

Abstract
Key points
Small‐conductance Ca2+‐activated K+ (SK) channels expressed in ventricular myocytes are dormant in health, yet become functional in cardiac disease.
SK channels are voltage independent and their gating is controlled by intracellular [Ca2+] in a biphasic manner. Submicromolar [Ca2+] activates the channel via constitutively‐bound calmodulin, whereas higher [Ca2+] exerts inhibitory effect during depolarization.
Using a rat model of cardiac hypertrophy induced by thoracic aortic banding, we found that functional upregulation of SK2 channels in hypertrophic rat ventricular cardiomyocytes is driven by protein kinase A (PKA) phosphorylation. Using site‐directed mutagenesis, we identified serine‐465 as the site conferring PKA‐dependent effects on SK2 channel function.
PKA phosphorylation attenuates I SK rectification by reducing the Ca2+/voltage‐dependent inhibition of SK channels without changing their sensitivity to activating submicromolar [Ca2+]i.
This mechanism underlies the functional recruitment of SK channels not only in cardiac disease, but also in normal physiology, contributing to repolarization under conditions of enhanced adrenergic drive.
Abstract
Small‐conductance Ca2+‐activated K+ (SK) channels expressed in ventricular myocytes (VMs) are dormant in health, yet become functional in cardiac disease. We aimed to test the hypothesis that post‐translational modification of SK channels under conditions accompanied by enhanced adrenergic drive plays a central role in disease‐related activation of the channels. We investigated this phenomenon using a rat model of hypertrophy induced by thoracic aortic banding (TAB). Western blot analysis using anti‐pan‐serine/threonine antibodies demonstrated enhanced phosphorylation of immunoprecipitated SK2 channels in VMs from TAB rats vs. Shams, which was reversible by incubation of the VMs with PKA inhibitor H89 (1 μmol L–1). Patch clamped VMs under basal conditions from TABs but not Shams exhibited outward current sensitive to the specific SK inhibitor apamin (100 nmol L–1), which was eliminated by inhibition of PKA (1 μmol L–1). Beta‐adrenergic stimulation (isoproterenol, 100 nmol L–1) evoked I SK in VMs from Shams, resulting in shortening of action potentials in VMs and ex vivo optically mapped Sham hearts. Using adenoviral gene transfer, wild‐type and mutant SK2 channels were overexpressed in adult rat VMs, revealing serine‐465 as the site that elicits PKA‐dependent phosphorylation effects on SK2 channel function. Concurrent confocal Ca2+ imaging experiments established that PKA phosphorylation lessens rectification of I SK via reduction Ca2+/voltage‐dependent inhibition of the channels at high [Ca2+] without affecting their sensitivity to activation by Ca2+ in the submicromolar range. In conclusion, upregulation of SK channels in diseased VMs is mediated by hyperadrenergic drive in cardiac hypertrophy, with functional effects on the channel conferred by PKA‐dependent phosphorylation at serine‐465.
Keywords: Small‐conductance Ca2+‐activated K+ channels, Ventricular arrhythmia, PKA, Calcium transients, Cardiac electrophysiology
Key points
Small‐conductance Ca2+‐activated K+ (SK) channels expressed in ventricular myocytes are dormant in health, yet become functional in cardiac disease.
SK channels are voltage independent and their gating is controlled by intracellular [Ca2+] in a biphasic manner. Submicromolar [Ca2+] activates the channel via constitutively‐bound calmodulin, whereas higher [Ca2+] exerts inhibitory effect during depolarization.
Using a rat model of cardiac hypertrophy induced by thoracic aortic banding, we found that functional upregulation of SK2 channels in hypertrophic rat ventricular cardiomyocytes is driven by protein kinase A (PKA) phosphorylation. Using site‐directed mutagenesis, we identified serine‐465 as the site conferring PKA‐dependent effects on SK2 channel function.
PKA phosphorylation attenuates I SK rectification by reducing the Ca2+/voltage‐dependent inhibition of SK channels without changing their sensitivity to activating submicromolar [Ca2+]i.
This mechanism underlies the functional recruitment of SK channels not only in cardiac disease, but also in normal physiology, contributing to repolarization under conditions of enhanced adrenergic drive.
Introduction
Sudden cardiac death as a result of ventricular tachyarrhythmias remains a major cause of mortality worldwide (Benjamin et al. 2018). Enhanced triggered activity for arrhythmia under conditions such as heart failure at the cellular level has been largely ascribed to reduced repolarizing K+ currents, increased L‐type Ca2+ current (I Ca) and Na+/Ca2+ exchanger (NCX1)‐mediated depolarization, caused by untimely releases of sarcoplasmic reticulum (SR) Ca2+ by hyperactive ryanodine receptors (RyR2s) (Pogwizd & Bers, 2004; Qu & Weiss, 2006; Zima et al. 2014). Recently, small conductance Ca2+‐activated K+ (SK) channels have emerged as a promising therapeutic target because of their ability to offset depolarizing force of I Ca and I NCX and mitigate disease‐associated loss of repolarization reserve (Chang et al. 2015a; Clements et al. 2015; Terentyev et al. 2014). However, the exact role of SK channels in arrhythmogenesis and the mechanisms that regulate their function in the heart remain poorly understood.
All three SK channel isoforms (SK1‐3) encoded by the genes KCNN1, KCNN2 and KCNN3 are detected in cardiac tissue (Skibsbye et al. 2014; Tuteja et al. 2005; Xu et al. 2003). Our previous report demonstrated that two of these isoforms are present in sarcolemma of rat ventricular myocytes (VMs): SK2 and SK3 (Kim et al. 2017). SK channels exist as heterotetrameric multicomplex proteins with six transmembrane domains and they display small single channel conductances of ∼10–20 pS (Adelman et al. 2012; Tuteja et al. 2010). Because SK channels lack a classical voltage sensor, their gating is controlled primarily by intracellular [Ca2+] ([Ca2+]i) in a biphasic manner (Maylie et al. 2003; Soh & Park, 2002; Xia et al. 1998). Channel activation is conferred by calmodulin (CaM) constitutively bound to the C‐terminus, with an EC50 of ∼0.3‐1 μmol L–1 (Li N et al. 2009; Li W et al. 2009; Schumacher et al. 2001; Schumacher et al. 2004; Xia et al. 1998). In addition, higher [Ca2+]i has been reported to inhibit SK channels in a voltage‐dependent manner (Soh & Park, 2002), providing the basis for rectification of I SK.
Unlike in atria (Diness et al. 2011; Li N et al. 2009; Skibsbye et al. 2014), SK channels in VMs are dormant in health and become active in cardiac disease both in animal models and human patients (Chang et al. 2013b; Chua et al. 2011; Clements et al. 2015; Bonilla et al. 2014; Lee et al. 2013; Mahida, 2014; Ni et al. 2013). Functional recruitment of plasmalemmal SK channels can occur very rapidly. For example, contribution of SK channels to repolarization became obvious 10 min after the induction of acute myocardial infraction or 30 min of ischaemia in rat hearts (Gui et al. 2012; Tenma et al. 2018). Several hypotheses were proposed to explain this phenomenon including: (i) increased expression levels (Chang et al. 2013a; Ni et al. 2013); (ii) increased activity as a result of CaMKII phosphorylation (Mizukami et al. 2015; Tenma et al. 2018); and (iii) an increase in sensitivity to activating Ca2+ as a result of dephosphorylation of SK‐bound CaM given changes in the activities of SK‐associated protein casein kinase 2 (CK2) and protein phosphatase 2A (PP2A) (Allen et al. 2007; Bildl et al. 2004; Yang et al. 2015; Zhang et al. 2014). We previously reported that functional upregulation of plasmalemmal SK channels in VMs from hypertrophic rat hearts was not paralleled by increased expression levels (Kim et al. 2017), suggesting that post‐translational modifications may instead underlie enhanced channel activity.
Phosphoproteomic studies revealed that SK channels can be directly phosphorylated by protein kinase A (PKA) at N‐terminal serine‐136, within the CaM‐binding domain at serine‐465, and at C‐terminal serine‐568 to serine‐570 (for rat SK2) (Blom et al. 1999; Ren et al. 2006). Interestingly, recent studies suggest that β‐adrenergic stimulation evokes apamin‐sensitive repolarizing current in ventricles of optically mapped ex vivo rat and rabbit hearts. (Chen et al. 2018; Kamada et al. 2018). By contrast, earlier experiments using heterologous systems suggest that PKA phosphorylation negatively affects SK function either by reducing channel activity in HEK cells (SK3) (Clarysse et al. 2014) or by interfering with surface localization of SK2 in COS7 cells (Ren et al. 2006).
To gain insight into the mechanisms of upregulation of SK channels in VMs from diseased hearts and to determine the role of PKA in this process in particular, we used a clinically relevant rat model of cardiac arrhythmia with hypertrophy induced by thoracic aortic banding (TAB) (Kim et al. 2017; del Monte et al. 2002). Using ex vivo optical mapping and single cell electrophysiology in conjunction with confocal Ca2+ imaging, we found that PKA‐dependent phosphorylation is the major determinant of functional upregulation of the channels via attenuation of voltage‐dependent inhibition by [Ca2+]i. Furthermore, using cultured rat VMs overexpressing wild‐type (WT) and mutant SK2 channels, we have identified the site within the channel responsible for functional upregulation driven by PKA‐mediated phosphorylation, namely serine‐465.
Methods
Ethical approval
All procedures involving animals were approved by The Rhode Island Hospital Institutional Animal Care and Use Committee and conformed with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85‐23, revised 2011) and the policies and regulations set out in the editorial in The Journal of Physiology and Experimental Physiology by Grundy (2015). During procedures, all steps were taken to minimize animals pain and suffering.
Male Sham and TAB Sprague–Dawley rats (RGD catalogue no. 10395233, RRID:RGD_10395233) were purchased from Charles River Laboratories (Wilmington, MA, USA). Animals were shipped 5–7 days after surgery and acclimatized for 3–4 weeks in the Rhode Island Hospital animal facility. Experiments were performed 4–5 weeks after aortic banding procedure. Animals were fed ad libitum. In total, 29 Sham rats and 30 TAB rats were used for the present study.
In vivo cardiac function
Male Sham and TAB Sprague–Dawley rats were sedated with continuous isoflurane (1–3%) via induction chamber and nose cone and then the chest was shaved. Transthoracic M‐mode and two‐dimensional echocardiography was performed on a Vevo® 2100 Imaging System (Fujifilm VisualSonics, Inc., Toronto, ON, Canada). The analysis included recording the dimensions of the left ventricle, as well as the total heart weight/body weight ratio. The entire procedure took 15–30 min and resulted in no pain to the animal. During the procedure, animals were closely monitored for any signs of distress, temperature and heart rate changes via an ECG. Rats were then monitored during recovery from anaesthesia for 1–2 h to ensure normal movement and activity before being returned to normal housing.
Myocyte isolation and cell culture
Whole hearts and VMs were isolated from male Sham and TAB Sprague–Dawley rats. Rats were injected with 120 mg kg–1 pentobarbital i.p. as a terminal procedure. The heart was removed from the rats via bilateral thoracotomy, mounted on optical mapping set up or a Langendorff apparatus and retrogradely perfused with Tyrode solution containing collagenase II (Worthington Biochemical Corp. Lakewood, NJ, USA) at 37°C. VMs were isolated as described previously (Terentyev et al. 2014), before plating onto laminin‐coated coverslips. VMs were used within 8 h of isolation. For experiments with rat SK2 (rSK2) channel overexpression, adult rat VMs were isolated from 9–12‐week old Sprague–Dawley male rats (RGD catalogue no. 70508; RRID:RGD_70508) from Harlan Laboratories (Indianapolis, IN, USA), as described previously (Terentyev et al. 2014). Myocytes were plated in 24‐well plates on laminin‐coated glass coverslips, cultured in serum‐free medium 199 (Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 25 mmol L–1 NaHCO3, 10 mmol L–1 Hepes, 5 mmol L–1 creatine, 5 mmol L–1 taurine, 10 U mL–1 penicillin, 10 μg mL–1 streptomycin and 10 μg mL–1 gentamycin (pH 7.3). Unattached cells were removed after 1 h and the remaining VMs were infected with adenoviruses at a multiplicity of infection of 10 for SK channel and dominant‐negative mutant phospholamban virus (dnPLB) constructs. Myocytes were cultured at 37°C in 95% air/5% CO2 for 36–48 h before the analysis.
Construction of WT and mutant SK2 adenoviruses
Adenovirus carrying recombinant WT rSK2 sequence was constructed as described previously (Terentyev et al. 2014; Kim et al. 2017), utilizing the ViraPower Gateway expression system (Thermo Fisher Scientific). Briefly, the coding region of rat SK2 sequence was cloned into pENTR™ 1A vector, and then recombined into pAd/CMV/V5‐DEST™ with the LR recombination reaction. Sequence‐verified plasmid was digested with restriction enzyme PacI, before transfection into HEK293A cells (RRID:CVCL_6910) using Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA). Titre of amplified adenoviral stocks was determined using the Adeno‐X qPCR Titration Kit (Takara Bio USA, Inc., Mountain View, CA, USA). To introduce S136D into rSK2 construct, we applied site‐directed mutagenesis using the Quik Change Site‐Directed Mutagenesis Kit (Agilent Technologies Inc., Santa Clara, CA, USA). To introduce phosphomimetic mutation S465D and S465A into rat SK2 construct, we applied site‐directed mutagenesis using the Q5® Site‐Directed Mutagenesis Kit (New England Biolabs Inc., Ipswich, MA, USA) in accordance with the manufacturer's instructions. dnPLB with K3E/R14E mutations was used to enhance SERCa2a function and restore Ca2+ transient amplitude (a kind gift from Dr M. Ziolo, The Ohio State University, Columbus, OH, USA) (Ziolo et al. 2005). A virus with non‐coding sequence following CMV was used as a control.
Western blotting and immunoprecipitation
The antibodies used were SK2 (Sigma, St Louis, MO, USA; catalogue no. SAB2501396, Lot#9678P1, RRID:AB_10961767; dilution 1:2000; Alomone Labs, Jerusalem, Israel; catalogue no. APC‐028, Lot#APC028AN1702, RRID:AB_2040126; dilution 1:1000), GAPDH (Abcam, Cambridge, MA, USA; catalogue no. ab8245, Lot#GR232949‐15, RRID:AB_2107448; dilution 1:5000), calmodulin (CaM) (Abcam; catalogue no. ab45689, Lot#gr291267‐5, RRID:AB_1946552; dilution 1:1000), phospho‐calmodulin (phosphoCaM) (Abcam; catalogue no. ab61001, Lot#GR10051348, RRID:AB_942205; dilution 1:1000), casein kinase 2a E‐2 (CK2) (Santa Cruz Biotechnology, Dallas, TX, USA; catalogue no. sc‐365787, Lot#E2411, RRID:AB_10844012; dilution 1:1000), protein phosphatase 2A C subunit (PP2A‐C) (Millipore, Burlington, MA, USA; catalogue no. 05‐421, Lot#JBC1856024, RRID:AB_309726; dilution 1:1000), Ca2+/calmodulin‐dependent kinase II (CaMKII) (Thermo Fisher Scientific; catalogue no. MA1‐048, Lot#CVP107, RRID:AB_325403; dilution 1:1000), phospho‐Ca2+/calmodulin‐dependent kinase II (phospho‐CaMKII) (Thermo Fisher Scientific; catalogue no. PA1‐4614, Lot#196‐120, RRID:AB_2259386; dilution 1:1000 dilution), phospho‐PKA substrate (Cell Signaling Technology, Danvers, MA, USA; catalogue no. 9621, Lot#20, RRID:AB_330304; dilution 1:2000), phospho‐serine/threonine (Abcam; catalogue no. ab17464, Lot#GR3209832‐1, RRID:AB_443891; dilution 1:1000) and phospholamban [PLB; (2D12)] (a kind gift from Dr Z. Chen, Indiana University School of Medicine, Indianapolis, IN, USA; Chen et al. 2007; dilution 1:1000). Custom polyclonal rat phospho‐S465‐SK2 site‐specific antibodies were generated by YenZym Antibodies, LLC (San Francisco, CA, USA) by immunization of rabbits with the peptide LRpSVKMEQRKLNDQC. Antibodies were affinity‐purified by enzyme‐linked immunosorbent assay.
Freshly isolated or cultured rat VMs were lysed in lysis buffer from Cell Signaling (catalogue no. 9803S), supplemented with phosphatase (Calbiochem, San Diego, CA, USA; catalogue no. 524625) and protease inhibitor cocktails (Sigma; catalogue no. P8340) as described previously (Terentyev et al. 2014). Samples (20–30 μg of proteins) were resolved on a 4–20% gel via SDS‐PAGE, transferred onto nitrocellulose membranes, and probed with antibodies specific for these proteins and subsequently probed with a goat anti‐mouse secondary (Promega, Madison, WI, USA; catalogue no. W4021, Lot#0000292575, RRID:AB_430834; dilution 1:10,000 ), goat anti‐rabbit secondary (Promega; catalogue no. W4011, Lot#0000292577, RRID:AB_430833; dilution 1:10,000) or donkey anti‐goat secondary antibodies (Promega; catalogue no. V805A, Lot#0000265854, RRID:AB_430838; dilution 1:10,000). Blots were developed with ECL (Bio‐Rad Laboratories, Hercules, CA, USA; catalogue no. 1705061) and quantified and analysed using ImageJ (NIH, Bethesda, MD, USA; RRID: SCR_003070) and Origin, version 8 (OriginLab Corp., Northampton, MA, USA; RRID:SCR_014212).
For immunoprecipitation, freshly isolated rat VMs were lysed using cell lysis buffer from Cell Signaling (catalogue no. 9803S), supplemented with phosphatase (Calbiochem; catalogue no. 524625) and protease inhibitor cocktails (Sigma; catalogue no. P8340). A 2 h long immunoprecipitation of SK2 was performed at 4°C using a Catch and Release v2.0 Kit (Millipore; catalogue no. 17‐500) in accordance with the manufacturer instructions using anti‐SK2 antibody (Sigma; catalogue no. SAB2501396, Lot#9678P1, RRID:AB_10961767; 5 μg of antibody) and a negative control antibody comprising normal mouse IgG (Santa Cruz Biotechnology; catalogue no. sc‐2025, Lot#J2015, RRID:AB_737182; 5 μg of antibody). Samples were analysed by immunoblotting.
For the assessment of native protein complexes using blue native polyacrylamide gel electrophoresis (BN‐PAGE), we used freshly isolated VMs suspended in buffer containing 225 mmol L–1 mannitol, 70 mmol L–1 sucrose, 10 mmol L–1 Hepes and 1 mmol L–1 EGTA (pH 7.4). The whole‐cell suspension was placed in a pre‐cooled 5 mL Wheaton™ Potter‐Elveheim Tissue Grinder (Fisher Scientific, Hampton, NH, USA; catalogue no. 22‐290067). The cells were homogenized and homogenate was centrifuged at 700 g for 10 min. The pellet consisting of nuclei and cell debris was discarded and the supernatant was then centrifuged at 17,000 g for 15 min. The brown pellet was considered as mitochondrial fraction and discarded. The supernatant was used to precipitate membrane fraction at 100,000 g for 1 h. Samples were then solubilized using the NativePAGE™ Sample Prep Kit (Invitrogen; catalogue no. BN2008, Lot#1619815) in accordance with the manufacturer's instructions. Processed samples were resolved on NativePAGE™ 4–16% Bis‐Tris Protein Gels, 1.0 mm, 15‐well (Invitrogen; catalogue no. BN1004BOX, Lot#18061260) by SDS‐PAGE at 150 V for 1 h and 250 V for 1.5 h. Samples were transferred onto nitrocellulose membranes before being probed with antibodies, as described above. The primary antibodies used were SK2 (Sigma; catalogue no. SAB2501396, Lot#9678P1, RRID:AB_10961767; dilution 1:2000), Cav1.2α1c L‐type Ca2+ channel (LTCC) subunit (Alomone Labs; catalogue no. ACC‐003, Lot#ACC013AN0502, RRID:AB_2039771; dilution 1:1000), Na+/K+‐ATPase (NKA; Abcam; catalogue no. ab76020, Lot#GR3184452‐8, RRID:AB_1310695; dilution 1:1000). The secondary antibodies used are described above.
In experiments utilizing site‐specific phospho‐SK2‐S465 antibodies (dilution 1:500), maximum phosphorylation for normalization was achieved by incubation of TAB and Sham VMs with β‐adrenergic agonist isoproterenol (ISO) (1 μmol L–1) and phosphatase inhibitor (1 μmol L–1) calyculin A for 15 min.
Proximity ligation assay and immunofluorescence
The Duolink® proximity ligation assay (PLA) (Duolink® In Situ Detection Reagents Orange, Sigma; catalogue no. DUO92007, Lot#SLBV3905) allows for the detection of proteins that are colocalized <40 nm of each other. Oligonucleotide labelled secondary antibodies or PLA probes generate signal only when bound in close proximity to two primary antibodies that have bound to the sample in close proximity.
Freshly isolated VMs were plated on laminin‐coated coverslips and prepared for the PLA and immunofluorescence by fixing with 4% paraformaldehyde and permeabilized with 0.2% Triton X‐100/PBS (pH 7.2) containing 1% BSA. The PLA probe protocol was followed in accordance with the manufacturer's instructions. Briefly, samples were blocked and washed before incubation with primary antibodies for 1 h at room temperature. Primary antibodies used were SK2 (Sigma; catalogue no. SAB2501396, Lot#9678P1, RRID:AB_10961767; dilution 1:2000) and Cav1.2α1c subunit (Alomone Labs; catalogue no. ACC‐003, Lot#ACC013AN0502, RRID:AB_2039771; dilution 1:1000) or RyR2 (Thermo Fisher Scientific; catalogue no. MA3‐916, Lot#SD241387, RRID:AB_2183054; dilution 1:1000). Next, samples were washed and PLA probes added and for ligation they were incubated for 1 h at 37 °C. PLA probes used were Duolink® In Situ PLA® Probe Anti‐Goat PLUS (Sigma; catalogue no. DUO92003, Lot# SLBW7564; dilution in accordance with the manufacturer's instructions) and Duolink® In Situ PLA® Probe Anti‐Rabbit MINUS (Sigma; catalogue no. DUO92005, Lot#SLBZ4516; dilution in accordance with the manufacturer's instructions). PLA probes were then amplified in accordance with the manufacturer's instructions using a DNA ligase at 37 °C, and then samples were immediately processed for imaging. Samples were also probed for RyR2. Primary antibody used was RyR2 (Thermo Fisher Scientific; catalogue no. MA3‐916, Lot#SD241387, RRID:AB_2183054; dilution 1:1000) or anti‐calsequestrin (CSQ) (Affinity Bioreagents, Golden, CO, USA; catalogue no. PA1‐913, Lot#387‐112, RRID:AB_2071461; dilution 1:5000). Secondary antibody used was rabbit anti‐mouse IgG (H+L) cross‐adsorbed secondary antibody, Alexa Fluor 488 (Thermo Fisher Scientific; catalogue no. A‐11059, Lot#1567256, RRID:AB_2534106; dilution 1:1000).
Images were acquired using a SP5 II confocal system (Leica Microsystems, Wetzlar, Germany) equipped with a 63 × 1.4 numerical aperture oil objective using the 488 nm line of the argon ion laser and the 546 nm line of the HeNe laser for excitation. Emitted fluorescence was collected at the wavelengths 500–530 nm and 560–660 nm. The signal from each detected pair of PLA probes was visualized as a fluorescent red spot, and was quantified as PLA puncta μm–2 in ImageJ (NIH; RRID: SCR_003070). Manders overlap coefficients were calculated to assess the level of colocalization between SK2‐LTCC PLA puncta and RyR2 in ImageJ (NIH; RRID: SCR_003070), with an M1 coefficient of 1.0 indicating complete colocalization of red to green images and an M2 coefficient of 1.0 indicating complete colocalization of green to red images.
Whole‐cell patch clamp of VMs
Whole‐cell patch clamp recordings of currents and membrane potential were carried out using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA) filtered at 2 kHz and digitized at a sampling rate of 5 kHz as described previously (Terentyev et al. 2014). To record SK channel currents from rat VMs, depolarizing voltage steps from a holding potential of –40 mV at 10 mV intervals were applied at 2 s intervals under voltage clamp at room temperature. For recordings from rat VMs overexpression SK2, voltage steps were from a holding potential of –45 mV. Action potentials (APs) were elicited by short current pulses applied at 1.2× threshold under current clamp. Bath solution (pH 7.3) contained 140 mmol L–1 NaCl, 5.4 mmol L–1 KCl, 1 mmol L–1 MgCl2, 1 mmol L–1 CaCl2, 10 mmol L–1 Hepes and 5.6 mmol L–1 glucose. Recording electrodes were 2–4 MΩ containing nominally Ca2+ free pipette solution (pH 7.2): 90 mmol L–1 K‐aspartate, 50 mmol L–1 KCl, 5 mmol L–1 Mg‐ATP, 5 mmol L–1 NaCl, 1 mmol L–1 MgCl2, 0.1 mmol L–1 Tris‐GTP, 10 mmol L–1 Hepes and 0.1 mmol L–1 Rhod‐2 K+‐salt (Thermo Fisher Scientific). Free [Mg2+] was 1.37 mmol L–1 (Maxchelator) (Bers et al. 2010). Usually, the series resistance was compensated 40–60%. Apamin (APA), a selective SK1, 2 and 3 polypeptide inhibitor (IC50 < 10 nmol L–1; Alomone Labs) was used to identify SK currents. UCL‐1684 is a non‐peptidic voltage independent blocker of SK channels (IC50 < 10 nmol L–1) (Hosseini et al. 2001; Rosa et al. 1998). For β‐adrenergic stimulation, VMs were treated with ISO (100 nmol L–1). To inhibit PKA, synthetic peptide PKA Inhibitor 14–22 Amide (PKI) (Calbiochem; catalogue no. 476485) was added to pipette solution (1 μmol L–1).
Confocal imaging and estimation of [Ca2+]i
During whole‐cell voltage clamp experiments, intracellular Ca2+ imaging was simultaneously performed at room temperature using a SP5 II confocal microscope (Leica) equipped with 63× 1.4 numerical aperture oil objective in line‐scan mode at a rate of 5 ms per line, synchronized with the electrophysiological setup. Rhod‐2 was excited using the 543 nm line of the HeNe laser and fluorescence emission was collected at the wavelengths 560–660 nm. Dynamical Rhod‐2 fluorescence signal during Ca2+ transient was converted to [Ca2+]i (Cheng et al. 1993), using the equation; [Ca2+]i = K d × (F – F min)/(F max – F), where K d Rhod‐2 = 1.58 μmol L–1 (Escobar et al. 1997; Trafford et al. 1995) and F min = F max/15. F max was determined by breaking the patch pipette and measuring the Rhod‐2 fluorescence as the dye was exposed to the 1 mmol L–1 Ca2+ bath solution. When there was a change in baseline fluorescence during the experiment, F/F 0 was used in place of F in the calculation of [Ca2+]i. Submembrane [Ca2+] ([Ca2+]sm) was calculated based on (Weber et al. 2002). Once [Ca2+]i was determined, the data were smoothed by the Savitzky–Golay method with 5 points of window. Then the data were differentiated and multiplied by a diffusion constant γ of 110 ms. These data were added to the [Ca2+]i data to obtain the rise and peak of [Ca2+]sm. [Ca2+]i and [Ca2+]i + γ × d[Ca2+]i/dt were plotted and the decay fit with a single exponential such that it followed the decay of [Ca2+]i (Fig. 13).
Figure 13.
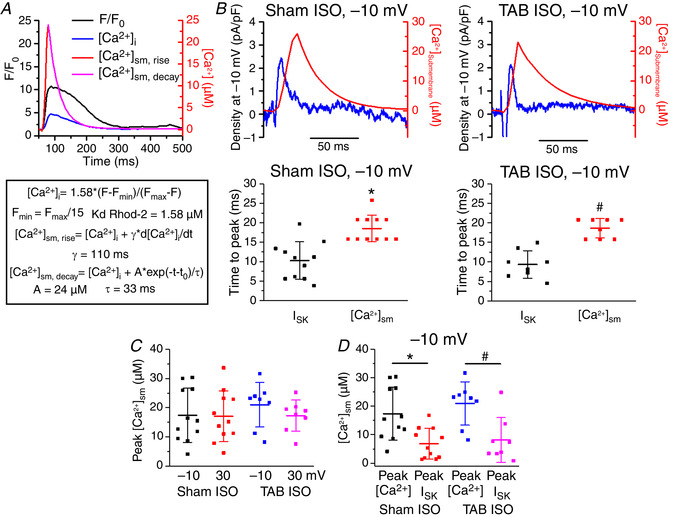
ISK peaks, whereas submembrane [Ca2+] continues to rise, indicating an inhibition of ISK by Ca2+
A, sample traces and equations for calculating submembrane Ca2+ concentration ([Ca2+]sm) based on calculations used previously (Weber et al. 2002). B, top: representative traces of I SK (blue) and [Ca2+]sm (red) at a –10 mV voltage step under ISO stimulation (100 nmol L–1). Bottom: plots of the time to peak for I SK and [Ca2+]sm in Sham and TAB myocytes. Line indicates mean ± SD. * P = 1.7 × 10−4, n = 11, N = 8, Student's t test. #P = 2.4 × 10−5, n = 8, N = 7, Student's t test. C, plot of peak [Ca2+]sm at −10 and 30 mV voltage steps under ISO stimulation (100 nmol L–1 for 3 min). Line indicates mean ± SD. There is no significant difference in any of the peak [Ca2+]sm values, P = 1, one‐way ANOVA, with a Bonferroni post hoc test. D, plot comparing the [Ca2+]sm at the peak [Ca2+] to [Ca2+]sm at peak I SK for Sham and TAB myocytes under ISO stimulation. Line indicates mean ± SD. * P = 0.001, n = 11, N = 8, paired Student's t test. #P = 0.003, n = 8, N = 7, paired Student's t test.
Ex vivo optical mapping
Beating hearts were harvested from anaesthetized Sham and TAB rats via thoracotomy and were retrogradely perfused through the aorta in a Langendorff perfusion system (Radnoti Glass Technology, Monrovia, CA, USA) with 130 mmol L–1 NaCl, 24 mmol L–1 NaHCO3, 1.0 mmol L–1 MgCl2, 5.0 mmol L–1 KCl, 1.2 mmol L–1 NaH2PO4, 5 mmol L–1 dextrose and 1 mmol L–1 CaCl2 (pH 7.4), gassed with 95% O2 and 5% CO2. Constant flow perfusion was set to 10 mL min–1 with a peristaltic pump. Hearts were placed in a water‐heated chamber to maintain temperature at 37 ± 0.2°C and then 5 μmol L–1 blebbistatin was added to perfusate to reduce movement artefact. Hearts were stained with voltage sensitive indicator di‐4‐ANNEPS, using 20 μL of stock solution (1 mg mL–1 of DMSO) delivered through a bubble trap, above the aortic cannula. The ECGs were continuously monitored with a Powerlab system (AD Instruments, Sydney, NSW, Australia; RRID:SCR_001620). The optical apparatus has been described previously (Kim et al. 2015). Fluorescence images of APs were recorded from the anterior surface of the heart using a CMOS camera (100 × 100 pixels, 2000 frames s–1, 1.5 × 1.5 cm2 field of view; Ultima‐L; SciMedia, Costa Mesa, CA, USA). Hearts were stimulated with 150 ms cycle length and perfused with 50 nmol L–1 ISO and/or 10 nmol L–1 APA. Action potential durations (APDs) and conduction velocities were measured using dF/dt for activation and 75% of AP amplitude for repolarization by means of digital image analysis routines.
Statistical analysis
Statistical analysis of electrophysiological, biochemical and Ca2+ imaging data was performed using Origin 8.0 (OriginLab Corp.; RRID:SCR_014212). Data are presented as the mean ± SD. Statistical significance between groups were performed using Student's t test (paired and unpaired) and one‐way ANOVA with a Bonferroni post hoc test where appropriate. P < 0.05 was considered statistically significant.
Results
Serine/threonine phosphorylation of SK2 channels is enhanced in hypertrophic rat ventricular myocytes
To investigate the regulatory mechanisms that control SK function, we used rats with hypertrophy induced by TAB. In this well‐established model, ligation of the ascending aorta induces pressure overload and the development of cardiac hypertrophy (del Monte et al. 2002; Wei et al. 2010). As determined by echocardiography, Fig. 1A–D shows that the left ventricular posterior wall thickness is significantly increased in the hearts of TAB rats 4 weeks after surgery, both in systole and diastole. The total heart weight/body weight ratio was 3.81 ± 0.09 (n = 29) and 4.61 ± 0.11* (n = 30) mg g–1 for Sham and TAB respectively (* P < 0.05, Student's t test, indicating significant heart enlargement in TAB animals. We have also previously demonstrated that, when challenged with β‐adrenergic agonist ISO (50 nmol L–1), 100% of ex vivo TAB rat hearts develop ventricular tachycardia/ventricular fibrillation compared to 15% of Shams (Kim et al. 2017).
Figure 1.
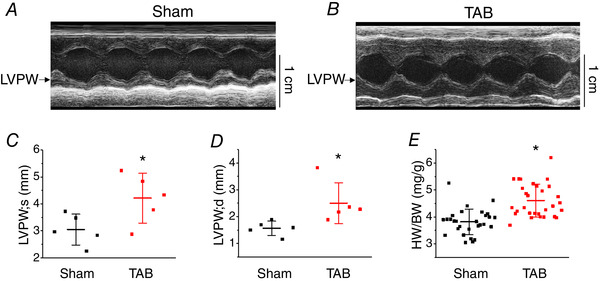
Echocardiographic properties of Sham and TAB rat hearts
Representative echocardiographic M‐mode images in age‐matched Sham (A) and TAB rats (B) at the level of mitral valve. Left ventricular posterior wall (LVPW) dimensions of TAB rats are significantly increased in comparison to Sham, both in systole (C) and diastole (D). Mean ± SD of data indicated by line. N = 5 per group, * P = 0.04 (LVPW; s) and * P = 0.05 (LVPW; d), Student's t test. (E) Heart weight to body weight ratio of rats used in the present study. Mean ± SD of data indicated by line, Sham, N = 29; TAB, N = 30. * P < 0.001, Student's t test.
To gain insights into potential post‐translational changes in hypertrophy, we performed western blot analysis using immunoprecipitated SK2 complexes from freshly isolated Sham and TAB VMs (Fig. 2). Consistent with our previous report where anti‐SK2 antibodies were validated using shRNAs (Kim et al. 2017), SK2 protein bands were detected at ∼100 kDa (Fig. 2A ), which is higher than the predicted molecular weight (62.2 KDa). Furthermore, protein bands at ∼100 kDa were not visible after application of anti‐SK2 peptide.
Figure 2.

Enhanced serine/threonine phosphorylation of SK2 in VMs from TAB rat hearts
A, SK2 channels immunoprecipitated from Sham and TAB VMs, with a band appearing at ∼100 kDa, as indicated by an arrow. This band is not present when anti‐SK2 and SK2 peptide are applied. B, SK2 channels immunoprecipitated from TAB VMs demonstrate increased serine/threonine phosphorylation. C, plot of optical density (OD) normalized to SK2 levels for phosphorylated‐serine/threonine SK2. Mean ± SD of data indicated by line. * P = 0.008 (P‐serine/threonine SK2), Student's t test. D and E, representative western blots and plots of OD normalized to SK2 levels for CK2, and PP2A‐C. Phosphorylated‐CaM (P‐CaM) was normalized to CaM in Sham and TAB rats, respectively. Mean ± SD of data indicated by line.
Probing with anti‐pan‐phospho‐serine/threonine antibody revealed significantly increased phosphorylation of SK2 channels in TABs vs. Shams (normalized optical density of Sham 100% ± 79.20% vs. TAB 1128% ± 602.18%, n = 6 per group) (Fig. 2B and C ).
It was suggested that Ca2+ sensitivity of SK channels increases with dephosphorylation of associated CaM at the threonine‐79 site as a result of decreased levels of SK‐bound kinase CK2, or increased levels of phosphatase PP2A (Bildl et al. 2004; Yang et al. 2015; Zhang et al. 2014). Using anti‐phospho‐T79 CaM antibody, we did not find significant differences in phosphorylation of SK2‐bound CaM from TAB VMs compared to that from Sham VMs (Fig. 2D and E ). Furthermore, we did not find significant changes in the abundance of SK‐tethered kinase CK2, or opposing PP2A‐C. These results imply CaM dephosphorylation probably does not serve as a mechanism for enhanced SK activity in our model of cardiac hypertrophy.
Next, we assessed potential changes in activities of two major serine/threonine kinases CaMKII and PKA in TABs vs. Shams using whole‐cell lysates from freshly isolated VMs. Experiments with anti‐phospho‐PKA substrate antibody (Fig. 3A and B ) supported enhanced activity of the kinase in TABs vs. Shams. Figure 3C and D shows enhanced phosphorylation of CaMKII at threonine‐286, an indicator of CaMKII activation in hypertrophy (Tenma et al. 2018). To test which of these two kinases is responsible for SK hyperphosphorylation in hypertrophy, we incubated myocytes from TABs with pharmacological inhibitors of CaMKII, KN93 (500 nmol L–1) and PKA, H89 (1 μmol L–1) for 30 min. Subsequent probing of immunoprecipitated SK2 channels with anti‐serine/threonine antibodies (Fig. 3E and F ) revealed a significant H89‐induced reduction of phosphorylation, whereas KN93 was ineffective. This suggests SK2 phosphorylation at serine/threonine sites is PKA‐ and not CaMKII‐dependent, which prompted us to next focus on the functional effects of PKA‐mediated phosphorylation on the channels.
Figure 3.

Serine/threonine phosphorylation of SK2 in TAB rat hearts is PKA‐dependent
A, representative western blot demonstrating enhanced PKA‐dependent phosphorylation in TAB CMs. B, plot of optical density (OD) for phosphorylated‐PKA substrate normalized to GAPDH in Sham and TAB rat hearts, respectively. * P = 0.048, Student's t test. C, representative western blots demonstrating enhanced phosphorylation of CaMKII in VMs form TAB rats. D, plot of OD for phosphorylated‐CaMKII normalized to CaMKII in Sham and TAB rats. * P = 0.01, Student's t test. E, serine/threonine phosphorylation of immunoprecipitated SK2 in TAB hearts is reversible by inhibition of PKA (H89, 1 μmol L–1) but not inhibition of CaMKII (KN93, 500 nmol L–1). F, plot of OD for phosphorylated‐serine/threonine SK2 in TAB rats normalized to SK2 under normal conditions or treated with KN93 to block CaMKII activity or H89 to block PKA activity. * P = 0.03, one‐way ANOVA. BL, baseline. For all plots, mean ± SD of data indicated by line.
SK current in hypertrophic rat ventricular myocytes with preserved Ca2+ cycling
To isolate the SK current in TAB VMs, patch clamped VMs were held at −40 mV to inactivate Na+ and most of voltage‐dependent K+ currents. Intracellular [Ca2+] was measured simultaneously using line‐scan laser confocal imaging with 100 μmol L–1 Ca2+ indicator Rhod‐2 in the pipette solution. Under these conditions, integral currents upon depolarizing wsteps at 10 mV increments are predominantly carried by LTCCs and SK channels that lack voltage‐dependent inactivation. Figure 4A demonstrates representative recordings of integral current before (black traces) and after application of specific peptide SK inhibitor APA (100 nmol L–1 for 3 min, red) (Stocker, 2004). The SK current (I SK) was obtained by subtraction of traces before and after channel inhibition by APA. Figure 4B shows superimposed traces of I SK (blue), I Ca (black, i.e. residual integral current after APA) and I Ca‐induced cytosolic Ca2+ transient derived from confocal line‐scan imaging recording of Rhod‐2 signal (in red) evoked by a depolarizing step to −20 mV. As seen in Fig. 4B , the APA‐sensitive current rapidly activates and inactivates when [Ca2+]i still continues to rise, confirming biphasic regulation of SK channels by Ca2+ in native myocytes. Furthermore, the current–voltage (I–V) relationship data presented in Fig. 4C demonstrates that I SK peaks at −20 mV (left, black) and declines at higher voltages, whereas amplitudes of both I Ca and Ca2+ transients continue to rise (Fig. 4C , centre and right, respectively). These data suggest that, in native VMs, SK channels activated by submicromolar [Ca2+] during Ca2+ transient can be effectively inhibited in a voltage‐dependent manner when [Ca2+]i reaches supramicromolar concentrations.
Figure 4.
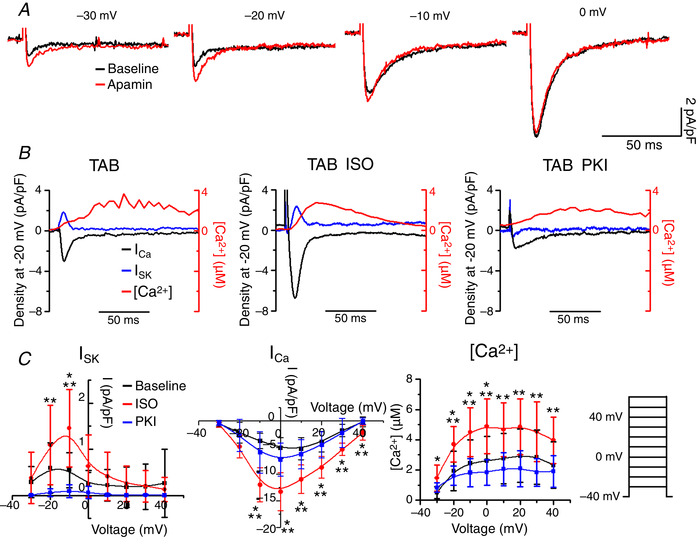
PKA inhibition alleviates ISK in TAB rat cardiomyocytes
A, representative traces of integral current before and after APA (100 nmol L–1) recorded at depolarizing steps with 10 mV intervals from HP ‐40 mV. B, representative superimposed traces of I SK (blue), I Ca (black) and [Ca2+]i transient (red) recorded in TAB cells depolarized to −20 mV. I SK is enhanced by β‐adrenergic stimulation with ISO (100 nmol L–1 for 3 min, centre) and decreased by PKA inhibitor PKI (1 μmol L–1) in the pipette solution. C, pooled mean ± SD I–V and peak [Ca2+]/V relationships for (B), with TAB cells at baseline (black), treated with ISO (red) and PKI (blue). n = 6–9, N = 5–8. I SK: * P = 0.01 (−10 mV) vs. TAB baseline. ** P = 0.02 (−20 mV); P = 7 × 10−4 (−10 mV) vs. TAB PKI. I Ca: * P = 5.9 × 10−5 (−10 mV); P = 6.1 × 10−5 (0 mV); P = 5.7 × 10−4 (10 mV); P = 6.3 × 10−4 (20 mV), P = 0.002 (30 mV); P = 0.004 (40 mV) vs. TAB baseline. ** P = 0.003 (−10 mV); P = 0.003 (0 mV); P = 0.007 (10 mV); P = 0.004 (20 mV); P = 0.01 (30 mV); P = 0.004 (40 mV) vs. TAB PKI. [Ca2+]: * P = 0.006 (−30 mV); P = 0.009 (−20 mV); P = 0.02 (−10 mV); P = 0.01 (0 mV) vs. TAB baseline. ** P = 0.004 (−20 mV); P = 0.004 (−10 mV); P = 0.002 (0 mV); P = 0.01 (10 mV); P = 0.01 (20 mV); P = 0.02 (30 mV); P = 0.03 (40 mV) vs. TAB PKI, one‐way ANOVA with a Bonferroni post hoc test.
Next, we aimed to test whether changes in PKA activity can modulate I SK. Incubation of TAB VMs with the β‐adrenergic agonist ISO (100 nmol L–1 for 3 min) increased I SK amplitude and shifted the I–V curve to the right in parallel to an increase in I Ca and Ca2+ transients (Fig. 4B , centre, and Fig. 4C , red lines). By contrast, inhibition of PKA by the specific peptide inhibitor PKI (1 μmol L–1) introduced into patch‐pipette solution diminished I SK in TAB VMs without significantly altering I Ca and Ca2+ transient amplitudes (Fig. 4B , right, and Fig. 4C , blue lines). These data strongly suggest that PKA phosphorylation of SK channels in VMs from TABs positively modulates their activity, possibly lessening the voltage‐dependent inhibition of the channels that occurs at higher concentrations of intracellular Ca2+.
Although Yu et al. 2014 previously demonstrated that apamin at concentrations of up to 500 nmol L–1 does not affect Na+, Ca2+ and other major K+ currents, concern remains that 100 nmol L–1 apamin can exert non‐specific effects. To address this issue, we used the non‐peptidic inhibitor of SK channels UCL‐1684 (Hosseini et al. 2001; Rosa et al. 1998) to isolate I SK in TAB myocytes. Using 1 μmol L–1 UCL‐1684, we obtained results similar to those with 100 nmol L–1 APA (Fig. 5).
Figure 5.

SK current in TAB rat VMs obtained by application of UCL‐1684
A, representative superimposed traces of I SK (blue), I Ca (black) and [Ca2+]i transient (red) recorded in TAB VMs depolarized to −25 mV from −45 holding potential. I SK was obtained after subtraction of the current after application of 1 μmol L–1 UCL‐1684. Stimulus voltage protocol (right). B, pooled mean ± SD I–V and peak [Ca2+]/V relationships for (A). n = 5, N = 5.
Current clamp experiments demonstrated that APA prolongs APD in myocytes from TABs paced at stimulation frequency 0.5 Hz under basal conditions and in the presence of ISO (100 nmol L–1) (Fig. 6A ). These data confirm that, in disease, SK channels contribute to repolarization (Chua et al. 2011; Skibsbye et al. 2014) Similarly, in optically mapped ex vivo TAB hearts stained with voltage sensitive dye di‐4‐ANNEPS, 10 nmol L–1 APA prolonged APD under basal conditions (Fig. 6B ). Unfortunately, we were unable to assess potential APA‐dependent changes in APD under β‐adrenergic stimulation because, in all of the hearts studied, perfusion with ISO evoked ventricular tachycardia/ventricular fibrillation, which is consistent with our previous report (Kim et al. 2017).
Figure 6.
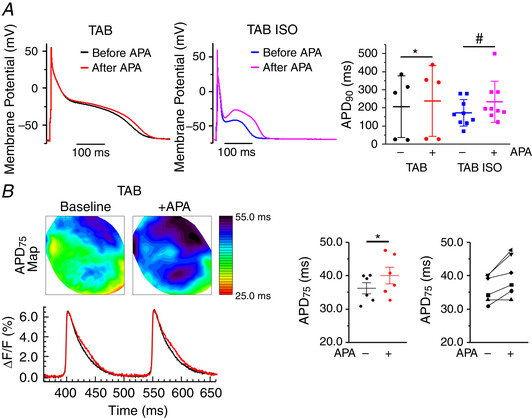
Apamin prolongs action potentials in TAB rat VMs and TAB rat whole heart
A, representative current clamp traces in TAB rat VMs under baseline (left) or ISO (100 nmol L–1 for 3 min) (right) before and after treatment with apamin (100 nmol L–1 for 3 min). Pooled mean ± SD APD90. n = 5–9, N = 4–7, * P = 0.04, #P = 0.03, Student's t test. B, representative APD maps before and after APA (10 nmol L–1) recorded under basal conditions and in the presence of β‐adrenergic agonist ISO (50 nmol L–1) and the corresponding APD profile. Right, Pooled data for APD75. * P < 0.05, paired Student's t test, N = 6.
β‐adrenergic stimulation evokes SK current in Sham rat VMs
It has remained unclear why APD and currents in healthy VMs from humans and various animal models are insensitive to APA, despite immunodetection of endogenous SK channels (Bonilla et al. 2014; Chua et al. 2011; Gui et al. 2012; Hsieh et al. 2013; Nagy et al. 2011). We reasoned that, if PKA‐dependent phosphorylation does indeed play a major role in the modulation of SK channel function, we should be able to evoke SK current to affect APD by β‐adrenergic stimulation in freshly isolated VMs from Sham hearts. As shown in Fig. 7A and B , application of ISO (100 nmol L–1 for 3 min) increased I Ca and Ca2+ transients and evoked I SK in VMs from Sham rat hearts, which was otherwise undetectable under basal conditions. The I–V curve in Fig. 7B (left) reveals an ISO‐evoked APA‐sensitive current with characteristics resembling I SK from ISO‐stimulated TAB VMs (Fig. 4C , left, red), with a peak at −10 mV. Figure 7C demonstrates representative recordings of APs in current clamped VMs from Shams before and after APA application (100 nmol L–1 for 3 min). In line with previous studies in healthy VMs under basal conditions (Chua et al. 2011; Gui et al. 2012), application of APA produced no effects. On the other hand, in the presence of ISO (100 nmol L–1), APA significantly prolonged APD.
Figure 7.
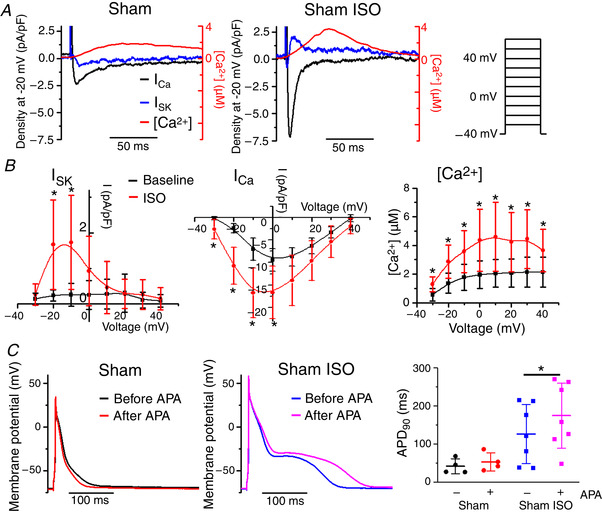
β‐adrenergic stimulation evokes ISK in Sham rat VMs
A, representative superimposed traces of I SK (blue), I Ca (black) and [Ca2+]i transient recorded in TAB VMs depolarized to –20 mV. I SK is enhanced by β‐adrenergic stimulation with ISO (100 nmol L–1). B, pooled I–V and peak [Ca2+]/V relationships for (A), with Sham VMs at baseline (black) and treated with ISO (red). Mean ± SD, n = 5–11, N = 5–8. I SK: * P = 0.03 (−20 mV), P = 0.03 (−10 mV) vs. baseline; I Ca: * P = 0.03 (−30 mV), P = 7.7 × 10−4 (−20 mV), P = 0.003 (−10 mV), P = 0.02 (0 mV) vs. baseline. [Ca2+]: * P = 0.006 (−30 mV), P = 0.004 (−20 mV), P = 0.007 (−10 mV), P = 0.01 (0 mV), P = 0.01 (10 mV), P = 0.01 (20 mV), P = 0.01 (30 mV), P = 0.03 (40 mV) vs. baseline, one‐way ANOVA with a Bonferroni post hoc test. C, representative APD traces recorded before and after application of apamin (APA, 100 nmol L–1 for 3–6 min) under baseline conditions and in the presence of ISO (100 nmol L–1). Right: pooled data for APD90. Mean ± SD of data indicated by line, n = 4–7, N = 4–6. * P = 0.002, paired Student's t test.
The currents derived using 100 nmol L–1 APA or 1 μmol L–1 UCL‐1684 are relatively small: ∼1–4 pA pF–1 (Figs 4 and 5, respectively). Figure 8 depicts additional control experiments corroborating our findings. Figure 8 A demonstrates representative traces and pooled data for I Ca, Ca2+ transients and I SK derived using 1 nmol L–1 APA in Sham myocytes challenged with ISO (100 nmol L–1). A lower concentration of APA produces similar results to those when 100 nmol L–1 APA is used (Fig. 7), which is consistent with the results of a study by Yu et al. 2014 showing that the latter APA concentration does not affect other major ionic currents in the cardiomyocytes.
Figure 8.
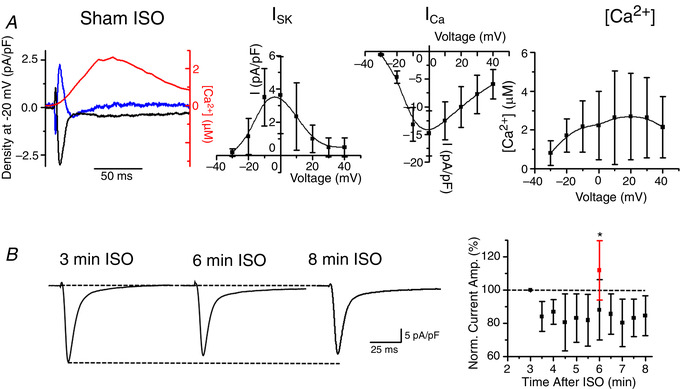
SK current in Sham rat VMs with application of ISO
A, representative superimposed traces of I SK (blue), I Ca (black) and [Ca2+]i transient (red) recorded in Sham VMs depolarized to −20 mV (left). I SK was obtained by application of 1 nmol L–1 APA. Pooled I–V and peak [Ca2+]/V relationships. B, representative inward current traces recorded in Sham VMs at 3, 6 and 8 min following application of 100 nmol L–1 ISO at −10 mV (HP −40 mV). Right: plot of normalized inward current amplitude vs. time after application of ISO. Mean ± SD, n = 5, N = 2. The normalized inward current after 6 min of ISO and 3 min of 100 nmol L–1 apamin is plotted in red. Mean ± SD, n = 8, N = 7, * P = 0.04 vs. ISO only, Student's t test.
Figure 8B demonstrates time‐dependent changes in inward current of ISO‐treated myocytes depolarized to −10 mV from a holding potential of −40 mV. The time point used to apply SK channel antagonists APA or UCL‐1684 was 3 min after ISO and I SK was derived from recordings obtained 3–5 min after application of these drugs. Therefore, inward current amplitude recorded 3 min after application of ISO was considered as 100%. Between 6 and 8 min after application of the ISO time window, there was a ∼20% decrease in inward current, probably as a result of I Ca rundown. Importantly, pooled data show that, in the presence of 100 nmol L–1 APA, integral inward current amplitude increases, which is consistent with the block of outward component (i.e. I SK).
To extend our cell studies to the tissue level, as shown in Fig. 6, we performed optical mapping experiments in ex vivo hearts from Sham rats stained with the voltage sensitive indicator di‐4‐ANNEPS. Representative APD maps of Langendorff‐perfused Sham hearts under basal conditions and after incubation with 50 nmol L–1 ISO are shown in Fig. 9A and B . APD maps and corresponding AP traces (Fig. 9C and D ) demonstrate that 30 min of perfusion with APA (10 nmol L–1) has no effects under baseline but evokes significant prolongation of repolarization in the presence of ISO (Fig. 9E and F ). Of note, APA did not affect conduction velocity (Fig. 9G ).
Figure 9.
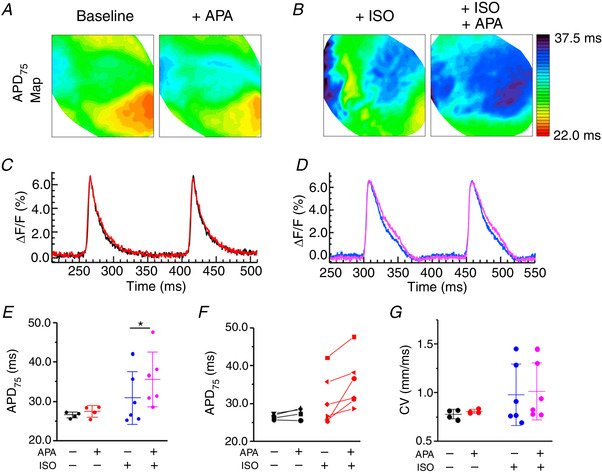
ISK inhibition prolongs APD in ex vivo Sham rat hearts under β‐adrenergic‐stimulation
A and B, representative APD maps before and after APA (10 nmol L–1) recorded under basal conditions and in the presence of β‐adrenergic agonist ISO (50 nmol L–1) and the corresponding APD profiles C–F, pooled data for APD75. G, pooled data for conduction velocity (CV). * P < 0.05, paired Student's t test, N = 4–6.
Biphasic regulation of native SK channels by [Ca2+]
Previous studies using heterologous expression systems demonstrated that, in addition to being activated by Ca2+ in the submicrocmolar range, SK channels are sensitive to voltage‐dependent inhibition by Ca2+ (IC50 ∼20 μmol L–1) (Soh & Park, 2002). During Ca2+ release from the SR, [Ca2+] rapidly increases from ∼100 nmol L–1 up to 100 μmol L–1 in specific subcellular compartments in close proximity to RyR clusters (i.e. dyads) (Antoons et al. 2011; Cannell et al. 2013). Therefore, localization of SK channels relative to the sources of Ca2+ is expected to determine the I SK kinetics of activation and inactivation. Assessment of the exact localization of SKs on surface membrane using imaging techniques is confounded by the quality of existing antibodies and the presence of these channels in other cellular compartments including mitochondria (Kim et al. 2017). Lu et al. (2007) demonstrated that SK channels in mouse myocytes exist in complexes with LTCCs. Using the BN‐PAGE technique, where mild processing of samples allows many protein complexes to remain intact, we confirmed that, in Sham and TAB rat VMs, SK2 and the α1c subunit of LTCC can be detected in the same band, indicative of complex formation (Fig. 10, SK2‐LTTC α1c complex indicated by black arrow). Samples for BN‐PAGE were prepared from the membrane fraction of freshly isolated VMs and Na+/K+‐ATPase was used as the loading control. These results supported the specific tagging of SK2 channels present in plasmalemma in complex with LTCCs using PLA (Fig. 11). To stain fixed VMs from Shams and TAB rats, we used primary antibodies against SK2, LTCC α1c and RyR2. Secondary antibodies for RyR2 were labelled with fluorescein (green, Alexa‐488). Secondary antibodies for SK2 and LTCC were conjugated with complementary DNA strands that form a red fluorescent tag after processing if in close proximity (i.e. less than 40 nm). Technical control experiments included samples processed for the entire PLA and colocalization assay but without incubation with primary antibodies (Fig. 11B ). The density of SK2‐LTCC complexes visualized using PLA was not statistically different in Shams vs. TABs (Fig. 11C ). Importantly, analysis of confocal immunofluorescence images indicated that colocalization of SK2‐LTCC complexes with RyR2 clusters in TABs and Shams is low (Fig. 11D ), suggesting that the majority of plasmalemmal SK channels are situated outside the dyad. To test this further, we tagged SK2 channels in complex with RyR2 using PLA, and assessed the colocalization of these protein pairs with calsequestrin to visualize junctional SR (CSQ) (Fig. 12). The overall density of SK2‐RyR2 pairs was significantly lower than density of SK2‐LTCC pairs (0.005 ± 0.005 SK2‐RyR2 vs. 0.063 ± 0.043 SK2‐LTCC PLA puncta μm–2 in Sham VMs, P < 0.005, Student's t test; and 0.023 ± 0.020 SK2‐RyR2 vs. 0.101 ± 0.067 SK2‐LTCC PLA puncta μm–2 in TAB VMs, P < 0.005, Student's t test). Manders coefficients of colocalization were also low, suggesting that the very few SK2‐RyR2 pairs visualized by the PLA technique do not reside in the dyad. Figure 12C shows representative control images of VMs stained by anti‐SK2, anti‐Cav1.2a1c, anti‐RyR2 and anti‐CSQ antibodies without PLA.
Figure 10.
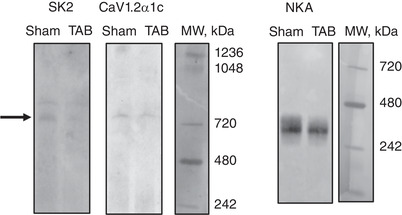
BN‐PAGE reveals a physical interaction of SK2 and the α1c subunit of LTCC in Sham and TAB rat VMs
Samples for BN‐PAGE were prepared from the membrane fraction of freshly isolated Sham and TAB rat VMs. SK2 and the α1c subunit of LTCC can be detected at the same band, indicative of a physical interaction between these two proteins. NKA was used as a loading control. N = 3 per group.
Figure 11.

PLA reveals low level colocalization of SK2‐Cav1.2α1c complexes with RyR2 in Sham and TAB rat VMs
A, PLA of SK2 and Cav1.2α1c, and colocalization with RyR2. Left: each red fluorescent dot represents a site of interaction of proteins that are within 40 nm proximity. Centre: cells were probed with anti‐RyR2 antibody. Right: merged image showing colocalization of SK2 protein pairs with RyR2. B, PLA and RyR2 secondary antibodies only, as a technical control. C, quantification of the number of PLA puncta per μm2 in Sham and TAB VMs. No significant differences were observed between groups, Student's t test, Sham n = 29, TAB n = 28, N = 4 per group. D, Manders coefficients M1 and M2 reveals a low level of colocalization of SK2‐Cav1.2α1c with RyR2. No significant differences were observed between groups Student's t test: Sham, n = 31; TAB, n = 26; N = 4 per group.
Figure 12.

PLA reveals low level colocalization of SK2‐RyR2 complexes with CSQ in Sham and TAB rat VMs
A, PLA of SK2 and RyR2 and colocalization with CSQ. Left: each red fluorescent dot represents a site of interaction of proteins that are within 40 nm proximity. Centre: cells were probed with anti‐CSQ antibody. Right: merged image showing colocalization of SK2 protein pairs with RyR2. B, Manders coefficients M1 and M2 reveals a low level of colocalization of SK2‐RyR2 with CSQ. No significant differences were observed between groups. Student's t test: Sham, n = 8; TAB, n = 15; N = 3 per group. C, SK2, RyR2, Cav1.2α1c and CSQ primary antibodies without PLA protein pairing.
An absence of SK channels in the immediate proximity of SR Ca2+ release channel clusters allowed us to employ mathematical apparatus developed by Trafford et al. (1995) and Weber et al. (2002) for estimation of [Ca2+] in the subsarcolemmal compartment from the fluorescence signal of Ca2+ indicator Rhod‐2 during the Ca2+ transient (Fig. 13A ). Fluorescence was converted into bulk [Ca2+]i. To assess the rapid rising phase, we differentiated the [Ca2+]i and multiplied by a diffusion constant γ of 110 ms (Weber et al., 2002). The result was then added to [Ca2+]i to determine the rise and peak of submembrane Ca2+ ([Ca2+]sm). To estimate the decay of [Ca2+]sm, the [Ca2+]i + γ*d[Ca2+]i/dt trace was fit with a single exponential curve using the decay of [Ca2+]i as a guide to fitting (Fig. 13A ). Figure 13B depicts superimposed representative traces of I SK and [Ca2+]sm in ISO‐treated voltage clamped Sham and TAB VMs recorded during depolarization from holding potential of −40 mV to −10 mV. I SK was obtained from current recording before and after application 100 nmol L–1 APA as described above. Importantly, I SK at −10 mV reaches the peak significantly earlier than [Ca2+]sm, which confirms that Ca2+ not only activates native SK channels, but also inhibits SK channels at higher concentrations. The pooled data for I SK and [Ca2+]sm time to peak are shown in Fig. 13B . Also, at higher 30 mV voltage pulses, I SK current is negligible despite little change in [Ca2+]sm amplitude (Fig. 13C ). Peak [Ca2+]sm amplitude and [Ca2+]sm at peak I SK are presented in Fig. 13C and D. The data suggest that, in the presence of ISO, I SK amplitude reaches a maximum when [Ca2+]sm on average rises to ∼10 μmol L–1 in both Sham and TAB VMs. The continuing rise of [Ca2+]sm above this level effectively elicits I SK inactivation. The absence of I SK at higher voltages at similar amplitudes of [Ca2+]sm suggests strong voltage‐dependence of SK channel block by Ca2+. Notably, in the absence of ISO, [Ca2+]sm in Sham VMs at −10 mV was 10.4 ± 6.5 μmol L–1 (mean ± SD, n = 10, N = 6) and TAB VMs (10 mV) was 7.6 ± 5.1 μmol L–1 (mean ± SD, n = 8, N = 6) which is within the previously reported activation range for SK channels (Li N et al. 2009; Li W et al. 2009; Schumacher et al. 2001; Schumacher et al. 2004; Soh & Park, 2002; Xia et al. 1998). These data suggest that β‐adrenergic stimulation evokes I SK in freshly isolated VMs by lessening Ca2+ dependent block.
β‐adrenergic stimulation reduces Ca2+/voltage‐dependent inhibition of SK2 channels without affecting activation by [Ca2+]
To gain mechanistic insights into the roles of PKA in modulation of SK function, we used the well‐established experimental system of cultured rat VMs. In line with our previous report (Terentyev et al. 2014), adenovirus‐mediated overexpression of rSK2 in cultured VMs produced a robust I SK between 36 and 48 h after infection with a multiplicity of infection of 10. As seen in Fig. 14A and B , application of ISO enhanced I SK in parallel with the increase in Ca2+ transient amplitudes in voltage clamped VMs (holding potential −45 mV). This effect was more pronounced at higher depolarizations resulting in ISO‐mediated loss of rectification, as seen in respective current‐voltage relationships (Fig. 14B , left, red vs. black). To distinguish between the possible effect of increasing [Ca2+]i and the direct effect of PKA‐mediated phosphorylation of SK channels, rSK2 was coexpressed with dnPLB (K3E/R14E mutation) (Ziolo et al. 2005). Adenoviral‐mediated expression of each protein was confirmed by western blot analysis using VMs infected with adenovirus carrying an empty vector as a control (Fig. 14C ). Coexpression of dnPLB relieves inhibition of SERCa2a resulting in higher Ca2+ transients during voltage pulses of the experiment, without increasing PKA activity. Importantly, despite the dnPLB‐mediated increase in intracellular Ca2+ transient amplitude, this was not accompanied by an increase in I SK. Increased Ca2+ transient amplitude alone did not increase I SK at 5 mV compared to baseline (Fig. 14A , left vs. right) and did not lessen the rectification of I SK as observed in the ISO‐treated group (Fig. 14B , blue vs. red line). These results are consistent with the hypothesis that PKA phosphorylation enhances I SK predominantly via reduction of voltage‐dependent inhibition of the channels by [Ca2+]i.
Figure 14.
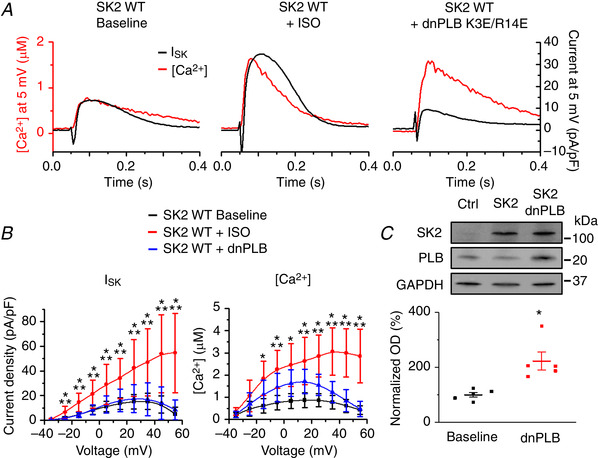
Effects of β‐adrenergic stimulation on ISK in cultured rat VMs overexpressing rSK2
A, Representative traces of [Ca2+]i transients and I SK before and after application of ISO (100 nmol L–1) in control rSK2‐overexpressing VMs and VMs co‐expressing dnPLB. VMs were cultured with adenoviral expression vectors for 48 h. B, pooled I–V and [Ca2+]/V relationships for (A). Mean ± SD, n = 8–10, N = 4–5. I SK: * P = 0.002 (−35 mV), P = 0.003 (−25 mV), P = 2.6 × 10−4 (−15 mV), P = 0.002 (−5 mV), P = 0.003 (5 mV), P = 0.002 (15 mV), P = 0.002 (25 mV), P = 0.003 (35 mV), P = 0.001 (45 mV), P = 6.3 × 10−5 (55 mV) vs. baseline. ** P = 0.002 (−35 mv), P = 0.002 (−25 mV), P = 1.6 × 10−4 (−15 mV), P = 0.002 (−5 mV), P = 0.006 (5 mV), P = 0.005 (15 mV), P = 0.004 (25 mV), P = 0.004 (35 mV), P = 8.7 × 10−4 (45 mV), P = 8.5 × 10−5 (55 mV) vs. dnPLB. [Ca2+]: * P = 0.001 (−15 mV), P = 3.2 × 10−4 (−5 mV), P = 2.3 × 10−4 (5 mV), P = 1.7 × 10−4 (15 mV), P = 4.3 × 10−6 (25 mV), P = 2.3 × 10−6 (35 mV), P = 1.6 × 10−6 (45 mV), P = 1.8 × 10−6 (55 mV) vs. baseline. ** P = 0.04 (−5 mV), P = 0.04 (15 mV), P = 5.8 × 10−4 (25 mV), P = 2.7 × 10−5 (35 mV), P = 3.6 × 10−6 (45 mV), P = 1.1 × 10−6 (55 mV) vs. dnPLB, one‐way ANOVA with a Bonferroni post hoc test. C, representative western blots from VMs probed for SK2 and PLB. The control lane (Ctrl) represents VMs infected with virus carrying an empty vector, the SK2 lane represents VMs infected with a WT SK2 virus, and the SK2 dnPLB lane represents VMs infected with WT SK2 and dnPLB viruses. Mean ± SD optical density normalized to GAPDH for SK2 at baseline is 100 ± 58.9 and SK2 + dnPLB is 150.3 ± 22.6. Mean ± SD optical density for PLB at baseline is 99.99 ± 19.8 and for SK2 + dnPLB 222.6 ± 73.04, which is significantly different compared to baseline, P = 0.02, Student's t test, N = 5.
Previous studies ascribed I SK enhancement in VMs from diseased hearts to the leftward shift of sensitivity of channels to activating [Ca2+]i with a decrease in EC50 from ∼600 nmol L–1 to 200 nmol L–1 (Chang et al. 2013b; Gui et al. 2012). To assess the possible effects of PKA on SK sensitivity to activating [Ca2+], we used Ca2+ transients evoked by depolarization to 5 mV as a ramp of [Ca2+] during simultaneous recording of I SK. Figure 15A shows superimposed representative traces of I SK recorded before and after application of ISO (100 nmol L–1 for 3 min, black and red, respectively) and corresponding Ca2+ transients (Fig. 15A , lower). The analysis that followed used the decaying phase of Ca2+ transient where [Ca2+]i matches [Ca2+]sm after the peak of I SK, which also allowed us to avoid contamination with I Ca. In Fig. 15B normalized SK currents from the same VM are plotted against [Ca2+]i where the peak amplitude of I SK under ISO was considered as I Max. Importantly, curve fitting using Hill's equation produced similar results for data obtained under baseline conditions and after challenge with ISO showing no change in EC50 to [Ca2+] (baseline EC50 = 460 nmol L–1, h = 2.6; ISO EC50 = 480 nmol L–1, h = 2.9). To validate our approach, we performed similar analysis in VMs overexpressing an rSK2 mutant with enhanced sensitivity to [Ca2+]i (R396E/K397E, EC50 = 200 nmol L–1, Li & Aldrich, 2011). Representative current recordings (Fig. 15A , blue and magenta) demonstrated a visibly slower decay kinetics of the RE/KE mutant, which translated into a significant shift in the current/[Ca2+] relationship to the left (baseline EC50 = 230 nmol L–1, h = 2.5; ISO EC50 = 240 nmol L–1, h = 2.5) (Fig. 15B ). Pooled data for EC50 are presented in Fig. 15C . These results further confirm that PKA phosphorylation does not change the affinity of SK channels to activating submicromolar [Ca2+] but, instead, reduces Ca2+/voltage‐dependent inhibition at higher levels of intracellular Ca2+.
Figure 15.
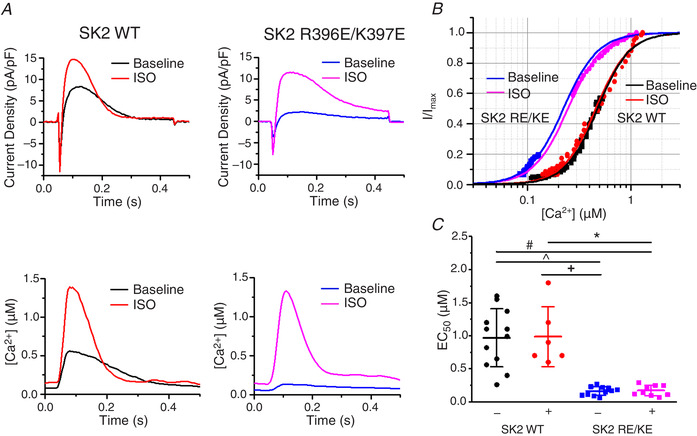
β‐adrenergic stimulation of ISK in cultured rat VMs does not shift Ca2+ sensitivity
A, representative traces of I SK and [Ca2+]i transients before and after application of ISO (100 nmol L–1) in WT rSK2‐overexpressing VMs and VMs expressing the rSK2 double mutant R396E/K397E. VMs were cultured with adenoviral expression vectors for 48 h. B, SK current dependence on [Ca2+] for one WT rSK2 expressing VM and one rSK2 double mutant expressing VM. Current was normalized to the peak current in the presence of the β‐adrenergic agonist ISO (100 nmol L–1, I max) and plotted from the peak through decay to minimum. Data was fitted with a Hill equation I/I max = 1/[1 + (EC50/[Ca2+])h] where h is the Hill coefficient. C, plot of average EC50 values for WT rSK2 and the rSK2 double mutant recorded under basal conditions and in the presence of the β‐adrenergic agonist ISO (100 nmol L–1). Mean ± SD of data indicated by line, n = 6–12, N = 4‐5. ^P = 2.4 × 10−6, #P = 7.9 × 10−6, +P = 4.9 × 10−5, * P = 1.0 × 10−4, one‐way ANOVA with a Bonferroni post hoc test.
Serine‐465 confers PKA‐mediated regulation of rSK2 activity
A phosphoproteomics study revealed that, out of >40 serine/threonine residues in rSK2, only five can be phosphorylated by PKA (Blom et al. 1999). Based on experiments in a heterologous system, Ren et al. (2006) proposed that phosphorylation of three adjacent C‐terminal serines (S568‐570) in HEK293 cells reduced the incorporation of channels into the surface membrane. These results are not easy to reconcile with the enhanced SK activity in VMs from diseased hearts. Also, it was shown that another SK isoform, SK3, is inhibited by cAMP‐PKA (Clarysse et al. 2014). The SK3 isoform encompasses two out of three homologous C‐terminus serines (S719‐720) and an N‐terminus serine‐285 homologous to rSK2 serine‐136 (S136). Sequence comparison shows that the remaining SK2 PKA phosphorylation site, serine‐465 (S465), is absent in rat SK3 (Fig. 16). Therefore, we focused on delineating the possible functional relevance of C‐terminus S465, which resides in CaM‐binding domain of SK2 (Li W et al. 2009; Ren et al. 2006; Schumacher et al. 2001). We produced an adenoviral vector carrying the phosphomimetic mutants rSK2‐S465A and rSK2‐S465D constructs for functional tests in cultured adult rat VMs (Figs 17 and 18). In addition, we generated another phosphomimetic mutant of rSK2, S136D, for comparison (Fig. 19). This N‐terminus serine is conserved between all three SK isoforms (Fig. 16). Similar to rSK2 WT overexpression studies (Fig. 14), experiments in rSK2‐S465D and rSK2‐S136D expressing VMs demonstrated ISO‐mediated relief of I SK rectification paralleled by an increase in Ca2+ transient amplitudes (Figs 18 and 19, respectively). No SK current was observed in VMs expressing the dephosphomimetic mutant rSK2‐S465A (Fig. 17). To determine the direct effects of PKA‐mediated phosphorylation at S465 and S136 as opposed to secondary effects of ISO‐induced increased [Ca2+]i, we coexpressed rSK2 phosphomimetic mutants with dnPLB. The expression of dnPLB increases [Ca2+]i in the absence of PKA‐mediated effects. Importantly, the current density was significantly increased in VMs coexpressing rSK2‐S465D and dnPLB but not rSK2‐S136D (Figs 18B and 19B , respectively). Moreover, in the presence of dnPLB, rectification of I SK that occurs at higher voltages and [Ca2+] was significantly relieved only in the rSK2‐S465D group (Fig. 18B , blue vs. black line) as opposed to rSK2‐WT (Fig. 14) or rSK2‐S136D (Fig. 19), providing evidence that phosphorylation at this particular site underlies functional upregulation, reducing voltage‐dependent channel inhibition by high [Ca2+]i. Importantly, the sensitivity to [Ca2+] activation of rSK2 was not changed by S465D mutation (Fig. 18C ).
Figure 16.

Protein sequence alignment of rat and human SK isoforms showing putative PKA phosphorylation sites
Potential PKA phosphorylation sites of SK2, as suggested in Ren et al. (2006) are highlighted in red.
Figure 17.
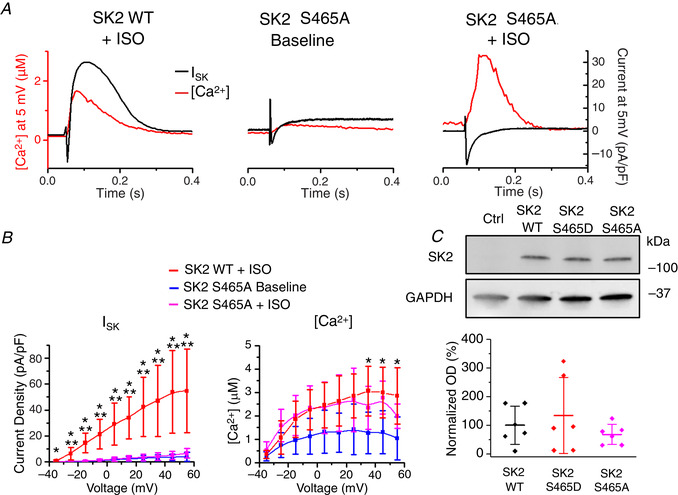
Cultured rat VMs overexpressing SK2 phosphomimetic mutant S465A show no SK current
A, representative traces of [Ca2+]i transients and I SK at baseline and after incubation with ISO (100 nmol L–1, 3 min) in VMs expressing the rSK2‐S465A mutant or WT rSK2 (from Fig. 10) B, pooled mean ± SD I–V and peak [Ca2+]/V relationships for (A), n = 5–6, N = 5 and rSK2 WT + ISO data from Fig. 6. I SK: * P = 0.02 (−35 mV), P = 0.005 (−25 mV), P = 2.2 × 10−4 (−15 mV), P = 4 × 10−4 (−5 mV), P = 6.6 × 10−4 (5 mV), P = 1.8 × 10−4 (15 mV), P = 5.7 × 10−4 (25 mV), P = 0.001 (35 mV), P = 0.001 (45 mV), P = 0.001 (55 mV) WT ISO vs. S465A baseline. ** P = 0.01 (−25 mV), P = 4.8 × 10−4 (−15 mV), P = 7.6 × 10−4 (−5 mV), P = 0.001 (5 mV), P = 3.9 × 10−4 (15 mV), P = 0.001 (25 mV), P = 0.002 (35 mV), P = 0.003 (45 mV), P = 0.004 (55 mV) rSK2 WT ISO vs. rSK2‐S465A ISO. [Ca2+]: * P = 0.02 (35 mV), P = 0.03 (45 mV), P = 0.002 (55 mV) WT ISO vs. S465A baseline, one‐way ANOVA with a Bonferroni post hoc test. C, representative western blots from VMs expressing rSK2 WT, rSK2‐S465D and rSK2‐S465A.
Figure 18.
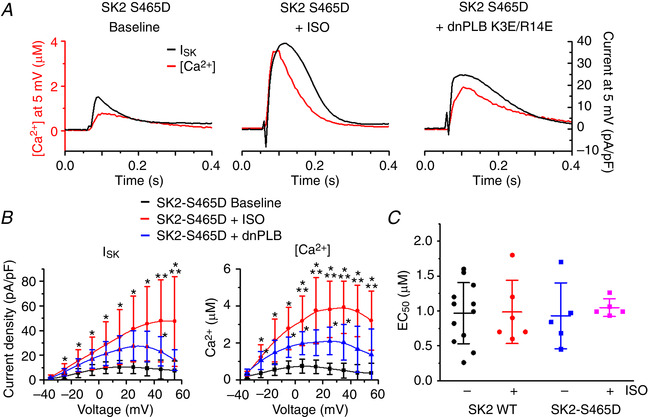
SK2 phosphomimetic mutation S465D alleviates voltage‐dependent inhibition of SK channels by [Ca2+]
A, representative traces of [Ca2+]i transients and I SK at baseline and after incubation with ISO (100 nmol L–1 for 3 min) in VMs expressing the rSK2‐S465D mutant and dnPLB. B, pooled mean ± SD I–V and peak [Ca2+]/V relationships for (A), n = 7–8, N = 6. I SK: * P = 0.01 (−25 mV), P = 0.02 (−15 mV), P = 0.03 (−5 mV), P = 0.03 (5 mV), P = 0.02 (15 mV), P = 0.008 (25 mV), P = 0.006 (35 mV), P = 0.02 (45 mV, dnPLB), P = 0.008 (55 mV) vs. baseline. ** P = 0.02 (45 mV), P = 0.03 (55 mV) vs. dnPLB. [Ca2+]: * P = 0.01 (−25 mV), P = 0.04 (−25 mV, dnPLB), P = 0.004 (−15 mV), P = 1.4 × 10−4 (−5 mV), P = 0.03 (−5 mV, dnPLB), P = 1.4 × 10−4 (5 mV), P = 0.04 (5 mV, dnPLB), P = 1.7 × 10−4 (15 mV), P = 2.2 × 10−5 (25 mV), P = 0.02 (25 mV, dnPLB), P = 1.5 × 10−5 (35 mV), P = 0.03 (35 mV, dnPLB), P = 6.4 × 10−5 (45 mV), P = 0.001 (55 mV) vs. baseline. ** P = 0.047 (5 mV), P = 0.02 (15 mV), P = 0.01 (25 mV), P = 0.006 (35 mV), P = 0.006 (45 mV), P = 0.03 (55 mV) vs. dnPLB, one‐way ANOVA with a Bonferroni post hoc test. C, plot of EC50 values for rSK2 WT and the rSK2‐S465D recorded under basal conditions and in the presence of the β‐adrenergic agonist ISO (100 nmol L–1). Mean ± SD of data indicated by line. P = 1 for all comparisons, n = 5‐12, N = 4–5, one‐way ANOVA.
Figure 19.
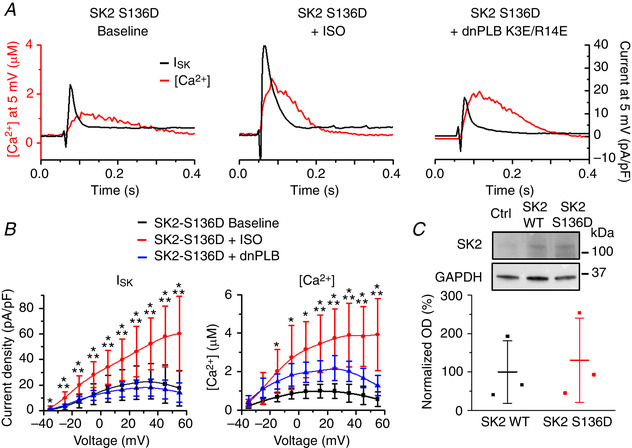
N‐terminus S136 does not confer PKA‐dependent changes in SK2 function
A, representative traces of [Ca2+]i transients and I SK at baseline and after incubation with ISO (100 nmol L–1) in VMs expressing the rSK2‐S136D mutant and dnPLB. B, pooled mean ± SD I–V and peak [Ca2+]/V relationships for (A). n = 6–8, N = 5–6 per group. I SK: * P = 0.02 (−35 mV), P = 0.001 (−25 mV), P = 5.8 × 10−4 (−15 mV), P = 0.005 (−5 mV), P = 0.02 (5 mV), P = 0.02 (15 mV), P = 0.01 (25 mV), P = 0.004 (35 mV), P = 0.002 (45 mV), P = 0.001 (55 mV) vs. baseline. ** P = 0.01 (−25 mV), P = 0.003 (−15 mV), P = 0.006 (−5 mV), P = 0.01 (5 mV), P = 0.01 (15 mV), P = 0.007 (25 mV), P = 0.002 (35 mV), P = 0.001 (45 mV), P = 0.001 (55 mV) vs. dnPLB. [Ca2+]: * P = 0.01 (−15 mV), P = 0.01 (−5 mV), P = 0.001 (5 mV), P = 5.1 × 10−4 (15 mV), P = 2.5 × 10−4 (25 mV), P = 1.7 × 10−4 (35 mV), P = 7.4 × 10−5 (45 mV), P = 1.5 × 10−4 (55 mV) vs. baseline. ** P = 0.049 (15 mV), P = 0.04 (25 mV), P = 0.02 (35 mV), P = 0.004 (45 mV), P = 0.002 (55 mV) vs. dnPLB, one‐way ANOVA with a Bonferroni post hoc test. C, representative western blots from VMs expressing rSK2 WT or rSK2‐S136D.
Next, we obtained custom antibodies that specifically recognize phospho‐S465. The western blot analysis presented in Fig. 20 confirmed that there is phosphorylation of this site in TAB VMs, which can be reduced in the presence of PKA inhibitor H89 (1 μmol L–1 for 30 min) but not CaMKII inhibitor KN93 (500 nmol L–1 for 30 min) (for comparison, see Fig. 3). Incubation of Sham VMs with ISO (100 nmol L–1 for 5 min) enhanced phosphorylation of S465.
Figure 20.

Enhanced phosphorylation of SK2‐S465 in TAB rats VMs is PKA‐dependent
A, representative western blot of SK2 and phosphorylated S465 (left) and plot of p‐S465 band optical density (OD) normalized to SK2 levels for Sham and TAB rats (right). Each lane represents a sample from a separate rat. * P = 0.005, paired Student's t test. B, representative western blot of SK2 and phosphorylated S465 (top) and plots of p‐S465 band OD normalized to SK2 levels for Sham and TAB rats under baseline conditions, or treated with ISO (* P = 0.01, paired Student's t test), KN93 or H89 (* P = 0.04, one‐way ANOVA with a Bonferroni post hoc test) (bottom).
Discussion
SK channels are rapidly gaining recognition as a novel therapeutic target to treat cardiac arrhythmias (Chiamvimonvat et al. 2017; Clements et al. 2015; Heijman & Dobrev, 2017). However, their anti‐arrhythmic potential cannot be fully appreciated because of an incomplete understanding of mechanisms regulating SK channel function. Using a rat model of pressure‐overload‐induced hypertrophy, TAB, we aimed to delineate the molecular mechanisms underlying functional recruitment of SK channels in the VMs from diseased hearts. The results of the present study demonstrate that, under conditions of enhanced adrenergic drive such as hypertrophy, enhanced PKA phosphorylation of SK channels at a specific C‐terminus serine within the CaM‐binding domain (S465 for rat SK2) attenuates the inhibition of the channels that normally occurs at higher voltages and concentrations of intracellular Ca2+. The results provide the first evidence that, under β‐adrenergic stimulation, PKA phosphorylation of SK channels evokes I SK that contributes to repolarization in healthy VMs and ex vivo hearts from Sham animals.
PKA phosphorylation as a mechanism for functional recruitment of SK channels in ventricular myocytes from healthy and diseased hearts
It is well established that SK channels expressed in VMs are dormant in health but become functional in cardiac disease (Bonilla et al. 2014; Chang et al. 2013b; Chua et al. 2011; Lee et al. 2013; Mahida et al. 2014; Ni et al. 2013). The mechanism underlying this process remains unclear. Originally, it was considered that the enhanced activity of SK channels in cardiac disease could be explained by an increase in expression levels (Bonilla et al. 2014; Chang et al. 2013b; Ni et al. 2013). However, in our recent work, we found that the levels of SK channels in samples from the membrane fraction of isolated TAB VMs are significantly decreased compared to levels of SK channels in Sham VMs, eliminating such a possibility (Kim et al. 2017). Yang et al. (2015) proposed that decreased CK2 abundance leads to dephosphorylation of SK‐bound CaM at Thr79, thereby increasing the sensitivity of SK channels to activating [Ca2+]. Other studies attributed dephosphorylation of CaM to the increase of activity/levels of phosphatase PP2A tethered to the complex (Allen et al. 2007; Bildl et al. 2004). The results of western blot analysis of immunoprecipitated SK2 complexes (Fig. 2D ) suggest that this is not the case for hypertrophic VMs from TAB rats because there are no changes in the phosphorylation of CaM and the levels of CK2 and the catalytic subunit of PP2A‐C in SK channel complex (Fig. 2D and E ). Two recent reports implicate CaMKII‐mediated phosphorylation of the channel in this process (Mizukami et al. 2015; Tenma et al. 2018). Indeed, our experiments with phospho‐specific antibodies confirm that SK2 channels from TAB VMs are phosphorylated by serine/threonine kinases (Fig. 2 and 3) and both CaMKII and PKA are more active in myocytes from hypertrophic hearts compared to controls (Fig. 3A–D ). However, SK2 phosphorylation was not reversed by the pharmacological CaMKII inhibitor KN93, in contrast to the PKA inhibitor H89, which was effective (Fig. 3E and F ). Similar results were obtained in experiments using a specific anti‐phospho‐SK2‐S465 antibody (Fig. 20). The major role of PKA in modulation of SK activity as assessed in our functional studies is supported by the following findings: (i) specific PKA inhibitor PKI diminishes I SK in VMs from TABs (Fig. 4); (ii) application of β‐adrenergic agonist ISO to activate PKA evokes I SK in myocytes from Shams (Figs 7A and B and 8A ), which prompts apparent contribution of SK channels to repolarization unmasked by APA both in VMs (Fig. 7C ) and ex vivo optically mapped hearts from healthy animals (Fig. 9); (iii) a similarity of the effects of phosphomimetic mutation rSK2‐S465D and ISO on I SK rectification in adult rat cultured VMs SK2‐overexpression experimental system (Figs 14 and 18); and (iv) alleviation of I SK by introducing dephosphomimetic mutation S456A (Fig. 17). In line with our data, a recent report demonstrated an enhancement of I SK in rabbit hearts challenged with ISO (Chen et al. 2018). Taken together, these results support the central role of PKA as a regulator of SK channel activity in cardiac hypertrophy. Furthermore, these data provide an insight into the role of SK channels in normal physiology as an integral part of the response to catecholaminergic surge during stress, providing additional repolarization to mitigate the depolarizing force of increased I Ca and I NCX to limit proarrhythmic incidences such as triggered activity.
Notably, previous studies using overexpressed SKs in heterologous experimental cell systems demonstrated PKA‐mediated functional downregulation of the channels (Clarysse et al. 2014; Ren et al. 2006), which contradicts our data obtained in native VMs. Ren et al. (2006) showed that activation of PKA in COS7 cells evoked almost complete translocation of SK2 from the surface membrane to the cytosol. Our recent report (Kim et al. 2017) confirms that, in TAB VMs, the levels of sarcolemmal SK2 and SK3 are lower than in Sham VMs. However, 20% downregulation of SK2 in VMs was not nearly as dramatic as the rapid and complete loss of plasmalemmal channels in COS7 cells. This suggests that strong association with other proteins such as LTCCs and α‐actinin (Lu et al. 2009; Zhang et al. 2017; Zhang et al. 2018) effectively interferes with retrograde transport of SK channels from the plasmalemma of VMs, which results in net functional upregulation of the channels by PKA phosphorylation. This may explain the well‐established presence of I SK under conditions accompanied by enhanced catecholaminergic drive, such as hypertrophy or heart failure.
Biphasic response of SK channels to [Ca2+]i and its modulation by PKA in ventricular myocytes
Detailed knowledge of the control of SK channel gating by [Ca2+] has accumulated over the last 20 years of research. Submicromolar [Ca2+]i effectively activates SK channels via constitutively bound CaM with an EC50 of ∼0.3–1 μmol L–1 (Adelman et al. 2012; Li N et al. 2009; Soh & Park, 2002; Tuteja et al. 2010; Xia et al. 1998). Because, in the majority of cell types, higher elevations of [Ca2+]i are rare, the fact that SK channels can be inhibited by [Ca2+]i in a voltage‐dependent manner with an IC50 of ∼20 μmol L–1 (Soh & Park, 2002) is often overlooked. The latter cannot be discounted in VMs where submembrane [Ca2+] has been estimated to reach 20 or even 100 μmol L–1 in the dyad during the peak of Ca2+ transient (Antoons et al. 2011; Cannell et al. 2013; Shannon et al. 2004). Recent work using super‐resolution microscopy places SK channels within a nanodomain also occupied by LTCCs and RyR2s in VMs (Zhang et al. 2018). The close proximity to these major sources of [Ca2+] further highlights the potential role of Ca2+‐dependent inhibition of SK channels during Ca2+ cycling. Our BN‐PAGE experiments confirm the physical interaction of SK2 with α1c subunit of LTCC (Fig. 10). Further immunolocalization studies using PLA showed that plasmalemmal SK2‐LTCC complexes are not precisely aligned with clusters of SR Ca2+ release channels RyR2s (Figs 11 and 12). The absence of SK channels in the dyad probably prevents ultrarapid exposure to very high dyadic [Ca2+] during Ca2+ release, which is estimated to be 100–200 μmol L–1 (Antoons et al. 2011; Cannell et al. 2013; Shannon et al. 2004) and is 5‐ to 10‐fold higher that the reported IC50 (20 μmol L–1; Soh & Park, 2002).
The presence of functional SK channels in diseased VMs usually is inferred from the effect of APA on APD, and currents are routinely measured before and after APA under conditions where [Ca2+]i is clamped in the submicromolar range with Ca2+ chelators (Chang et al. 2013b; Gui et al. 2012; Yu et al. 2014; Zhang et al. 2014). By contrast, our cell electrophysiology experiments in conjunction with confocal Ca2+ imaging and analysis were specifically designed to assess SK function within the full physiological range of [Ca2+] in the vicinity of the channel. Outward I SK (i.e. SK inhibitor‐sensitive component) measured in voltage clamped VMs from TABs (Figs 4 and 5) and Shams under ISO stimulation (Figs 7 and 8A ) exhibited very fast activation kinetics followed by a rapid drop, presumably because [Ca2+]i rising during the Ca2+ transient reaches concentrations sufficient to inhibit the channel. Analysis of dynamics of submembrane Ca2+ during the Ca2+ transient in voltage clamped TAB and Sham VMs at −10 mV revealed that, in the presence of ISO, I SK reaches its maximum amplitude within an average of 10 ms (Fig. 13B ) when [Ca2+]sm is ∼10 μmol L–1 (Fig. 13D ). A further increase in [Ca2+]sm, which lasts for additional 10–12 ms, evokes robust I SK decay. Because of the dual action of Ca2+ (i.e. activation and inactivation), the dynamics of the I SK response differs from the response of NCX1, another plasmamembrane Ca2+‐regulated transporter with similar EC50 ∼400 nmol L–1, which closely follows both rise and decay of [Ca2+]sm (Weber et al. 2002).
Importantly, I SK in native VMs was detectable only under conditions where PKA was active and it exhibited rectification with peak amplitude reached at –10 to −20 mV followed by a robust decrease at higher voltages (Figs 4C , 5, 7B and 8A ). In cultured VMs overexpressing rSK2 channels, ISO completely alleviated rectification, whereas the I–V curve of I SK under baseline showed a peak at ∼10 mV (Fig. 14). Nevertheless, freshly isolated VMs and the overexpression system both produced qualitatively similar results, implying that the main effect of PKA phosphorylation is to reduce the Ca2+/voltage‐dependent inhibition of the channels. Indeed, our experiments using intrinsic Ca2+ handling machinery to produce a ramp of [Ca2+]i to assess the sensitivity of SK channels within its activation range demonstrated no change in EC50 ∼1 μmol L–1 before and after challenge with ISO (Fig. 15). This is in contrast to previous findings in VMs from failing hearts where enhanced sensitivity to activation by Ca2+ was proposed to underlie disease‐associated upregulation of SK channels (Bildl et al. 2004; Chang et al. 2013b; Chua et al. 2011; Ni et al. 2013; Yang et al. 2015). Most probably, this discrepancy stems from an underestimation of the role of the Ca2+/voltage‐dependent channel inhibition as a result of challenges in the assessment of I Max in the previous studies. Future detailed experimental studies in conjunction with spatially resolved computational modelling are needed to help settle this controversy.
Notably, the abundant divalent cation Mg2+ was also reported to inhibit SK channels in heterologous system (Soh & Park, 2001; Soh & Park, 2002). However, this inhibition was largely relieved in the presence of [Ca2+] at 2–20 μmol L–1 (Soh & Park, 2001) when [Mg2+] was close to its physiological range which is ∼1 mmol L–1 in VMs (Bers, 2002). Furthermore, the inhibitory action of Mg2+ may be substantially reduced in the presence of physiological levels of ATP, as the case of RyR2 (Bers, 2002). The free [Mg2+] in our pipette solution for cell electrophysiology experiments was 1.37 mmol L–1 (Maxchelator; Bers et al. 2010). Increasing it to supra‐physiological 5 mmol L–1 did not produce apparent effects on I SK parameters in VMs overexpressing rSK2 (data not shown), suggesting that the role of [Mg2+]i in dynamic regulation of SK channels in cardiac myocytes is relatively small compared to [Ca2+]i. Unfortunately, we were unable to assess sensitivity of SK channels to inhibition by Ca2+ in native Sham rat VMs with preserved Ca2+ cycling in the absence of PKA activation. Under these conditions, we were unable to record discernible I SK despite of sufficient [Ca2+]sm for activation of the channels. Future single channel patch clamp studies from native VMs using an inside–out configuration are warranted to address this question.
Functional upregulation of SK channels can be viewed as an adaptive mechanism to reduce risk of Ca2+‐dependent arrhythmia in diseased hearts (Bonilla et al. 2014; Chang et al. 2013b; Clements et al. 2015; Kim et al. 2017). It is clear that this protection is limited because arrhythmia still persists under conditions such as heart failure. It is tempting to hypothesize that, if indeed SK channel sensitivity to activating [Ca2+] is not changed under disease‐related remodelling, there is a reasonable rationale for using pharmacological agents that act by enhancing SK Ca2+ sensitivity (Hougaard et al. 2004; Kim et al. 2017; Strobaek et al. 2004). This could potentially further facilitate repolarization and thereby reduce the risk of Ca2+‐dependent ventricular tachyarrhythmias.
In conclusion, the results of the present study show that the functional upregulation of SK2 channels in hypertrophic rat ventricular cardiomyocytes is driven by enhanced PKA‐dependent phosphorylation at S465. PKA phosphorylation attenuates I SK rectification by reducing the Ca2+/voltage‐dependent inhibition of SK channels without changing their sensitivity to activating submicromolar [Ca2+]. This mechanism underlies the functional recruitment of SK channels not only in cardiac disease, but also in normal physiology, contributing to repolarization under conditions of enhanced adrenergic drive.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
DT, BRC and GK participated in the study design. SH and DT wrote first draft of manuscript. SH, IP, RT, PB, TYK, KR and RTC performed the experiments. SH, IP, RT, PB, TYK, KR, RTC, BRC and DT conducted data interpretation and analyses. SH, IP, RT, PB, TYK, KR, RTC, BRC, GK and DT reviewed the manuscript submitted for publication. All authors revised and approved the final version of the manuscript. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by American Heart Association Grant #18POST33960456 (SH); National Heart, Lung, and Blood Institute at the National Institutes of Health (NIH) NIH RO1HL135236 (RTC); NIH R01HL110791 (GK); NIH R01HL096669 (BRC); and American Heart Association Grant in Aid 15GRNT25650002 and NIH R01HL121796 (DT).
Biographies
Shanna Hamilton is currently a postdoctoral researcher at The Ohio State University in Ohio, USA. She received a PhD in Medicine and Biophysics at Cardiff University in Wales, UK, studying mutations in the cardiac ryanodine receptor associated with disease. In the laboratory of Dr Dmitry Terentyev, she now studies the regulation of Ca2+ dynamics and mechanisms of arrhythmia in multiple cardiac disease models.

Iuliia Polina is currently a postdoctoral scholar at the Medical University of South Carolina (MUSC) in Charleston. She received a PhD in Physiology at the Sechenov Institute in Saint‐Petersburg, Russia. She now studies the cellular and molecular mechanisms underlying hypertension and diabetes, using confocal microscopy, patch clamp and molecular biology techniques.
Radmila Terentyeva obtained her Master's degree in 1992 at St Petersburg State University, Russia, and is now a Senior Research Associate at The Ohio State University in Ohio, USA. For almost 20 years, she has been involved in studies of cardiac physiology and disease with specific expertise in biochemistry, cell imaging, channel biophysics and molecular biology.
Edited by: Don Bers & Bjorn Knollmann
References
- Adelman JP, Maylie J & Sah P (2012). Small‐conductance Ca2+‐activated K+ channels: form and function. Annu Rev Physiol 74, 245–269. [DOI] [PubMed] [Google Scholar]
- Allen D, Fakler B, Maylie J & Adelman JP (2007). Organization and regulation of small conductance Ca2+‐activated K+ channel multiprotein complexes. J Neurosci 27, 2369–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoons G, Livshitz L, Rudy Y & Sipido KR (2011). Microdomain [Ca²⁺] near ryanodine receptors as reported by L‐type Ca²⁺ and Na+/Ca²⁺ exchange currents. J Physiol (Lond) 589, 2569–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O'Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS & Muntner P; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee (2018). Heart disease and stroke statistics – 2018 update: a report from the American Heart Association. Circulation 137, e67–e492. [DOI] [PubMed] [Google Scholar]
- Bers DM, Patton CW & Nuccitelli R (2010). A practical guide to the preparation of Ca(2+) buffers. Methods Cell Biol 99, 1–26. [DOI] [PubMed] [Google Scholar]
- Bildl W, Strassmaier T, Thurm H, Andersen J, Eble S, Oliver D, Knipper M, Mann M, Schulte U, Adelman JP & Fakler B (2004). Protein kinase CK2 is coassembled with small conductance Ca(2+)‐activated K+ channels and regulates channel gating. Neuron 43, 847–858. [DOI] [PubMed] [Google Scholar]
- Blom N, Gammeltoft S & Brunak S (1999). Sequence and structure‐based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294, 1351–1362. [DOI] [PubMed] [Google Scholar]
- Bonilla IM, Long VP, Vargas‐Pinto P, Wright P, Belevych A, Lou Q, Mowrey K, Yoo J, Binkley PF, Fedorov VV, Györke S, Janssen PM, Kilic A, Mohler PJ & Carnes CA (2014). Calcium‐activated potassium current modulates ventricular repolarization in chronic heart failure. PLoS ONE 9, e108824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell MB, Kong CH, Imtiaz MS & Laver DR (2013). Control of sarcoplasmic reticulum Ca2+ . Biophys J 104, 2149–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PC, Hsieh YC, Hsueh CH, Weiss JN, Lin SF & Chen PS (2013. a). Apamin induces early afterdepolarizations and torsades de pointes ventricular arrhythmia from failing rabbit ventricles exhibiting secondary rises in intracellular calcium. Heart Rhythm 10, 1516–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PC, Turker I, Lopshire JC, Masroor S, Nguyen BL, Tao W, Rubart M, Chen PS, Chen Z & Ai T (2013b). Heterogeneous upregulation of apamin‐sensitive potassium currents in failing human ventricles. J Am Heart Assoc 2, e004713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Yin D, Guo S, Xu DZ, Wang Z, Chen Z, Rubart‐von der Lohe M, Lin SF, Everett TH, Weiss JN & Chen PS (2018). Sex‐specific activation of SK current by isoproterenol facilitates action potential triangulation and arrhythmogenesis in rabbit ventricles. J Physio l 596, 4299–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Akin BL & Jones LR (2007). Mechanism of reversal of phospholamban inhibition of the cardiac Ca2+‐ATPase by protein kinase A and by anti‐phospholamban monoclonal antibody 2D12. J Biol Chem 282, 20968‐20976. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ & Cannell MB (1993). Calcium sparks: elementary events underlying excitation‐contraction coupling in heart muscle. Science 262, 740–744. [DOI] [PubMed] [Google Scholar]
- Chiamvimonvat N, Chen‐Izu Y, Clancy CE, Deschenes I, Dobrev D, Heijman J, Izu L, Qu Z, Ripplinger CM, Vandenberg JI, Weiss JN, Koren G, Banyasz T, Grandi E, Sanguinetti MC, Bers DM & Nerbonne JM (2017). Potassium currents in the heart: functional roles in repolarization, arrhythmia and therapeutics. J Physiol 595, 2229–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua SK, Chang PC, Maruyama M, Turker I, Shinohara T, Shen MJ, Chen Z, Shen C, Rubart‐von der Lohe M, Lopshire JC, Ogawa M, Weiss JN, Lin SF, Ai T & Chen PS (2011). Small‐conductance calcium‐activated potassium channel and recurrent ventricular fibrillation in failing rabbit ventricles. Circ Res 108, 971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarysse L, Guéguinou M, Potier‐Cartereau M, Vandecasteele G, Bougnoux P, Chevalier S, Chantôme A & Vandier C (2014). cAMP‐PKA inhibition of SK3 channel reduced both Ca2+ entry and cancer cell migration by regulation of SK3‐Orai1 complex. Pflugers Arch 466, 1921–1932. [DOI] [PubMed] [Google Scholar]
- Clements RT, Terentyev D & Sellke FW (2015). Ca(2+)‐activated K(+) channels as therapeutic targets for myocardial and vascular protection. Circ J 79, 455–462. [DOI] [PubMed] [Google Scholar]
- del Monte F, Butler K, Boecker W, Gwathmey JK & Hajjar RJ (2002). Novel technique of aortic banding followed by gene transfer during hypertrophy and heart failure. Physiol Genomics 9, 49–56. [DOI] [PubMed] [Google Scholar]
- Diness JG, Skibsbye L, Jespersen T & Bartels ED (2011). Effects on atrial fibrillation in aged hypertensive rats by Ca2+‐activated K+ channel inhibition. Hypertension 57, 1129. [DOI] [PubMed] [Google Scholar]
- Escobar AL, Velez P, Kim AM, Cifuentes F, Fill M & Vergara JL (1997). Kinetic properties of DM‐nitrophen and calcium indicators: rapid transient response to flash photolysis. Pflugers Arch 434, 615–631. [DOI] [PubMed] [Google Scholar]
- Gui L, Bao Z, Jia Y, Qin X, Cheng ZJ, Zhu J & Chen Q‐H (2012). Ventricular tachyarrhythmias in rats with acute myocardial infarction involves activation of small‐conductance Ca2+‐activated K+ channels. Am J Physiol Heart Circ Physiol 304, H118–H130. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijman J & Dobrev D (2017). Inhibition of small‐conductance Ca2+‐activated K+ channels: the long‐awaited breakthrough for antiarrhythmic drug therapy of atrial fibrillation? Circ Arrhythm Electrophysiol 10, e005776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini R, Benton DC, Dunn PM, Jenkinson DH & Moss GW (2001). SK3 is an important component of K(+) channels mediating the afterhyperpolarization in cultured rat SCG neurones. J Physiol 535, 323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hougaard C, Eriksen BL, Jorgensen S, Johansen TH, Dyhring T, Madsen LS, Strobaek D & Christophersen P (2004). Selective positive modulation of the SK3 and SK2 subtypes of small conductance Ca2+‐activated K+ channels. Br J Pharmacol 151, 655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh YC, Chang PC, Hsueh CH, Lee YS, Shen C, Weiss JN, Chen Z, Ai T, Lin SF & Chen PS (2013). Apamin‐sensitive potassium current modulates action potential duration restitution and arrhythmogenesis of failing rabbit ventricles. Circ Arrhythm Electrophysiol 6, 410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada R, Yokoshiki H, Mitsuyama H, Watanabe M, Mizukami K, Tenma T, Takahashi M, Takada S & Anzai T (2018). Arrhythmogenic β‐adrenergic signaling in cardiac hypertrophy: the role of small‐conductance calcium‐activated potassium channels via activation of CaMKII. Eur J Pharmacol 844, 110–117. [DOI] [PubMed] [Google Scholar]
- Kim TY, Kunitomo Y, Pfeiffer Z, Patel D, Hwang J, Harrison K, Patel B, Jeng P, Ziv O, Lu Y, Peng X, Qu Z, Koren G & Choi BR (2015). Complex excitation dynamics underlie polymorphic ventricular tachycardia in a transgenic rabbit model of long QT syndrome type 1. Heart Rhythm 12, 220–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TY, Terentyeva R, Roder KHF, Li W, Liu M, Greener I, Hamilton S, Polina I, Murphy KR, Clements RT, Jr Dudley SC, Koren G, Choi BR & Terentyev D (2017). SK channel enhancers attenuate Ca2+‐dependent arrhythmia in hypertrophic hearts by regulating mito‐ROS‐dependent oxidation and activity of RyR. Cardiovasc Res 113, 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Chang PC, Hsueh CH, Maruyama M, Park HW, Rhee KS, Hsieh YC, Shen C, Weiss JN, Chen Z, Lin SF & Chen PS (2013). Apamin‐sensitive calcium‐activated potassium currents in rabbit ventricles with chronic myocardial infarction. J Cardiovasc Electrophysiol 24, 1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Timofeyev V, Tuteja D, Xu D, Lu L, Zhang Q, Zhang Z, Singapuri A, Albert TR, Rajagopal AV, Bond CT, Periasamy M, Adelman J & Chiamvimonvat N (2009). Ablation of a Ca2+‐activated K+ channel (SK2 channel) results in action potential prolongation in atrial myocytes and atrial fibrillation. J Physiol (Lond) 587, 1087–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W & Aldrich RW (2011). Electrostatic influences of charged inner pore residues on the conductance and gating of small conductance Ca2+ activated K+ channels. Proc Natl Acad Sci U S A 108, 5946–5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Halling DB, Hall AW & Aldrich RW (2009). EF hands at the N‐lobe of calmodulin are required for both SK channel gating and stable SK‐calmodulin interaction. J Gen Physiol 134, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Timofeyev V, Li N, Rafizadeh S, Singapuri A, Harris TR & Chiamvimonvat N (2009). Alpha‐actinin2 cytoskeletal protein is required for the functional membrane localization of a Ca2+‐activated K+ channel (SK2 channel). Proc Natl Acad Sci U S A 106, 18402–18407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Zhang Q, Timofeyev V, Zhang Z, Young JN, Shin HS, Knowlton AA, Chiamvimonvat N (2007). Molecular coupling of a Ca2+‐activated K+ channel to L‐type Ca2+ channels via alpha‐actinin2. Circ Res 100, 112–120. [DOI] [PubMed] [Google Scholar]
- Mahida S (2014). Expanding role of SK channels in cardiac electrophysiology. Heart Rhythm 11, 1233–1238. [DOI] [PubMed] [Google Scholar]
- Maylie J, Bond CT, Herson PS, Lee W‐S & Adelman JP (2003). Small conductance Ca2+‐activated K+ channels and calmodulin. J Physiol (Lond) 554, 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizukami K, Yokoshiki H, Mitsuyama H, Watanabe M, Tenma T, Takada S & Tsutsui H (2015). Small‐conductance Ca2+‐activated K+ current is upregulated via the phosphorylation of CaMKII in cardiac hypertrophy from spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 309, H1066–H1074. [DOI] [PubMed] [Google Scholar]
- Nagy N, Márton Z, Kiss L, Varró A, Nánási PP & Tóth A (2011). Role of Ca²+‐sensitive K+ currents in controlling ventricular repolarization: possible implications for future antiarrhytmic drug therapy. Curr Med Chem 18, 3622–3639. [DOI] [PubMed] [Google Scholar]
- Ni Y, Wang T, Zhuo X, Song B, Zhang J, Wei F, Bai H, Wang X, Yang D, Gao L & Ma A (2013). Bisoprolol reversed small conductance calcium‐activated potassium channel (SK) remodeling in a volume‐overload rat model. Mol Cell Biochem 384, 95–103. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM & Bers DM (2004). Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med 14, 61–66. [DOI] [PubMed] [Google Scholar]
- Ren Y, Barnwell LF, Alexander JC, Lubin FD, Adelman JP, Pfaffinger PJ, Schrader LA & Anderson AE (2006). Regulation of surface localization of the small conductance Ca2+‐ activated potassium channel, SK2, through direct phosphorylation by cAMP‐dependent protein kinase. J Biol Chem 281, 11769–11779. [DOI] [PubMed] [Google Scholar]
- Rosa JC, Galanakis D, Ganellin CR, Dunn PM & Jenkinson DH (1998). Bis‐quinolinium cyclophanes: 6,10‐diaza‐3(1,3),8(1,4)‐dibenzena‐1,5(1,4)‐ diquinolinacyclodecaphane (UCL 1684), the first nanomolar, non‐peptidic blocker of the apamin‐sensitive Ca(2+)‐ activated K+ channel. J Med Chem 41, 2–5. [DOI] [PubMed] [Google Scholar]
- Schumacher MA, Crum M & Miller MC (2004). Crystal structures of apocalmodulin and an apocalmodulin/SK potassium channel gating domain complex. Structure 12, 849–860. [DOI] [PubMed] [Google Scholar]
- Schumacher MA, Rivard AF, Bächinger HP & Adelman JP (2001). Structure of the gating domain of a Ca2+‐activated K+ channel complexed with Ca2+/calmodulin. Nature 410, 1120–1124. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Wang F, Puglisi J, Weber C & Bers DM (2004). A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J 87, 3351–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibsbye L, Poulet C, Diness JG, Bentzen BH, Yuan L, Kappert U, Matschke K, Wettwer R, Ravens U, Grunnet M, Christ T & Jespersen T (2014). Small‐conductance calcium‐ activated potassium (SK) channels contribute to action potential repolarization in human atria. Cardiovasc Res 103, 156–167. [DOI] [PubMed] [Google Scholar]
- Soh H & Park CS (2001). Inwardly rectifying current‐voltage relationship of small‐conductance Ca2+‐activated K+ channels rendered by intracellular divalent cation blockade. Biophys J 80, 2207–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh H & Park CS (2002). Localization of divalent cation‐binding site in the pore of a small conductance Ca(2+)‐activated K(+) channel and its role in determining current‐voltage relationship. Biophys J 83, 2528–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobaek D, Teuber L, Jorgensen TD, Ahring PK, Kjaer K, Hansen RS, Olesen SP, Christophersen P & Skaaning‐Jensen B (2004). Activation of human IK and SK Ca2+ ‐ activated K+ channels by NS309 (6,7‐dichloro‐1H‐indole‐2,3‐dione 3‐oxime). Biochim Biophys Acta 1665, 1–5. [DOI] [PubMed] [Google Scholar]
- Stocker M (2004). Ca(2+)‐activated K+ channels: molecular determinants and function of the SK family. Nat Rev Neurosci 5, 758–770. [DOI] [PubMed] [Google Scholar]
- Tenma T, Mitsuyama H, Watanabe M, Kakutani N, Otsuka Y, Mizukami K, Kamada R, Takahashi M, Takada S, Sabe H, Tsutsui H & Yokoshiki H (2018). Small‐conductance Ca2+‐activated K+ channel activation deteriorates hypoxic ventricular arrhythmias via CaMKII in cardiac hypertrophy. Am J Physiol Heart Circ Physiol 315, H262–H272. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Rochira JA, Terentyeva R, Roder K, Koren G & Li W (2014). Sarcoplasmic reticulum Ca²⁺ release is both necessary and sufficient for SK channel activation in ventricular myocytes. Am J Physiol Heart Circ Physiol 306, H738–H746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trafford AW, Díaz ME, O'Neill SC & Eisner DA (1995). Comparison of subsarcolemmal and bulk calcium concentration during spontaneous calcium release in rat ventricular myocytes. J Physiol 488, 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuteja D, Rafizadeh S, Timofeyev V, Wang S, Zhang Z, Li N, Mateo RK, Singapuri A, Young JN, Knowlton AA & Chiamvimonvat N (2010). Cardiac small conductance Ca2+‐ activated K+ channel subunits form heteromultimers via the coiled‐coil domains in the C termini of the channels. Circ Res 107, 851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuteja D, Timofeyev V, Lu L, Sharma D, Zhang Z, Xu Y, Nie L, Vázquez AE, Young JN, Glatter KA & Chiamvimonvat N (2005). Differential expression of small‐conductance Ca2+‐activated K+ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am J Physiol Heart Circ Physiol 289, H2714–H2723. [DOI] [PubMed] [Google Scholar]
- Qu Z & Weiss JN (2006). Dynamics and cardiac arrhythmias. J Cardiovasc Electrophysiol 17, 1042–1049. [DOI] [PubMed] [Google Scholar]
- Weber CR, 3rd Piacentino V, Ginsburg KS, Houser SR & Bers DM (2002). Na(+)‐Ca(2+) exchange current and submembrane [Ca(2+)] during the cardiac action potential. Circ Res 90, 182–189. [DOI] [PubMed] [Google Scholar]
- Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, Weiss RM, Anderson ME, Cheng H & Song LS (2010). T‐tubule remodeling during transition from hypertrophy to heart failure. Circ Res 107, 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Fakler B, Rivard A, Wayman G, Johnson‐Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J & Adelman JP (1998). Mechanism of calcium gating in small‐conductance calcium‐activated potassium channels. Nature 395, 503–507. [DOI] [PubMed] [Google Scholar]
- Xu Y, Tuteja D, Zhang Z, Xu D, Zhang Y, Rodriguez J, Nie L, Tuxson HR, Young JN, Glatter KA, Vazquez AE, Yámoah EN & Chiamvimonvat N (2003). Molecular identification and functional roles of a Ca(2+)‐activated K+ channel in human and mouse hearts. J Biol Chem 278, 49085–49094. [DOI] [PubMed] [Google Scholar]
- Yang D, Wang T, Ni Y, Song B, Ning F, Hu P, Luo L, Wang Y & Ma A (2015). Apamin‐sensitive K+ current upregulation in volume‐overload heart failure is associated with the decreased interaction of CK2 with SK2. J Membr Biol 248, 1181–1189. [DOI] [PubMed] [Google Scholar]
- Yu CC, Ai T, Weiss JN & Chen PS (2014). Apamin does not inhibit human cardiac Na+ current, L‐type Ca2+ current or other major K+ currents. PLoS ONE 9, e96691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Meng XY, Cui M, Pascal JM, Logothetis DE & Zhang JF (2014). Selective phosphorylation modulates the PIP2 sensitivity of the CaM‐SK channel complex. Nat Chem Biol 10, 753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XD, Coulibaly ZA, Chen WC, Ledford HA, Lee JH, Sirish P, Dai G, Jian Z, Chuang F, Brust‐Mascher I, Yamoah EN, Chen‐Izu Y, Izu LT & Chiamvimonvat N (2018). Coupling of SK channels, L‐type Ca2+ channels, and ryanodine receptors in cardiomyocytes. Sci Rep 8, 4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XD, Timofeyev V, Li Ning, Myers RE, Zhang DM, Singapuri A, Lau VC, Bond CT, Adelman J, Lieu DK & Chiamvimonvat N (2014). Critical roles of a small conductance Ca2+‐activated K+ channel (SK3) in the repolarization process of atrial myocytes. Cardiovasc Res 101, 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Ledford HA, Park S, Wang W, Rafizadeh S, Kim HJ, Xu W, Lu L, Lau VC, Knowlton AA, Zhang XD, Yamoah EN & Chiamvimonvat N (2017). Distinct subcellular mechanisms for the enhancement of the surface membrane expression of SK2 channel by its interacting proteins, α‐actinin2 and filamin A. J Physiol 595, 2271–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Bovo E, Mazurek SR, Rochira JA, Li W & Terentyev D (2014). Ca handling during excitation‐contraction coupling in heart failure. Pflugers Arch 466, 1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziolo MT, Martin JL, Bossuyt J, Bers DM & Pogwizd SM (2005). Adenoviral gene transfer of mutant phospholamban rescues contractile dysfunction in failing rabbit myocytes with relatively preserved SERCA function. Circ Res 96, 815–817. [DOI] [PubMed] [Google Scholar]