged mechanical ventilation during acute EN because of pulmonary complications has been described as risk factor. Involvement of HLA genes has been suggested in relation to heterotopic ossification, which could explain its rareness. 143 , 144
Actions taken to limit dermatological sequelae include the promotion of re‐epithelialization by removing unviable skin and covering the denuded areas with dressings. 12 Management of dermatologic sequelae focuses on protecting the vulnerable re‐epithelialized skin by for instance avoiding sun exposure and improving skin elasticity with silicone gels in hypertrophic scarring. 11 , 138
Ocular sequelae
Mucosal lesions require more time to re‐epithelialize than skin and healing often involves scar formation. 2 Of patients experiencing acute mucosal involvement during EN, 73% showed persistent mucosal lesions. 145 Chronic ocular complications are most frequent, occurring in up to 90% of EN patients, and can even cause blindness (Table 4). 2 , 146 Inflammation appears to play an important role in ocular sequelae, and risk factors related to innate immunity‐related have been identified. 147 , 148 , 149 , 150 , 151 The ocular surface milieu shows pro‐inflammatory, profibrotic and antiapoptotic characteristics. In short, the inflammatory process during acute EN damages the mucin‐producing goblet cells and limbal corneal stem cells, which hinders re‐epithelization. Meanwhile, the inflammation induces hyperproliferation of conjunctival keratinocytes and fibrosis, forming hyperkeratinization and scar tissue. The scar tissue subsequently obstructs ductal openings of lacrimal glands, which – especially combined with goblet cell deficiency – promotes eye dryness. 146 , 152 , 153 , 154 , 155 , 156 , 157 Scarring also forms aberrant adhesions within the eye (e.g. symblepharon) and causes abnormal eyelid positioning (i.e. entropion, ectropion) and misdirected eye lashes (i.e. trichiasis). As trichiasis and eye dryness are triggers themselves for conjunctival inflammation, a self‐enhancing process has constituted involving persistent conjunctival inflammation. 158 , 159 The role of inflammation in ocular sequelae is emphasized by the finding that HIV patients show less severe ocular involvement. 160
Acute ocular symptoms increase the risk but are not required for the development of eye sequelae, which can arise up to decades after acute EN. 11 , 12 , 161 , 162 Late development of ocular complications could be explained by exhaustion of the transient amplifying cells backing up for the limbal stem cell deficiency induced during acute EN. When these cells cannot maintain the corneal epithelium anymore, the stem cell deficiency becomes clear. Stem cell failure might also be induced at a later time by prolonged ocular inflammation after acute EN. 158
Acute ocular care to minimalize chronic symptoms involves topical corticosteroids to dampen inflammation, topical antibiotics to prevent infections and artificial tears to prevent dryness. 39 Moreover, amniotic membrane transplantation (AMT) to the ocular surface during acute EN has been shown to prevent ocular sequelae. AMT forms a physical barrier to protect against infections, has an anti‐inflammatory and anti‐fibrotic effect and promotes re‐epithelialization. 163 , 164 , 165 After the acute phase, topical corticosteroids should be administrated for several months. 39 To improve eye dryness, artificial tears, occlusion of the tear drainage duct and smaller scleral contact lenses can be applied. Oral mucous membrane transplantations might be required to treat conjunctival sequelae, whereas eye lash depilation is used to treat trichiasis. 11 , 159
Other mucosal lesions
Although less common and less described, other mucosa present long‐term complications as well (Table 4). Similar to ocular lesions, these are characterized by occlusions and strictures due to adherence of denuded surfaces and fibrosis after mucosal inflammation and epithelial sloughing. 11 , 133 , 166 Restenosis often occurs after surgical opening of strictures, probably due to the underlying persistent inflammation. 166 , 167
Options to prevent or control long‐term mucosal complications are limited. Intravaginal glucocorticoids, vaginal moulds and menstrual suppression can be utilized during acute EN as preventive measures. 168 Management of oral sequelae focuses on oral hygiene, 12 as salivary gland involvement leads to reduced saliva activity which stimulates caries, gingival inflammation and periodontitis. 11 For pulmonary sequelae, there is no curative treatment, but steroids, antibiotics and bronchodilators might improve respiration. Monitoring is essential as lung transplantation is the only cure, but could be contraindicated because of other complications or use of mechanical ventilation. 167 , 169
Hepatic and renal sequelae
Chronic complications of the liver and kidney (Table 4) seem to establish themselves differently from mucosal sequelae. Systemic symptoms such as fluid loss and toxic effects of the drug might induce acute hepatic or renal dysfunction rather than EN pathology per se. Cholestasis and hepatitis usually resolve after the acute phase of EN, but cases with chronic cholestasis presented as a vanishing bile duct syndrome have been described. 11 , 133 This syndrome is thought to be caused by hepatocellular necrosis and ischaemic hepatitis induced by fluid loss, but involvement of a TCR‐HLA interaction involving an antigen present in/on bile duct epithelium has also been hypothesized. 133 Hence, immunosuppression and TNFα blockers have been suggested as therapeutic options, but results have been inconsistent. 170 , 171 , 172 Of patients with acute kidney injury, 5% require long‐term dialysis. 11 Moreover, studies have shown tubular damage and fibrotic glomerular alterations, probably due to high cytokine levels and nephrotoxic substances in addition to fluid loss. 133
Psychological sequelae
It is becoming increasingly clear that EN also results in a psychological scar (Table 4). In 2011, the first article demonstrating the persistent psychological impact of EN was published. Interviews showed that patients had experienced their condition as avoidable and mistaken by healthcare professionals, who were insufficiently aware of EN. The majority of patients became afraid of taking medicines in general. 173 Analysis of internet messages from EN survivors indicated that unanswered questions and concerns remained after the acute phase. 174 Clinical questionnaires confirmed post‐traumatic stress disorder (PTSD) in 23%–26% of EN survivors. 134 , 175 In a study among 17 EN survivors, 65% showed symptoms related to PTSD, 71% experienced overall psychological distress and 71% were unemployed. Questionnaires on health‐related quality of life pointed out that skin conditions had a very to extremely large effect on life quality in half of the patients. 134 Given the large psychological impact, evaluation for depression, anxiety, PTSD and fear of taking medicines should be implicated in EN management. Clear communication on disease progression and prognosis is important both during the acute and chronic phase to reduce questions and insecurities. 173 Furthermore, individual and/or group support should be offered. 39
IRTEN
The International Registry for Toxic Epidermal Necrolysis (IRTEN) Registry (https://www.irten.org) was established to investigate clinical features, prognostic predictors and outcome of SJS and TEN patients worldwide. The aim of the IRTEN register is to further our understanding of the causes, predisposing factors, clinical characteristics, medical management including therapy, and pathogenesis of SJS and TEN with the long‐term objective of identifying means to reduce the medical and economic burden of these two severe cutaneous adverse reactions (SCAR) on public health and improve the safety of medication use.
In practice, the IRTEN Registry enables: (i) high‐quality prospective anonymized clinical data collection and continuous surveillance of drug causality including newly registered drugs with adequate pharmacoepidemiologic methodology; (ii) easy online access to reference information on SJS and TEN; (iii) the constitution of an international cohort of at least 300 documented SJS/TEN patients for Europe, Asia and America in order to further study clinical and biological characteristics of SJS and TEN including drug causality, outcome, prognostic factors, ethnic factors, sequelae and impact on quality of life; and (iv) the decentralized collection of biological samples (plasma, lymphocytes, DNA, RNA and skin biopsy samples) with prior informed consent for research purposes including high‐quality studies on pharmacogenetics, transcriptomics and pathomechanisms of SJS and TEN.
Conclusion
The severe cutaneous adverse reaction EN is mediated by an abnormal immune response most likely resulting from HLA interactions with the causal drug and subsequent T‐cell activation. The resulting cytotoxic response against keratinocytes induces widespread keratinocyte apoptosis/necroptosis as well as a progressive local inflammatory process, which can cause systemic symptoms and contributes to long‐term sequelae. Genetic risk factors have been identified at several steps in EN pathogenesis, involving drug metabolism, drug‐HLA and TCR‐HLA interaction and thus the shaping of the immune response. The majority of genetic associations still require validation and functional studies to show their role in disease pathogenesis and use for genetic testing, a crucial step in prevention of EN. Until genetic tests are widely implemented, awareness during the use of EN‐associated drugs is essential to quickly stop drug administration upon disease development. Long‐term symptoms seem to be ameliorated by stimulating re‐epithelialization during the acute phase with adequate treatment. To that end, conclusive evidence on therapeutic strategies is required, as a consensus is currently still lacking. Survivors should be informed of the chronic phase of EN, and long‐term support is needed to reduce development of psychological sequelae. International collaboration in data collection is key to improve understanding, management and outcome of this rare disease.
Conflict of interest
The authors have nothing to disclose.
Funding source
None.
References
- 1. Lyell A. Toxic epidermal necrolysis: an eruption resembling scalding of the skin. Br J Dermatol 1956; 68: 355–361. [DOI] [PubMed] [Google Scholar]
- 2. Letko E, Papaliodis DN, Papaliodis GN, Daoud YJ, Ahmed AR, Foster CS. Stevens‐Johnson syndrome and toxic epidermal necrolysis: a review of the literature. Ann Allergy Asthma Immunol 2005; 94: 419–436; quiz 436–418, 456. [DOI] [PubMed] [Google Scholar]
- 3. Bastuji‐Garin S, Rzany B, Stern RS, Shear NH, Naldi L, Roujeau JC. Clinical classification of cases of toxic epidermal necrolysis, Stevens‐Johnson syndrome, and erythema multiforme. Arch Dermatol 1993; 129: 92–96. [PubMed] [Google Scholar]
- 4. Heng YK, Lee HY, Roujeau JC. Epidermal necrolysis: 60 years of errors and advances. Br J Dermatol 2015; 173: 1250–1254. [DOI] [PubMed] [Google Scholar]
- 5. Estrella‐Alonso A, Aramburu JA, Gonzalez‐Ruiz MY, Cachafeiro L, Sanchez MS, Lorente JA. Toxic epidermal necrolysis: a paradigm of critical illness. Rev Bras Ter Intensiva 2017; 29: 499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sekula P, Dunant A, Mockenhaupt M et al Comprehensive survival analysis of a cohort of patients with Stevens‐Johnson syndrome and toxic epidermal necrolysis. J Invest Dermatol 2013; 133: 1197–1204. [DOI] [PubMed] [Google Scholar]
- 7. Stern RS, Divito SJ. Stevens‐Johnson syndrome and toxic epidermal necrolysis: associations, outcomes, and pathobiology‐thirty years of progress but still much to be done. J Invest Dermatol 2017; 137: 1004–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kohanim S, Palioura S, Saeed HN et al Stevens‐johnson syndrome/toxic epidermal necrolysis–a comprehensive review and guide to therapy. I. systemic disease. Ocul Surf 2016; 14: 2–19. [DOI] [PubMed] [Google Scholar]
- 9. Pirmohamed M, Ostrov DA, Park BK. New genetic findings lead the way to a better understanding of fundamental mechanisms of drug hypersensitivity. J Allergy Clin Immunol 2015; 136: 236–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vercueil A, Walsh S. Extracutaneous manifestations and long‐term sequelae of Stevens‐Johnson syndrome/toxic epidermal necrolysis. Br J Dermatol 2015; 172: 312. [DOI] [PubMed] [Google Scholar]
- 11. Lee HY, Walsh SA, Creamer D. Long‐term complications of Stevens‐Johnson syndrome/toxic epidermal necrolysis (SJS/TEN): the spectrum of chronic problems in patients who survive an episode of SJS/TEN necessitates multidisciplinary follow‐up. Br J Dermatol 2017; 177: 924–935. [DOI] [PubMed] [Google Scholar]
- 12. Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: Part II. Prognosis, sequelae, diagnosis, differential diagnosis, prevention, and treatment. J Am Acad Dermatol 2013; 69: 87 e181‐116; quiz 203‐184. [DOI] [PubMed] [Google Scholar]
- 13. Bastuji‐Garin S, Fouchard N, Bertocchi M, Roujeau JC, Revuz J, Wolkenstein P. SCORTEN: a severity‐of‐illness score for toxic epidermal necrolysis. J Invest Dermatol 2000; 115: 149–153. [DOI] [PubMed] [Google Scholar]
- 14. Trent JT, Kirsner RS, Romanelli P, Kerdel FA. Use of SCORTEN to accurately predict mortality in patients with toxic epidermal necrolysis in the United States. Arch Dermatol 2004; 140: 890–892. [DOI] [PubMed] [Google Scholar]
- 15. Vaishampayan SS, Das AL, Verma R. SCORTEN: does it need modification? Indian J Dermatol Venereol Leprol 2008; 74: 5–37. [DOI] [PubMed] [Google Scholar]
- 16. Cartotto R, Mayich M, Nickerson D, Gomez M. SCORTEN accurately predicts mortality among toxic epidermal necrolysis patients treated in a burn center. J Burn Care Res 2008; 29: 41–146. [DOI] [PubMed] [Google Scholar]
- 17. Sekula P, Liss Y, Davidovici B et al Evaluation of SCORTEN on a cohort of patients with Stevens‐Johnson syndrome and toxic epidermal necrolysis included in the RegiSCAR study. J Burn Care Res 2011; 32: 37–245. [DOI] [PubMed] [Google Scholar]
- 18. Beck A, Quirke KP, Gamelli RL, Mosier MJ. Pediatric toxic epidermal necrolysis: using SCORTEN and predictive models to predict morbidity when a focus on mortality is not enough. J Burn Care Res 2015; 36: 67–177. [DOI] [PubMed] [Google Scholar]
- 19. Hague JS, Goulding JM, Long TM, Gee BC. Respiratory involvement in toxic epidermal necrolysis portends a poor prognosis that may not be reflected in SCORTEN. Br J Dermatol 2007; 157: 294–1296. [DOI] [PubMed] [Google Scholar]
- 20. Torres‐Navarro I, Briz‐Redon A, Botella‐Estrada R. Accuracy of SCORTEN to predict the prognosis of Stevens‐Johnson syndrome/toxic epidermal necrolysis: a systematic review and meta‐analysis. J Eur Acad Dermatol Venereol 2019. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 21. Noe MH, Hubbard RA, Micheletti RG. The ABCD‐10 risk prediction model for in‐hospital mortality among patients with Stevens‐Johnson syndrome/toxic epidermal necrolysis‐reply. JAMA Dermatol 2019; 155(9): 088. [DOI] [PubMed] [Google Scholar]
- 22. Noe MH, Rosenbach M, Hubbard RA et al Development and validation of a risk prediction model for in‐hospital mortality among patients with Stevens‐Johnson syndrome/toxic epidermal necrolysis‐ABCD‐10. JAMA Dermatol 2019; 155: 48–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mockenhaupt M. The current understanding of Stevens‐Johnson syndrome and toxic epidermal necrolysis. Expert Rev Clin Immunol 2011; 7: 03–813; quiz 814–805. [DOI] [PubMed] [Google Scholar]
- 24. Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis: Part I. Introduction, history, classification, clinical features, systemic manifestations, etiology, and immunopathogenesis. J Am Acad Dermatol 2013; 69: 73 e171‐113; quiz 185‐176. [DOI] [PubMed] [Google Scholar]
- 25. Teo YX, Walsh SA. Severe adverse drug reactions. Clin Med (Lond) 2016; 16: 9–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mustafa SS, Ostrov D, Yerly D. Severe cutaneous adverse drug reactions: presentation, risk factors, and management. Curr Allergy Asthma Rep 2018; 18: 6. [DOI] [PubMed] [Google Scholar]
- 27. Sukasem C, Katsila T, Tempark T, Patrinos GP, Chantratita W. Drug‐induced Stevens‐Johnson syndrome and toxic epidermal necrolysis call for optimum patient stratification and theranostics via pharmacogenomics. Annu Rev Genomics Hum Genet 2018; 19: 29–353. [DOI] [PubMed] [Google Scholar]
- 28. Mockenhaupt M, Viboud C, Dunant A et al Stevens‐Johnson syndrome and toxic epidermal necrolysis: assessment of medication risks with emphasis on recently marketed drugs. The EuroSCAR‐study. J Invest Dermatol 2008; 128: 5–44. [DOI] [PubMed] [Google Scholar]
- 29. Collins LK, Chapman MS, Carter JB, Samie FH. Cutaneous adverse effects of the immune checkpoint inhibitors. Curr Probl Cancer 2017; 41: 25–128. [DOI] [PubMed] [Google Scholar]
- 30. Doesch J, Debus D, Meyer C et al Afatinib‐associated Stevens‐Johnson syndrome in an EGFR‐mutated lung cancer patient. Lung Cancer 2016; 95: 5–38. [DOI] [PubMed] [Google Scholar]
- 31. Jha P, Himanshu D, Jain N, Singh AK. Imatinib‐induced Stevens‐Johnsons syndrome. BMJ Case Rep 2013; 2013: bcr2012007926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scheinfeld N. Imatinib mesylate and dermatology part 2: a review of the cutaneous side effects of imatinib mesylate. J Drugs Dermatol 2006; 5: 28–231. [PubMed] [Google Scholar]
- 33. Vivar KL, Deschaine M, Messina J et al Epidermal programmed cell death‐ligand 1 expression in TEN associated with nivolumab therapy. J Cutan Pathol 2017; 44: 81–384. [DOI] [PubMed] [Google Scholar]
- 34. Sassolas B, Haddad C, Mockenhaupt M et al ALDEN, an algorithm for assessment of drug causality in Stevens‐Johnson Syndrome and toxic epidermal necrolysis: comparison with case‐control analysis. Clin Pharmacol Ther 2010; 88: 0–68. [DOI] [PubMed] [Google Scholar]
- 35. Lee HY, Tey HL, Pang SM, Thirumoorthy T. Systemic lupus erythematosus presenting as Stevens‐Johnson syndrome and toxic epidermal necrolysis: a report of three cases. Lupus 2011; 20: 47–652. [DOI] [PubMed] [Google Scholar]
- 36. Chaby G, Ingen‐Housz‐Oro S, De Prost N, Wolkenstein P, Chosidow O, Fardet L. Idiopathic Stevens‐Johnson syndrome and toxic epidermal necrolysis: prevalence and patients characteristics. J Am Acad Dermatol 2019; 80: 1453–1455. [DOI] [PubMed] [Google Scholar]
- 37. Chung WH, Shih SR, Chang CF et al Clinicopathologic analysis of coxsackievirus a6 new variant induced widespread mucocutaneous bullous reactions mimicking severe cutaneous adverse reactions. J Infect Dis 2013; 208: 968–1978. [DOI] [PubMed] [Google Scholar]
- 38. Creamer D, Walsh SA, Dziewulski P et al U.K. guidelines for the management of Stevens–Johnson syndrome/toxic epidermal necrolysis in adults 2016. Br J Dermatol 2016; 174: 194–1227. [DOI] [PubMed] [Google Scholar]
- 39. White KD, Abe R, Ardern‐Jones M et al SJS/TEN 2017: building multidisciplinary networks to drive science and translation. J Allergy Clin Immunol Pract 2018; 6: 8–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schneider JA, Cohen PR. Stevens‐Johnson syndrome and toxic epidermal necrolysis: a concise review with a comprehensive summary of therapeutic interventions emphasizing supportive measures. Adv Ther 2017; 34: 235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wolkenstein P, Latarjet J, Roujeau JC et al Randomised comparison of thalidomide versus placebo in toxic epidermal necrolysis. Lancet 1998; 352: 586–1589. [DOI] [PubMed] [Google Scholar]
- 42. Wang CW, Yang LY, Chen CB et al Randomized, controlled trial of TNF‐alpha antagonist in CTL‐mediated severe cutaneous adverse reactions. J Clin Invest 2018; 128: 85–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ng QX, De Deyn M, Venkatanarayanan N, Ho CYX, Yeo WS. A meta‐analysis of cyclosporine treatment for Stevens‐Johnson syndrome/toxic epidermal necrolysis. J Inflamm Res 2018; 11: 35–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Poizeau F, Gaudin O, Le Cleach L et al Cyclosporine for epidermal necrolysis: absence of beneficial effect in a retrospective cohort of 174 patients‐exposed/unexposed and propensity score‐matched analyses. J Invest Dermatol 2018; 138: 293–1300. [DOI] [PubMed] [Google Scholar]
- 45. Correia O, Delgado L, Ramos JP, Resende C, Torrinha JA. Cutaneous T‐cell recruitment in toxic epidermal necrolysis. Further evidence of CD8+ lymphocyte involvement. Arch Dermatol 1993; 129: 66–468. [PubMed] [Google Scholar]
- 46. Miyauchi H, Hosokawa H, Akaeda T, Iba H, Asada Y. T‐cell subsets in drug‐induced toxic epidermal necrolysis. Possible pathogenic mechanism induced by CD8‐positive T cells. Arch Dermatol 1991; 127: 51–855. [PubMed] [Google Scholar]
- 47. Le Cleach L, Delaire S, Boumsell L et al Blister fluid T lymphocytes during toxic epidermal necrolysis are functional cytotoxic cells which express human natural killer (NK) inhibitory receptors. Clin Exp Immunol 2000; 119: 25–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nassif A, Bensussan A, Dorothee G et al Drug specific cytotoxic T‐cells in the skin lesions of a patient with toxic epidermal necrolysis. J Invest Dermatol 2002; 118: 28–733. [DOI] [PubMed] [Google Scholar]
- 49. Chung WH, Hung SI, Yang JY et al Granulysin is a key mediator for disseminated keratinocyte death in Stevens‐Johnson syndrome and toxic epidermal necrolysis. Nat Med 2008; 14: 343–1350. [DOI] [PubMed] [Google Scholar]
- 50. Paquet P, Paquet F, Al Saleh W, Reper P, Vanderkelen A, Pierard GE. Immunoregulatory effector cells in drug‐induced toxic epidermal necrolysis. Am J Dermatopathol 2000; 22: 13–417. [DOI] [PubMed] [Google Scholar]
- 51. Paquet P, Nikkels A, Arrese JE, Vanderkelen A, Pierard GE. Macrophages and tumor necrosis factor alpha in toxic epidermal necrolysis. Arch Dermatol 1994; 130: 05–608. [PubMed] [Google Scholar]
- 52. Redwood AJ, Pavlos RK, White KD, Phillips EJ. HLAs: Key regulators of T‐cell‐mediated drug hypersensitivity. Hla 2018; 91: 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. White KD, Chung WH, Hung SI, Mallal S, Phillips EJ. Evolving models of the immunopathogenesis of T cell‐mediated drug allergy: The role of host, pathogens, and drug response. J Allergy Clin Immunol 2015; 136: 19–234; quiz 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Padovan E, Mauri‐Hellweg D, Pichler WJ, Weltzien HU. T cell recognition of penicillin G: structural features determining antigenic specificity. Eur J Immunol 1996; 26: 2–48. [DOI] [PubMed] [Google Scholar]
- 55. Naisbitt DJ, Gordon SF, Pirmohamed M et al Antigenicity and immunogenicity of sulphamethoxazole: demonstration of metabolism‐dependent haptenation and T‐cell proliferation in vivo. Br J Pharmacol 2001; 133: 95–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zanni MP, von Greyerz S, Schnyder B et al HLA‐restricted, processing‐ and metabolism‐independent pathway of drug recognition by human alpha beta T lymphocytes. J Clin Invest 1998; 102: 591–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Norcross MA, Luo S, Lu L et al Abacavir induces loading of novel self‐peptides into HLA‐B*57: 01: an autoimmune model for HLA‐associated drug hypersensitivity. AIDS 2012; 26:F21–F29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ostrov DA, Grant BJ, Pompeu YA et al Drug hypersensitivity caused by alteration of the MHC‐presented self‐peptide repertoire. Proc Natl Acad Sci USA 2012; 109: 959–9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Illing PT, Vivian JP, Dudek NL et al Immune self‐reactivity triggered by drug‐modified HLA‐peptide repertoire. Nature 2012; 486: 54–558. [DOI] [PubMed] [Google Scholar]
- 60. Wolkenstein P, Charue D, Laurent P, Revuz J, Roujeau JC, Bagot M. Metabolic predisposition to cutaneous adverse drug reactions. Role in toxic epidermal necrolysis caused by sulfonamides and anticonvulsants. Arch Dermatol 1995; 131: 44–551. [PubMed] [Google Scholar]
- 61. Sharma AM, Uetrecht J. Bioactivation of drugs in the skin: relationship to cutaneous adverse drug reactions. Drug Metab Rev 2014; 46: –18. [DOI] [PubMed] [Google Scholar]
- 62. Yin Y, Mariuzza RA. The multiple mechanisms of T cell receptor cross‐reactivity. Immunity 2009; 31: 49–851. [DOI] [PubMed] [Google Scholar]
- 63. Tohyama M, Hashimoto K. Immunological mechanisms of epidermal damage in toxic epidermal necrolysis. Curr Opin Allergy Clin Immunol 2012; 12: 76–382. [DOI] [PubMed] [Google Scholar]
- 64. Posadas SJ, Padial A, Torres MJ et al Delayed reactions to drugs show levels of perforin, granzyme B, and Fas‐L to be related to disease severity. J Allergy Clin Immunol 2002; 109: 55–161. [DOI] [PubMed] [Google Scholar]
- 65. Nassif A, Bensussan A, Boumsell L et al Toxic epidermal necrolysis: effector cells are drug‐specific cytotoxic T cells. J Allergy Clin Immunol 2004; 114: 209–1215. [DOI] [PubMed] [Google Scholar]
- 66. Knox PG, Milner AE, Green NK, Eliopoulos AG, Young LS. Inhibition of metalloproteinase cleavage enhances the cytotoxicity of Fas ligand. J Immunol 2003; 170: 77–685. [DOI] [PubMed] [Google Scholar]
- 67. Viard‐Leveugle I, Gaide O, Jankovic D et al TNF‐alpha and IFN‐gamma are potential inducers of Fas‐mediated keratinocyte apoptosis through activation of inducible nitric oxide synthase in toxic epidermal necrolysis. J Invest Dermatol 2013; 133: 89–498. [DOI] [PubMed] [Google Scholar]
- 68. Nickoloff BJ. Saving the skin from drug‐induced detachment. Nat Med 2008; 14: 311–1313. [DOI] [PubMed] [Google Scholar]
- 69. Viard I, Wehrli P, Bullani R et al Inhibition of toxic epidermal necrolysis by blockade of CD95 with human intravenous immunoglobulin. Science 1998; 282: 90–493. [DOI] [PubMed] [Google Scholar]
- 70. Abe R, Shimizu T, Shibaki A, Nakamura H, Watanabe H, Shimizu H. Toxic epidermal necrolysis and Stevens‐Johnson syndrome are induced by soluble Fas ligand. Am J Pathol 2003; 162: 515–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nassif A, Moslehi H, Le Gouvello S et al Evaluation of the potential role of cytokines in toxic epidermal necrolysis. J Invest Dermatol 2004; 123: 50–855. [DOI] [PubMed] [Google Scholar]
- 72. Viard‐Leveugle I, Bullani RR, Meda P et al Intracellular localization of keratinocyte Fas ligand explains lack of cytolytic activity under physiological conditions. J Biol Chem 2003; 278: 6183–16188. [DOI] [PubMed] [Google Scholar]
- 73. Gaultier F, Ejeil AL, Igondjo‐Tchen S et al Possible involvement of gelatinase A (MMP2) and gelatinase B (MMP9) in toxic epidermal necrolysis or Stevens‐Johnson syndrome. Arch Dermatol Res 2004; 296: 20–225. [DOI] [PubMed] [Google Scholar]
- 74. Su SC, Mockenhaupt M, Wolkenstein P et al Interleukin‐15 is associated with severity and mortality in Stevens‐Johnson syndrome/toxic epidermal necrolysis. J Invest Dermatol 2017; 137: 065–1073. [DOI] [PubMed] [Google Scholar]
- 75. Nakajima S, Watanabe H, Tohyama M et al High‐mobility group box 1 protein (HMGB1) as a novel diagnostic tool for toxic epidermal necrolysis and Stevens‐Johnson syndrome. Arch Dermatol 2011; 147: 110–1112. [DOI] [PubMed] [Google Scholar]
- 76. Tohyama M, Watanabe H, Murakami S et al Possible involvement of CD14+ CD16+ monocyte lineage cells in the epidermal damage of Stevens‐Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol 2012; 166: 22–330. [DOI] [PubMed] [Google Scholar]
- 77. Saito N, Qiao H, Yanagi T et al An annexin A1‐FPR1 interaction contributes to necroptosis of keratinocytes in severe cutaneous adverse drug reactions. Sci Transl Med 2014; 6: 45ra295. [DOI] [PubMed] [Google Scholar]
- 78. Takahashi R, Kano Y, Yamazaki Y, Kimishima M, Mizukawa Y, Shiohara T. Defective regulatory T cells in patients with severe drug eruptions: timing of the dysfunction is associated with the pathological phenotype and outcome. J Immunol 2009; 182: 071–8079. [DOI] [PubMed] [Google Scholar]
- 79. Mawson AR, Eriator I, Karre S. Stevens‐Johnson syndrome and toxic epidermal necrolysis (SJS/TEN): could retinoids play a causative role? Med Sci Monit 2015; 21: 33–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chung WH, Hung SI, Hong HS et al Medical genetics: a marker for Stevens‐Johnson syndrome. Nature 2004; 428: 86. [DOI] [PubMed] [Google Scholar]
- 81. Hung SI, Chung WH, Jee SH et al Genetic susceptibility to carbamazepine‐induced cutaneous adverse drug reactions. Pharmacogenet Genomics 2006; 16: 97–306. [DOI] [PubMed] [Google Scholar]
- 82. Wu XT, Hu FY, An DM et al Association between carbamazepine‐induced cutaneous adverse drug reactions and the HLA‐B*1502 allele among patients in central China. Epilepsy Behav 2010; 19: 05–408. [DOI] [PubMed] [Google Scholar]
- 83. Zhang Y, Wang J, Zhao LM et al Strong association between HLA‐B*1502 and carbamazepine‐induced Stevens‐Johnson syndrome and toxic epidermal necrolysis in mainland Han Chinese patients. Eur J Clin Pharmacol 2011; 67: 85–887. [DOI] [PubMed] [Google Scholar]
- 84. Wang Q, Zhou JQ, Zhou LM et al Association between HLA‐B*1502 allele and carbamazepine‐induced severe cutaneous adverse reactions in Han people of southern China mainland. Seizure 2011; 20: 46–448. [DOI] [PubMed] [Google Scholar]
- 85. Kim SH, Lee KW, Song WJ et al Carbamazepine‐induced severe cutaneous adverse reactions and HLA genotypes in Koreans. Epilepsy Res 2011; 97: 90–197. [DOI] [PubMed] [Google Scholar]
- 86. Khor AH, Lim KS, Tan CT et al HLA‐A*31: 01 and HLA‐B*15: 2 association with Stevens‐Johnson syndrome and toxic epidermal necrolysis to carbamazepine in a multiethnic Malaysian population. Pharmacogenet Genomics 2017; 27: 75–278. [DOI] [PubMed] [Google Scholar]
- 87. Hung SI, Chung WH, Liu ZS et al Common risk allele in aromatic antiepileptic‐drug induced Stevens‐Johnson syndrome and toxic epidermal necrolysis in Han Chinese. Pharmacogenomics 2010; 11: 49–356. [DOI] [PubMed] [Google Scholar]
- 88. An DM, Wu XT, Hu FY, Yan B, Stefan H, Zhou D. Association study of lamotrigine‐induced cutaneous adverse reactions and HLA‐B*1502 in a Han Chinese population. Epilepsy Res 2010; 92: 26–230. [DOI] [PubMed] [Google Scholar]
- 89. Shi YW, Min FL, Liu XR et al Hla‐B alleles and lamotrigine‐induced cutaneous adverse drug reactions in the Han Chinese population. Basic Clin Pharmacol Toxicol 2011; 109: 2–46. [DOI] [PubMed] [Google Scholar]
- 90. Koomdee N, Pratoomwun J, Jantararoungtong T et al Association of HLA‐A and HLA‐B alleles with lamotrigine‐induced cutaneous adverse drug reactions in the Thai population. Front Pharmacol 2017; 8: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lonjou C, Borot N, Sekula P et al A European study of HLA‐B in Stevens‐Johnson syndrome and toxic epidermal necrolysis related to five high‐risk drugs. Pharmacogenet Genomics 2008; 18: 9–107. [DOI] [PubMed] [Google Scholar]
- 92. Park HJ, Kim YJ, Kim DH et al HLA allele frequencies in 5802 Koreans: varied allele types associated with SJS/TEN according to culprit drugs. Yonsei Med J 2016; 57: 18–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chang CC, Ng CC, Too CL et al Association of HLA‐B*15: 3 and HLA‐B*15: 2 with phenytoin‐induced severe cutaneous adverse reactions in a Malay population. Pharmacogenomics J 2017; 17: 70–173. [DOI] [PubMed] [Google Scholar]
- 94. Hung SI, Chung WH, Liou LB et al HLA‐B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci USA 2005; 102: 134–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Tohkin M, Kaniwa N, Saito Y et al A whole‐genome association study of major determinants for allopurinol‐related Stevens‐Johnson syndrome and toxic epidermal necrolysis in Japanese patients. Pharmacogenomics J 2013; 13: 0–69. [DOI] [PubMed] [Google Scholar]
- 96. Kang HR, Jee YK, Kim YS et al Positive and negative associations of HLA class I alleles with allopurinol‐induced SCARs in Koreans. Pharmacogenet Genomics 2011; 21: 03–307. [DOI] [PubMed] [Google Scholar]
- 97. Goncalo M, Coutinho I, Teixeira V et al HLA‐B*58: 1 is a risk factor for allopurinol‐induced DRESS and Stevens‐Johnson syndrome/toxic epidermal necrolysis in a Portuguese population. Br J Dermatol 2013; 169: 60–665. [DOI] [PubMed] [Google Scholar]
- 98. Tassaneeyakul W, Jantararoungtong T, Chen P et al Strong association between HLA‐B*5801 and allopurinol‐induced Stevens‐Johnson syndrome and toxic epidermal necrolysis in a Thai population. Pharmacogenet Genomics 2009; 19: 04–709. [DOI] [PubMed] [Google Scholar]
- 99. Sukasem C, Jantararoungtong T, Kuntawong P et al HLA‐B (*) 58: 1 for allopurinol‐induced cutaneous adverse drug reactions: implication for clinical interpretation in Thailand. Front Pharmacol 2016; 7: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Cristallo AF, Schroeder J, Citterio A et al A study of HLA class I and class II 4‐digit allele level in Stevens‐Johnson syndrome and toxic epidermal necrolysis. Int J Immunogenet 2011; 38: 03–309. [DOI] [PubMed] [Google Scholar]
- 101. Ueta M, Kaniwa N, Sotozono C et al Independent strong association of HLA‐A*02: 6 and HLA‐B*44: 3 with cold medicine‐related Stevens‐Johnson syndrome with severe mucosal involvement. Sci Rep 2014; 4: 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ueta M, Kannabiran C, Wakamatsu TH et al Trans‐ethnic study confirmed independent associations of HLA‐A*02: 6 and HLA‐B*44: 3 with cold medicine‐related Stevens‐Johnson syndrome with severe ocular surface complications. Sci Rep 2014; 4: 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kongpan T, Mahasirimongkol S, Konyoung P et al Candidate HLA genes for prediction of co‐trimoxazole‐induced severe cutaneous reactions. Pharmacogenet Genomics 2015; 25: 02–411. [DOI] [PubMed] [Google Scholar]
- 104. Carr DF, Chaponda M, Jorgensen AL et al Association of human leukocyte antigen alleles and nevirapine hypersensitivity in a Malawian HIV‐infected population. Clin Infect Dis 2013; 56: 330–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tempark T, Satapornpong P, Rerknimitr P et al Dapsone‐induced severe cutaneous adverse drug reactions are strongly linked with HLA‐B*13: 01 allele in the Thai population. Pharmacogenet Genomics 2017; 27: 29–437. [DOI] [PubMed] [Google Scholar]
- 106. Kim SH, Kim M, Lee KW et al HLA‐B*5901 is strongly associated with methazolamide‐induced Stevens‐Johnson syndrome/toxic epidermal necrolysis. Pharmacogenomics 2010; 11: 79–884. [DOI] [PubMed] [Google Scholar]
- 107. Genin E, Chen DP, Hung SI et al HLA‐A*31: 1 and different types of carbamazepine‐induced severe cutaneous adverse reactions: an international study and meta‐analysis. Pharmacogenomics J 2014; 14: 81–288. [DOI] [PubMed] [Google Scholar]
- 108. Ozeki T, Mushiroda T, Yowang A et al Genome‐wide association study identifies HLA‐A*3101 allele as a genetic risk factor for carbamazepine‐induced cutaneous adverse drug reactions in Japanese population. Hum Mol Genet 2011; 20: 034–1041. [DOI] [PubMed] [Google Scholar]
- 109. Mockenhaupt M, Wang CW, Hung SI et al HLA‐B*57: 1 confers genetic susceptibility to carbamazepine‐induced SJS/TEN in Europeans. Allergy 2019; 74: 227–2230. [DOI] [PubMed] [Google Scholar]
- 110. Ferrell PB Jr, McLeod HL. Carbamazepine, HLA‐B*1502 and risk of Stevens‐Johnson syndrome and toxic epidermal necrolysis: US FDA recommendations. Pharmacogenomics 2008; 9: 543–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Karnes JH, Miller MA, White KD et al Applications of immunopharmacogenomics: predicting, preventing, and understanding immune‐mediated adverse drug reactions. Annu Rev Pharmacol Toxicol 2019; 59: 63–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wei CY, Chung WH, Huang HW, Chen YT, Hung SI. Direct interaction between HLA‐B and carbamazepine activates T cells in patients with Stevens‐Johnson syndrome. J Allergy Clin Immunol 2012; 129: 562–1569.e1565. [DOI] [PubMed] [Google Scholar]
- 113. Zhou P, Zhang S, Wang Y, Yang C, Huang J. Structural modeling of HLA‐B*1502/peptide/carbamazepine/T‐cell receptor complex architecture: implication for the molecular mechanism of carbamazepine‐induced Stevens‐Johnson syndrome/toxic epidermal necrolysis. J Biomol Struct Dyn 2016; 34: 806–1817. [DOI] [PubMed] [Google Scholar]
- 114. Pan RY, Chu MT, Wang CW et al Identification of drug‐specific public TCR driving severe cutaneous adverse reactions. Nat Commun 2019; 10: 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Yang CW, Hung SI, Juo CG et al HLA‐B*1502‐bound peptides: implications for the pathogenesis of carbamazepine‐induced Stevens‐Johnson syndrome. J Allergy Clin Immunol 2007; 120: 70–877. [DOI] [PubMed] [Google Scholar]
- 116. Elsheikh A, Lavergne SN, Castrejon JL et al Drug antigenicity, immunogenicity, and costimulatory signaling: evidence for formation of a functional antigen through immune cell metabolism. J Immunol 2010; 185: 448–6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Naisbitt DJ, Farrell J, Gordon SF et al Covalent binding of the nitroso metabolite of sulfamethoxazole leads to toxicity and major histocompatibility complex‐restricted antigen presentation. Mol Pharmacol 2002; 62: 28–637. [DOI] [PubMed] [Google Scholar]
- 118. Pavlos R, McKinnon EJ, Ostrov DA et al Shared peptide binding of HLA Class I and II alleles associate with cutaneous nevirapine hypersensitivity and identify novel risk alleles. Sci Rep 2017; 7: 653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ko TM, Chung WH, Wei CY et al Shared and restricted T‐cell receptor use is crucial for carbamazepine‐induced Stevens‐Johnson syndrome. J Allergy Clin Immunol 2011; 128: 266–1276.e1211. [DOI] [PubMed] [Google Scholar]
- 120. Nicoletti P, Bansal M, Lefebvre C et al ABC transporters and the proteasome complex are implicated in susceptibility to Stevens‐Johnson syndrome and toxic epidermal necrolysis across multiple drugs. PLoS One 2015; 10:e0131038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Sijts EJ, Kloetzel PM. The role of the proteasome in the generation of MHC class I ligands and immune responses. Cell Mol Life Sci 2011; 68: 491–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Carr DF, Bourgeois S, Chaponda M et al Genome‐wide association study of nevirapine hypersensitivity in a sub‐Saharan African HIV‐infected population. J Antimicrob Chemother 2017; 72: 152–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Dainichi T, Uchi H, Moroi Y, Furue M. Stevens‐Johnson syndrome, drug‐induced hypersensitivity syndrome and toxic epidermal necrolysis caused by allopurinol in patients with a common HLA allele: what causes the diversity? Dermatology 2007; 215: 6–88. [DOI] [PubMed] [Google Scholar]
- 124. Carr DF, Chaponda M, Cornejo Castro EM et al CYP2B6 c.983T>C polymorphism is associated with nevirapine hypersensitivity in Malawian and Ugandan HIV populations. J Antimicrob Chemother 2014; 69: 329–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Chung WH, Chang WC, Lee YS et al Genetic variants associated with phenytoin‐related severe cutaneous adverse reactions. JAMA 2014; 312: 25–534. [DOI] [PubMed] [Google Scholar]
- 126. Ciccacci C, Di Fusco D, Marazzi MC et al Association between CYP2B6 polymorphisms and Nevirapine‐induced SJS/TEN: a pharmacogenetics study. Eur J Clin Pharmacol 2013; 69: 909–1916. [DOI] [PubMed] [Google Scholar]
- 127. Ciccacci C, Latini A, Politi C et al Impact of glutathione transferases genes polymorphisms in nevirapine adverse reactions: a possible role for GSTM1 in SJS/TEN susceptibility. Eur J Clin Pharmacol 2017; 73: 253–1259. [DOI] [PubMed] [Google Scholar]
- 128. Ng CY, Yeh YT, Wang CW et al Impact of the HLA‐B(*)58: 1 allele and renal impairment on allopurinol‐induced cutaneous adverse reactions. J Invest Dermatol 2016; 136: 373–1381. [DOI] [PubMed] [Google Scholar]
- 129. Tassaneeyakul W, Prabmeechai N, Sukasem C et al Associations between HLA class I and cytochrome P450 2C9 genetic polymorphisms and phenytoin‐related severe cutaneous adverse reactions in a Thai population. Pharmacogenet Genomics 2016; 26: 25–234. [DOI] [PubMed] [Google Scholar]
- 130. Uitto J. The gene family of ABC transporters–novel mutations, new phenotypes. Trends Mol Med 2005; 11: 41–343. [DOI] [PubMed] [Google Scholar]
- 131. Wu X, Liu W, Zhou W. Association of CYP2C9*3 with phenytoin‐induced Stevens‐Johnson syndrome and toxic epidermal necrolysis: a systematic review and meta‐analysis. J Clin Pharm Ther 2018; 43: 08–413. [DOI] [PubMed] [Google Scholar]
- 132. Gibson A, Ogese M, Pirmohamed M. Genetic and nongenetic factors that may predispose individuals to allergic drug reactions. Curr Opin Allergy Clin Immunol 2018; 18: 25–332. [DOI] [PubMed] [Google Scholar]
- 133. Saeed H, Mantagos IS, Chodosh J. Complications of Stevens‐Johnson syndrome beyond the eye and skin. Burns 2016; 42: 0–27. [DOI] [PubMed] [Google Scholar]
- 134. Dodiuk‐Gad RP, Olteanu C, Feinstein A et al Major psychological complications and decreased health‐related quality of life among survivors of Stevens‐Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol 2016; 175: 22–424. [DOI] [PubMed] [Google Scholar]
- 135. Olteanu C, Shear NH, Chew HF et al Severe physical complications among survivors of Stevens‐Johnson syndrome and toxic epidermal necrolysis. Drug Saf 2018; 41: 77–284. [DOI] [PubMed] [Google Scholar]
- 136. (IRTEN) IRfTEN . https://www.irten.org/home.html.
- 137. Paquet P, Jacob E, Quatresooz P, Jacquemin D, Pierard GE. Delayed reepithelialization and scarring deregulation following drug‐induced toxic epidermal necrolysis. Burns 2007; 33: 00–104. [DOI] [PubMed] [Google Scholar]
- 138. Kavanagh GM, Page P, Hanna MM. Silicone gel treatment of extensive hypertrophic scarring following toxic epidermal necrolysis. Br J Dermatol 1994; 130: 40–541. [DOI] [PubMed] [Google Scholar]
- 139. Gelfer A, Rivers JK. Long‐term follow‐up of a patient with eruptive melanocytic nevi after Stevens‐Johnson syndrome. Arch Dermatol 2007; 143: 555–1557. [DOI] [PubMed] [Google Scholar]
- 140. Ardakani NM, Tiwari SM, Wood BA. Pseudomelanomas following Stevens‐Johnson syndrome. Am J Dermatopathol 2017; 39: 53–856. [DOI] [PubMed] [Google Scholar]
- 141. Yang CW, Cho YT, Chen KL, Chen YC, Song HL, Chu CY. Long‐term sequelae of Stevens‐Johnson syndrome/toxic epidermal necrolysis. Acta Derm Venereol 2016; 96: 25–529. [DOI] [PubMed] [Google Scholar]
- 142. Gibson CJ, Poduri KR. Heterotopic ossification as a complication of toxic epidermal necrolysis. Arch Phys Med Rehabil 1997; 78: 74–776. [DOI] [PubMed] [Google Scholar]
- 143. Samanci N, Balci N, Alpsoy E. Heterotopic ossification related to toxic epidermal necrolysis in a patient with Behcet's disease. J Dermatol 2005; 32: 69–473. [DOI] [PubMed] [Google Scholar]
- 144. Garland DE, Alday B, Venos KG. Heterotopic ossification and HLA antigens. Arch Phys Med Rehabil 1984; 65: 31–532. [PubMed] [Google Scholar]
- 145. Oplatek A, Brown K, Sen S, Halerz M, Supple K, Gamelli RL. Long‐term follow‐up of patients treated for toxic epidermal necrolysis. J Burn Care Res 2006; 27: 6–33. [DOI] [PubMed] [Google Scholar]
- 146. Chang VS, Chodosh J, Papaliodis GN. Chronic ocular complications of Stevens‐Johnson syndrome and toxic epidermal necrolysis: the role of systemic immunomodulatory therapy. Semin Ophthalmol 2016; 31: 78–187. [DOI] [PubMed] [Google Scholar]
- 147. Ueta M, Sawai H, Sotozono C et al IKZF1, a new susceptibility gene for cold medicine‐related Stevens‐Johnson syndrome/toxic epidermal necrolysis with severe mucosal involvement. J Allergy Clin Immunol 2015; 135: 538–1545.e1517. [DOI] [PubMed] [Google Scholar]
- 148. Ueta M, Tamiya G, Tokunaga K et al Epistatic interaction between Toll‐like receptor 3 (TLR3) and prostaglandin E receptor 3 (PTGER3) genes. J Allergy Clin Immunol 2012; 129: 413–1416.e1411. [DOI] [PubMed] [Google Scholar]
- 149. Ueta M, Sotozono C, Nakano M et al Association between prostaglandin E receptor 3 polymorphisms and Stevens‐Johnson syndrome identified by means of a genome‐wide association study. J Allergy Clin Immunol 2010; 126: 218–1225.e1210. [DOI] [PubMed] [Google Scholar]
- 150. Ueta M, Sotozono C, Inatomi T, Kojima K, Hamuro J, Kinoshita S. Association of IL4R polymorphisms with Stevens‐Johnson syndrome. J Allergy Clin Immunol 2007; 120: 457–1459. [DOI] [PubMed] [Google Scholar]
- 151. Ueta M, Sotozono C, Inatomi T et al Toll‐like receptor 3 gene polymorphisms in Japanese patients with Stevens‐Johnson syndrome. Br J Ophthalmol 2007; 91: 62–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Ueta M. Results of detailed investigations into Stevens‐Johnson syndrome with severe ocular complications. Invest Ophthalmol Vis Sci 2018; 59:DES183–DES191. [DOI] [PubMed] [Google Scholar]
- 153. Nishida K, Yamanishi K, Yamada K et al Epithelial hyperproliferation and transglutaminase 1 gene expression in Stevens‐Johnson syndrome conjunctiva. Am J Pathol 1999; 154: 31–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Saito T, Nishida K, Sugiyama H et al Abnormal keratocytes and stromal inflammation in chronic phase of severe ocular surface diseases with stem cell deficiency. Br J Ophthalmol 2008; 92: 04–410. [DOI] [PubMed] [Google Scholar]
- 155. Ueta M. Innate immunity of the ocular surface and ocular surface inflammatory disorders. Cornea 2008; 27: S31–S40. [DOI] [PubMed] [Google Scholar]
- 156. Ueta M, Nishigaki H, Sotozono C, Kinoshita S. Downregulation of interferon‐gamma‐induced protein 10 in the tears of patients with Stevens‐Johnson syndrome with severe ocular complications in the chronic stage. BMJ Open Ophthalmol 2017; 1:e000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Gurumurthy S, Iyer G, Srinivasan B, Agarwal S, Angayarkanni N. Ocular surface cytokine profile in chronic Stevens‐Johnson syndrome and its response to mucous membrane grafting for lid margin keratinisation. Br J Ophthalmol 2018; 102: 69–176. [DOI] [PubMed] [Google Scholar]
- 158. De Rojas MV, Dart JK, Saw VP. The natural history of Stevens Johnson syndrome: patterns of chronic ocular disease and the role of systemic immunosuppressive therapy. Br J Ophthalmol 2007; 91: 048–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Sotozono C, Ueta M, Severe YN. Dry Eye with combined mechanisms is involved in the ocular sequelae of SJS/TEN at the chronic stage. Invest Ophthalmol Vis Sci 2018; 59:Des80–Des86. [DOI] [PubMed] [Google Scholar]
- 160. Jongkhajornpong P, Lekhanont K, Siriyotha S, Kanokrungsee S, Chuckpaiwong V. Factors contributing to long‐term severe visual impairment in Stevens‐Johnson syndrome and toxic epidermal necrolysis. J Ophthalmol 2017; 2017: 087578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Lopez‐Garcia JS, Rivas Jara L Garcia‐Lozano CI, Conesa E, de Juan IE, Murube del Castillo J. Ocular features and histopathologic changes during follow‐up of toxic epidermal necrolysis. Ophthalmology 2011; 118: 65–271. [DOI] [PubMed] [Google Scholar]
- 162. Gueudry J, Roujeau JC, Binaghi M, Soubrane G, Muraine M. Risk factors for the development of ocular complications of Stevens‐Johnson syndrome and toxic epidermal necrolysis. Arch Dermatol 2009; 145: 57–162. [DOI] [PubMed] [Google Scholar]
- 163. Hsu M, Jayaram A, Verner R, Lin A, Bouchard C. Indications and outcomes of amniotic membrane transplantation in the management of acute stevens‐johnson syndrome and toxic epidermal necrolysis: a case‐control study. Cornea 2012; 31: 394–1402. [DOI] [PubMed] [Google Scholar]
- 164. Shammas MC, Lai EC, Sarkar JS, Yang J, Starr CE, Sippel KC. Management of acute Stevens‐Johnson syndrome and toxic epidermal necrolysis utilizing amniotic membrane and topical corticosteroids. Am J Ophthalmol 2010; 149: 203–213.e202. [DOI] [PubMed] [Google Scholar]
- 165. Tseng SC. Acute management of Stevens‐Johnson syndrome and toxic epidermal necrolysis to minimize ocular sequelae. Am J Ophthalmol 2009; 147: 49–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Hotaling JM, Hotaling AJ. Otologic complications of Stevens‐Johnson syndrome and toxic epidermal necrolysis. Int J Pediatr Otorhinolaryngol 2014; 78: 408–1409. [DOI] [PubMed] [Google Scholar]
- 167. Xu N, Chen X, Wu S et al Chronic pulmonary complications associated with toxic epidermal necrolysis: a case report and literature review. Exp Ther Med 2018; 16: 027–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Kaser DJ, Reichman DE, Laufer MR. Prevention of vulvovaginal sequelae in Stevens‐Johnson syndrome and toxic epidermal necrolysis. Rev Obstet Gynecol 2011; 4: 1–85. [PMC free article] [PubMed] [Google Scholar]
- 169. Woo T, Saito H, Yamakawa Y et al Severe obliterative bronchitis associated with Stevens‐Johnson syndrome. Intern Med 2011; 50: 823–2827. [DOI] [PubMed] [Google Scholar]
- 170. Momen S, Dangiosse C, Wedgeworth E, Walsh S, Creamer D. A case of toxic epidermal necrolysis and vanishing bile duct syndrome, requiring liver transplantation. J Eur Acad Dermatol Venereol 2017; 31:e450–e452. [DOI] [PubMed] [Google Scholar]
- 171. White JC, Appleman S. Infliximab/Plasmapheresis in vanishing bile duct syndrome secondary to toxic epidermal necrolysis. Pediatrics 2014; 134:e1194–e1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Karnsakul W, Arkachaisri T, Atisook K, Wisuthsarewong W, Sattawatthamrong Y, Aanpreung P. Vanishing bile duct syndrome in a child with toxic epidermal necrolysis: an interplay of unbalanced immune regulatory mechanisms. Ann Hepatol 2006; 5: 16–119. [PubMed] [Google Scholar]
- 173. Butt TF, Cox AR, Lewis H, Ferner RE. Patient experiences of serious adverse drug reactions and their attitudes to medicines: a qualitative study of survivors of Stevens‐Johnson syndrome and toxic epidermal necrolysis in the UK. Drug Saf 2011; 34: 19–328. [DOI] [PubMed] [Google Scholar]
- 174. Butt TF, Cox AR, Oyebode JR, Ferner RE. Internet accounts of serious adverse drug reactions: a study of experiences of Stevens‐Johnson syndrome and toxic epidermal necrolysis. Drug Saf 2012; 35: 159–1170. [DOI] [PubMed] [Google Scholar]
- 175. Hefez L, Zaghbib K, Sbidian E et al Post‐traumatic stress disorder in Stevens‐Johnson syndrome and toxic epidermal necrolysis: prevalence and risk factors. A prospective study of 31 patients. Br J Dermatol 2019; 180: 1206–1213. [DOI] [PubMed] [Google Scholar]