

Summary
Environmental monitoring of bacteria using phage‐based biosensors has been widely developed for many different species. However, there are only a few available methods to detect specific bacteriophages in raw environmental samples. In this work, we developed a simple and efficient assay to rapidly monitor the phage content of a given sample. The assay is based on the bistable expression of the Salmonella enterica opvAB operon. Under regular growth conditions, opvAB is only expressed by a small fraction of the bacterial subpopulation. In the OpvABON subpopulation, synthesis of the OpvA and OpvB products shortens the O‐antigen and confers resistance to phages that use LPS as a receptor. As a consequence, the OpvABON subpopulation is selected in the presence of such phages. Using an opvAB::gfp fusion, we could monitor LPS‐binding phages in various media, including raw water samples. To enlarge our phage‐biosensor panoply, we also developed biosensors able to detect LPS, as well as protein‐binding coliphages. Moreover, the combination of these tools allowed to identify the bacterial receptor triggering phage infection. The epigenetic opvAB::gfp biosensor thus comes in different flavours to detect a wide range of bacteriophages and identify the type of receptor they recognize.
Introduction
Bacteriophages, the viruses that infect bacteria, are ubiquitous on Earth, very abundant and highly diverse (Wommack and Colwell, 2000; Roux et al., 2015; Beller and Matthijnssens, 2019; Lawrence et al., 2019). They participate in the daily turnover of bacterial communities and are hypothesized to be major drivers of carbon recycling (Jover et al., 2014). Their abundance and diversity have long been acknowledged in the oceans and more recently associated to numerous microbiomes as a major part of the viromes (Abeles and Pride, 2014; Allen and Abedon, 2014; Hurwitz et al., 2014; Roux et al., 2015). Bacteriophages are considered a vast reservoir of genes and a major vector for horizontal gene transfer that allows the emergence of new biological functions (Rinke et al., 2013; Hatfull, 2015). Furthermore, phages have been at the origin of many discoveries and concepts in molecular biology (Rohwer and Segall, 2015; Salmond and Fineran, 2015; Ofir and Sorek, 2018).
Among the applications resulting from a century of phage research, phage therapy stands out by receiving more and more attention nowadays. Although experimented since the 1920s by F. d'Hérelle and subsequently abandoned in Western countries following the development of antibiotherapies (Peitzman, 1969), phage therapy has a long continuous history and usage in Eastern Europe (Chanishvili, 2016). Undoubtfully, the major threat to public health represented by antibiotic resistance constitutes a breeding ground for modern phage therapy (Abedon et al., 2017; Rohde et al., 2018; Djebara et al., 2019). Subsequently, many academic groups and biotech companies are hunting for more phage resources in diverse environments, thus highlighting the need for novel detection and quantification methods. Another popular application is the use of bacteriophages to specifically detect bacteria in environmental samples for diagnosis purposes of pathogens (Farooq et al., 2018). Examples of such biosensors able to detect enterobacteria using different readouts such as fluorescence or luminescence have been described recently (Vinay et al., 2015; Franche et al., 2016).
A key aspect of bacteriophage infection, which is relevant for characterization as well as detection, is the receptor‐binding step. Phages recognize a variety of receptors located at the surface of bacteria including lipopolysaccharide (LPS), outer membrane proteins, pili and flagella (Rakhuba et al., 2010; Bertozzi Silva et al., 2016; Letarov and Kulikov, 2017). Extracellular appendages are used by bacteriophages to specifically recognize their target cells and irreversible binding to the appropriate surface receptor triggers genome ejection from the capsid and injection in the host cytoplasm (Andres et al., 2010; Hu et al., 2015; Arnaud et al., 2017; Wang et al., 2019). Thus, bacteriophage binding to receptors constitutes the primary and mandatory step to a successful infection. Highlighting this importance, many studies aim at characterizing these interactions at the molecular level to understand and predict the host‐range and specificity of bacteriophages.
In enterobacteria, the LPS plays an important role in host recognition by phages that belong to two major categories: (i) phages that use LPS as a receptor and genome ejection trigger (e.g., Salmonella P22) and (ii) phages that bind loosely to LPS and then need an outer membrane protein for genome injection (e.g., Escherichia coli T5). For both categories, different types of modification of the O‐antigen can affect recognition such as the length or the glycan composition of the O‐antigen (Davies et al., 2013; Cota et al., 2015; Wahl et al., 2019). The Salmonella enterica opvAB operon encodes two inner membrane proteins that shorten the O‐antigen chain length. Transcription of opvAB undergoes phase variation under the control of Dam methylation and OxyR (Cota et al., 2012). Phase‐variable synthesis of OpvA and OpvB proteins results in bacterial subpopulation mixtures of standard (OpvABOFF) and shorter (OpvABON) O‐antigen chains in the LPS exposed at the cell surface. The dramatic change in LPS structure caused by opvAB expression renders S. enterica resistant to bacteriophages that recognize and need the O‐antigen for successful infection such as 9NA, Det7 and P22 (Walter et al., 2008; Casjens et al., 2014; Cota et al., 2015). As a result, in the presence of these bacteriophages, the OpvABON subpopulation is enriched, which can be monitored using an opvAB::gfp fusion (Cota et al., 2015). This property was used in the present study to design an epigenetic, highly sensitive bacteriophage biosensor, allowing the detection of tiny amounts of phages and the identification of the bacterial receptor they use. We describe two different biosensor versions able to detect phages using either the LPS or the FhuA protein as a receptor. Additional variants of this biosensor could be engineered depending on the type of phage one would like to detect and quantify.
Results
Proof of concept using characterized S. enterica bacteriophages
Based on the results published by Cota and collaborators (Cota et al., 2015), we foresaw that the opvAB operon, a bacterial locus under epigenetic control, could be used as a bacteriophage biosensor. Indeed, the OpvABOFF subpopulation was shown to be killed by bacteriophages that belonged to different families and used LPS as receptor for infection, leading to enrichment of the OpvABON subpopulation. This population is insensitive to such infections due to the shortening of its LPS by the opvAB gene products (Cota et al., 2015). Thus, if an opvAB::gfp fusion was used, enrichment of OpvABON cells could be detected by increased fluorescence intensity (Cota et al., 2015). To start with, we compared the efficiency of two different methods of fluorescence detection, flow cytometry and plate reader monitoring, to detect three different bacteriophages known to use LPS for infection. As shown in Fig. 1A, flow cytometry turned out to be not only very sensitive but also descriptive of the bacterial population heterogeneity. In contrast, fluorescence detection using a plate reader only provided a rough assessment of the population structure (Fig. 1B). On the other hand, fluorescence detection using a plate reader does not necessitate a high‐cost machine such as a flow cytometer and has the advantage of giving a measure according to the OD600 of the culture. Our standard protocol for phage detection by flow cytometry includes 8 h of incubation to ensure that phages kill the vast majority of OpvABOFFcells. Such cells are the predominant type in the initial population and are steadily generated during growth as a consequence of bistable expression of the opvAB operon, thus leading to both the enrichment of the OpvABON population and amplification of the number of virions. However, thanks to the sensitivity of this technique and because fluorescence increases as a function of time, measurements can be done earlier than 8 h. As depicted in Fig. 1C, incubation for 1 or 2 h after dilution into fresh medium in the presence of P22_H5 led to 2.6% and 11.3% of positive cells respectively. Since flow cytometry is highly reproducible, these levels are sufficient to discriminate an OpvABON subpopulation from the background level of cells that passed the fluorescence threshold in the absence of phage (0.2%) (Fig. 1C). The OpvAB biosensor mutated on the GATC sites (SV8578) was also able to detect wild‐type P22, a temperate phage (Fig. S2). As expected, the fact that only a fraction of the bacterial population was lysed, probably due to lysogenization, slowed down fluorescence increase. Detection of temperate phages may thus require longer incubation times. These experiments thus validated the proof‐of‐concept of a fluorescent biosensor for the detection of LPS‐using bacteriophages based on a modified opvAB operon.
Fig 1.
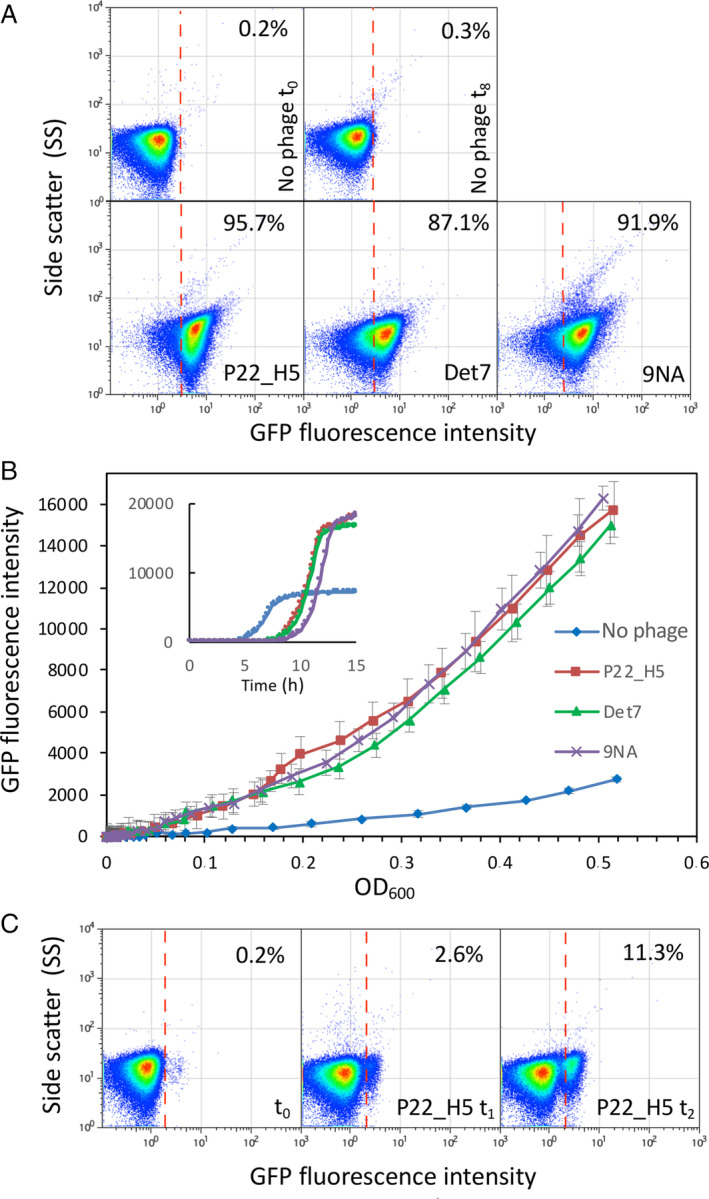
Detection of increased fluorescence intensity upon selection of the OpvABON subpopulation in the presence of bacteriophages.
A. GFP fluorescence distribution in an S. enterica strain carrying an opvAB::gfp fusion (SV6727) before (t = 0 h) and after growth in LB without phage, or in the presence of P22 H5, 9NA or Det7 (t = 8 h). Data are represented by a dot plot (side scatter versus fluorescence intensity [ON subpopulation size]). All data were collected for 100,000 events per sample.
B. Growth curves of strain SV6727 in contact with P22 H5, Det7 or 9NA phage. Data are represented by growth curves (fluorescence intensity) versus growth (OD600) or [time] (insert).
C. GFP fluorescence distribution of strain SV6727 before (t = 0 h) and after growth in LB containing P22_H5 (t = 1 or 2 h). [Color figure can be viewed at wileyonlinelibrary.com]
Detection of uncharacterized S. enterica bacteriophages
To go further with the phage detection tool, we decided to isolate and purify S. enterica‐infecting bacteriophages and test whether they could be detected without previous characterization of the phage receptor. Indeed, a phage that would need a functional and extended LPS should kill the OpvABOFF subpopulation as the known phages did. In contrast, a phage that would not need (or that could bypass) LPS to infect should not discriminate the two subpopulations. Various phages were enriched, isolated and partially characterized from different environments in the Seville area as described in Experimental procedures section. Without going deeper into the characterization of those phages, the plaque morphologies (not shown), and even more the images obtained after negative staining by TEM indicated that the six isolated phages were all different (Fig. 2A). This was further confirmed by the sequence determination of the six phage genomes (D. R. Olivenza et al., in preparation). Detection of the phages was assayed by flow cytometry using the opvAB::gfp fusion. Interestingly, all six were perfectly detected 8 h post‐infection with more than 60% of the cells in the ON state (Fig. 2B). This proportion reached up to 90% for three individual phages (Se_F1, Se_F6 and Se_AO). In contrast, Se_F3 and Se_ML killed the OpvABOFF subpopulation only partially. A conclusion from these experiments was that the opvAB::gfp fusion provides an efficient tool to detect unknown LPS‐dependent bacteriophages of S. enterica. The variations observed between the different flow cytometry profiles could be due to different phage‐specific usage of the LPS as receptor or to partial resistance of the OFF subpopulation that could reduce OpvABON subpopulation enrichment.
Fig 2.
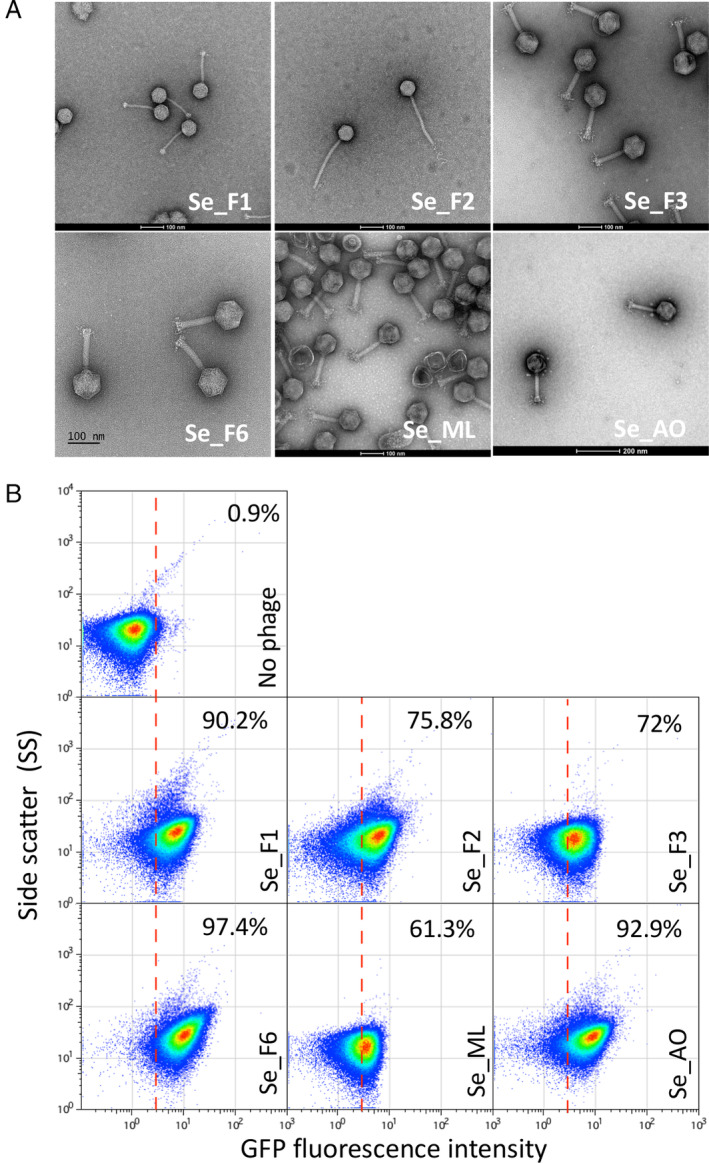
Detection of new bacteriophages.
A. Negative staining of bacteriophages visualized by electron microscopy.
B. GFP fluorescence distribution in strain SV6727 (opvAB::gfp) before (t = 0) and after growth in LB containing new purified bacteriophages (Se_F1, Se_F2, Se_F3, Se_F6, Se_ML and Se_AO) after 8 h. Data are represented by a dot plot [side scatter versus fluorescence intensity (ON subpopulation size)]. The percentage of ON cells in each sample is indicated. All data were collected for 100,000 events per sample. [Color figure can be viewed at wileyonlinelibrary.com]
An especially appealing objective of the present tool was to use the opvAB::gfp fusion to detect bacteriophages in crude samples. Crude water samples collected in the Seville area were assayed for the presence of phages in the presence of the biosensor strain for 10 h before analysing the resulting populations by flow cytometry. Among the four samples tested, three (EM1, EM2 and EM3) displayed a significant increase in GFP activity, indicating that these samples contained indeed bacteriophages that depend on LPS for infection (Fig. 3). The presence of LPS‐depending phages in the samples was confirmed by plaque assay after enrichment (Fig. S1). One sample (AO) did not appear to contain phages able to infect S. enterica. Taken together, these results prove that our opvAB‐based fluorescent biosensor can efficiently detect the presence of LPS‐depending bacteriophages in environmental samples without any preprocessing other than incubation with the biosensor.
Fig 3.
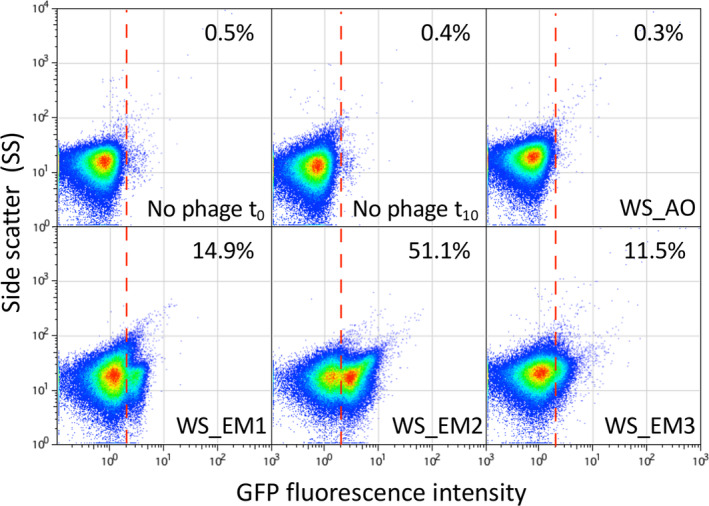
Detection of bacteriophages from crude water samples.
GFP fluorescence distribution in strain SV6727 (opvAB::gfp) before and after growth in LB for 10 h in the presence of crude water previously filtered. Data are represented by a dot plot [side scatter versus fluorescence intensity (ON subpopulation size)]. The ON subpopulation percentage is shown for each sample. Data were collected for 100,000 events per sample. The presence of bacteriophages was previously verified by a plaque assay (20–30 pfu/ml) on a S. enterica lawn. [Color figure can be viewed at wileyonlinelibrary.com]
Sensitivity and optimization of the detection tool
To challenge the limits of the phage detection tool, 9NA was used (Fig. 1). We first determined the detection limit in a fluorescence plate reader by using serial dilutions of 9NA. As shown in Fig. 4A, only the most diluted sample, equalling to 10 PFU/ml, corresponding to a MOI of ≈1 × 10−5 PFU/cell, could not be distinguished from the control experiment with no phage. Therefore, using a fluorescence plate reader, as little as 100 PFU/ml could be detected. Interestingly, the curves obtained in the presence of sufficient amounts of phage (at least 102 PFU/ml) showed a peculiar profile. In the first 5 h, the global tendency was an increase of the whole population (OpvABON and OpvABOFF), followed by a rapid decrease (Fig. 4A, inset). Five hours post‐infection, the bacterial growth resumed more or less rapidly depending on the amount of phage initially present in the culture. As expected, the fluorescence intensity correlated with growth only in samples infected with 9NA (≥100 PFU/ml) (Fig. 4A), indicating that only the ON subpopulation grew under these conditions. This was confirmed by the fact that the highest fluorescence intensity was reached in the presence of the highest amount of phage, which killed the OpvABOFF subpopulation more efficiently. As expected, in the absence of phage (or in the presence of a low number of phages), bacterial growth did not correlate with fluorescence increase, indicating that the OpvABOFF population mainly accounts for bacterial growth. Based on this experiment, we tentatively concluded that strong, dose‐dependent fluorescence intensity was reached 10 h post infection (Fig. 4A, inset). In order to test those parameters using flow cytometry, the same serial dilutions of phages were applied for 10 h and enrichment of the OpvABON subpopulation was monitored. In contrast with the plate reader experiment, all the phage concentrations tested above 1 PFU/ml were able to enrich the ON subpopulation (Fig. 4B). Moreover, a linear correlation was observed between the phage concentration and the OpvABON subpopulation size up to 102 PFU/ml, which could be used as a calibration curve to determine the phage concentration in a given sample. Hence, the phage biosensor appears to be highly sensitive and can detect as little as 10 PFU/ml.
Fig 4.
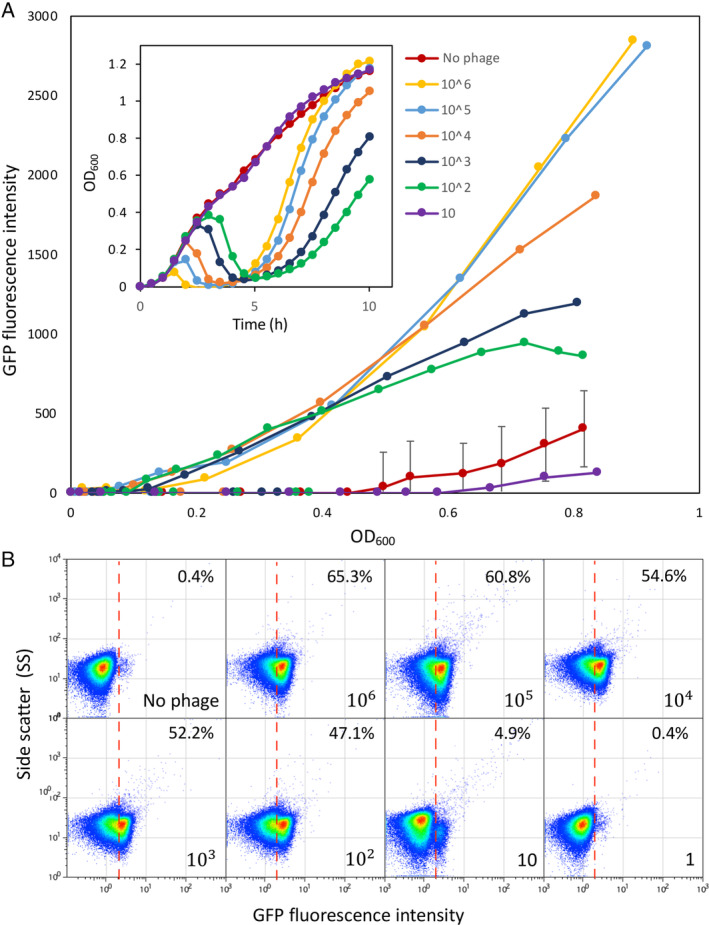
OpvAB phage‐biosensor sensitivity.
A. Growth curves of SV6727 (opvAB::gfp) in contact with serial dilutions of phage 9NA, from 106 up to 101 PFU/ml. Data are represented by growth curves showing OD600 versus time (h) (inset) and [fluorescence intensity (ON subpopulation size) versus growth (OD600)].
B. GFP fluorescence distribution in SV6727 before (t = 0) and after growth (t = 10 h) in LB containing serial dilutions of 9NA phage starting from 106 up to 100 PFU/ml. Data are represented by a dot plot [side scatter versus fluorescence intensity (ON subpopulation size)]. All data were collected for 100,000 events per sample. [Color figure can be viewed at wileyonlinelibrary.com]
Our next objective was to improve the detection limits of our biosensor. As we noticed that killing of the OpvABOFF subpopulation (and thus selection of the OpvABON subpopulation) was more efficient in the presence of a high number of 9NA PFU (Fig. 4A), we aimed at improving the method by adding less cells to increase the ratio phage/biosensor. Indeed, dilution of the cells lowered the number of phages that could be detected (Fig. 5A). It is remarkable that for the smallest dilution of the overnight culture the concentration of phages detected using a microplate reader was around 4 × 105 PFU/ml, whereas as little as 4 PFU/ml were detected using a higher dilution of the biosensor (equivalent to 8.8 × 104 cells/ml, corresponding to about 17,600 ON cells), thus increasing 5‐log the detection limit while the number of cells decreased 3‐log only.
Fig 5.
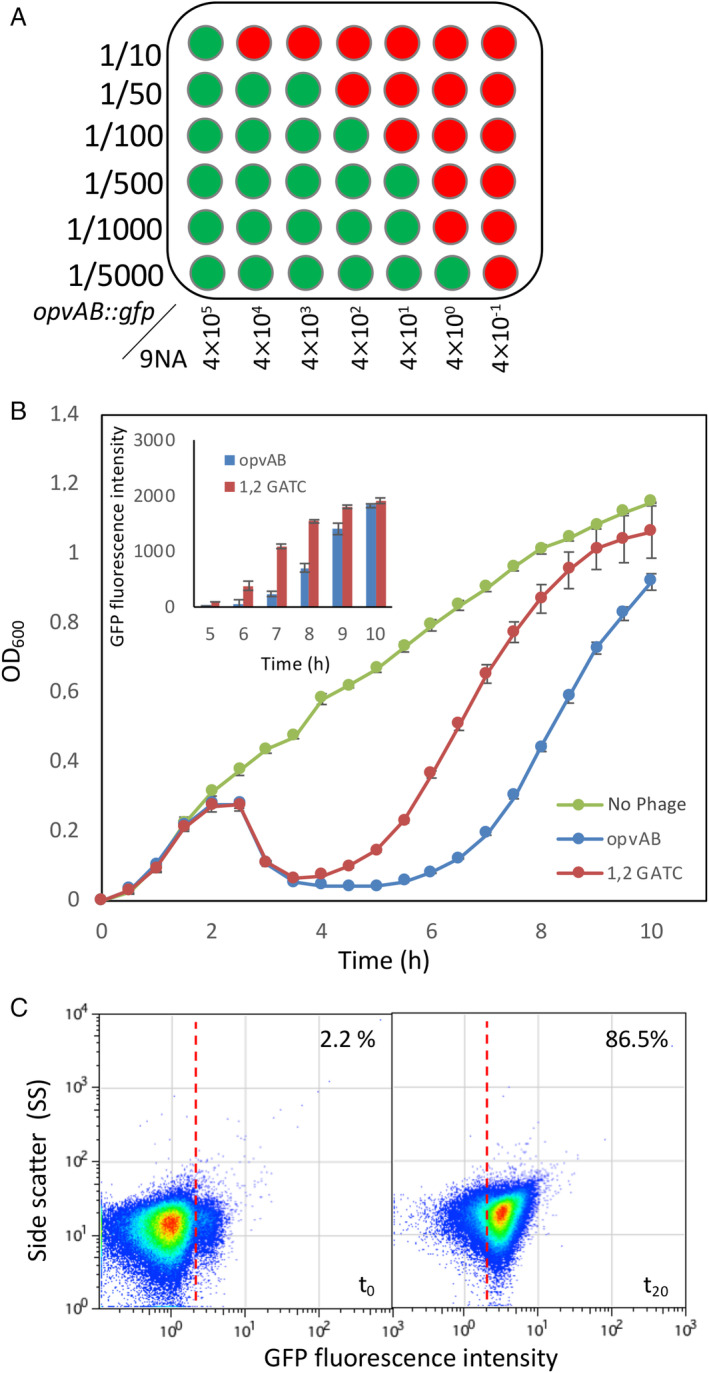
Improving the opvAB biosensor sensitivity.
A. Bacterial dilutions (10−1 up to 2.10−4) of strain SV6727 grown in the presence of serial dilutions of bacteriophage 9NA. Bacterial growth was monitored in a microplate reader. Green wells are fluorescence‐positive, and red wells are fluorescence‐negative.
B. Growth curves obtained for a strain expressing the gfp fusion under the control of a wild‐type promoter (SV6727) in the absence of phage (green) and in the presence of 9NA (blue), and for a derivative strain carrying point mutations in two GATC sites at the opvAB promoter region (SV8578) in the presence of phage 9NA (red). Fluorescence intensity data were obtained at several time points of the growth curve (inclusion).
C. GFP fluorescence distribution in SV8578 (GATC1,2 opvAB::gfp) before (t = 0 h) and after growth in LB containing 9NA phage (t = 20 h). Data are represented by a dot plot [side scatter versus fluorescence intensity (ON subpopulation size)]. All data were collected for 100,000 events per sample. [Color figure can be viewed at wileyonlinelibrary.com]
Another improvement originated from earlier work showing that the OpvABON subpopulation size increased when specific GATC sites present in the promoter region were mutated (Cota et al., 2012; Cota et al., 2015). Indeed, when the mut1,2GATC opvAB::gfp construct was used as a biosensor for phage 9NA, the fluorescence increased faster than with the construct bearing the wild‐type promoter and reached a plateau in about 10 h (Fig. 5B, inset). Interestingly, the largest difference was observed around 7 h post‐infection.
We then combined both improvements (low cell density and mutations in GATC sites) to reach an even lower detection limit. In the experiment described in Fig. 5C, we used as little as eight 9NA phages diluted into 500 ml of LB containing a dilution (1/10) from an overnight culture of the biosensor strain and phage enrichment was allowed to proceed for 15 h. Then the mixture was diluted again into fresh medium to let the OpvABON subpopulation enrich for 5 h before the flow cytometry assay. It is remarkable that an extremely small initial number of phages was able to enrich the ON subpopulation up to 86.5% under the conditions described (Fig. 5C), thus lowering the detection level to about 1.6 phages per 100 ml. As a reminder, before optimization the detection limit with flow cytometry was around 1000 phages per 100 ml.
Design of an opvAB::gfp biosensor able to detect coliphages
Up to this point, the design and improvement of the phage biodetection method was performed using S. enterica as a chassis. To widen our capacity to detect phages in environmental samples, we decided to adapt the biosensor to coliphages. To do so, we first had to restore a full length LPS as E. coli K12 MG1655 is well known to carry an IS5‐interrupted version of the wbbL gene encoding a rhamnosyltranferase (Browning et al., 2013). This is clearly evidenced by the length and profile of the LPS following extraction, and separation by SDS‐PAGE and silver staining is affected in MG1655 (Fig. 6A, Lane 1). Indeed, the MG1655 LPS profile did not show the typical bands in the upper part of the gel, which correspond to the large concatemers of oligosaccharide units. The interrupted wbbL gene was complemented either by adding a plasmid‐based copy of wbbL or by integrating ectopically a single copy of the wild‐type gene. In both cases, the full‐length LPS structure was restored as indicated by the multiple bands in the upper part of the gel (Fig. 6A, Lanes 4 and 5). Engineering of strains carrying an opvAB::gfp fusion on the chromosome allowed us to examine the consequences of opvAB expression in E. coli. A wild‐type opvAB control region only caused a subtle alteration of the LPS profile, an observation consistent with the small size of the ON subpopulation (Fig. 6A, Lane 6). Modification of the length distribution of glycan chains in the O antigen was however unambiguous when the LPS+ E. coli strain harboured a GATC‐less opvAB::gfp fusion (Fig. 6A, Lanes 6 and 7). In this background, opvAB expression is constitutive and phase variation is abolished, thus rendering a population that is entirely OpvABON (Cota et al., 2015).
Fig 6.
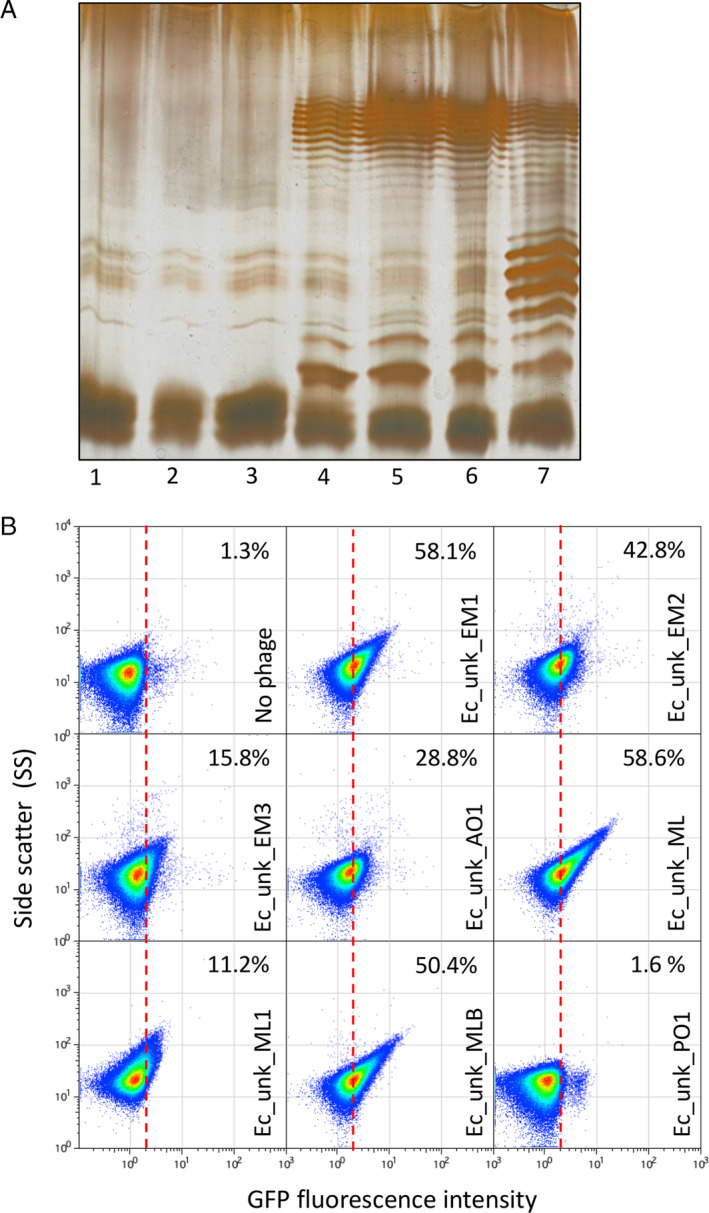
Restoration of E. coli MG1655 O‐Antigen and detection of unknown coliphages.
A. Electrophoretic visualization of LPS profiles from different MG1655 derivatives, as follows: 1, Wild‐type Escherichia coli MG1655 strain containing an IS5‐inactivated version of the wbbl gene; 2, E. coli MG1655 with a deletion of the lactose operon (DR3); 3, E. coli DR3 carrying the empty pETb vector; 4, E. coli DR3 carrying a pETb derivative containing a wild type wbbL gene; 5, E. coli DR3 carrying the wbbL gene integrated into the genome, replacing the altered IS5‐wbbl gene (LPS+ strain); 6, E. coli MG1655 LPS+ carrying the opvAB::gfp construction (DR29); 7, E. coli MG1655 LPS+ opvAB::gfp GATC‐less (DR30).
B. GFP fluorescence distribution in E. coli DR29 before (t = 0 h) and after growth in LB, or LB containing purified uncharacterized bacteriophages (Ec_Unk_EM1, Ec_Unk_EM2, Ec_Unk_EM3, Ec_Unk_AO1, Ec_Unk_ML, Ec_Unk_ML1, Ec_Unk_MLB and Ec_Unk_PO1 (t = 8 h). Data are represented by a dot plot [side scatter versus fluorescence intensity (ON subpopulation size)]. ON subpopulation sizes (percentages) are shown for each sample. All data were collected for 100,000 events per sample. [Color figure can be viewed at wileyonlinelibrary.com]
Using the opvAB::gfp‐carrying LPS+ E. coli strain, we isolated and purified seven coliphages from water samples from the Seville area. As shown in Fig. 6B, seven of the coliphages were able to select the OpvABON subpopulation with an enrichment efficacy ranging from 11.2% to 58.6%, far higher than the control without phage (1.27%). Again, this difference could account to the use of the LPS as a primary receptor or as a full receptor for the isolated coliphages except for Ec_unk_PO1. Whatever the case, this experiment shows that the method works alike in E. coli and in S. enterica, and that the opvAB::gfp fusion is a versatile tool that could be used with different enterobacterial chassis.
Expanding the opvAB::gfp biosensor to the detection of phages that use proteins as receptors: bacteriophage T5 detection as a proof‐of‐concept
As coliphage detection showed some variation (Fig. 6B), perhaps depending on the receptor molecule used for recognition at the bacterial surface, we decided to design an E. coli biosensor that would specifically detect a phage known to bind a protein. Phage T5 is an appropriate model: although it uses LPS for initial adsorption, binding to the ferrichrome transporter FhuA on the outer membrane constitutes the trigger for DNA injection (Flayhan et al., 2012; Arnaud et al., 2017). We first thought to make a simple PopvAB::fhuA‐gfp fusion, but that would have provided us with a biosensor working in the opposite way from the opvAB::gfp biosensor. Indeed, in this case, the presence of T5 would select for the OFF (non‐fluorescent) subpopulation, and detection of a decrease in fluorescence can be expected to be less reliable than monitoring an increase of fluorescence intensity. Interestingly, T5 encodes a lipoprotein, encoded by the llp gene, which is an inhibitor of T5 infection (Braun et al., 1994). Llp is synthesized upon T5 infection and prevents superinfection of the host by other T5 virions by interacting with the FhuA receptor, resulting in its inactivation (Pedruzzi et al., 1998). Moreover, the llp phage gene is expressed in the early stage of T5 infection, which not only prevents superinfection but also protects progeny phages from being inactivated by the receptor present in envelope fragments of lysed host cells (Decker et al., 1994). We thus reasoned that placing the llp gene under the control of the opvAB promoter might confer resistance to T5 only in the ON state. As expected from the literature, a ΔfhuA strain proved resistant to T5, but not to T4, which uses OmpC as receptor (Washizaki et al., 2016) (Fig. 7A). In turn, ectopic expression of llp from a plasmid also confers resistance to the E. coli MG1655 WT strain as in the ΔfhuA mutant. We thus integrated a PopvAB::llp‐gfp fusion at the lac locus in E. coli MG1655 and assayed this new biosensor with T5 for 10 h on a plate reader, periodically monitoring the OD600 and the florescence intensity (Fig. 7B). As predicted, enrichment of the ON subpopulation was detected after a few (Lawrence et al., 2019) hours and GFP fluorescence intensity increased, indicating that this fusion performed well as a T5 biosensor. We then assayed the biosensor using flow cytometry to estimate the efficiency of ON subpopulation enrichment, which ranged from 82% to almost 90% in 10 h for the four different phages isolated on E. coli MG1655 (not complemented) (Fig. 7C). Interestingly, addition of colicin M led to a similar enrichment of the ON subpopulation, thus confirming that production of the FhuA inhibitor Llp was also able to prevent colicin M binding. We also performed an additional control, using one of the LPS‐binding phages assayed in Fig. 6. As predicted based on its effect on the Ec_opvAB::gfp biosensor, this specific phage (Ec_unk_EM1) was unable to select the PopvAB::llp‐gfp ON subpopulation (Fig. 7C). Conversely, Ec_unk_PO1 (selected to turn on the PopvAB::llp‐gfp fusion, and thus likely recognizing FhuA) was not detected using the Ec_opvAB::gfp biosensor (Fig. 6C). Together, these experiments show that the method can accommodate not only different bacterial chassis (Salmonella and E. coli) but also different types of receptors, thus providing primary indication on the type of receptor used by a given phage.
Fig 7.
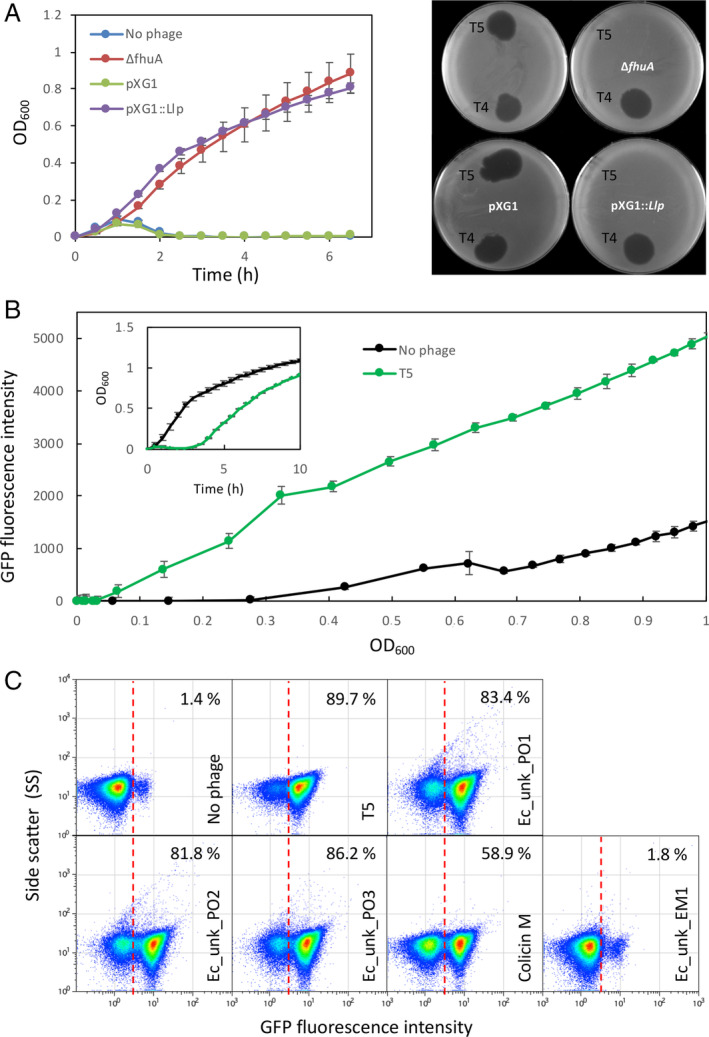
Bacteriophage T5 detection.
A. Left. Bacterial growth curves in the presence of phage T5 (2.5 × 107). In blue, E. coli MG1655 wild type. In red, a strain with a deletion of the fhuA gene (DR42). In green, Escherichia coli carrying an empty pXG1 plasmid, used as a control. In purple, E. coli carrying plasmid pXG1 containing the llp gene from phage T5. Right. Plaque assay using the same strains and bacteriophages T5 and T4.
B. Growth curves of an E. coli derivative strain DR41 carrying a PopvAB::llp‐gfp fusion in the presence or absence of T5 phage for 10 h. Inlet, the same data are represented as a function of time.
C. GFP fluorescence distribution in DR41 (PopvAB ::llp‐gfp) before (t = 0) and after growth in LB containing T5, unknown E. coli phages (Ec_Unk_PO1, Ec_Unk_PO2, Ec_Unk_PO3 isolated on E. coli MG1655 and Ec_Unk_EM1 isolated on DR28), or colicin M (3 μg/ml, t = 8 h). Data are represented by a dot plot [side scatter versus fluorescence intensity (ON subpopulation size)]. All data were collected for 100,000 events per sample. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
The challenge of detecting and accurately counting bacteriophages has been around for some time as more and more researchers are interested in characterizing bacteriophages for ecological or biotechnological purposes. Beside the gold standard plaque assay method, a variety of techniques have been developed, each with pros and cons (Baran et al., 2018; Morella et al., 2018). These techniques are based on methodologies such as quantitative PCR (ddPCR or regular qPCR), FISH, electron microscopy and flow cytometry, and they can be classified into three types: culture‐dependent, sequence‐based and particle‐based (Baran et al., 2018). Another way to classify these techniques is to consider the characteristics of the enumeration and the additional infection parameters that can be obtained (Ackermann, 2009). For example, the classical plaque assay can discriminate infectious versus non‐infectious particles, gives information on the adsorption rate, is doable without any particular equipment, does not need genome sequencing, but it is difficult to perform in a high‐throughput manner. In contrast, a ddPCR‐based method allows high throughput analysis but requires knowledge of the phage genome sequence and highly specialized equipment. The information provided is also different as there is no possibility to discriminate infectious versus non‐infectious particles while it can discern between different phages in a single assay (Morella et al., 2018). In the spectra of current methods, flow cytometry–based approaches are among the most sensitive and most published methods. They rely on labelling of the phage genome with a fluorescent dye (Brussaard, 2004; Carreira et al., 2015; de la Cruz Peña et al., 2018), although some label‐free protocols have been developed recently (Ma et al., 2016).
In the present work, we combine culture‐based methods with flow cytometry, which allows to quantify with high sensitivity infectious particles only (Fig. 4). The method is based on opvAB, a locus present S. enterica but neither in Salmonella bongori nor in other enteric bacteria (Cota et al., 2012). Phase‐variable expression of opvAB produces subpopulations of phage‐sensitive (OpvABOFF) and phage‐resistant (OpvABON) cells (Cota et al., 2015). Cloning of a gfp gene downstream of the opvAB operon provides a simple and efficient sensor to monitor the increase of the OpvABON subpopulation by flow cytometry. This increase provides indirect evidence that the OpvABOFF subpopulation undergoes lysis, which in turn indicates the presence of a phage that uses the O antigen as receptor. An advantage of this type of sensor is that lysis of OpvABOFF cells and concomitant increase of OpvABON cells produce amplification of the signal over time. This feature makes the sensor highly sensitive: 109 phages/ml can be detected in 1 h, and an amount of phage as low as 2 PFU in 100 ml can be detected in less than 20 h.
Starting with the original biosensor construct in S. enterica (Figs. 1, 2) able to detect purified or unpurified phages present in raw water samples (Fig. 3), we expanded our collection of biosensors to detect coliphages. In an MG1655 derivative with a complete (reconstructed) LPS, a constitutively expressed opvAB operon integrated at the lac locus shortens the E. coli O‐antigen (Fig. 6A). As a consequence, the engineered E. coli strain is able to detect LPS‐binding phages in a way similar to the S. enterica strain (Figs. 2 and 6). Of note, the enrichment of the OpvABON subpopulation by killing of the OFF subpopulation was not as effective with coliphages as for phages active against S. enterica. Nevertheless, as the detection by flow cytometry is very sensitive and reproducible, the enrichment levels achieved were sufficient for a proper detection. The sensitivity was however decreased, perhaps indicating that the coliphages used in the study required an additional component of the outer membrane as receptor, rendering LPS binding less crucial for successful infection. This hypothesis is strengthened by the fact that in the case of the FhuA‐binding phages detected using the PopvAB‐llp::gfp biosensor, the enrichment of the ON subpopulation was much more efficient (Fig. 7). Indeed, it is known that T5 relies essentially on FhuA for a productive lytic cycle, whereas the polymannose decoration of the O‐antigen is used as a primary receptor important for primary binding but dispensable for the overall cycle (Fig. 7) (Braun and Wolff, 1973; Heller and Braun, 1982).
An important add‐on value of the method described here is its capacity to discriminate bacteriophages infecting the same host but using different types of receptors (Figs. 6 and 7). This property is relevant in the frame of phage therapy when isolation of phages using different receptors is a must to overcome resistance due to mutations in the receptor (Bai et al., 2019). This work, allowing a rapid discrimination between phages using LPS or FhuA as receptors, could thus be systematically used prior to the assembly of phage cocktails. Moreover, this method could be implemented for other receptors used by bacteriophages as long as a specific inhibitor is available (like Llp for FhuA‐binding phage: Fig. 7). For T‐even coliphages, for example, a trick might be to use the outer‐membrane protein TraT, encoded by the F plasmid, that masks or modifies the conformation of outer‐membrane protein A (OmpA), which is the receptor for many T‐even‐like phages (Riede and Eschbach, 1986; Labrie et al., 2010; Bertozzi Silva et al., 2016). If an inhibitor‐encoding gene does not exist for a dedicated receptor, an alternative would be to use the receptor gene itself fused to the gfp and controlled by the opvAB promoter. A potential drawback of this strategy is that the biosensor might be less sensitive for phage enumeration as the presence of phage would be detected as a decrease in fluorescence. Nevertheless, this alternative procedure to determine the host receptor remains open.
Experimental procedures
Bacterial strains
Bacterial strains used in this study are listed in Table 1. Strains of S. enterica belong to serovar Typhimurium and derive from the mouse‐virulent strain ATCC 14028. For simplicity, S. enterica serovar Typhimurium is routinely abbreviated as S. enterica. Strain SV6727, harbouring an opvAB::gfp transcriptional fusion downstream of the opvB stop codon (OpvAB+), was described in Cota et al. (2015). Escherichia coli K12 MG1655 was provided by the E. coli Genetic Stock Center. DR3, an MG1655 derivative with the lactose operon deleted (Olivenza et al., 2019), was used as an intermediate strain for the construction of DR29 and DR30. For construction of DR29, the opvAB::gfp fusion was amplified from SV6727 using the oligonucleotides MG‐opvA and MG‐opvB (Table 2). For construction of DR30, the opvAB::gfp mutGATC construction was amplified from SV6729 using MG‐opvA and MG‐opvB. PCR products were recombined into the chromosome of DR3 at the lac locus (Datsenko and Wanner, 2000).
Table 1.
Bacterial strains and plasmids used in this study.
| Strain name | Genotype | Reference |
|---|---|---|
| ATCC 14028 a | Wild type | ATCC |
| SV6727 a | ATCC 14028 opvAB::gfp | (Cota et al., 2015) |
| SV6729 a | ATCC 14028 mut 1,2,3,4 GATC opvAB::gfp | (Cota et al., 2015) |
| SV8578 a | ATCC 14028 mut 1,2 GATC opvAB::gfp | (Cota et al., 2015) |
| WG1 b | Wild type | (Browning et al., 2013) |
| MG1655 b | Wild type | E. coli Genetic Stock Center |
| CC118 b | phoA20 thi‐1 rspE rpoB argE(Am) recA1 (lambda pir) | (Manoil and Beckwith, 1985) |
| S17 b | recA pro hsdR RP4‐2‐Tc::Mu‐Km::Tn7 (lambda pir) | (Simon et al., 1983) |
| DR3 b | E. coli MG1655 ∆lacZY | This work |
| DR28 b | E. coli MG1655 ∆lacZY LPS+ | This work |
| DR29 b | E. coli MG1655 ∆lacZY opvAB::gfp LPS+ | This work |
| DR30 b | E. coli MG1655 ∆lacZY mutGATC opvAB::gfp LPS+ | This work |
| DR34 b | E. coli TD2158 ∆ypjA::km | This work |
| DR40 b | E. coli MG1655 ∆IS5::km | This work |
| DR41 b | E. coli MG1655 P opvAB::llp::gfp | This work |
| DR42 b | E. coli MG1655 ∆fhuA | KEIO collection |
| Plasmids | Characteristics | Reference |
|---|---|---|
| pKD46 | Lambda Red recombinase, AmpR | (Datsenko and Wanner, 2000) |
| pKD4 | Kanamycin resistance gene flanked by FRT sites. | (Datsenko and Wanner, 2000) |
| pCP20 | FLP recombinase, temperature sensitive replicon at 37°C, AmpR, CmR | (Datsenko and Wanner, 2000) |
| pET20b | T7 promoter, AmpR | (Datsenko and Wanner, 2000) |
| pET20b‐wbbl | wbbL gene under constitutive expression | (Browning et al., 2013) |
| pDMS197 | Contains λ pir‐dependent R6K replication origin; requires lambda pir‐containing bacteria strain. SacB (sucrose sensitivity), TcR | (Edwards et al., 1998) |
| pDMS197‐wbbl | TcR | This work |
| pXG1 | PLtetO promoter, gfp fusion cloning vector | (Urban and Vogel, 2007) |
| pXG1::llp | T5 llp gene under constitutive expression | This work |
Salmonella enterica.
Escherichia coli.
Table 2.
Oligonucleotides used in this study.
| Oligo | Sequence (5´→3′) |
|---|---|
| Operonlac‐PS1 | ATGATAGCGCCCGGAAGAGAGTCAATTCAGGGTGGTGAATGTGTAGGCTGGAGATGCTTC |
| Operonlac‐PS2 | TAGGCCTGATAAGCGCAGCGTATCAGGCAATTTTTATAATCATATGAATATCCTCCTTAG |
| MG‐opvA | ATGATAGCGCCCGGAAGAGAGTCAATTCAGGGTGGTGAATTCATTTGGTTATAAATAGAG |
| MG‐opvB | TAGGCCTGATAAGCGCAGCGTATCAGGCAATTTTTATAATGAGTTTATCTCTGCGCAATGT |
| ypjA‐PS1 | CCTTGATATATGCCCCGCCCGGGATGGCTGCCTTCACTACGTGTAGGCTGGAGCTGCTTC |
| ypjA‐PS2 | TTGCGCACAGCCGCCTTCAGCCACGGCTCAACTTCCATACCATATGAATATCCTCCTTAG |
| ypjA‐opvA‐F | CCTTGATATATGCCCCGCCCGGGATGGCTGCCTTCACTACTTCATTTGGTTATAAATAGAG |
| ypjA‐opvB‐R | TTGCGCACAGCCGCCTTCAGCCACGGCTCAACTTCCATACGAGTTTATCTCTGCGCAATG |
| IS50‐PS1 | CATGAAGCATGATGATTTGCTGACATATATTAAATATGTCGTGTAGGCTGGAGCTGCTTC |
| IS50‐PS2 | GATCCTGCGCACCAATCAACAACCGTATCAGAATAGATACCATATGAATATCCTCCTTAG |
| SacI‐wbbl‐F | AAAGAGCTCATGGTATATATAATAATCGTTTCCCACGGAC |
| XbaI‐wbbl‐R | AAATCTAGATTACGGGTGAAAAACTGATGAAATTCGATC |
| IS5‐PS1 | CATGAAGCATGATGATTTGCTGACATATATTAAATATGTCGTGTAGGCTGGAGCTGCTTC |
| IS5‐PS2 | GATCCTGCGCACCAATCAACAACCGTATCAGAATAGATACCATATGAATATCCTCCTTAG |
| pXG1‐Llp‐F | TAAGAAGGAGATATACATATGGCTAGCAAAGGAGAAGAAC |
| pXG1‐Llp‐R | ATTTTTTCATGGTACCTTTCTCCTCTTTAATG |
| Llp‐F | GAAAGGTACCATGAAAAAATTATTTTTAGCTATGGC |
| Llp‐R | TGCTAGCCATATGTATATCTCCTTCTTATTAGAAAACTCCCTCGCATG |
| opvA‐Llp‐F | TTATGTGTGGGTTTTATCTTATGAAGAAATATACGTTCGCTAAGGAGGTTTTCTAATGAAAAAATTATTTTTAGC |
| gfp‐Llp‐R | AAAGTTCTTCTCCTTTACTCATATGTATATCTCCTTCTTATTAGAAAACTCCCTCGCATG |
Restoration of O‐antigen synthesis in E. coli MG1655
Escherichia coli MG1655 is unable to synthesize O‐antigen due to a disruption of the wbbL gene caused by an IS5 element (Blattner et al., 1997). The WbbL protein is a rhamnose transferase necessary for O‐antigen synthesis (Stevenson et al., 1994). For complementation assays, a pETb vector containing a copy of the wild‐type wbbl gene (kindly provided by Browning et al., 2013), was transformed into strain DR3 as a positive control of O‐antigen restoration upon gene complementation. In order to replace the mutant wbbl gene on the MG1655 chromosome, the wild‐type wbbl gene was amplified from E. coli WG1 using oligonucleotides SacI‐wbbl1 and XbaI‐wbbl2 (Table 2). The resulting PCR fragment was cloned onto suicide plasmid pDMS197 (Hautefort et al., 2003) after restriction enzyme digestion to obtain plasmid pDMS::wbbl. This plasmid was propagated in E. coli CC118 λ pir. Plasmids derived from pDMS197 were transformed into E. coli S17 λ pir. The resulting strain was then used as a donor for mating with DR40 harbouring a KmR cassette, which was integrated into the chromosome using IS5‐PS1 and IS5‐PS2 oligos and pKD4 as a DNA template (Datsenko and Wanner, 2000). TcR transconjugants were selected on E plates supplemented with tetracycline. Several TcR transconjugants were grown in nutrient broth (NB) and plated on NB supplemented with 5% sucrose. Individual tetracycline‐sensitive segregants were then screened for kanamycin sensitivity, and the affected chromosome region was sequenced after PCR amplification with external oligonucleotides. The resulting DR28 strain is an E. coli MG1655 derivative with the lac operon deleted and the O‐antigen restored.
Construction of an E. coli strain expressing T5 lipoprotein
The T5 lipoprotein llp gene was amplified by PCR and cloned into pXG1 to obtain pXG1::Llp. For the construction of strain DR41, the T5 llp and gfp genes were amplified and linked together by a fusion PCR using opvAB‐llp‐F/llp‐R and gfp‐F/gfp‐R oligos (Table 2). The resulting PCR product (llp::gfp) was integrated at the lac locus of DR3. A synthetic ribosome‐binding site named BI‐RBS used in a previous study (Olivenza et al., 2019) was inserted upstream of the llp gene to optimize Llp production.
Bacteriophages
Bacteriophages 9NA (Wilkinson et al., 1972; Casjens et al., 2014) and Det7 (Walter et al., 2008) were kindly provided by Sherwood Casjens, University of Utah, Salt Lake City. Bacteriophage P22_H5 is a virulent derivative of bacteriophage P22 that carries a mutation in the c2 gene (Smith and Levine, 1964) and was kindly provided by John R. Roth, University of California, Davis. T5 bacteriophage was provided by Pascale Boulanger, I2BC, Orsay, France. Bacteriophages Se_F1, Se_F2, Se_F3 and Se_F6 infecting S. enterica were isolated and purified from waste water samples in Seville. Genome sequences are under submission. Other waste water bacteriophages were isolated upon infection of either DR28 (E. coli MG1655 ΔlacZY LPS+) or MG1655. A list of all the phages isolated in this study is included in Table 3.
Table 3.
Bacteriophages used in this study.
| Bacteriophage | Host | Origin | Reference or accession number |
|---|---|---|---|
| P22 | Salmonella enterica | Sherwood Casjens | NC_002371 |
| Det7 | S. enterica | Sherwood Casjens | NC_027119 |
| 9NA | S. enterica | Sherwood Casjens | NC_025443 |
| T5 | Escherichia coli F | Pascale Boulanger | AY692264 |
| Se_F1 | S. enterica | Sevilla, Spain (Waste water) | This work |
| Se_F2 | S. enterica | Sevilla, Spain (Waste water) | This work |
| Se_F3 | S. enterica | Sevilla, Spain (Waste water) | This work |
| Se_F6 | S. enterica | Sevilla, Spain (Waste water) | This work |
| Se_ML1 | S. enterica | Sevilla, Spain (Maria Luisa Park) | This work |
| Se_AO1 | S. enterica | Sevilla, Spain (Sheep farm) | This work |
| Ec_Unk_EM | DR28 | Sevilla, Spain (Waste water) | This work |
| Ec_Unk_EM1 | DR28 | Sevilla, Spain (Waste water) | This work |
| Ec_Unk_EM2 | DR28 | Sevilla, Spain (Waste water) | This work |
| Ec_Unk_AO1 | DR28 | Sevilla, Spain (Sheep farm) | This work |
| Ec_Unk_ML | DR28 | Sevilla, Spain (Maria Luisa Park) | This work |
| Ec_Unk_ML1 | DR28 | Sevilla, Spain (Maria Luisa Park) | This work |
| Ec_Unk_MLB | DR28 | Sevilla, Spain (Maria Luisa Park) | This work |
| Ec_Unk_PO1 | MG1655 | Cáceres, Spain (chicken farm) | This work |
| Ec_Unk_PO2 | MG1655 | Cáceres, Spain (chicken farm) | This work |
| Ec_Unk_PO3 | MG1655 | Cáceres, Spain (chicken farm) | This work |
Culture media
Bertani's lysogeny broth (LB) was used as standard liquid medium. NB was used to recover cultures after transduction or transformation. Solid media contained agar at 1.5% final concentration. Green plates (Chan et al., 1972) contained methyl blue (Sigma‐Aldrich, St. Louis, MO, USA) instead of aniline blue. Antibiotics were used at the concentrations described previously (Torreblanca and Casadesús, 1996). E50X salts [H3C6H5O7 · H2O (300 g/l), MgSO4 (14 g/l), K2HPO4 · 3H2O (1965 g/l), NaNH4HPO4 · H2O (525 g/l)] were used to prepare E minimal plates [E50 × (20 ml/l), glucose (0.2%), agar 20 g/l].
Electrophoretic visualization of LPS profiles
To investigate LPS profiles as described in Buendía‐Clavería et al. (2003), bacterial cultures were grown overnight in LB. Bacterial cells were harvested and washed with 0.9% NaCl. Around 3 × 108 cells were pelleted by centrifugation and resuspended in lysis buffer [60 mM Tris/HCl (pH 6.8) containing 2% (w/v) SDS, 1 mM EDTA]. The mixture was then heated at 100°C for 5 min, then diluted eightfold in the same buffer without SDS. The extracts were further treated with Benzonase (200 U) and proteinase K (10 μg/ml) at 37°C for 5 h. Electrophoresis of the crude samples was then performed on a 16.5% (wt/vol) Tris/Tricine buffer. Gels were then fixed and silver‐stained in a silver solution according to Tsai et al. (1981).
Bacteriophage challenge
Bacterial cultures were grown at 37°C in LB (5 ml) containing phages [100 μl of phage lysate (108–1010pfu)]. Cultures were diluted 1:100 in LB + phage and incubated until exponential phase (OD600 ~ 0.3) before flow cytometry analysis. To monitor bacterial growth, overnight bacterial cultures were diluted 1:100 in 200 μl of LB. Five microliters of a bacteriophage lysate were added (108–1010 pfu). OD600 and fluorescence intensity were subsequently measured at 30‐min intervals using a Synergy™ HTX Multi‐Mode Microplate Reader from Biotek.
Flow cytometry
Bacterial cultures were grown at 37°C in LB or LB containing phage until exponential (OD600 ~0.3) or stationary phase (OD600 ~4). Cells were then diluted in phosphate‐buffered saline (PBS) to obtain a final concentration of ~107 cfu/ml. Data acquisition and analysis were performed using a Cytomics FC500‐MPL cytometer (Beckman Coulter, Brea, CA, USA). Data were collected for 100,000 events per sample, and analysed with CXP and FlowJo 8.7 software.
Phage isolation from water samples
Samples of water were filtered using a Millex‐GS filter, 0.22 μm filter pore. Each sample was divided into two samples; chloroform was added to one of the parts. The filtered sample of water (1 ml) was added to an overnight culture of host bacterial strain (0.5 ml). The mixture was supplemented with 3.5 ml of LB broth and was incubated overnight at 37°C without shaking. The culture was then centrifuged at 5000 rpm during 5 min. The supernatant was filtered using a filter, 0.22 μm pore size, in order to discard bacterial cells and debris.
Fifty microliters of an overnight culture of the bacterial target was spread on LB plates. Lysates were dropped onto the agar surface and left to dry. The plates were inspected for lysis zones after overnight incubation at 37°C. The spot assay was used to assess the bactericidal ability of different phage lysates. Isolation of phage was performed by the double agar layer method using Salmonella or E. coli as a host system. Isolated plaques were suspended in LB and were streaked in plates which contained a bacterial layer in order to isolate pure phage. Phages were characterized on the basis of plaque morphology and host range. Moreover, certain phages were characterized by electron microscopy and their genomes were sequenced.
Electron microscopy
Electron microscopy was performed at the Mediterranean Institute of Microbiology (IMM) facility. Phage suspensions (5 μl) were dropped onto a copper grid covered with Formvar and carbon and left 3 min at 25°C, after what the excess of liquid was removed. Phage particles were stained according to Ackermann (2009) with a 2% uranyl acetate solution. Dried grids were then observed using a transmission microscope FEI Tecnai 200 kV coupled to an Eagle CCS 2k × 2k camera. Images were then analysed using the ImageJ software.
Detection of phage in a crude water sample
A tube with 3.5 ml of LB, 0.5 ml of an overnight culture of SV6727 strain (opvAB::gfp) and 1 ml of a 0.22 μm filtered water sample was incubated for 7 h without shaking. The culture was diluted at a ratio of 1/200 in LB medium and incubated at 37°C with shaking (200 rpm) for 3 h before dilution in PBS buffer. All water samples were collected in the Sevilla area.
Data acquisition and analysis were performed using a Cytomics FC500‐MPL cytometer (Beckman Coulter). Data were collected for 100,000 events per sample and analysed with CXP and FlowJo 8.7 software. The presence of phages in water samples was checked simultaneously by plating 1 ml of water on a plate with S. enterica poured into soft agar.
Supporting information
Fig. S1 Plaque assay performed with the crude samples tested in Fig. 3 on S. enterica displaying a full‐length (WT) or a short (wzz) LPS.
Fig. S2: GFP fluorescence distribution in strain SV6727 (opvAB::gfp) after growth in LB in the absence (no phage) or presence of P22_wt for 8 h Data are represented by a dot plot (side scatter versus fluorescence intensity [ON subpopulation size]). ON subpopulation sizes (percentages) are shown for each sample. Data were collected for 30,000 events.
Acknowledgements
This study was supported by grant BIO2016‐75235‐P from the Ministerio de Ciencia, Innovación y Universidades of Spain to J.C. and grant ANR‐16‐CE12‐0029‐01 from the Agence Nationale de la Recherche to M.A. We are grateful to Elena Puerta‐Fernández for advice and discussions, to Rocío Fernández for her help in sample processing, and to Modesto Carballo, Laura Navarro and Cristina Reyes from the Servicio de Biología, CITIUS, Universidad de Sevilla, for help with experiments performed at the facility. We are also very thankful to Artemis Kosta and Hugo Le Guenno at the imaging facility of the Mediterranean Institute of Microbiology (IMM) for TEM imaging, and Denis Duché, LISM Marseille, for the kind gift of colicin M. M.A. is grateful to Nicolas Ginet for critical reading of the manuscript and to the whole phage group @LCB for constant support regarding the phage project at the University of Sevilla.
References
- Abedon, S.T. , García, P. , Mullany, P. , and Aminov, R. (2017) Editorial: phage therapy: past, present and future. Front Microbiol 8: 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abeles, S.R. , and Pride, D.T. (2014) Molecular bases and role of viruses in the human microbiome. J Mol Biol 426: 3892–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackermann, H.‐W. (2009) Basic phage electron microscopy. Methods Mol Biol 501: 113–126. [DOI] [PubMed] [Google Scholar]
- Allen, H.K. , and Abedon, S.T. (2014) Virus ecology and disturbances: impact of environmental disruption on the viruses of microorganisms. Front Microbiol 5: 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres, D. , Hanke, C. , Baxa, U. , Seul, A. , Barbirz, S. , and Seckler, R. (2010) Tailspike interactions with lipopolysaccharide effect DNA ejection from phage P22 particles in vitro. J Biol Chem 285: 36768–36775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud, C.‐A. , Effantin, G. , Vivès, C. , Engilberge, S. , Bacia, M. , Boulanger, P. , et al (2017) Bacteriophage T5 tail tube structure suggests a trigger mechanism for Siphoviridae DNA ejection. Nat Commun 8: 1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, J. , Jeon, B. , and Ryu, S. (2019) Effective inhibition of Salmonella typhimurium in fresh produce by a phage cocktail targeting multiple host receptors. Food Microbiol 77: 52–60. [DOI] [PubMed] [Google Scholar]
- Baran, N. , Goldin, S. , Maidanik, I. , and Lindell, D. (2018) Quantification of diverse virus populations in the environment using the polony method. Nat Microbiol 3: 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beller, L. , and Matthijnssens, J. (2019) What is (not) known about the dynamics of the human gut virome in health and disease. Curr Opin Virol 37: 52–57. [DOI] [PubMed] [Google Scholar]
- Bertozzi Silva, J. , Storms, Z. , and Sauvageau, D. (2016) Host receptors for bacteriophage adsorption. FEMS Microbiol Lett 363: fnw002. [DOI] [PubMed] [Google Scholar]
- Blattner, F.R. , Plunkett, G. , Bloch, C.A. , Perna, N.T. , Burland, V. , Riley, M. , et al (1997) The complete genome sequence of Escherichia coli K‐12. Science 277: 1453–1474. [DOI] [PubMed] [Google Scholar]
- Braun, V. , and Wolff, H. (1973) Characterization of the receptor protein for phage T5 and colicin M in the outer membrane of E. coli B. FEBS Lett 34: 77–80. [DOI] [PubMed] [Google Scholar]
- Braun, V. , Killmann, H. , and Herrmann, C. (1994) Inactivation of FhuA at the cell surface of Escherichia coli K‐12 by a phage T5 lipoprotein at the periplasmic face of the outer membrane. J Bacteriol 176: 4710–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning, D.F. , Wells, T.J. , França, F.L.S. , Morris, F.C. , Sevastsyanovich, Y.R. , Bryant, J.A. , et al (2013) Laboratory adapted Escherichia coli K‐12 becomes a pathogen of Caenorhabditis elegans upon restoration of O antigen biosynthesis. Mol Microbiol 87: 939–950. [DOI] [PubMed] [Google Scholar]
- Brussaard, C.P.D. (2004) Optimization of procedures for counting viruses by flow cytometry. Appl Environ Microbiol 70: 1506–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buendía‐Clavería, A.M. , Moussaid, A. , Ollero, F.J. , Vinardell, J.M. , Torres, A. , Moreno, J. , et al (2003) A purL mutant of Sinorhizobium fredii HH103 is symbiotically defective and altered in its lipopolysaccharide. Microbiology 149: 1807–1818. [DOI] [PubMed] [Google Scholar]
- Carreira, C. , Staal, M. , Middelboe, M. , and Brussaard, C.P.D. (2015) Counting viruses and bacteria in photosynthetic microbial mats. Appl Environ Microbiol 81: 2149–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casjens, S.R. , Leavitt, J.C. , Hatfull, G.F. , and Hendrix, R.W. (2014) Genome sequence of Salmonella phage 9NA. Genome Announc 2: e00531‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, R.K. , Botstein, D. , Watanabe, T. , and Ogata, Y. (1972) Specialized transduction of tetracycline resistance by phage P22 in Salmonella typhimurium. II properties of a high‐frequency‐transducing lysate. Virology 50: 883–898. [DOI] [PubMed] [Google Scholar]
- Chanishvili, N. (2016) Bacteriophages as therapeutic and prophylactic means: summary of the soviet and post soviet experiences. Curr Drug Deliv 13: 309–323. [DOI] [PubMed] [Google Scholar]
- Cota, I. , Blanc‐Potard, A.B. , and Casadesús, J. (2012) STM2209‐STM2208 (opvAB): a phase variation locus of Salmonella enterica involved in control of O‐antigen chain length. PLoS One 7: e36863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota, I. , Sánchez‐Romero, M.A. , Hernández, S.B. , Pucciarelli, M.G. , García‐del Portillo, F. , and Casadesús, J. (2015) Epigenetic control of Salmonella enterica O‐antigen chain length: a tradeoff between virulence and bacteriophage resistance. PLoS Genet 11: e1005667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz Peña, M.J. , Martinez‐Hernandez, F. , Garcia‐Heredia, I. , Lluesma Gomez, M. , Fornas, Ò. , and Martinez‐Garcia, M. (2018) Deciphering the human virome with single‐virus genomics and metagenomics. Viruses 10: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko, K.A. , and Wanner, B.L. (2000) One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc Natl Acad Sci U S A 97: 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, M.R. , Broadbent, S.E. , Harris, S.R. , Thomson, N.R. , and van der Woude, M.W. (2013) Horizontally acquired glycosyltransferase operons drive salmonellae lipopolysaccharide diversity. PLoS Genet 9: e1003568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker, K. , Krauel, V. , Meesmann, A. , and Heller, K.J. (1994) Lytic conversion of Escherichia coli by bacteriophage T5: blocking of the FhuA receptor protein by a lipoprotein expressed early during infection. Mol Microbiol 12: 321–332. [DOI] [PubMed] [Google Scholar]
- Djebara, S. , Maussen, C. , De Vos, D. , Merabishvili, M. , Damanet, B. , Pang, K.W. , et al (2019) Processing phage therapy requests in a Brussels military hospital: lessons identified. Viruses 11: 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, R.A. , Keller, L.H. , and Schifferli, D.M. (1998) Improved allelic exchange vectors and their use to analyze 987P fimbria gene expression. Gene 207: 149–157. [DOI] [PubMed] [Google Scholar]
- Farooq, U. , Yang, Q. , Ullah, M.W. , and Wang, S. (2018) Bacterial biosensing: recent advances in phage‐based bioassays and biosensors. Biosens Bioelectron 118: 204–216. [DOI] [PubMed] [Google Scholar]
- Flayhan, A. , Wien, F. , Paternostre, M. , Boulanger, P. , and Breyton, C. (2012) New insights into pb5, the receptor binding protein of bacteriophage T5, and its interaction with its Escherichia coli receptor FhuA. Biochimie 94: 1982–1989. [DOI] [PubMed] [Google Scholar]
- Franche, N. , Vinay, M. , and Ansaldi, M. (2016) Substrate‐independent luminescent phage‐based biosensor to specifically detect enteric bacteria such as E. coli . Environ Sci Pollut Res Int 24: 42–51. [DOI] [PubMed] [Google Scholar]
- Hatfull, G.F. (2015) Dark matter of the biosphere: the amazing world of bacteriophage diversity. J Virol 89: 8107–8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hautefort, I. , Proença, M.J. , and Hinton, J.C.D. (2003) Single‐copy green fluorescent protein gene fusions allow accurate measurement of Salmonella gene expression in vitro and during infection of mammalian cells. Appl Environ Microbiol 69: 7480–7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller, K. , and Braun, V. (1982) Polymannose O‐antigens of Escherichia coli, the binding sites for the reversible adsorption of bacteriophage T5+ via the L‐shaped tail fibers. J Virol 41: 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, B. , Margolin, W. , Molineux, I.J. , and Liu, J. (2015) Structural remodeling of bacteriophage T4 and host membranes during infection initiation. Proc Natl Acad Sci U S A 112: E4919–E4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz, B.L. , Westveld, A.H. , Brum, J.R. , and Sullivan, M.B. (2014) Modeling ecological drivers in marine viral communities using comparative metagenomics and network analyses. Proc Natl Acad Sci U S A 111: 10714–10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jover, L.F. , Effler, T.C. , Buchan, A. , Wilhelm, S.W. , and Weitz, J.S. (2014) The elemental composition of virus particles: implications for marine biogeochemical cycles. Nat Rev Microbiol 12: 519–528. [DOI] [PubMed] [Google Scholar]
- Labrie, S.J. , Samson, J.E. , and Moineau, S. (2010) Bacteriophage resistance mechanisms. Nat Rev Microbiol 8: 317–327. [DOI] [PubMed] [Google Scholar]
- Lawrence, D. , Baldridge, M.T. , and Handley, S.A. (2019) Phages and human health: more than idle hitchhikers. Viruses 11: 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letarov, A.V. , and Kulikov, E.E. (2017) Adsorption of bacteriophages on bacterial cells. Biochem Biokhimiia 82: 1632–1658. [DOI] [PubMed] [Google Scholar]
- Ma, L. , Zhu, S. , Tian, Y. , Zhang, W. , Wang, S. , Chen, C. , et al (2016) Label‐free analysis of single viruses with a resolution comparable to that of electron microscopy and the throughput of flow cytometry. Angew Chem Int Ed Engl 55: 10239–10243. [DOI] [PubMed] [Google Scholar]
- Manoil, C. , and Beckwith, J. (1985) TnphoA: a transposon probe for protein export signals. Proc Natl Acad Sci U S A 82: 8129–8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morella, N.M. , Yang, S.C. , Hernandez, C.A. , and Koskella, B. (2018) Rapid quantification of bacteriophages and their bacterial hosts in vitro and in vivo using droplet digital PCR. J Virol Methods 259: 18–24. [DOI] [PubMed] [Google Scholar]
- Ofir, G. , and Sorek, R. (2018) Contemporary phage biology: from classic models to new insights. Cell 172: 1260–1270. [DOI] [PubMed] [Google Scholar]
- Olivenza, D.R. , Nicoloff, H. , Antonia Sánchez‐Romero, M. , Cota, I. , Andersson, D.I. , and Casadesús, J. (2019) A portable epigenetic switch for bistable gene expression in bacteria. Sci Rep 9: 11261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedruzzi, I. , Rosenbusch, J.P. , and Locher, K.P. (1998) Inactivation in vitro of the Escherichia coli outer membrane protein FhuA by a phage T5‐encoded lipoprotein. FEMS Microbiol Lett 168: 119–125. [DOI] [PubMed] [Google Scholar]
- Peitzman, S.J. (1969) Felix d'Herelle and bacteriophage therapy. Trans Stud Coll Physicians Phila 37: 115–123. [PubMed] [Google Scholar]
- Rakhuba, D.V. , Kolomiets, E.I. , Dey, E.S. , and Novik, G.I. (2010) Bacteriophage receptors, mechanisms of phage adsorption and penetration into host cell. Pol J Microbiol 59: 145–155. [PubMed] [Google Scholar]
- Riede, I. , and Eschbach, M.L. (1986) Evidence that TraT interacts with OmpA of Escherichia coli . FEBS Lett 205: 241–245. [DOI] [PubMed] [Google Scholar]
- Rinke, C. , Schwientek, P. , Sczyrba, A. , Ivanova, N.N. , Anderson, I.J. , Cheng, J.‐F. , et al (2013) Insights into the phylogeny and coding potential of microbial dark matter. Nature 499: 431–437. [DOI] [PubMed] [Google Scholar]
- Rohde, C. , Resch, G. , Pirnay, J.‐P. , Blasdel, B.G. , Debarbieux, L. , Gelman, D. , et al (2018) Expert opinion on three phage therapy related topics: bacterial phage resistance, phage training and prophages in bacterial production strains. Viruses 10: 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohwer, F. , and Segall, A.M. (2015) In retrospect: a century of phage lessons. Nature 528: 46–48. [DOI] [PubMed] [Google Scholar]
- Roux, S. , Hallam, S.J. , Woyke, T. , and Sullivan, M.B. (2015) Viral dark matter and virus‐host interactions resolved from publicly available microbial genomes. eLife 4: e08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmond, G.P.C. , and Fineran, P.C. (2015) A century of the phage: past, present and future. Nat Rev Microbiol 13: 777–786. [DOI] [PubMed] [Google Scholar]
- Simon, R. , Priefer, U. , and Pühler, A. (1983) A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Bio/Technology 1: 784–791. [Google Scholar]
- Smith, H.O. , and Levine, M. (1964) Two sequential repressions of DNA synthesis in the establishment of LYSOGENY by phage P22 and its mutants. Proc Natl Acad Sci U S A 52: 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson, G. , Neal, B. , Liu, D. , Hobbs, M. , Packer, N.H. , Batley, M. , et al (1994) Structure of the O antigen of Escherichia coli K‐12 and the sequence of its rfb gene cluster. J Bacteriol 176: 4144–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torreblanca, J. , and Casadesús, J. (1996) DNA adenine methylase mutants of Salmonella typhimurium and a novel dam‐regulated locus. Genetics 144: 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, C.M. , Frasch, C.E. , and Mocca, L.F. (1981) Five structural classes of major outer membrane proteins in Neisseria meningitidis . J Bacteriol 146: 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban, J.H. , and Vogel, J. (2007) Translational control and target recognition by Escherichia coli small RNAs in vivo. Nucleic Acids Res 35: 1018–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinay, M. , Franche, N. , Grégori, G. , Fantino, J.‐R. , Pouillot, F. , and Ansaldi, M. (2015) Phage‐based fluorescent biosensor prototypes to specifically detect enteric bacteria such as E. coli and Salmonella enterica typhimurium. PLoS One 10: e0131466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl, A. , Battesti, A. , and Ansaldi, M. (2019) Prophages in Salmonella enterica: a driving force in reshaping the genome and physiology of their bacterial host? Mol Microbiol 111: 303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, M. , Fiedler, C. , Grassl, R. , Biebl, M. , Rachel, R. , Hermo‐Parrado, X.L. , et al (2008) Structure of the receptor‐binding protein of bacteriophage det7: a podoviral tail spike in a myovirus. J Virol 82: 2265–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. , Tu, J. , Liu, J. , and Molineux, I.J. (2019) Structural dynamics of bacteriophage P22 infection initiation revealed by cryo‐electron tomography. Nat Microbiol 4: 1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washizaki, A. , Yonesaki, T. , and Otsuka, Y. (2016) Characterization of the interactions between Escherichia coli receptors, LPS and OmpC, and bacteriophage T4 long tail fibers. Microbiologyopen 5: 1003–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson, R.G. , Gemski, P. , and Stocker, B.A. (1972) Non‐smooth mutants of Salmonella typhimurium: differentiation by phage sensitivity and genetic mapping. J Gen Microbiol 70: 527–554. [DOI] [PubMed] [Google Scholar]
- Wommack, K.E. , and Colwell, R.R. (2000) Virioplankton: viruses in aquatic ecosystems. Microbiol Mol Biol Rev 64: 69–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Plaque assay performed with the crude samples tested in Fig. 3 on S. enterica displaying a full‐length (WT) or a short (wzz) LPS.
Fig. S2: GFP fluorescence distribution in strain SV6727 (opvAB::gfp) after growth in LB in the absence (no phage) or presence of P22_wt for 8 h Data are represented by a dot plot (side scatter versus fluorescence intensity [ON subpopulation size]). ON subpopulation sizes (percentages) are shown for each sample. Data were collected for 30,000 events.