

Abstract
An imbalance of cellular oxidants and reductants causes redox stress, which must be rapidly detected to restore homeostasis. In bacteria, the Firmicutes encode conserved Spx‐family transcriptional regulators that modulate transcription in response to redox stress. SpxA1 is an Spx‐family orthologue in the intracellular pathogen Listeria monocytogenes that is essential for aerobic growth and pathogenesis. Here, we investigated the role of SpxA1 in growth and virulence by identifying genes regulated by SpxA1 in broth and during macrophage infection. We found SpxA1‐activated genes encoding heme biosynthesis enzymes and catalase (kat) were required for L. monocytogenes aerobic growth in rich medium. An Spx‐recognition motif previously defined in Bacillus subtilis was identified in the promoters of SpxA1‐activated genes and proved necessary for the proper activation of two genes, indicating this regulation by SpxA1 is likely direct. Together, these findings elucidated the mechanism of spxA1 essentiality in vitro and demonstrated that SpxA1 is required for basal expression of scavenging enzymes to combat redox stress generated in the presence of oxygen.
Keywords: heme, peroxide, redox, regulation, respiration
Listeria monocytogenes is a model intracellular pathogen that requires the redox‐responsive transcriptional regulator SpxA1 for aerobic growth and virulence. Here, we identified the genes regulated by SpxA1 and found that SpxA1‐dependent expression of genes encoding catalase and heme biosynthesis enzymes was necessary for aerobic growth in vitro. These results demonstrated that SpxA1 regulation is required to combat peroxide stress in oxygen‐rich environments.
1. INTRODUCTION
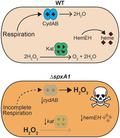
Bacterial pathogens are challenged with abundant redox stressors present in the environment and during infection of a mammalian host. The innate immune system attempts to eliminate invading pathogens after phagocytosis by activating the respiratory burst, bombarding pathogens with reactive oxygen species (ROS) such as superoxide, hydrogen peroxide and hypochlorous acid in the phagosome (Winterbourn and Kettle, 2013). In addition to these exogenous ROS, bacteria must also manage the endogenous ROS generated during aerobic respiration from the reaction of oxygen with free metals, quinones, or flavoproteins (Imlay, 2008; Reniere, 2018). To combat these assaults, bacteria produce many enzymes that detoxify ROS as well as regulators to sense ROS and induce the appropriate responses as redox stress arises (Ruhland and Reniere, 2018).
Listeria monocytogenes is a low G + C Gram‐positive bacterium that lives in nature as a saprophyte and in mammals as an intracellular pathogen. The virulence factors employed by L. monocytogenes to cause disease have been extensively studied (Freitag et al., 2009). After a host cell phagocytoses L. monocytogenes, the bacteria secrete the pore‐forming toxin listeriolysin O, which damages the phagosomal membrane and enables the bacteria to escape into the host cytosol. In the cytosol, L. monocytogenes produces ActA, which recruits host actin and propels the bacteria throughout the cell and into neighboring cells (Tilney and Portnoy, 1989). This intracellular lifecycle requires that L. monocytogenes adapt quickly to changing conditions, including the drastic change from the oxidizing phagosome to the reduced cytosolic environment. While genetic data suggest a link between redox sensing and virulence gene regulation (Reniere et al., 2015; Reniere et al., 2016), the molecular mechanisms by which L. monocytogenes senses the host environment and adapts appropriately are not well understood.
One transcriptional regulator that controls the response to oxidative stress is the ArsC‐family protein Spx, which is conserved in low G + C Firmicutes (Zuber, 2004; Zuber, 2009). Spx function has been most well characterized in Bacillus subtilis, where it regulates hundreds of genes by interacting with the alpha‐C‐terminal domain (αCTD) of RNA polymerase (RNAP) (Rochat et al., 2012). Spx senses redox stress via an amino‐terminal cysteine‐x‐x‐cysteine motif that forms an intramolecular disulfide bond upon oxidation. Oxidized Spx then activates genes encoding proteins required to resolve oxidative stress, such as thioredoxins and bacillithiol biosynthesis machinery (Nakano, Küster‐Schöck, et al., 2003; Zuber, 2004; Nakano et al., 2005; Rochat et al., 2012). Additionally, reduced Spx is important for basal expression of redox homeostasis genes in the absence of oxidative stress (Rochat et al., 2012).
L. monocytogenes encodes two Spx‐family proteins: spxA1 and spxA2. SpxA1 is essential for aerobic growth and pathogenesis, while SpxA2 is dispensable in both conditions (Whiteley et al., 2017). In this work, we identified SpxA1‐regulated genes in L. monocytogenes and ascertained those that are required for aerobic growth and virulence. We demonstrated that although SpxA1 regulates hundreds of genes, the severe growth defect of ∆spxA1 in broth can be rescued simply by supplementing the media with exogenous heme or catalase. While catalase and heme biosynthesis enzymes (hemEH) were required for aerobic growth in vitro, neither was necessary for intracellular growth in macrophages. Collectively, these results support a model in which SpxA1 directly activates genes required for aerobic growth and distinguishes its role in vitro from its role during infection.
2. RESULTS
2.1. Transcriptomics to identify SpxA1‐regulated genes
SpxA1 is essential for L. monocytogenes aerobic growth and pathogenesis (Whiteley et al., 2017). To investigate the mechanisms behind these phenotypes, we took a global approach and analyzed the SpxA1‐dependent transcriptome. RNA was harvested from L. monocytogenes wild type (wt) and ∆spxA1 strains grown anaerobically in rich broth (brain heart infusion, BHI) and from J774 macrophages infected with either strain for 8 hr. Following rRNA depletion, Illumina sequencing of the cDNA revealed hundreds of genes were changed in abundance in an SpxA1‐dependent manner (see Experimental Procedures for details). Genes that exhibited a greater than two‐fold difference in abundance between wt and ∆spxA1 samples (p < .001) were included for further analysis. Table 1 includes genes with the greatest reduction in ∆spxA1 compared to wt during infection as well as those most highly changed in BHI.
TABLE 1.
Top SpxA1‐activated genes
| LMRG | Lmo | Gene | Function | Fold change in ∆spxA1 | Spx Motif (Position relative to + 1 transcription) a | |
|---|---|---|---|---|---|---|
| BHI | J774 | |||||
| LMRG_01912 | lmo2785 | kat | Catalase | −66.8 | −11.5 |

|
| LMRG_01620 | lmo2212 | hemE, uroD | Uroporphyrinogen decarboxylase | −15.8 | −46.5 |

|
| LMRG_01621 | lmo2211 | hemH, cpfC | Ferrochelatase | −15.4 | −36.8 | |
| LMRG_01781 | lmo2467 | – | Chitin‐binding protein | −12.2 | −25.5 |

|
| LMRG_01954 | lmo2742 | – | Hypothetical protein, SH3 domains | −12.2 | −22.9 |

|
| LMRG_01953 | lmo2743 | – | Transaldolase | −8.2 | −20.6 | |
| LMRG_01977 | lmo2719 | – | tRNA‐adenosine deaminase | −11.1 | −19.8 |

|
| LMRG_00292 | lmo0609 | – | Rhodanese Homology Domain | −9.1 | −20.7 |

|
| LMRG_01978 | lmo2718 | cydA | Cytochrome bd oxidase subunit I | −9.4 | – | No motif |
| LMRG_01980 | lmo2716 | cydC | ABC transporter | −9.1 | – | |
| LMRG_01979 | lmo2717 | cydB | Cytochrome bd oxidase subunit II | −8.7 | – | |
| LMRG_01981 | lmo2715 | cydD | ABC transporter | −7.4 | – | |
| LMRG_00343 | lmo0656 | – | Uncharacterized membrane protein | −7.3 | −16.1 |

|
| LMRG_00478 | lmo0790 | – | Cys‐tRNA(Pro) deacylase | −6.6 | −11.6 | No motif |
| LMRG_00477 | lmo0789 | – | DAP‐epimerase Superfamily | −6.4 | −9.7 | |
| LMRG_01211 | lmo2061 | – | Hypothetical protein | −6.1 | −13.1 | No TSS |
| LMRG_01210 | lmo2060 | – | Hypothetical protein | −5.7 | −11.3 | |
| LMRG_01212 | lmo2062 | – | Copper transport protein | −4.7 | −16.3 | |
| LMRG_01925 | lmo2770 | gshF | Glutathione synthase | −4.9 | −11.8 | No TSS |
| LMRG_01656 | lmo2176 | – | TetR‐family transcriptional regulator | −4.8 | −27.9 | No TSS |
| LMRG_02247 | lmo0822 | – | SoxR‐family transcriptional regulator | −4.3 | −11.5 | No TSS |
| LMRG_00270 | lmo0588 | – | DNA photolyase | −4.2 | −12.1 |

|
| LMRG_00294 | lmo0611 | acpD | FMN‐dependent NADH‐azoreductase 1 | −2.3 | −27.0 |

|
During anaerobic growth in vitro, SpxA1 directly or indirectly activated 145 genes, and 87% of those also exhibited SpxA1‐dependent expression during infection (Table 1 and Table S1). There are numerous similarities between the genes regulated by SpxA1 in L. monocytogenes and the Spx regulon in B. subtilis. SpxA1 activated many homologous genes, including: fbp (fructose‐1,6‐bisphosphatase), hemH (ferrochelatase), lmo2256 (similar to yraA), tpx (thiol peroxidase), trxB (thioredoxin reductase), and the thioredoxins trxA and yjbH (Nakano, Küster‐Schöck, et al., 2003). Spx‐family proteins also regulate low molecular weight thiol biosynthesis in both organisms. In L. monocytogenes, SpxA1 activated gshF, which codes for glutathione synthase. B. subtilis does not produce glutathione and instead synthesizes bacillithiol, production of which is regulated by Spx (Gaballa et al., 2013).
In the absence of spxA1, 72 genes exhibited increased expression during anaerobic growth in vitro, suggesting that SpxA1 normally functions to directly or indirectly repress those genes (Table S2). Approximately 60% of the repressed genes showed SpxA1‐dependent expression during macrophage infection. SpxA1‐repressed genes encode proteins involved in diverse functions, including: transport of amino acids (e.g., ctaP and gltD), sugars (lmo0859‐61 operon), potassium (kdpAB) and zinc (znuA); phosphotransferase system components (ulaA, bvrB and fructose‐specific components); and nucleotide metabolism (guaA, guaB2 and purE). The genes with the greatest increase in expression in ∆spxA1 were lmo0437, encoding an uncharacterized protein with a predicted NAD(P)‐dependent oxidoreductase domain, and the mecA‐coiA operon, which is adjacent to spxA1 in the genome. MecA and CoiA are proteins involved in competence in other Gram‐positive bacteria (Grossman, 1995; Desai and Morrison, 2007), but their functions are unclear in L. monocytogenes, which is not known to be naturally competent.
2.2. Promoter analysis
Spx‐family proteins lack a canonical DNA‐binding domain and instead directly interact with the RNAP αCTD to guide positive or negative regulation of genes (Ruhland and Reniere, 2018). In B. subtilis, Spx‐activated promoters exhibit extended −35 boxes with G at position −44 and C at position −43 relative to the transcription start site (TSS) (Reyes and Zuber, 2008; Lin et al., 2013). Here, we refer to this sequence as the “GC‐motif”. T at position −44 is also associated with Spx‐activated expression, although the magnitude of regulation is reduced (Rochat et al., 2012). To determine which differentially expressed genes identified in our RNA‐seq analysis were directly regulated by SpxA1, the DNA sequences surrounding the TSSs of differentially expressed genes were analyzed in silico. Nine of the 15 operons (60%) in Table 1 contained the GC‐motif (or TC) at or near the −44/−43 position relative to the TSS. Several of the GC‐motifs were shifted one or two nucleotides from the −44/−43 position. However, the TSSs were mapped in a genome‐wide manner and have not been individually validated, so single‐nucleotide precision of TSS mapping would not be expected (Wurtzel et al., 2012). Four of the TSSs have not been mapped and, therefore, could not be analyzed for potential GC‐motifs. Rochat et al. found 71% of the B. subtilis Spx‐activated promoters contained C at position −43, which is similar to the frequency found here (Rochat et al., 2012). In contrast, the GC‐motif is not found in Spx‐repressed promoters in B. subtilis, but instead, A or T at position −43 is correlated with repression (Rochat et al., 2012). In our analysis, 29 of the 46 transcriptional units (63%) repressed by SpxA1 contained A or T at position −43 (Table S2), which is about the frequency expected by chance in a bacterium with 39% GC content. Together, these results suggested SpxA1 directly activates a majority of the genes identified by our transcriptomics, although SpxA1‐mediated repression may be direct or indirect.
2.3. The roles of SpxA1‐dependent genes during infection
We hypothesized that SpxA1‐dependent transcriptional changes enable aerobic growth and virulence and hence, in the absence of spxA1, appropriate gene induction does not occur. Therefore, the top SpxA1‐activated genes were examined to determine which may be required for virulence but were insufficiently activated in ∆spxA1 (Table 1). Each SpxA1‐dependent gene or operon was overexpressed from the constitutive HyPer promoter and stably integrated into the chromosome via the plasmid pPL2t, referred to here as pOE (Lauer et al., 2002; Reniere et al., 2016). Overexpression was verified by qPCR of six of the overexpressing strains and indeed, all of the target genes were expressed at or above wt levels when integrated into the ∆spxA1 mutant (Figure S1).
L. monocytogenes ∆spxA1 is able to replicate intracellularly and spread cell‐to‐cell, albeit less efficiently than wt (Whiteley et al., 2017). To identify the genes that contribute to these phenotypes in vivo, the overexpressing strains were evaluated in a plaque assay. Plaque assays measure cell‐to‐cell spread over 3 days, which requires L. monocytogenes be able to enter cells, escape the vacuolar compartment, replicate in the host cytosol, and use actin‐based motility to spread to neighboring cells (Sun et al., 1990; Reniere et al., 2016). Hence, the ability to form a plaque is highly correlated with pathogenicity in murine models of infection. L. monocytogenes ∆spxA1 formed a plaque approximately 23% the size of wt and the overexpressing strains formed similarly sized plaques, ranging from 15% to 37% the size of wt (Figure 1a).
FIGURE 1.
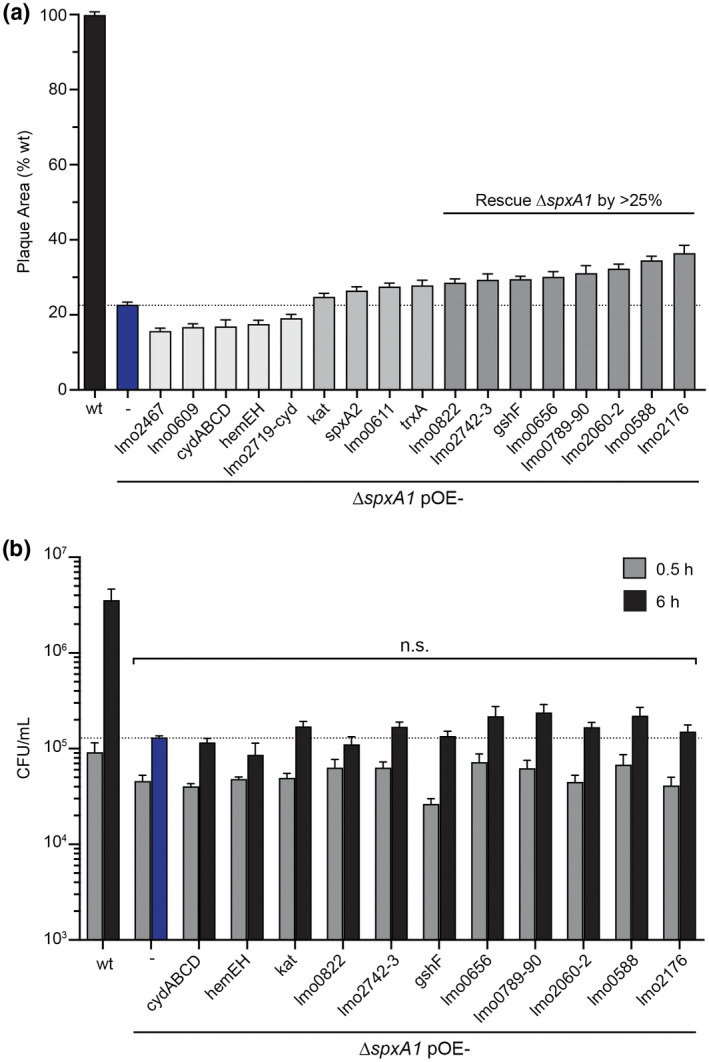
Intracellular replication and cell‐to‐cell spread of ∆spxA1 overexpressing strains. (a) Plaque area measured as a percentage of the wt strain. (b) Intracellular growth kinetics of ∆spxA1 overexpressing strains in BMDMs, measured at 0.5 and 6 hr post‐infection. Bacteria were plated anaerobically and CFU were enumerated after 24 hr. All ∆spxA1 overexpressing strains exhibited similar growth as the ∆spxA1 parental strain (n.s., p > .05). In both panels, data are the means and standard error of the means (SEM) of three independent experiments
In addition to the top SpxA1‐activated genes in Table 1, we investigated the roles of spxA2 and trxA in the ∆spxA1 plaque defect. SpxA1 and SpxA2 share 25% amino acid identity (56% similarity) and we hypothesized that SpxA2 may be able to compensate for the loss of spxA1 if overexpressed. In Staphylococcus aureus, Spx is essential but overexpressing trxA suppresses spx essentiality (Villanueva et al., 2016). However, overexpressing either spxA2 or trxA did not substantially increase the plaque size of the ∆spxA1 parental strain (Figure 1a).
The ∆spxA1 mutant is not impaired in vacuolar escape during infection of bone marrow‐derived macrophages (BMDMs) (Whiteley et al., 2017). Therefore, we presume that the ∆spxA1 plaque defect is primarily due to impaired intracellular growth. To test the roles of SpxA1‐dependent genes in intracellular growth, BMDMs were infected with the overexpressing strains and bacteria were plated anaerobically to enumerate colony forming units (CFU). For these experiments, strains that formed plaques at least 25% larger than the ∆spxA1 parental strain were included (Figure 1a). During a 6‐hr infection, wt increased approximately 40‐fold, while the ∆spxA1 strain increased threefold and the overexpressing strains grew similarly to the ∆spxA1 parental strain (Figure 1b). These results demonstrated the pleiotropic nature of SpxA1 regulation, as restoring expression of a single SpxA1‐dependent gene or operon was insufficient to restore virulence to the ∆spxA1 mutant.
2.4. The roles of SpxA1‐dependent genes during aerobic growth
The panel of overexpressing strains were next grown in the presence of oxygen to evaluate aerobic growth. The only overexpressing strains that formed colonies on BHI agar incubated aerobically were ∆spxA1 pOE‐cydABCD and ∆spxA1 pOE‐hemEH (Figure 2). The ∆spxA1 pOE‐cydABCD strain constitutively expressed the operon encoding cytochrome bd oxidase, resulting in a few tiny colonies that were visible after several days on solid media, but did not grow larger. In contrast, ∆spxA1 pOE‐hemEH grew more robustly. This strain constitutively expressed the genes encoding uroporphyrinogen decarboxylase (HemE, also named UroD) and ferrochelatase (HemH, also named CpfC), which are two enzymes in the heme biosynthesis pathway (Dailey et al., 2017). However, neither pOE‐cydABCD nor pOE‐hemEH rescued ∆spxA1 aerobic growth in a shaking flask, as measured by optical density, nor did overexpression of any other gene or operon in Table 1 (data not shown).
FIGURE 2.

Overexpressing hemEH rescued ∆spxA1 aerobic growth on solid media. All L. monocytogenes ∆spxA1 overexpressing strains were streaked for growth aerobically and anaerobically on BHI. Only ∆spxA1 pOE‐cydABCD and ∆spxA1 pOE‐hemEH grew on solid media in the presence of oxygen. Colonies were visible after 3–5 days
Overexpressing hemEH rescued growth of ∆spxA1 on solid media but not in aerobic liquid culture. Hence, we hypothesized that heme is necessary for growth in the presence of oxygen, but reasoned that overexpressing only two of the nine genes encoding enzymes in the heme biosynthetic pathway was insufficient. The remaining seven genes are encoded in three loci elsewhere in the chromosome, making it difficult to engineer a strain overexpressing all nine genes. Therefore, we next supplemented the growth media with exogenous heme in an attempt to restore ∆spxA1 aerobic growth. Addition of 5 or 10 µM heme partially rescued ∆spxA1 growth in a shaking flask, although it did not fully restore growth to wt levels (Figure 3a). We did not observe any effect of adding exogenous iron, suggesting that this phenotype is specific to the heme moiety (data not shown).
FIGURE 3.

Exogenous heme partially rescued ∆spxA1 aerobic growth. (a) Aerobic growth kinetics measured by optical density (OD600) of L. monocytogenes grown in TSB alone or supplemented with heme to a final concentration of 5 or 10 µM. (b) Aerobic growth of L. monocytogenes strains in TSB or TSB supplemented with heme 24 hr post‐inoculation with 5 × 107 CFU/ml. The ∆spxA1 strain was engineered to over‐express genes encoding catalase (pOE‐kat) or the cytochrome bd operon (pOE‐cydABCD), as indicated. In both panels, data are the mean and SEM of three independent experiments. p values were calculated using a heteroscedastic Student's t test. *p < .05
Heme is a redox‐active molecule that is required for the function of proteins involved in electron transport, oxygen carrying, and as an enzyme cofactor (Reniere et al., 2007). In addition, excess free heme is toxic due to its ability to generate ROS and cause oxidative damage to proteins and DNA (Anzaldi and Skaar, 2010; Wakeman et al., 2012). Surprisingly, heme rescued ∆spxA1 growth rather than inducing additional redox stress, suggesting heme was being used as a protein cofactor. In L. monocytogenes, heme‐binding proteins include cytochrome oxidases, catalase, heme import and biosynthesis proteins, and enzymes in the cobalamin synthesis pathway. The gene with the greatest reduction in expression in the ∆spxA1 mutant grown in broth was kat, encoding the only catalase produced by L. monocytogenes (Table 1). L. monocytogenes produces two terminal cytochrome oxidases: a cytochrome bd oxidase (encoded by cydABCD) and a cytochrome aa3‐type menaquinol oxidase (encoded by qoxABCD). However, only the cydABCD operon transcript, which is required for respiration and intracellular growth (Corbett et al., 2017), was significantly less abundant in ∆spxA1 (Table 1). The cydABCD operon also had a small effect on ∆spxA1 growth when constitutively expressed (Figure 2). We, therefore, postulated that exogenous heme partially rescued aerobic growth via its ability to act as a cofactor for catalase and/or cytochrome bd oxidase. To test this, kat or the cydABCD operon was overexpressed in ∆spxA1 in the presence of exogenous heme and CFU were measured after 24 hr of aerobic growth. Surprisingly, overexpressing kat significantly increased ∆spxA1 survival in the absence of heme (Figure 3b). However, in the presence of exogenous heme there was no effect of overexpressing kat or cydABCD (Figure 3b). These data suggested that L. monocytogenes ∆spxA1 is unable to survive aerobically due to insufficient heme and catalase production. Moreover, the effect of exogenous heme on ∆spxA1 aerobic growth was either independent of catalase and cytochrome bd or requires overexpression of multiple genes.
2.5. Catalase is required for aerobic growth in vitro, but not intracellular growth
Overexpressing kat had a small but significant effect on ∆spxA1 aerobic growth (Figure 3b). The kat overexpressing strain exhibited 10‐fold higher kat expression than wt in anaerobic broth (Figure S1), although we were unable to verify the protein was over‐produced in this strain, as anaerobically grown bacteria did not produce detectable catalase (Figure S2). Together, these data led to the hypothesis that additional catalase may be required to completely detoxify the endogenous ROS produced aerobically in the ∆spxA1 mutant. Indeed, addition of catalase to the media rescued growth of ∆spxA1 in aerobic shaking flasks (Figure 4a). Concentrations of catalase as low as 0.01 mg/ml of restored ∆spxA1 growth over 100,000‐fold (Figure 4b). Importantly, boiled catalase (0.01 mg/ml) did not rescue aerobic growth, indicating that enzymatic activity was required (data not shown). These data suggested that hydrogen peroxide toxicity resulting from insufficient production of catalase is the primary reason ∆spxA1 cannot grow aerobically.
FIGURE 4.

Exogenous catalase rescued ∆spxA1 aerobic growth. (a) Aerobic growth kinetics of wt and ∆spxA1 in TSB or TSB supplemented with 1 mg/ml of catalase. (b) Aerobic growth 24 hr post‐inoculation in TSB alone or supplemented with various concentrations of catalase, as measured by plating for CFU and incubating anaerobically. p values were calculated using a heteroscedastic Student's t test. n.s., not significant (p > .05); *p < .05; ***p < .001
The rescue of ∆spxA1 growth by catalase suggests that toxic levels of peroxide were either present in the media or produced by the bacteria. If not specifically treated, rich media contains hydrogen peroxide from metal‐catalyzed glucose oxidation. Hydrogen peroxide is also generated over time as media is exposed to light (Li and Imlay, 2018). To determine if ∆spxA1 aerobic growth was inhibited by peroxide present in the media, we performed two important controls. First, TSB was degassed overnight in an anaerobic chamber prior to aerobic subculture of bacteria. Second, media was treated with catalase and then autoclaved to inactivate it. The media in both of these scenarios had the same low concentration of peroxide as the catalase‐containing media at the start of the experiment (Figure S3a), but did not contain active catalase during bacterial growth. Importantly, all media and culture flasks were kept in strict darkness to eliminate photochemical generation of hydrogen peroxide (Li and Imlay, 2018). The only medium which supported ∆spxA1 aerobic growth contained active catalase throughout the experiment (Figure S3b), demonstrating that the toxic peroxide was generated by the bacteria.
To further explore the role of catalase in L. monocytogenes, a ∆kat mutant was constructed under anaerobic conditions and tested for growth in the presence of oxygen. To delete kat it was also necessary to remove the neighboring gene lmo2784, a putative transcriptional antiterminator, to prevent toxicity of the plasmid in Escherichia coli during cloning (see Experimental Procedures). All phenotypes were verified to be kat‐dependent and independent of lmo2784 (Figure S4a). The L. monocytogenes ∆kat strain grown anaerobically overnight and then subcultured into aerobic shaking flasks exhibited normal growth for the first 4 hr. However, after 4 hr, no additional growth was observed (Figure 5a). This defect was complemented by expressing kat from its native promoter at an ectopic site in the chromosome (∆kat pPL2.kat). To determine if ∆kat was killed by oxygen or if growth was merely inhibited after 4 hr, we tracked growth over time by enumerating live bacteria plated anaerobically. This experiment revealed that L. monocytogenes lacking kat replicated for approximately 4 hr, but then died over time in aerobic culture (Figure 5b). Moreover, supplementing the media with exogenous catalase fully restored the aerobic growth of ∆kat (Figure S4b), confirming that death is due to lack of functional catalase. These results demonstrated that catalase is required for L. monocytogenes aerobic replication.
FIGURE 5.

Catalase is required for aerobic growth, but not intracellular growth. (a) and (b) Aerobic growth kinetics of wt, ∆kat and the complemented strain, as measured by OD600 and by enumerating CFU after growth for 24 hr. (c) Co‐culture of wt or ∆kat with ∆spxA1 at the indicated inoculum ratios. For clarity, only ∆spxA1 growth is shown. Wt was grown overnight aerobically or anaerobically, as indicated. (d) Intracellular growth kinetics in BMDMs of wt, ∆kat and the complemented strain. L. monocytogenes strains were grown overnight anaerobically at 37°C before infecting BMDMs with an MOI of 0.1. Bacteria were plated anaerobically and CFU were enumerated. In all panels, data are the means and SEM of three independent experiments. Student's t test. ** p < 0.01
Our results showed L. monocytogenes ∆spxA1 aerobic replication requires exogenous catalase, likely for its ability to efficiently detoxify peroxide generated by the bacteria. We therefore hypothesized that catalase produced by wt L. monocytogenes may detoxify the media and enable ∆spxA1 growth. To test this, bacteria were washed and then co‐cultured in aerobic shaking flasks at a ratio of 1:1 or 10:1 (wt:∆spxA1) and differentially plated over time to enumerate CFU. The ∆spxA1 mutant grew aerobically in the presence of equal numbers of wt bacteria if wt was grown aerobically overnight before co‐culture (Figure 5c, open circles). Co‐culture with equal wt L. monocytogenes grown anaerobically overnight diminished ∆spxA1 death but did not enable growth of the mutant (Figure 5c, closed circles). Increasing the ratio of wt‐to‐mutant bacteria enhanced ∆spxA1 replication in both cases (Figure 5c, squares). Furthermore, the ∆kat mutant was unable to rescue ∆spxA1 even when co‐cultured at a 10:1 ratio (∆kat:∆spxA1), indicating that ∆spxA1 replication required functional catalase made by the co‐cultured strain for aerobic growth. These data demonstrated that ∆spxA1 can replicate aerobically if peroxide is detoxified by catalase, whether from purified enzyme added exogenously or bacteria in co‐culture. Additionally, these results supported our finding that anaerobically grown L. monocytogenes does not produce measurable catalase activity (Figure S2) and indicate oxygen‐dependent regulation of catalase that we are currently investigating.
Considering the importance of catalase to L. monocytogenes growth and that mammalian host cells attack invading pathogens with ROS during the respiratory burst, we postulated that catalase might also be important for intracellular growth. BMDMs were infected with L. monocytogenes wt, ∆kat, and the complemented strain that were grown overnight anaerobically, and intracellular growth was measured by plating bacteria anaerobically to enumerate CFU. Surprisingly, the ∆kat mutant was able to replicate in BMDMs at the same rate as wt (Figure 5d), demonstrating that while kat is required for growth in vitro, it is completely dispensable for intracellular growth.
2.6. The Spx GC‐motif is required for hemEH expression and aerobic growth
Having observed the importance of heme and catalase for ∆spxA1 aerobic growth, we sought to leverage the phenotypes of mutants in these pathways to assess direct SpxA1 activation. To this end, we anaerobically generated a ∆hemEH mutant, which formed small colonies on solid agar and did not replicate aerobically in broth in the absence of exogenous heme (Figure 6a). This was unsurprising, as S. aureus heme biosynthesis mutants are known to form small colonies on agar and exhibit a reduced growth rate when incubated in the presence of oxygen (Proctor et al., 2006). Aerobic growth of L. monocytogenes ∆hemEH was rescued by addition of exogenous heme or by expressing hemEH from the native promoter at a neutral locus in the chromosome (Figure 6a, pPL2.hemEH). Although hemEH was required for aerobic growth, it was dispensable for BMDM infection (Figure 6b).
FIGURE 6.
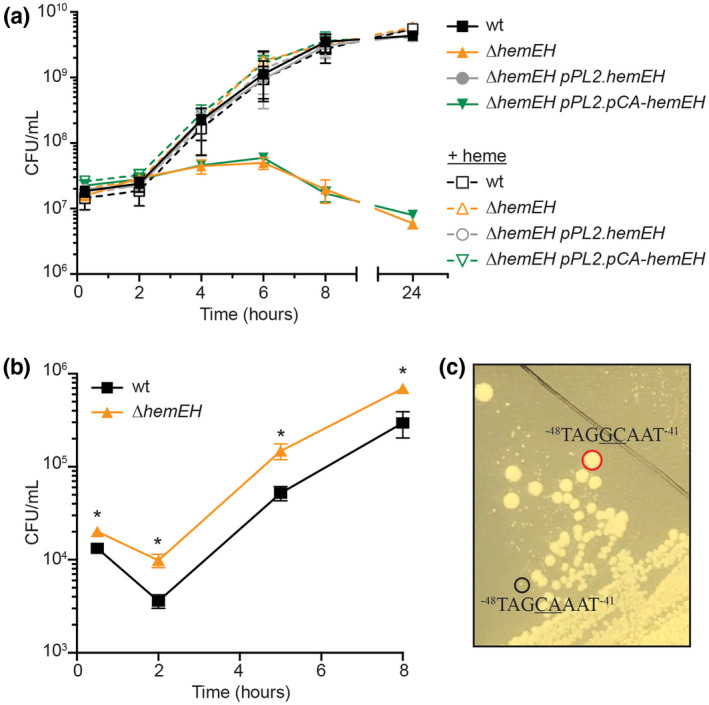
The GC‐motif is required for SpxA1‐activation of hemEH. (a) Aerobic growth kinetics in the absence (solid lines) or presence of 5 µM heme (dotted lines), as measured by enumerating CFU. (b) Intracellular growth kinetics in BMDMs, as in Fig. 5D. In panels a and b, data are the means and SEMs of three independent experiments. p values were calculated using a heteroscedastic Student's t test. *p < .05. (c) Image of suppressor mutants (red circle) on rich agar incubated aerobically. Large and small colonies were sequenced and a portion of the extended −35 box of each is shown with the putative GC‐motif underlined
The B. subtilis Spx‐RNAP complex recognizes a GC‐motif at the −44/−43 positions relative to TSSs (Reyes and Zuber, 2008; Rochat et al., 2012), and many of the genes we identified as SpxA1‐activated contain similar motifs (Table 1 and Table S1). To test the role of the GC‐motif in SpxA1 regulation in L. monocytogenes, promoter regions were engineered with GC at the −44/−43 positions mutated to CA, the least represented nucleotides at those positions in B. subtilis Spx‐activated promoters (Rochat et al., 2012). The GC‐motifs were disrupted in the native promoters in pPL2 and integrated into the chromosomes of the respective deletion strains. Mutation of the hemEH GC‐motif to CA at positions ‐45/‐44 abolished aerobic growth (Figure 6a, pPL2.pCA‐hemEH), suggesting that SpxA1‐mediated activation of hemEH transcription was required for aerobic replication. Consistent with this, transcript of hemEH was reduced approximately 20‐fold when the GC‐motif was mutated to CA (Figure S5a). Similarly, lmo2743 expression required the GC‐motif (Figure S5b). Surprisingly, we did not see an effect of disrupting the GC‐motif in the kat promoter (data not shown), indicating that either SpxA1‐mediated kat regulation is indirect or that kat regulation is more complex. Indeed, kat has two annotated TSS, only one of which has a GC‐motif (Wurtzel et al., 2012), and is also transcriptionally regulated by PerR (Rea et al., 2005).
Strains lacking hemEH expression (both ∆hemEH and the GC‐motif mutant) formed small colonies on solid media in the presence of oxygen (Figure 6c, black circle), and were therefore, routinely cultured anaerobically to promote optimal growth. However, upon incubating the ∆hemEH GC‐motif mutant in the presence of oxygen, large colonies spontaneously appeared after a few days at room temperature (Figure 6c, red circle). We hypothesized that these large colonies were suppressor mutants and sequenced the hemEH promoters of four large colonies and four small colonies from the mixed regions. All four large colonies had undergone a double substitution that reverted the disrupted Spx‐motif (CA) back to the wt sequence of GC at positions −45/−44 relative to the TSS and no other mutations were observed in the promoter region (Figure 6c). The remaining small colonies retained the CA at these positions. These results validated that the GC‐motif observed in Spx‐activated genes in B. subtilis is conserved in L. monocytogenes. Moreover, SpxA1‐dependent activation of hemEH and lmo2743 required the GC‐motif and the transcriptional activation of hemEH was required for aerobic growth of L. monocytogenes.
3. DISCUSSION
Redox stress is a common danger that all bacteria must be prepared to defend against. In addition to exogenous redox stress encountered during infection from the immune response or in the environment from competing bacteria, endogenous oxidative stress is generated continuously in the presence of oxygen (Imlay, 2008; Reniere, 2018). To combat these stressors, bacteria basally express detoxifying enzymes and are poised to rapidly respond to an increase in oxidative stress via redox‐sensing transcriptional regulators. Gram‐positive bacteria employ Spx‐family proteins to sense oxidative stress and regulate cognate genes necessary to detoxify ROS and survive. Herein, we identified L. monocytogenes genes regulated by SpxA1 in vitro and during macrophage infection. This study demonstrated that SpxA1 is required to activate genes for production of heme and catalase to detoxify endogenous ROS generated in the presence of oxygen. These results highlight the pleiotropic nature of SpxA1, as the top genes activated by SpxA1 (kat and hemEH) were each individually required for aerobic growth in vitro. Conversely, kat and hemEH were not required during macrophage infection, suggesting an alternative role for SpxA1 in vivo.
Based on the similarities of Spx proteins and DNA recognition sequences, we propose a model of SpxA1‐mediated transcriptional regulation in L. monocytogenes congruent with what has been described for B. subtilis Spx (Zuber, 2009; Rojas Tapias and Helmann, 2019). Spx was originally described as an “anti‐α factor” that negatively regulates gene expression by binding RNAP and disrupting activator‐stimulated transcription (Nakano, Nakano, et al., 2003). The mechanism by which Spx‐family proteins activate gene expression is less well understood. Our data are consistent with the model that SpxA1 directs RNAP to promoters with a GC‐motif near the −44/−43 positions relative to the TSS (Lin et al., 2013). Furthermore, our results suggest the GC‐motif is necessary for direct SpxA1‐mediated activation in those promoters that contain it, but the minimal sequence sufficient for SpxA1 recognition is not yet known. Disrupting the GC‐motif in the lmo2743 and hemEH promoters decreased expression of these genes to levels similar to that of the ∆spxA1 mutant. Further, mutation of the hemEH promoter GC‐motif abolished aerobic growth in broth and resulted in a small colony phenotype on solid media similar to the ∆hemEH mutant. These small colonies spontaneously reverted to the native sequence and normal colony phenotype, underscoring the strong selective pressure for SpxA1‐mediated hemEH expression during aerobic growth. In the hemEH promoter region, the GC‐motif is shifted to the −45/−44 position relative to the TSS, rather than the canonical −44/−43 position (Rochat et al., 2012). Our data suggest that either the actual TSS is one nucleotide upstream of the annotated TSS (Wurtzel et al., 2012), or that the L. monocytogenes SpxA1 recognition sequence is more flexible than that of B. subtilis Spx. Ongoing studies are aimed at disentangling these possibilities.
While the mechanism of Spx function appears to be conserved, there are also several key differences between the regulons of B. subtilis Spx and L. monocytogenes SpxA1. For example, Spx represses katA, encoding one of three catalases produced by B. subtilis (Nakano, Nakano, et al., 2003; Rochat et al., 2012). In contrast, our results show that L. monocytogenes SpxA1 activates expression of kat, the only catalase encoded in its genome. Additionally, we did not observe SpxA1‐dependent changes in sodA expression, although it is one of the most induced genes of the B. subtilis Spx regulon (Nakano, Küster‐Schöck, et al., 2003). Overall, our transcriptome and mutagenesis analyses supported that L. monocytogenes SpxA1 plays a similar role to B. subtilis Spx in regulating a response to oxidative stress, although with some important differences.
Our data provide strong evidence that heme production is directly regulated by SpxA1. Heme is a tetrapyrrole ring complexed to a central iron atom, making it a versatile redox‐active molecule used as a cofactor by many enzymes, including catalase, cytochrome oxidase and heme peroxidase. Heme can be taken up from the environment or synthesized de novo via the Hem proteins (HemALBCDEYHQ) that have recently been renamed to reflect their enzymatic activity (Choby and Skaar, 2016; Dailey et al., 2017). Regulation of heme biosynthesis has been extensively studied in S. aureus, and in that organism, the majority of genes encoding hemEH appear to be constitutively expressed. The exception is HemA/GtrA, the first committed enzyme in the biosynthetic pathway, which is post‐transcriptionally regulated, acting as a switch to increase or decrease heme synthesis (Choby et al., 2018). Given that we observed changes in hemEH expression in the ∆spxA1 strain and that control of hemEH expression requires the GC‐motif, we hypothesize that hemEH is the switch that is directly regulated by SpxA1 to control heme synthesis in response to redox stress in L. monocytogenes.
In addition to its stringent requirement for heme, bacteria also employ many survival strategies to detoxify ROS in their environments. We demonstrated that kat expression is dependent on SpxA1 and is absolutely required for aerobic growth of L. monocytogenes. Aerobic growth of ∆spxA1 was rescued by addition of exogenous catalase or co‐culture with wt L. monocytogenes. Together, these results suggested that ∆spxA1 is unable to replicate in the presence of oxygen due to hydrogen peroxide‐mediated toxicity. Control experiments suggested the predominant source of peroxide is generated by the bacteria rather than exogenous peroxide present in the media. To our knowledge, this is the first demonstration that a facultative aerobic bacterium requires catalase for aerobic replication. Interestingly, our results also suggested that catalase is not produced when L. monocytogenes is grown anaerobically. We were unable to detect catalase activity from bacteria grown anaerobically even though transcript levels of kat were unaffected by oxygen. Furthermore, wt L. monocytogenes grown anaerobically rescued ∆spxA1 replication to a lesser extent than aerobically grown bacteria in co‐culture. Future work will investigate the oxygen‐dependent regulation of catalase production.
Bacterial pathogens are exposed to 5–10 µM hydrogen peroxide in the host phagosome (Mishra and Imlay, 2012). Counterintuitively, catalase‐deficient L. monocytogenes transposon mutants are fully virulent in both a chicken embryo model and an intravenous murine model of infection, and catalase‐deficient strains have been isolated in the clinic (Leblond‐Francillard et al., 1989; Cepeda et al., 2006; Azizoglu and Kathariou, 2010). Together with the results presented here, these data suggest that L. monocytogenes catalase is necessary to detoxify endogenous peroxide generated during aerobic growth but is dispensable in vivo. Similarly, hemEH was required for aerobic replication but expendable during intracellular growth. These results demonstrate that the ∆spxA1 growth phenotype in vitro can be genetically uncoupled from the virulence defect observed in host cells, suggesting that SpxA1 may sense and respond to distinct stressors in each environment.
SpxA1 positively regulates kat and hemEH and both catalase and heme are required for aerobic growth. We initially hypothesized that heme was required solely as a cofactor for catalase and/or cytochrome bd oxidase; however the data did not support this theory. Specifically, individually overexpressing kat or cydABCD in the presence of exogenous heme did not restore ∆spxA1 growth compared to adding heme alone. Conversely, overexpressing kat in the absence of exogenous heme rescued a small amount of ∆spxA1 death. These data indicated ∆spxA1 is capable of producing sufficient heme for some catalase to be functional when kat is overexpressed. The mechanism by which exogenous heme rescued ∆spxA1 aerobic growth remains to be determined. We speculate that exogenous heme is bound by a protein or proteins that relieve oxidative stress in ∆spxA1 and the identity of these proteins is under investigation.
Prior studies demonstrated that L. monocytogenes lacking cydAB uses fermentative metabolism and thus, displays only a modest reduction in aerobic growth (Corbett et al., 2017). While we therefore do not predict that the lack of cytochrome bd plays a significant role in the ∆spxA1 aerobic growth defect, it may be contributing to ROS production. Indeed, Enterococcus faecalis strains with non‐functional cytochrome bd produce extracellular superoxide due to the nonenzymatic reaction of quinones with oxygen (Huycke et al., 2001). Like E. faecalis, the primary quinone utilized by L. monocytogenes is menaquinone, which is more prone to autoxidation than ubiquinone (Korshunov and Imlay, 2006). Taken together, we propose a model in which L. monocytogenes ∆spxA1 produces less cytochrome bd than wt, generating ROS from the incomplete electron transport chain. While these ROS would be readily detoxified by wt bacteria, ∆spxA1 is deficient in catalase production and is thus more sensitive to peroxide‐mediated toxicity. Finally, the ∆spxA1 mutant is severely limited for heme production, which further contributes to decreased aerobic survival. In summary, our data show that the severe ∆spxA1 aerobic growth defect is the result of increased ROS in the absence of catalase and heme.
4. EXPERIMENTAL PROCEDURES
4.1. Bacterial strains and culture conditions
L. monocytogenes mutants were derived from wt 10403S (Bécavin et al., 2014), cultured in brain heart infusion (BHI) or tryptic soy broth (TSB) at 37°C in the dark, with shaking, unless otherwise stated. All chemicals were purchased from Sigma Aldrich unless otherwise stated. Catalase from bovine liver (2,000–5,000 units/mg) was dissolved in media and filtered. Boiled catalase (0.01 mg/ml) did not rescue ∆spxA1 aerobic growth. Hemin (heme) stocks were made fresh for each experiment by dissolving hemin in 0.1 N NaOH. Controls containing NaOH only were also included and showed no effect on growth (data not shown). Antibiotics were used at the following concentrations: streptomycin, 200 µg/ml; chloramphenicol, 10 µg/ml (E. coli), 7.5 µg/ml (L. monocytogenes); tetracycline, 2 µg/ml; carbenicillin, 100 µg/ml; and erythromycin, 1 µg/ml.
L. monocytogenes strains are listed in Table S3 and E. coli strains are listed in Table S4. Plasmids were introduced to E. coli via chemical competence and heat‐shock and introduced into L. monocytogenes wt via trans‐conjugation from E. coli SM10 (Simon et al., 1983). Transducing lysates were then prepared and used to infect the ∆spxA1 mutant, thereby generating the overexpressing strains.
4.2. Generalized Transductions
Transducing lysates were prepared by mixing donor strain with U153 phage, as described previously (Reniere et al., 2016). After overnight incubation at 30°C in LB soft agar, phage were eluted from the agar, filter sterilized and mixed with recipient L. monocytogenes (∆spxA1) for 30 min at room temperature. Transductants were selected on antibiotic‐containing agar at 37°C anaerobically.
4.3. L. monocytogenes strain construction
In‐frame deletions were carried out by allelic exchange using a conjugation‐proficient version of the suicide vector pKSV7 (Camilli et al., 1993). We were not able to generate a pKSV7 construct to delete kat (catalase, lmo2785) which we hypothesize was due to toxicity of the neighboring gene in E. coli. Therefore, we simultaneously deleted both kat and the neighboring unannotated gene lmo2784. pKSV7.kat‐2784 was constructed by amplifying a 5ʹ homologous region and a 3ʹ homologous region, followed by synthesis by over‐lapping extension (SOE) PCR to join the fragments together. This cassette was restriction‐digested and ligated into pKSV7‐oriT (Camilli et al., 1993). A vector with the mutant ∆kat‐2784 allele was introduced into L. monocytogenes via trans‐conjugation, integrated into the chromosome, colony‐purified on selective nutrient‐agar, and subsequently cured of the plasmid by conventional methods (Reniere et al., 2016). Chromosomal mutations were confirmed by PCR and Sanger DNA sequencing when necessary. The resulting ∆kat‐2784 mutant was then complemented with pPL2.kat, pPL2.lmo2784, or pPL2.kat‐2784 to ensure that the observed phenotypes were due to loss of kat and not lmo2784. We did not observe a role for lmo2784 in aerobic growth or intracellular growth (Figure S4a). Therefore, in this manuscript we have referred to ∆kat‐2784 pPL2.lmo2784 simply as “∆kat” and ∆kat‐2784 pPL2.kat‐2784 as the complemented strain, for simplicity.
Knock‐in of genes into L. monocytogenes was carried out using pPL2 and pPL2t integration plasmids (Lauer et al., 2002; Whiteley et al., 2015). The HyPer promoter used to generate overexpression vectors is a modified Pspac(hy) with a G‐to‐T mutation at the −1 position relative to the TSS (Quisel et al., 2001; Reniere et al., 2016). To generate complementation vectors, genome fragments containing native promoter regions (100–200 nucleotides upstream of the annotated TSS) and genes were ligated into pPL2 or pPL2t. Promoter GC‐motifs were constructed by inverse PCR of complementation vectors. Integration was confirmed by antibiotic resistance.
4.4. RNA isolation
Nucleic acids were purified from bacteria harvested from broth culture or from infected macrophages, as previously described (Reniere et al., 2015; Sigal et al., 2016). Briefly, bacteria were grown overnight anaerobically in BHI at 37°C and subcultured 1:20 into BHI. After 4 hr of anaerobic growth at 37°C, bacteria were mixed 1:1 with ice‐cold methanol, pelleted and stored at −80°C. For infections, J774 cells were plated at a density of 2 × 107 cells in 150 mm tissue culture (TC)‐treated dishes and infected with an MOI of 20. After 30 min, the cells were washed twice with sterile PBS and media containing gentamicin was added. Eight hours post‐infection, cells were washed with PBS and lysed by addition of ice‐cold nuclease‐free water. Cells were collected by scraping, vortexed quickly, and pelleted. The bacteria were harvested by filtering the supernatant and freezing the filter at −80°C. RNA was then purified from the frozen bacterial samples via phenol‐chloroform extraction and DNase treatment according to published methods (Sigal et al., 2016).
4.5. Transcriptomics
Ribosomal RNA was removed from broth sample total RNA using the Ribo‐Zero rRNA Removal kit (Bacteria kit), according to manufacturer's recommendations (Illumina, Inc., San Diego, CA, USA). The Ribo‐Zero Gold rRNA Removal Kit (Epidemiology kit) was used to deplete both bacterial and mammalian rRNA from samples of infected cells. Depleted samples were then analyzed by the Genomics & Bioinformatics Shared Resources at Fred Hutchinson Cancer Research Center. Ribosomal‐depleted RNA integrity was confirmed using an Agilent 4200 TapeStation (Agilent Technologies, Inc., Santa Clara, CA) and quantified using a Trinean DropSense96 spectrophotometer (Caliper Life Sciences, Hopkinton, MA).
RNA‐seq libraries were prepared from rRNA‐depleted using the TruSeq RNA Sample Prep Kit v2, omitting the poly‐A selection step, (Illumina, Inc.) and a Sciclone NGSx Workstation (PerkinElmer, Waltham, MA, USA). Library size distributions were validated using an Agilent 4200 TapeStation. Additional library QC, blending of pooled indexed libraries and cluster optimization were performed using Life Technologies' Invitrogen Qubit® 2.0 Fluorometer (Life Technologies‐Invitrogen, Carlsbad, CA, USA). RNA‐seq libraries were pooled (10‐plex) and clustered onto a flow cell lane. Sequencing was performed using an Illumina HiSeq 2500 in rapid mode employing a paired‐end, 50 base read length (PE50) sequencing strategy. Image analysis and base calling were performed using Illumina's Real Time Analysis v1.18 software, followed by “demultiplexing” of indexed reads and generation of FASTQ files, using Illumina's bcl2fastq Conversion Software v1.8.4.
Reads of low quality were filtered prior to alignment to the reference genome (L. monocytogenes 10403S) using TopHat v2.1.0 (Trapnell et al., 2009). Counts were generated from TopHat alignments for each gene using the Python package HTSeq v0.6.1 (Anders et al., 2015). Genes with low counts across all samples were removed, prior to identification of differentially expressed genes using the Bioconductor package edgeR v3.12.1 (Robinson et al., 2010). A false discovery rate (FDR) method was employed to correct for multiple testing (Reiner et al., 2003), with differential expression defined as |log2 (ratio) | ≥ 1 (± 2‐fold) with the FDR set to 5%. Results were then evaluated using CLC Genomics Workbench (Qiagen) and transcripts that were changed >2‐fold (p < .001) were included in our analysis. In addition, the data were technically validated by measuring expression of 10 genes via quantitative RT‐PCR (qPCR) as described (Lobel and Herskovits, 2016), and an excellent correlation was confirmed (R 2 > .92).
4.6. Growth curves
For anaerobic growth, colonies were inoculated into broth and incubated at 37°C in closed containers containing anaerobic gas‐generating pouches (GasPak EZ; BD). In general, anaerobic overnight cultures were normalized to OD600 0.02 in 25 ml of rich broth in 250 ml of flasks and grown aerobically with shaking (250 rpm) for 24 hr. OD600 was measured every hour. For certain experiments, ∆spxA1 bacterial cultures were plated 24 hr post‐inoculation and incubated anaerobically to enumerate CFU.
Co‐culture experiments were performed by first growing bacteria overnight at 37°C in shaking tubes or anaerobically, as indicated. Cultures were washed with sterile PBS and normalized by OD600 to the indicated strain ratios. At each time point, bacteria were serially diluted and plated on BHI agar grown aerobically to enumerate wt or ∆kat L. monocytogenes, or plated on BHI agar containing chloramphenicol grown anaerobically to enumerate ∆spxA1, which contained integrated pPL2 for selection.
4.7. Intracellular growth curves
BMDMs were plated in 24‐well TC treated dishes at 6 × 105 cells per well. Generally, overnight cultures were incubated at 30°C anaerobically and static. After being washed three times and resuspended in PBS, bacterial suspensions were diluted 1:5000 into warmed BMDM media (Reniere et al., 2016). After the BMDMs were washed once with PBS, 1 ml of the bacterial suspension was added to each well. Thirty minutes post‐infection, cells were washed twice with PBS and 1 ml of BMDM media containing gentamicin (50 μg/ml) was added to each well. Time points were taken at 0.5, 2, 5 and 8 hr post‐infection. To measure bacterial growth, cells were lysed by addition of 250 µl of cold 0.1% NP40 and incubated for 5 min at room temperature, followed by serial dilutions and plating anaerobically. Experiments were performed with technical duplicates and experiments were repeated 3 times.
4.8. Plaque assays
Tissue culture‐treated 6‐well dishes were seeded with 1.2 × 106 L2 murine fibroblasts per well. L. monocytogenes ∆spxA1 strains were incubated anaerobically overnight at 30°C, stationary. Overnight cultures were diluted 1:10 in sterile PBS, and 10 µl was used to infect each well. One hour post‐infection, cells were washed twice with PBS, followed by addition of 3 ml of molten agarose‐Dulbecco modified Eagle Medium (DMEM) solution. This solution consisted of gentamicin at 10 µg/ml and a 1:1 mixture of 2X DMEM (Gibco) and 1.4% SuperPure agarose LE (U.S. Biotech Sources, LLC). Three days post‐infection, 2 ml of molten agarose‐DMEM solution containing neutral red was added to each well to visualize plaques. After 24 hr, the plaques were scanned and the area measured using ImageJ software (Schneider et al., 2012).
AUTHOR CONTRIBUTIONS
Monica R. Cesinger, Maureen K. Thomason, Mauna B. Edrozo, Cortney R. Halsey, and Michelle L. Reniere performed the experiments. Monica R. Cesinger, Maureen K. Thomason, and Michelle L. Reniere prepared the manuscript.
Supporting information
Supplementary Material
Acknowledgements
We thank Jacob Choby (Emory University), Aaron Whiteley (CU Boulder) and Joshua Woodward (University of Washington) for helpful discussions and Jeff Delrow and Alyssa Dawson (Fred Hutch) for technical assistance. The Genomics and Bioinformatics Shared Resources at Fred Hutchinson Cancer Research Center is partially funded from Cancer Center grant NCI 5P30CA015704‐43. M.R.C. was supported by the National Institutes of Health (NIH) under award number T32GM008268 and by the Howard Hughes Medical Institute through the James H. Gilliam Fellowships for Advanced Study program (#GT11030). C.R.H. was supported by NIH grant T32AI055396. Research in the Reniere Lab was supported by NIH grant RO1AI132356. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors do not have a conflict of interest to declare.
Cesinger MR, Thomason MK, Edrozo MB, Halsey CR, Reniere ML. Listeria monocytogenes SpxA1 is a global regulator required to activate genes encoding catalase and heme biosynthesis enzymes for aerobic growth. Mol Microbiol. 2020;114:230–243. 10.1111/mmi.14508
REFERENCES
- Anders, S. , Pyl, P.T. and Huber, W. (2015) HTSeq–a Python framework to work with high‐throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzaldi, L.L. and Skaar, E.P. (2010) Overcoming the heme paradox: heme toxicity and tolerance in bacterial pathogens. Infection and Immunity, 78, 4977–4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizoglu, R.O. and Kathariou, S. (2010) Temperature‐dependent requirement for catalase in aerobic growth of Listeria monocytogenes F2365. Applied and Environment Microbiology, 76, 6998–7003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bécavin, C. , Bouchier, C. , Lechat, P. , Archambaud, C. , Creno, S. , Gouin, E. , et al (2014) Comparison of widely used Listeria monocytogenes strains EGD, 10403S, and EGD‐e highlights genomic variations underlying differences in pathogenicity. MBio, 5, e00969‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilli, A. , Tilney, L.G. and Portnoy, D.A. (1993) Dual roles of plcA in Listeria monocytogenes pathogenesis. Molecular Microbiology, 8, 143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda, J.A. , Millar, M. , Sheridan, E.A. , Warwick, S. , Raftery, M. , Bean, D.C. , et al (2006) Listeriosis due to infection with a catalase‐negative strain of Listeria monocytogenes . Journal of Clinical Microbiology, 44, 1917–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choby, J. E. and Skaar, E. P. (2016). Heme synthesis and acquisition in bacterial pathogens. Journal of Molecular Biolology, 428(17), 3408–3428. 10.1016/j.jmb.2016.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choby, J.E. , Grunenwald, C.M. , Celis, A.I. , Gerdes, S.Y. , DuBois, J.L. and Skaar, E.P. (2018) Staphylococcus aureus HemX modulates glutamyl‐tRNA reductase abundance to regulate heme biosynthesis. MBio, 9, e02287–e2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett, D. , Goldrick, M. , Fernandes, V.E. , Davidge, K. , Poole, R.K. , Andrew, P.W. , et al (2017) Listeria monocytogenes has both a bd‐type and an aa3‐type terminal oxidase which allow growth in different oxygen levels and both are important in infection. Infection and Immunity, 85, e00354–e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey, H.A. , Dailey, T.A. , Gerdes, S. , Jahn, D. , Jahn, M. , O'Brian, M.R. , et al (2017) Prokaryotic heme biosynthesis: multiple pathways to a common essential product. Microbiology and Molecular Biology Reviews, 81, e00048–e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai, B.V. and Morrison, D.A. (2007) Transformation in Streptococcus pneumoniae: formation of eclipse complex in a coiA mutant implicates CoiA in genetic recombination. Molecular Microbiology, 63, 1107–1117. [DOI] [PubMed] [Google Scholar]
- Freitag, N.E. , Port, G.C. and Miner, M.D. (2009) Listeria monocytogenes ‐ from saprophyte to intracellular pathogen. Nature Reviews Microbiology, 7, 623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaballa, A. , Antelmann, H. , Hamilton, C.J. and Helmann, J.D. (2013) Regulation of Bacillus subtilis bacillithiol biosynthesis operons by Spx. Microbiology, 159, 2025–2035. [DOI] [PubMed] [Google Scholar]
- Grossman, A.D. (1995) Genetic networks controlling the initiation of sporulation and the development of genetic competence in Bacillus subtilis . Annual Review of Genetics, 29, 477–508. [DOI] [PubMed] [Google Scholar]
- Huycke, M.M. , Moore, D. , Joyce, W. , Wise, P. , Shepard, L. , Kotake, Y. , et al (2001) Extracellular superoxide production by Enterococcus faecalis requires demethylmenaquinone and is attenuated by functional terminal quinol oxidases. Molecular Microbiology, 42, 729–740. [DOI] [PubMed] [Google Scholar]
- Imlay, J.A. (2008) Cellular defenses against superoxide and hydrogen peroxide. Annual Review of Biochemistry, 77, 755–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korshunov, S. and Imlay, J.A. (2006) Detection and quantification of superoxide formed within the periplasm of Escherichia coli . Journal of Bacteriology, 188, 6326–6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer, P. , Chow, M.Y.N. , Loessner, M.J. , Portnoy, D.A. and Calendar, R. (2002) Construction, characterization, and use of two Listeria monocytogenes site‐specific phage integration vectors. Journal of Bacteriology, 184, 4177–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond‐Francillard, M. , Gaillard, J.L. and Berche, P. (1989) Loss of catalase activity in Tn1545‐induced mutants does not reduce growth of Listeria monocytogenes in vivo . Infection and Immunity, 57, 2569–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. and Imlay, J.A. (2018) Improved measurements of scant hydrogen peroxide enable experiments that define its threshold of toxicity for Escherichia coli . Free Radical Biology and Medicine, 120, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, A.A. , Walthers, D. and Zuber, P. (2013) Residue substitutions near the redox center of Bacillus subtilis Spx affect RNA polymerase interaction, redox control, and Spx‐DNA contact at a conserved cis‐acting element. Journal of Bacteriology, 195, 3967–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobel, L. and Herskovits, A.A. (2016) Systems level analyses reveal multiple regulatory activities of CodY controlling metabolism, motility and virulence in Listeria monocytogenes . PLoS Genetics, 12, e1005870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra, S. and Imlay, J. (2012) Why do bacteria use so many enzymes to scavenge hydrogen peroxide? Archives of Biochemistry and Biophysics, 525, 145–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano, S. , Erwin, K.N. , Ralle, M. and Zuber, P. (2005) Redox‐sensitive transcriptional control by a thiol/disulphide switch in the global regulator, Spx. Molecular Microbiology, 55, 498–510. [DOI] [PubMed] [Google Scholar]
- Nakano, S. , Küster‐Schöck, E. , Grossman, A.D. and Zuber, P. (2003) Spx‐dependent global transcriptional control is induced by thiol‐specific oxidative stress in Bacillus subtilis . Proceedings of the National Academy of Sciences of the United States of America, 100, 13603–13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano, S. , Nakano, M.M. , Zhang, Y. , Leelakriangsak, M. and Zuber, P. (2003) A regulatory protein that interferes with activator‐stimulated transcription in bacteria. Proceedings of the National Academy of Sciences of the United States of America, 100, 4233–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor, R.A. , von Eiff, C. , Kahl, B.C. , Becker, K. , McNamara, P. , Herrmann, M. , et al (2006) Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nature Reviews Microbiology, 4, 295–305. [DOI] [PubMed] [Google Scholar]
- Quisel, J.D. , Burkholder, W.F. and Grossman, A.D. (2001) In vivo effects of sporulation kinases on mutant Spo0A proteins in Bacillus subtilis . Journal of Bacteriology, 183, 6573–6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea, R. , Hill, C. and Gahan, C.G.M. (2005) Listeria monocytogenes PerR mutants display a small‐colony phenotype, increased sensitivity to hydrogen peroxide, and significantly reduced murine virulence. Applied and Environment Microbiology, 71, 8314–8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner, A. , Yekutieli, D. and Benjamini, Y. (2003) Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics, 19, 368–375. [DOI] [PubMed] [Google Scholar]
- Reniere, M.L. (2018) Reduce, induce, thrive: bacterial redox sensing during pathogenesis. Journal of Bacteriology, 200, 903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reniere, M.L. , Torres, V.J. and Skaar, E.P. (2007) Intracellular metalloporphyrin metabolism in Staphylococcus aureus . BioMetals, 20, 333–345. [DOI] [PubMed] [Google Scholar]
- Reniere, M.L. , Whiteley, A.T. and Portnoy, D.A. (2016) An in vivo selection identifies Listeria monocytogenes genes required to sense the intracellular environment and activate virulence factor expression. PLoS Path, 12, e1005741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reniere, M.L. , Whiteley, A.T. , Hamilton, K.L. , John, S.M. , Lauer, P. , Brennan, R.G. , et al (2015) Glutathione activates virulence gene expression of an intracellular pathogen. Nature, 517, 170–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes, D.Y. and Zuber, P. (2008) Activation of transcription initiation by Spx: formation of transcription complex and identification of a Cis‐acting element required for transcriptional activation. Molecular Microbiology, 69, 765–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, M.D. , McCarthy, D.J. and Smyth, G.K. (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochat, T. , Nicolas, P. , Delumeau, O. , Rabatinová, A. , Korelusová, J. , Leduc, A. , et al (2012) Genome‐wide identification of genes directly regulated by the pleiotropic transcription factor Spx in Bacillus subtilis . Nucleic Acids Research, 40, 9571–9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas Tapias, D. F. and Helmann, J. D. (2019) Roles and regulation of Spx family transcription factors in Bacillus subtilis and related species. Advances in Microbial Physiology, 75, 279–323. 10.1016/bs.ampbs.2019.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhland, B.R. and Reniere, M.L. (2018) Sense and sensor ability: redox‐responsive regulators in Listeria monocytogenes . Current Opinion in Microbiology, 47, 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, C.A. , Rasband, W.S. and Eliceiri, K.W. (2012) NIH image to imageJ: 25 years of image analysis. Nature Methods, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal, N. , Pasechnek, A. and Herskovits, A.A. (2016) RNA purification from intracellularly grown Listeria monocytogenes in macrophage cells. JoVE (Journal of Visualized Experiments), e54044–e54044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, R. , Priefer, U. and Pühler, A. (1983) A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram‐negative bacteria. Nature Biotechnology, 1(9), 784–791. [Google Scholar]
- Sun, A.N. , Camilli, A. and Portnoy, D.A. (1990) Isolation of Listeria monocytogenes small‐plaque mutants defective for intracellular growth and cell‐to‐cell spread. Infection and Immunity, 58, 3770–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney, L.G. and Portnoy, D.A. (1989) Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes . Journal of Cell Biology, 109, 1597–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Pachter, L. and Salzberg, S.L. (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva, M. , Jousselin, A. , Baek, K.T. , Prados, J. , Andrey, D.O. , Renzoni, A. , et al (2016) Rifampicin resistant rpoB alleles, or multicopy thioredoxin/thioredoxin reductase, suppress the lethality of the global stress regulator spx in Staphylococcus aureus . Journal of Bacteriology, 198, 2719–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeman, C.A. , Hammer, N.D. , Stauff, D.L. , Attia, A.S. , Anzaldi, L.L. , Dikalov, S.I. , et al (2012) Menaquinone biosynthesis potentiates haem toxicity in Staphylococcus aureus . Molecular Microbiology, 86, 1376–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteley, A.T. , Pollock, A.J. and Portnoy, D.A. (2015) The PAMP c‐di‐AMP Is essential for Listeria monocytogenes growth in rich but not minimal media due to a toxic increase in (p)ppGpp. Cell Host & Microbe, 17, 788–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteley, A.T. , Ruhland, B.R. , Edrozo, M.B. and Reniere, M.L. (2017) A redox‐responsive transcription factor is critical for pathogenesis and aerobic growth of Listeria monocytogenes . Infection and Immunity, 85, e00978–e1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbourn, C.C. and Kettle, A.J. (2013) Redox reactions and microbial killing in the neutrophil phagosome. Antioxidants & Redox Signaling, 18, 642–660. [DOI] [PubMed] [Google Scholar]
- Wurtzel, O. , Sesto, N. , Mellin, J.R. , Karunker, I. , Edelheit, S. , Bécavin, C. , et al (2012) Comparative transcriptomics of pathogenic and non‐pathogenic Listeria species. Molecular Systems Biology, 8, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber, P. (2004) Spx‐RNA polymerase interaction and global transcriptional control during oxidative stress. Journal of Bacteriology, 186, 1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber, P. (2009) Management of oxidative stress in Bacillus . Annual Review of Microbiology, 63, 575–597. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material