

Abstract
Alterations in the innate and adaptive immunity underpin psoriasis pathophysiology, with the Th17 cells subset now recognized as the fundamental cells in the key controlling pathway involved in its pathogenesis. Since psoriasis is a systemic disease with important comorbidity, further knowledge on the interleukin (IL)‐23/Th17 axis led to the hypothesis that there may be shared pathogenic pathways between primary skin disease and comorbidity. Psoriasis has been identified as a risk factor for cardiovascular and metabolic disease, and increasing evidence gives support to this epidemiological observation from the clinical‐pathologically field. As an example, increased levels of IL‐23 and IL‐23R have been found in human atherosclerotic plaque, and levels correlated with symptom duration and mortality. Also, upregulation of IL‐23/IL‐17 seems to play an important role in both myocardial damage and stroke, with interesting reports on deleterious effect neutralization after administration of related anti‐bodies in both associated conditions. In diabetic patients, increased levels of IL‐23/IL‐17 have also been observed and available data support a synergistic role of IL‐23/IL‐17 in β‐cells damage. In obesity, signs of an expansion of Th17 subset in adipose tissue have been reported, as well as elevated concentrations of IL‐23 in obese patients. In non‐alcoholic fatty liver disease, closely related to metabolic syndrome, but also in other mentioned cardiometabolic disorders, a predominance of IL‐23 and other related pro‐inflammatory factors has been identified as participating in their pathogenesis. Thus, the involvement of the IL‐23/Th17 axis in these shared psoriasis‐cardiometabolic pathogenic mechanisms is reviewed and discussed in the light of the existing preclinical and clinical evidence, including that from comorbid psoriasis patients.
Introduction
Plaque psoriasis is a prevalent chronic, immune‐mediated inflammatory disease classically characterized by typical skin lesions. Comorbidity is commonly present in psoriasis patients, consistent with the systemic nature of the disease. Cardiometabolic disease accounts for a substantial proportion of this comorbidity to the point of psoriasis being considered an independent risk factor for cardiovascular disease (CVD).1 Current evidence points to the role of Th17 cells and interleukin‐23 (IL‐23) on this condition.1, 2 Growing knowledge of its cardiometabolic comorbidity suggests this pathway may be a link between cutaneous and beyond‐the‐skin manifestations of psoriasis.
Cytokines are core mediators enabling communication, regulation and coordination between immune cells. Cytokines, along with antigens and antigen‐presenting cells (APCs) existing in the environment, are determinant for naïve CD4+ cells to differentiate in any of the known effector, memory or regulator subsets; in turn, the dominant cytokine profile secreted by each subset is responsible for their functional role.3, 4
The Th17 subset was the third effector subset discovered as a lineage of CD4+ T cells, along with the previously known Th1 and Th2 subsets.5, 6, 7 In humans, Th17 cells play a role in the defence against extracellular bacteria and fungi8, 9 and have a recognized role in development and maintenance of autoimmune and chronic inflammatory diseases.5, 10 Throughout the Th17 biologic cycle and function, a complex net of transcription factors and mediators are involved,8, 9, 10 among which IL‐23 and IL‐17A emerge as critical upstream and downstream CKs, respectively.8
Interleukin‐23 belongs to the IL‐12 cytokine family, along with IL‐12 and IL‐27 (involved in Th1 differentiation), and IL‐35 (involved in Treg differentiation and function). Dendritic cells (DCs), monocytes and macrophages (i.e. APCs) from activated skin and mucosae are the main IL‐23‐secreting cells.11 Structurally, IL‐23 is a heterodimer composed of a unique p19 subunit (IL‐23p19) and a p40 subunit, common to IL‐23 and IL‐12.12 IL‐23 is essential in the enhancement of memory T cells, regulation of antibody production, induction of IFN‐γ and proliferation of Th17 cells secreting IL‐17 and IL‐22, contributing to immune response against infection, but also attributed a pathogenic role in autoimmune diseases and cancer.8, 11, 13 IL‐23 is essential in Th17 differentiation because this CK is a strong inducer of STAT3, which mediates signalling along with retinoic acid orphan receptor C2 [RORC2 (RORγt in mice)].2, 11 Memory T cells upon activation by TGF‐β and IL‐6, but not naïve T cells, express the IL‐23 receptor, suggesting that IL‐23 plays a crucial role in Th17 expansion, survival and pathogenicity.5, 7, 14
Interleukin‐17A, along with IL‐17F and IL‐22 are the effector interleukins of Th17 cells.15 Although IL‐17A is preferentially produced by Th17 cells, they are not its exclusive source; other cells, like monocytes, DCs, natural killer (NK) T cells, neutrophils, innate lymphoid cells (ILCs) and γδ‐T cells also secrete IL‐17A. IL‐17A belongs to the IL‐17 cytokine family comprising five more isoforms (A–F), indeed IL‐17A and IL‐17F share similar functions.9, 16, 17 IL‐17A is positioned as a link between innate and adaptive immunity; it acts as an early mediator of the immune response at mucosal surfaces, by inducing the production of a variety of pro‐inflammatory molecules from tissues and activated cells, which result in recruiting neutrophils to tissues.9, 16 Interestingly, neutrophils can secrete IL‐17 themselves, acting as an amplifier and inducing the recruitment and activation of additional neutrophils. Thus, neutrophils seem to have a role not only in acute but also in chronic inflammation.12
Unlike other subsets, Th17 cells do not depend on its effector CK IL‐17 for differentiation. Rather, combinations of IL‐23 plus IL‐6, IL‐1β and TGF‐β are required for an efficient differentiation of Th17 subset (Fig. 1).9
Figure 1.
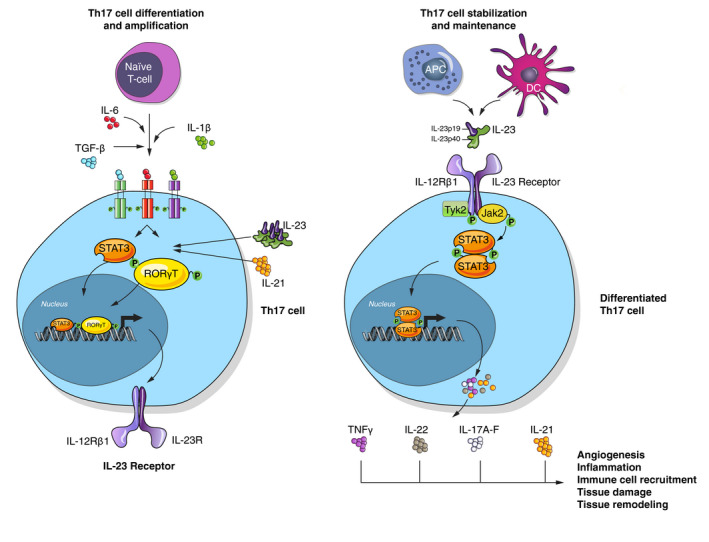
Schematic representation of Th17 cells developmental mechanism, regulators involved and effector functions.
Due to plasticity observed in CD4+ T cells, differentiation of Th17 in the presence of IL‐23 leads to tissue inflammation because of IL‐23‐induced downregulation of IL‐10 (involved in Treg cells function). Conversely, differentiation and function of Treg is mediated by TGF‐β, but, in the presence of IL‐6, Treg differentiation is inhibited in favour of Th17 (Fig. 2).9
Figure 2.

Schematic representation of Treg and Th17 cells regulation, stimulator and inhibitor molecules involved in development and ultimate effector action of each pathway.
The IL‐23/Th17 axis in psoriasis
Prevalence of psoriasis is estimated between 1.5% and 5% in most developed countries; it affects approximately 125 million people worldwide,1, 18 plaque psoriasis being the most frequent form of the disease.19 Despite the primary cause of psoriasis being unknown, some genetic factors are acknowledged risk factors for its development.20, 21 Further, a dysregulated immune response involving keratinocytes, vascular endothelial cells, Th17, Th1, Treg, γδ‐T cells, DCs, macrophages, neutrophils, mast cells and NK cells underlies the pathogenesis of psoriasis.19, 22, 23
The primary consideration of psoriasis as a Th1/Tc1‐mediated disease19, 24 was supported by efficacy of monoclonal antibodies targeting p40 subunit of IL‐12, the cytokine considered crucial in the Th1 differentiation.25 However, IL‐23 identification almost two decades ago,12 together with the fact that IL‐23 shared with IL‐12 the same p40 subunit, and the knowledge acquired on the pathway IL‐23/Th17 have actually changed the paradigm.2, 19, 22, 26
When any factor acts as a trigger on genetically predisposed skin, the inflammatory cascade and a dysregulated interaction between innate and adaptive immune components and cells of the skin are initiated. Stressed keratinocytes start an innate immune response by producing antimicrobial peptides (AMPs), ß‐defensins, cytokines and chemokines. These molecules attract neutrophils to and activate resident mast cells in the skin, and it is predominantly these two types of cells that contain IL‐17 in psoriasis. Also, keratinocytes release self‐DNA that forms complexes with cytokines and AMPs, inducing activation of DCs which migrate into the lymph nodes.15, 27 There, derived inflammatory milieu results in naïve T cells being exposed to specific patterns of cytokines inducing subset differentiation,28, 29 thus adding adaptive responses. Differentiated Th17 cells leave the nodes through the skin, where they produce IL‐17.22, 30 Some subsets of these activated Th17 cells, as well as other types of T cells, are programed to remain in the skin, so that populations of autoreactive tissue‐resident cells will persist and contribute to psoriasis pathology.31
The presence of TGF‐β IL‐6 and IL‐1β induces initial differentiation to Th17 cells and leads to upregulation of IL‐23R expression (required for IL‐23 signalling).7, 8, 29 Then, IL‐23 released by DCs and APCs can link its receptor, activating Th17 cells and contributing to their phenotype maintenance.2, 22 Therefore, IL‐23 plays a crucial role for expansion and survival of the Th17 subset.7 Its role might be even more critical in autoimmune inflammation considering that plasticity of Th17 cells may lead them to switch to a non‐classical phenotype (ex‐Th17) producing IFN‐γ but having lost its IL‐17 expression. These cells exhibit increased survival, and more active cytokine production are resistant to suppressive action of Treg cells and can elude treatments targeting IL‐17.32 Thus, Th17 lineage cells or key upstream mediators of their differentiation would need to be targeted instead.
Activated Th17 cells induce the production of the pro‐inflammatory cytokines IL‐17A‐F, IL‐22, IL‐21 and TNF‐α.2 Effects of IL‐17A are stimulation of neutrophils recruitment and activation, enhancement of angiogenesis, mediation of tissue remodelling, direct activation of keratinocytes and, synergistically with TNF‐α, enhancement of inflammation.22 IL‐22 induces keratinocyte hyperproliferation and AMPs secretion by keratinocytes, is involved in tissue remodelling and its levels have been showed to correlate with psoriasis severity.33, 34, 35 IL‐21 participates in expression of IL‐23R and RORγt.36 TNF‐α can increase IL‐23 synthesis, which in turn enhances synthesis of IL‐17 as well as of IFN‐γ by memory T cells, eventually contributing to perpetuation of skin inflammatory process.7, 13, 22
Animal studies have demonstrated that IL‐23, but not IL‐12, intradermal injections induce a psoriasis‐like disease in mice with elevated transcription of IL‐23/Th17‐related genes.37, 38 Further, a number of studies in psoriasis patients and healthy controls have shown increased levels of p40 and p19 subunits, but not p35 subunit (the unique IL‐12 subunit), and IFN‐γ in psoriatic lesions, compared with adjacent non‐lesioned or healthy skin.39, 40, 41 These findings may suggest that most of the pro‐inflammatory role attributed to IL‐12 would really arise from IL‐23. Histochemical studies also suggest that psoriatic keratinocytes could contribute to skin inflammation by secreting sufficient IL‐23 to amplify memory T‐cell‐produced IFN‐γ.39 Additionally, genetic studies have demonstrated associations between genes IL23A (encoding the IL‐23p19 subunit), IL23B (encoding the IL‐12/23p40 subunit), and IL23R (encoding the IL‐23R subunit), but not IL12A (encoding the IL12p35 subunit), and the presence of psoriasis.42
Not only cytokines and related molecules increased in psoriatic lesions but the result of lacking them, have been investigated. Deficiency in p40 results in a great decrease in plaque formation and markers of inflammation, according to results from animal studies. When the effect of lacking p35 or p19 subunits was studied separately, results showed the development of more severe or milder inflammation, respectively, than controls.43
All these findings highlight the divergent roles of IL‐12 and IL‐23 in psoriasis and consistently (i) point to the involvement of IL‐12 in pro‐inflammatory effects might be lower than classically believed and independent of IFN‐γ, and (ii) strengthen the predominant role of the IL‐23/Th17 axis in the current pathogenic model of psoriasis.
Psoriasis and cardiovascular diseases: what is the place for the IL‐23/Th17 axis?
Overview
Psoriasis is a disorder affecting, but not limited to, the skin. Thus, cardiac, metabolic, gastrointestinal, pulmonary and kidney disease, as well as malignancies, infections, or psychiatric disorders are associated conditions with variable incidence in psoriasis, which increase the disease burden and the mortality risk, particularly in severe psoriasis.1
Cardiovascular disease deserves special mention in psoriasis, as per results reported in large‐scale epidemiological studies. CVD constitutes the first or second cause of mortality (after malignancies) among psoriasis patients,44, 45, 46 and the risk is higher among severe patients.44, 45 Duration has also been associated with CVD risk.1, 47 In terms of excess mortality, CVD was attributed globally an excess rate of 1.44 (95% CI: 1.43–1.45) per 1000 person‐years;46 in absolute terms CVD is the main driver of excess mortality in psoriasis.45 Consistent with these findings, psoriasis patients are at higher risk of cardiovascular events, in particular those with severe disease (RR to general population ranged 1.70–3.04; 1.38–1.59; and 1.37–1.39, for myocardial infarction, stroke and CV mortality, respectively).48, 49, 50 These data support the hypothesis of psoriasis as an independent factor for CV events. In this regard, severe psoriasis was found to confer an additional 6.2% absolute risk of a 10‐year rate of CV events compared with the general population.51 Indeed, a study showed that a high percentage of patients at low or intermediate CV risk according to Framingham score must be reclassified as intermediate and high risk, respectively, when psoriasis was added as a scoring factor.52
These epidemiological data are consistent with findings about abnormalities in lipid profiles of psoriasis patients. An abnormal lipid pattern consisting of higher cholesterol concentrations in the very low‐density lipoproteins (LDL) and high‐density lipoproteins (HDL) fractions as well as higher levels of Apo A1 have been demonstrated at the onset of disease. This suggests psoriasis predisposition to lipid abnormalities, and hypothesizes a detrimental role of HDL‐c and Apo A1 caused by oxidative modifications.53 Of note, features of oxidative stress and impairment of the antioxidant system have been evidenced in psoriasis.54, 55, 56 Levels of lipoprotein(a) [Lp(a)] have been reported to be increased in psoriasis and positively correlated with markers of oxidative stress and negatively with markers of antioxidant activity.55, 57 Significantly lower levels of HDL‐c, and higher levels of total cholesterol, LDL cholesterol and/or triglycerides (TG) have been reported too.54, 57 Both lipid abnormalities and oxidative stress have been suggested to act along with inflammation in psoriasis to eventually induce atherosclerosis and increase the CVR in these patients.58, 59
Despite pathogenic links between psoriasis and CVD warranting further investigation, growing evidence shows the role of chronic inflammation and immune dysfunction, so that both could share some dysregulated pathways, such as increased oxidative stress, monocyte and neutrophil modulation, endothelial dysfunction and IL‐23/Th17 signalling (Fig. 3).1 In line with this, it has been proposed that psoriasis and atherosclerosis would share immunological mechanisms involving IL‐12/Th1 and IL‐23/Th17 pathways, leading either to the plaque growth promoted by TNF‐α and IFN‐γ from differentiated Th1, or to the plaque vulnerability caused by intraplaque angioneogenesis and haemorrhage, promoted by Th17 effector CKs. Further, Th1 and Th17 proliferation would be favoured by a decrease in Treg number and function, with subsequent lower levels of TGF‐β and IL‐10, both associated with anti‐inflammatory and CV protective effects.60
Figure 3.
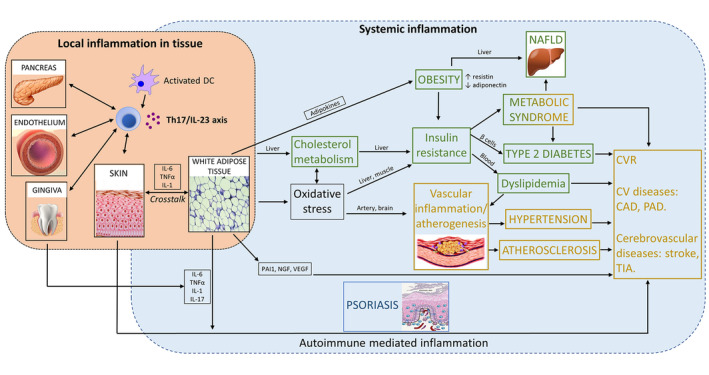
Scheme of involvement of Th17/IL‐23 axis in psoriasis and cardiometabolic diseases. CAD, coronary artery disease; CV, cardiovascular; CVR, cardiovascular risk; NAFLD, non‐alcoholic fatty liver disease; NGF, nerve growth factor; PAD, peripheral artery disease; PAI‐1, plasminogen activator inhibitor‐1; TIA, transient ischaemic attack; VEGF, vascular endothelial growth factor.. Yellow: CV diseases. Green: Metabolic diseases.
Several animal studies with either psoriasis or atherosclerosis models have found the IL‐23/Th17 axis to link psoriasis and vascular damage and dysfunction. Studies with mice overexpressing IL‐17A in keratinocytes and mimicking many hallmark skin features of severe psoriasis in humans, have demonstrated that alterations of IL‐17A and related downstream cytokines drove not only cutaneous but vascular inflammatory changes [increased reactive oxygen species (ROS) formation, oxidative stress, endothelial dysfunction, arterial hypertension and premature death].61, 62 Evidence from upstream cytokines have been reported as well. In models of atherosclerosis, an increase in IL‐23 secretion induced by the granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) has been shown to promote plaque instability by both increasing macrophages and DCs susceptibility to apoptosis and downregulating Bcl‐2, which triggers Th1 and Th17 responses and releases ROS.63 In patients with atherosclerosis, IL‐23 and IL23R were increased in atherosclerotic plaques, compared with non‐affected vessels and higher levels of IL‐23 that were observed in patients with more recent symptoms. Moreover, long‐term outcomes showed an adjusted association between higher IL‐23 plasma levels and mortality.64 Of note, blocking IL‐12 by vaccination reduced atherogenesis; however, involvement of IL‐23 in the effect could not be distinguished, because an anti‐p40 antibody was used.65
The beneficial effect of antibody treatment on both skin and CV outcomes is a promising line of investigation already yielding positive findings. As an example, antibodies targeting IL‐12/23p40, IL‐23p19 and IL‐17A/RA were administered to murine models of psoriasis developing CVD in response to systemic inflammation in a recent study. Treatment attenuated acanthosis in correlation with lengthening time to occlusive thrombus formation (with similar results to those from wild‐type controls), supports a common pathogenic pathway for both conditions.66
Coronary artery disease
Coronary artery disease (CAD) has been showed to have higher prevalence among psoriasis patients. Myocardial infarction (MI) may be the first manifestation of CAD, and its risk may be increased up to threefold, compared with non‐psoriatic controls.67, 68, 69 Interestingly, higher prevalence of psoriasis among CAD patients has also been reported.70
Certain polymorphisms of the human gene IL‐23 R have been associated with the risk and severity of atherosclerosis.71, 72 Investigations on myocardial injury, hypertrophy and remodelling mechanisms are providing growing knowledge on the role of the Th17 pathway. In murine models of MI, Ávalos et al. found increased post‐MI levels of IL‐6, IL‐23 and TGF‐β mRNA in the left ventricle. Surprisingly, levels of IL‐17A mRNA were not increased in the whole LV but only in the infarcted region, suggesting a role in the ventricular remodelling after MI.73
Remodelling, responsible for ventricular dysfunction and heart failure frequently developed after MI, is characterized by dilation and fibrosis phenomena in the myocardium and related to immune response after myocardial damage.74, 75, 76 Several studies addressing the potential role played by IL‐23 have showed a deleterious effect on MI models. Yan et al. demonstrated that IL‐23 rapidly increases after MI up to 3 days, whilst IL‐23R and IL‐17A upregulate progressively up to 7 days, and IL‐17R remains elevated until 14 days. The deficiency of IL‐23, IL‐17A and γδT cells resulted in a protective effect in mice, with higher survival after MI, less enlargement and less severe dysfunction of the LV, compared with wild‐type controls. Thus, IL‐23 from macrophages and neutrophils would act as an indispensable upstream regulator of IL‐17A, driving its production from γδT cells. Recruitment of γδT cells would also be favoured by IL‐23, whilst IL‐17A would promote neutrophil infiltration and fibrosis in myocardial tissue. These changes were evident from day 7 after MI, suggesting these mechanisms are crucial in remodelling.75 Additional studies with animal models of ischaemia/reperfusion injury consistently showed increases of IL‐23 in the injured myocardium. IL‐23 upregulation was also associated with enlargement of infarct size, high levels of typical biomarkers of myocardial damage (LDH and CK), pro‐inflammatory responses (with increase of IL‐17A, IL‐6 and TNF‐α releasing) and pro‐apoptotic effects (higher apoptotic index and lower Bcl‐2/Bax ratio). Through activation of JAK2/STAT3, IL‐23 induces secretion of IL‐17A, which eventually reinforces the inflammatory response and myocardial damage.77, 78 Importantly, neutralization of IL‐23 by administration of anti‐IL‐23p19 antibodies significantly reduced IL‐17A levels and ischaemia/reperfusion injury.77, 79
The inflammatory hypothesis of atherothrombosis has been present in the medical and research community for the last decade. In the field of rheumatology disease, this has led to explore a potential cardioprotective effect of biologic treatments targeting these inflammation pathways in psoriasis patients.80, 81, 82 Despite results from these retrospective studies being consistent with the overlapping mechanisms underlying both psoriasis and CVD, no studies to date have been conducted to prospectively assess the potential benefit of biologic therapy in psoriasis‐CVD patients. Nevertheless, the inflammatory hypothesis has been tested prospectively in two large randomized clinical trials involving around 25 000 CVD patients. The CANTOS study demonstrated the benefit of targeting IL‐1β to reduce CV events,83 whilst the CIRT study did not find this positive effect.84
Cerebrovascular disease
As mentioned earlier, stroke is a major cardiovascular event with a higher incidence among psoriasis population.47, 48, 49, 50
It is accepted that inflammation plays a crucial role within the complex pathophysiology of ischaemic stroke, particularly in exacerbation of brain damage. When ischaemia occurs, activation of microglia may result in secretion of both pro‐inflammatory CKs (from M1 phenotype, characterized by high expression of IL‐23 and IL‐12) and neuroprotective mediators (from M2 phenotype, characterized by high expression of IL‐10).85 Other cells, like macrophages, infiltrate the brain in the earlier phases of infarction, whilst neutrophils and lymphocytes join in later phases.86 Likewise, activated microglia is polarized to the M2 phenotype in the acute phase, switching to M1 later, mainly in the ischaemic penumbra.85, 87 IL‐23 secreted from macrophages and DCs promotes expansion of Th17 and γδT cells producing IL‐17, contributing to post‐stroke brain damage.88, 89
The importance of this pathway has been observed in several studies, which have shown elevations of the IL‐23/IL‐17 axis and IL‐23R associated with worsening of neuron damage, compared with controls in animal stroke models.86, 87, 90 In humans, increased levels of IL‐23 along with a markedly increased proportion of IL‐17A‐producing cells and elevation of IL‐17A levels, as well as other CKs, have been identified at several time points after stroke in comparison with controls.91, 92 Moreover, a positive correlation was found between IL‐23 levels and lesion volume.92 An increase of pro‐inflammatory mediators occurred simultaneously with a decrease in Treg cells and IL‐10 levels,91 supporting the hypothesis of a pro‐inflammatory/anti‐inflammatory imbalance as a mechanism involved in stroke and brain damage. Results from a study in mice are in line with this hypothesis, since immunomodulation exerted by bone marrow stem cells reduced IL‐23 and IL‐17 levels in serum and peri‐infarcted area.86
The effect of blocking IL‐23/IL‐17 has also been studied in stroke models. Interestingly, IL‐23 deficient animals showed significantly lower levels of γδT cells, subsequent lower secretion of IL‐17 and a decreased infarct size.90 Similar results were obtained after inhibition of IL‐12/IL‐23p40 subunit.93 Specific suppression of IL‐23p19 subunit resulted in lower levels of pro‐inflammatory IL‐23 and IL‐17 concurrent with upregulation of the Treg transcription factor FoxP3. Blocking of p19 subunit was, thus, associated with a less pronounced delayed phase of cerebral ischaemia and reduced infarction and neurological dysfunction.88
Peripheral artery disease
Lower limbs peripheral arterial disease (PAD) is a common syndrome among the adult population, mostly caused by atherosclerosis. In psoriatic patients, a 98% higher risk of PAD has been reported, compared with controls [OR: 1.98 (95% CI: 1.32–2.82)].94
Despite the scarce literature addressing the role of inflammation in PAD, the potential involvement of IL‐23 in this disease was assessed in a case‐control study, which first showed a significant increase of IL‐23 levels in PAD patients compared with controls.95
Hypertension
Data from epidemiological studies show that hypertension is more prevalent among psoriasis patients [OR: 1.58 (95% CI: 1.42–1.76)], and prevalence is associated with psoriasis severity.1 Furthermore, psoriasis patients are prone to suffer difficult‐to‐control hypertension, being 16.5 times and 19.9 times more likely to require three‐ or four‐drug treatment, respectively, than non‐psoriatic hypertensive patients.96
It is accepted that inflammation underlies the pathogenesis of hypertension, and activated immune cells are critical factors within this process.97, 98, 99 In particular, an involvement of IL‐23/IL‐17 pathway has been evidenced in this field too.
T cells and macrophages accumulate in kidneys and perivascular space. DCs are potent activators of T cells, which are polarized towards Th17 differentiation due to secretion of IL‐6, TNF‐α and IL‐23 secretion and STAT3 phosphorylation and signalling. IL‐17A produced by activated Th17 happened to be critical for vascular dysfunction and maintenance of hypertension,99, 100 and its production is increased in response to angiotensin‐II stimulus.101 It has been observed that increased stretch of hypertensive arteries activates endothelium, releasing IL‐6, IL‐23 and ROS whilst reducing nitric oxide (NO), and favouring STAT3 activation.100 Studies have also showed that isoketals (or isolevuglandins, derived from free radical‐mediated lipid peroxidation) accumulate in DCs in hypertension, promoting cytokine production and being able to act as neoantigens when adducted to proteins, activating DCs.98, 99 These isoketal‐modified proteins have been found to be elevated in circulating monocytes and DCs from hypertensive patients.98
Since an imbalance between Th17 and Treg is thought to underlie, at least in part, the pathophysiology of CV disorders, Liu et al.102 studied whether the use of common anti‐hypertensive and hypolipemiant drugs might have any effect on this pathway. Patients treated with a combination of telmisartan and rosuvastatin showed synergistic decrease of serum pro‐inflammatory components, including IL‐23, Th17 cells and IL‐17A, as well as increase of anti‐inflammatory components, including Treg, FoxP3 and IL‐10, and a reduction of carotid intima‐media thickness by ultrasound.102
Metabolic diseases and psoriasis: what is the place for the IL‐23/Th17 axis?
Overview
Metabolic disorders are more prevalent among patients with psoriasis. Different meta‐analyses found that psoriatic patients have 27% higher risk of diabetes (RR: 1.27; 95% CI, 1.16–1.40) and more than twofold risk of metabolic syndrome (OR: 2.26; 95% CI, 1.70–3.01). Indeed, psoriasis may act a as risk factor for difficult‐to‐control diabetes. Conversely, metabolic disorders may act as a risk factor for psoriasis development.1 Also, obesity is more frequent among psoriatic patients (OR: 1.66; 95% CI: 1.46–1.89) and may be a negative factor for systemic treatment response.1 A relationship between psoriasis severity and obesity and metabolic syndrome has also been observed.103, 104 Non‐alcoholic fatty liver disease (NAFLD), a disorder associated with metabolic syndrome, has also been shown to have a prevalence that is clearly higher among the psoriatic population.105 Fig. 3 represents the relationship between these diseases and psoriasis.
Obesity
Obesity is a central feature associated with increased risk of related metabolic dysfunction, including insulin resistance, diabetes, dyslipidaemia, hypertension and NAFLD, separately or together comprising metabolic syndrome.106, 107 In individuals with body mass index (BMI) above 25 kg/m2, each 5 kg/m2 increase has been associated with 30% increase in overall mortality, 40% increase in CV mortality and 60% increase in diabetic mortality.107 On the other hand, association between psoriasis and obesity has been largely supported by evidence. Epidemiological studies point to the risk of psoriasis being increased in the obese population; further, the risk increases with increasing BMI.108, 109 Obesity may negatively affect systemic treatment of psoriasis, including biologics; in psoriasis patients, a 12% (95% CI: 1.01–1.24) increased risk of treatment interruption because of lack of effectiveness and a 17% (95% CI: 1.02–1.36) increased risk of adverse event occurrence has been associated with each 5 kg/m2 increase in BMI.110 Conversely, improved treatment responses have been observed in patients after weight loss, with improved outcomes in both clinical (PASI) and quality of life (DLQI) variables.108
It is accepted that a close relationship between obesity and chronic inflammation exists. Macrophages and T cells with pro‐inflammatory effects accumulate in adipose tissue whilst anti‐inflammatory Treg cells are diminished.111 T cells can interact with DCs, regulating inflammatory response; in turn, adipose tissue can secrete multiple molecules, including cytokines (adipokines) with effects on inflammation signalling and endothelial dysfunction.112, 113 Interestingly, dysregulated levels of adipokines in psoriasis patients have been reported, suggesting these molecules may be a link between inflammation, obesity, psoriasis and conditions improving CV risk.113
Due to the current consideration of obesity as a chronic low‐inflammation state, a potential involvement of the IL‐23 pathway in obesity has been investigated. In obese mice, an increased level of DCs derived from adipose tissue (ATDCs) with an immature phenotype has been found infiltrating adipose tissue. These ATDCs secreted higher levels of IL‐6, TGF‐β and IL‐23 than those DCs derived from spleen. Higher levels of IL‐23p19 and IL‐12/IL‐23p40 mRNA, but not IL‐12p35, were also demonstrated. These findings suggest ATDCs promote Th17 differentiation in adipose tissue, mediated by IL‐23 and other CKs. This hypothesis was tested by administering anti‐IL‐6 or anti‐IL‐23 antibodies, resulting in a reduction of Th17 cells.112 Consistent results were observed in a study with obese women. Significantly increased serum concentrations of IL‐23, IL‐17, as well as leptin and macrophage migration inhibition factor (MIF), were evidenced, whilst IL‐12 and IFN‐γ levels remained unchanged. No correlation between IL‐17 or IL‐23 levels and leptin and MIF were observed, which support an IL‐23‐dependent increase of IL‐17 in obese patients.114 Another study showed that obese individuals′ intestine feature an increased jejunal mucosa surface as well as increased total and intra‐epithelial T‐cell density. Expression of IL‐23, IFN‐γ, TGF‐β and TNF‐α were also increased both in lamina propria and epithelium, as were IL‐17A levels, suggesting Th17 cells are expanded in obesity.111
Non‐alcoholic fatty liver disease
Non‐alcoholic fatty liver disease develops through a process of fat deposition on liver cells, in an alcohol consumption non‐dependent way. Mild forms of this disorder include steatosis, whilst severe forms are characterized by cell damage and include steatohepatitis, potentially progressing to cirrhosis. NAFLD is the leading cause of altered levels of hepatic enzymes in Western countries, and considered as the hepatic manifestation of metabolic syndrome. Prevalence of NAFLD is higher among patients with metabolic syndrome and those with obesity; a correlation between obesity severity and prevalence of NAFLD have been established.105, 115 The indolent development and subsequent underdiagnosis of NAFLD contrast with the dramatic fact that its aggressive form is the second most common cause of liver transplantation in the USA.116
Non‐alcoholic fatty liver disease is associated with psoriasis, with a higher frequency observed among these patients.105 Gisondi et al. found NAFLD was present in 47% of patients with plaque psoriasis, compared with a frequency of 28% in matched healthy controls. Some biomarkers (IL‐6, adiponectin) and PASI score were higher in psoriasis patients with NAFLD than in those without hepatic affectation.115 A later study found a similar prevalence (44% vs. 26%). In this study, both patients and controls presenting NAFLD were scored with the NAFLD fibrosis score. Psoriatic patients obtained higher scoring and were at higher risk of liver fibrosis than controls.117
The IL‐23/Th17 pathway and an imbalance between pro‐ and anti‐inflammatory signalling seem to play a role in NAFLD as well. Imbalanced levels of Th17/Treg has been demonstrated in animal models. An increase in proportion of Th17 cells and related cytokines IL‐6, IL‐23 and IL‐17, and RORγτ expression along with a decrease in Treg cells and FoxP3 expression have been identified in murine models118 but also in humans,119 and suggest a pro‐inflammatory predominance involved in the pathogenesis of NAFLD. Likewise, an increase in Th17 cells and related gene expression was observed in specimens of human livers affected with steatohepatitis.120 Some investigations using monoclonal antibodies and other agents with an inhibitory effect on Th17 differentiation have resulted in a decrease of Th17 and pro‐inflammatory molecules as well as reduced liver immune cell infiltration, steatosis, and hepatic injury.119
Insulin resistance and diabetes mellitus
Psoriasis patients are at higher risk of diabetes mellitus (DM), independently of other risk factors, and are more likely to require pharmacological treatment for their diabetes mellitus. Further, likelihood of insulin resistance and diabetic complications increase with psoriasis severity.1 Psoriasis patients have triple risk [HR: 3.02 (95% CI: 2.42; 3.63)] of dying because of endocrine, nutritional and metabolic disorders than controls, independently of DM contribution to CV mortality.121 A genome‐wide association study showed three different single‐nucleotide polymorphism, two of them located in the gene IL12B and 1 in the gene IL‐23R, associated with between 2.7‐ and 5.9‐fold higher risk of developing DM2 in patients with psoriasis.122
Immune cells, including T cells and DCs, are acknowledged to infiltrate pancreatic islets tissue, activating inflammatory and autoimmune responses in type 1 DM that eventually result in β‐cells destruction.123 Evidence on IL‐23 axis involvement has been reported. In a preclinical study, it was observed that both TGF‐β + IL‐6 and IL‐23 + IL‐6 can polarize naïve T cells to IL‐17‐producing Th17. However, Th17 cells polarized in the presence of IL‐23 secreted larger amounts of IL‐17, IL‐22 and expressed more IL‐23R on their surface, compared with those polarized in the presence of TGF‐β. When these cells were injected into non‐obese diabetic mice, all animals developed diabetes. On the contrary, TGF‐β‐polarized cells showed a Treg phenotype with significantly higher secretion of IL‐10; interestingly, these cells did not induce DM when injected into mice.124 This study suggest a diabetogenic potential for the IL‐23/Th17 axis and raises again the hypothesis of a Th17/Treg imbalance dependent on a predominant CK milieu.
Consistent with animal investigations, the involvement of the IL‐23/Th17 pathway in diabetic humans has also been reported. Fatima et al.125 found that IL‐23 and IL‐17A, among other pro‐inflammatory cytokines were significantly upregulated in type 1 diabetic patients, exhibiting a relationship with age and glycaemia and suggesting a synergistic interaction of IL‐23, IL‐17A and TNF‐α in β‐cells damage. Similarly, a later study showed an age‐dependent increase of IL‐23 mRNA and augmented levels of IL‐17A and IL‐22, among other CKs, in type 2 diabetic patients, confirming a role in DM onset.126
Conclusions
The IL‐23/Th17 axis has a prominent role in pathophysiology of psoriasis, in which the upregulation of this pathway, together with other inflammatory cytokines (e.g. TNF and type I IFN), contributes to develop a ‘pro‐inflammatory state’ in psoriasis patients. A growing body of recent evidence suggests inflammation, through multiple mediators and pathways, is mechanistically involved in most of cardiac and metabolic chronic diseases, including obesity and non‐alcoholic fat liver, known common psoriasis associated conditions. Here, we have reviewed the evidence on the IL‐23/Th17 pathway and suggest its upregulation (in addition to detrimental lifestyle) is a potential root of many cardiometabolic psoriasis comorbidity. This overlapping has also led to the hypothesis that highly specific monoclonal antibodies developed for systemic treatment of psoriasis might have an effect on the progression of its comorbidity, although at this stage the standard‐of‐care for psoriasis comorbidity is essential too and further evidence is needed to confirm or refute this supposition.
Acknowledgements
The authors would like to thank Esther Prieto, MD and Eva Mateu, PhD of TFS S.L. for editorial assistance and writing support. RBW is supported by the Manchester NIHR Biomedical Research Centre.
Conflict of interest
Dr. Carrascosa has received honoraria as consultant and/or speaker and/or consultant and/or participated as PI/SI from Pfizer, Eli Lilly, AbbVie, Almirall, Leo Pharma, Samsung Bioepis, Novartis, Celgene and Janssen Pharmaceuticals. Dr. Egeberg has received research funding from Pfizer, Eli Lilly, the Danish National Psoriasis Foundation, and the Kgl Hofbundtmager Aage Bang Foundation, and honoraria as consultant and/or speaker from AbbVie, Almirall, Leo Pharma, Samsung Bioepis, Pfizer, Eli Lilly, Novartis, Galderma, Dermavant, UCB, Bristol‐Myers Squibb, and Janssen Pharmaceuticals. Dr. Gisondi has received honoraria as consultant and/or speaker and/or consultant and/or participated as PI/SI from Pfizer, Eli Lilly, AbbVie, Almirall, Leo Pharma, Novartis, Celgene, UCB and Janssen Pharmaceuticals. Dr. Mrowietz has received honoraria, unrestricted educational grants, and has participated as investigator in clinical trials of products of the sponsor of this publication. Dr. Warren: Research Grants: AbbVie, Almirall, Amgen, Celgene, Janssen Pharmaceuticals, Eli Lilly, Leo Pharma, Novartis, Pfizer and UCB. Consulting Fees: AbbVie, Almirall, Amgen, Boehringer Ingelheim, Celgene, Janssen Pharmaceuticals, Leo Pharma, Eli Lilly, Novartis, Pfizer, Sanofi, Xenoport and UCB.
Funding sources
This publication was funded by Almirall R&D, Barcelona, Spain.
References
- 1. Takeshita J, Grewal S, Langan SM et al Psoriasis and comorbid diseases: epidemiology. J Am Acad Dermatol 2017; 76: 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Toussirot E. The IL23/Th17 pathway as a therapeutic target in chronic inflammatory diseases. Inflamm Allergy Drug Targets 2012; 11: 159–168. [DOI] [PubMed] [Google Scholar]
- 3. McInnes IB. Role of Cytokines in the Immune System. UpToDate Inc, Waltham, MA, 2015. URL http://www.uptodate.com (last accessed: 1 June 2018). [Google Scholar]
- 4. Ballas ZK. T Helper Subsets: Differentiation and Role in Disease. UpToDate Inc., Waltham, MA, 2016. URL http://www.uptodate.com (last accessed: 1 June 2018). [Google Scholar]
- 5. Langrish CL, Chen Y, Blumenschein WM et al IL‐23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 2005; 201: 233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dong C. Diversification of T‐helper‐cell lineages: finding the family root of IL‐17‐producing cells. Nat Rev Immunol 2006; 6: 329–333. [DOI] [PubMed] [Google Scholar]
- 7. McGeachy MJ, Cua DJ. The link between IL‐23 and Th17 cell‐mediated immune pathologies. Semin Immunol 2007; 19: 372–376. [DOI] [PubMed] [Google Scholar]
- 8. Wilson NJ, Boniface K, Chan JR et al Development, cytokine profile and function of human interleukin 17‐producing helper T cells. Nat Immunol 2007; 8: 950–957. [DOI] [PubMed] [Google Scholar]
- 9. Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature 2008; 453: 1051–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Louten J, Boniface K, de Waal Malefyt R. Development and function of TH17 cells in health and disease. J Allergy Clin Immunol 2009; 123: 1004–1011. [DOI] [PubMed] [Google Scholar]
- 11. Behzadi P, Behzadi E, Ranjbar R. IL‐12 family cytokines: general characteristics, pathogenic microorganisms, receptors, and signalling pathways. Acta Microbiol Immunol Hung 2016; 63: 1–25. [DOI] [PubMed] [Google Scholar]
- 12. Oppmann B, Lesley R, Blom B et al Novel p19 protein engages IL‐12p40 to form a cytokine, IL‐23, with biological activities similar as well as distinct from IL‐12. Immunity 2000; 13: 715–725. [DOI] [PubMed] [Google Scholar]
- 13. Tang C, Chen S, Qian H, Huang W. Interleukin‐23: as a drug target for autoimmune inflammatory diseases. Immunology 2012; 135: 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stritesky GL, Yeh N, Kaplan MH. IL‐23 promotes maintenance but not commitment to the Th17 lineage. J Immunol Baltim Md 1950 2008; 181: 5948–5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Prinz I, Sandrock I, Mrowietz U. Interleukin‐17 cytokines: effectors and targets in psoriasis‐A breakthrough in understanding and treatment. J Exp Med 2020; 217: e20191397 10.1084/jem.20191397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kolls JK, Lindén A. Interleukin‐17 family members and inflammation. Immunity 2004; 21: 467–476. [DOI] [PubMed] [Google Scholar]
- 17. Onishi RM, Gaffen SL. Interleukin‐17 and its target genes: mechanisms of interleukin‐17 function in disease. Immunology 2010; 129: 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Parisi R, Symmons DPM, Griffiths CEM, Ashcroft DM. Identification and Management of Psoriasis and Associated ComorbidiTy (IMPACT) project team. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol 2013; 133: 377–385. [DOI] [PubMed] [Google Scholar]
- 19. Boehncke W‐H. Etiology and pathogenesis of psoriasis. Rheum Dis Clin North Am 2015; 41: 665–675. [DOI] [PubMed] [Google Scholar]
- 20. Prieto‐Pérez R, Cabaleiro T, Daudén E, Ochoa D, Roman M, Abad‐Santos F. Genetics of psoriasis and pharmacogenetics of biological drugs. Autoimmune Dis 2013; 2013: 613086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ovejero‐Benito MC, Muñoz‐Aceituno E, Reolid A, Saiz‐Rodríguez M, Abad‐Santos F, Daudén E. Pharmacogenetics and pharmacogenomics in moderate‐to‐severe psoriasis. Am J Clin Dermatol 2018; 19: 209–222. [DOI] [PubMed] [Google Scholar]
- 22. Puig L. The role of IL 23 in the treatment of psoriasis. Expert Rev Clin Immunol 2017; 13: 525–534. [DOI] [PubMed] [Google Scholar]
- 23. Cai Y, Shen X, Ding C et al Pivotal role of dermal IL‐17‐producing γδ T cells in skin inflammation. Immunity 2011; 35: 596–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Austin LM, Ozawa M, Kikuchi T, Walters IB, Krueger JG. The majority of epidermal T cells in Psoriasis vulgaris lesions can produce type 1 cytokines, interferon‐gamma, interleukin‐2, and tumor necrosis factor‐alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol 1999; 113: 752–759. [DOI] [PubMed] [Google Scholar]
- 25. Leonardi CL, Kimball AB, Papp KA et al Efficacy and safety of ustekinumab, a human interleukin‐12/23 monoclonal antibody, in patients with psoriasis: 76‐week results from a randomised, double‐blind, placebo‐controlled trial (PHOENIX 1). Lancet Lond Engl 2008; 371: 1665–1674. [DOI] [PubMed] [Google Scholar]
- 26. Nograles KE, Zaba LC, Guttman‐Yassky E et al Th17 cytokines interleukin (IL)‐17 and IL‐22 modulate distinct inflammatory and keratinocyte‐response pathways. Br J Dermatol 2008; 159: 1092–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ganguly D, Chamilos G, Lande R et al Self‐RNA‐antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med 2009; 206: 1983–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aggarwal S, Ghilardi N, Xie M‐H, de Sauvage FJ, Gurney AL. Interleukin‐23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin‐17. J Biol Chem 2003; 278: 1910–1914. [DOI] [PubMed] [Google Scholar]
- 29. Bettelli E, Carrier Y, Gao W et al Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006; 441: 235–238. [DOI] [PubMed] [Google Scholar]
- 30. Keijsers RRMC, Joosten I, van Erp PEJ, Koenen HJPM, van de Kerkhof PCM. Cellular sources of IL‐17 in psoriasis: a paradigm shift? Exp Dermatol 2014; 23: 799–803. [DOI] [PubMed] [Google Scholar]
- 31. Clark RA. Resident memory T cells in human health and disease. Sci Transl Med 2015; 7: 269rv1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Basdeo SA, Cluxton D, Sulaimani J et al Ex‐Th17 (Nonclassical Th1) cells are functionally distinct from classical Th1 and Th17 cells and are not constrained by regulatory T cells. J Immunol Baltim Md 1950 2017; 198: 2249–2259. [DOI] [PubMed] [Google Scholar]
- 33. Wolk K, Witte E, Wallace E et al IL‐22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol 2006; 36: 1309–1323. [DOI] [PubMed] [Google Scholar]
- 34. Liang SC, Tan X‐Y, Luxenberg DP et al Interleukin (IL)‐22 and IL‐17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 2006; 203: 2271–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ekman A‐K, Bivik Eding C, Rundquist I, Enerbäck C. IL‐17 and IL‐22 promote keratinocyte stemness in the germinative compartment in psoriasis. J Invest Dermatol 2019; 139: 1564–1573.e8. [DOI] [PubMed] [Google Scholar]
- 36. Zhou L, Ivanov II, Spolski R et al IL‐6 programs T(H)‐17 cell differentiation by promoting sequential engagement of the IL‐21 and IL‐23 pathways. Nat Immunol 2007; 8: 967–974. [DOI] [PubMed] [Google Scholar]
- 37. Chan JR, Blumenschein W, Murphy E et al IL‐23 stimulates epidermal hyperplasia via TNF and IL‐20R2‐dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med 2006; 203: 2577–2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rizzo HL, Kagami S, Phillips KG, Kurtz SE, Jacques SL, Blauvelt A. IL‐23‐mediated psoriasis‐like epidermal hyperplasia is dependent on IL‐17A. J Immunol Baltim Md 1950 2011; 186: 1495–1502. [DOI] [PubMed] [Google Scholar]
- 39. Piskin G, Sylva‐Steenland RMR, Bos JD, Teunissen MBM. In vitro and in situ expression of IL‐23 by keratinocytes in healthy skin and psoriasis lesions: enhanced expression in psoriatic skin. J Immunol Baltim Md 1950 2006; 176: 1908–1915. [DOI] [PubMed] [Google Scholar]
- 40. Lee E, Trepicchio WL, Oestreicher JL et al Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med 2004; 199: 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen X, Tan Z, Yue Q, Liu H, Liu Z, Li J. The expression of interleukin‐23 (p19/p40) and interleukin‐12 (p35/p40) in psoriasis skin. J Huazhong Univ Sci Technolog Med Sci 2006; 26: 750–752. [DOI] [PubMed] [Google Scholar]
- 42. Nair RP, Duffin KC, Helms C et al Genome‐wide scan reveals association of psoriasis with IL‐23 and NF‐kappaB pathways. Nat Genet 2009; 41: 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kulig P, Musiol S, Freiberger SN et al IL‐12 protects from psoriasiform skin inflammation. Nat Commun 2016; 7: 13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Salahadeen E, Torp‐Pedersen C, Gislason G, Hansen PR, Ahlehoff O. Nationwide population‐based study of cause‐specific death rates in patients with psoriasis. J Eur Acad Dermatol Venereol 2015; 29: 1002–1005. [DOI] [PubMed] [Google Scholar]
- 45. Svedbom A, Dalén J, Mamolo C et al Increased cause‐specific mortality in patients with mild and severe psoriasis: a population‐based Swedish register study. Acta Derm Venereol 2015; 95: 809–815. [DOI] [PubMed] [Google Scholar]
- 46. Lee M‐S, Yeh Y‐C, Chang Y‐T, Lai M‐S. All‐cause and cause‐specific mortality in patients with psoriasis in Taiwan: a Nationwide Population‐based Study. J Invest Dermatol 2017; 137: 1468–1473. [DOI] [PubMed] [Google Scholar]
- 47. Egeberg A, Skov L, Joshi AA et al The relationship between duration of psoriasis, vascular inflammation, and cardiovascular events. J Am Acad Dermatol 2017; 77: 650–656.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Armstrong EJ, Harskamp CT, Armstrong AW. Psoriasis and major adverse cardiovascular events: a systematic review and meta‐analysis of observational studies. J Am Heart Assoc 2013; 2: e000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Samarasekera EJ, Neilson JM, Warren RB, Parnham J, Smith CH. Incidence of cardiovascular disease in individuals with psoriasis: a systematic review and meta‐analysis. J Invest Dermatol 2013; 133: 2340–2346. [DOI] [PubMed] [Google Scholar]
- 50. Raaby L, Ahlehoff O, de Thurah A. Psoriasis and cardiovascular events: updating the evidence. Arch Dermatol Res 2017; 309: 225–228. [DOI] [PubMed] [Google Scholar]
- 51. Mehta NN, Yu Y, Pinnelas R et al Attributable risk estimate of severe psoriasis on major cardiovascular events. Am J Med 2011; 124: 775. e1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mehta NN, Krishnamoorthy P, Yu Y et al The impact of psoriasis on 10‐year Framingham risk. J Am Acad Dermatol 2012; 67: 796–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mallbris L, Granath F, Hamsten A, Ståhle M. Psoriasis is associated with lipid abnormalities at the onset of skin disease. J Am Acad Dermatol 2006; 54: 614–621. [DOI] [PubMed] [Google Scholar]
- 54. Pietrzak A, Michalak‐Stoma A, Chodorowska G, Szepietowski JC. Lipid disturbances in psoriasis: an update. Mediators Inflamm 2010; 2010: 535612 10.1155/2010/535612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ferretti G, Bacchetti T, Campanati A, Simonetti O, Liberati G, Offidani A. Correlation between lipoprotein(a) and lipid peroxidation in psoriasis: role of the enzyme paraoxonase‐1. Br J Dermatol 2012; 166: 204–207. [DOI] [PubMed] [Google Scholar]
- 56. Peluso I, Cavaliere A, Palmery M. Plasma total antioxidant capacity and peroxidation biomarkers in psoriasis. J Biomed Sci 2016; 23: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pietrzak A, Kadzielewski J, Janowski K et al Lipoprotein (a) in patients with psoriasis: associations with lipid profiles and disease severity. Int J Dermatol 2009; 48: 379–387. [DOI] [PubMed] [Google Scholar]
- 58. Zhou Q, Mrowietz U, Rostami‐Yazdi M. Oxidative stress in the pathogenesis of psoriasis. Free Radic Biol Med 2009; 47: 891–905. [DOI] [PubMed] [Google Scholar]
- 59. Asha K, Singal A, Sharma SB, Arora VK, Aggarwal A. Dyslipidaemia & oxidative stress in patients of psoriasis: emerging cardiovascular risk factors. Indian J Med Res 2017; 146: 708–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Armstrong AW, Voyles SV, Armstrong EJ, Fuller EN, Rutledge JC. A tale of two plaques: convergent mechanisms of T‐cell‐mediated inflammation in psoriasis and atherosclerosis. Exp Dermatol 2011; 20: 544–549. [DOI] [PubMed] [Google Scholar]
- 61. Karbach S, Croxford AL, Oelze M et al Interleukin 17 drives vascular inflammation, endothelial dysfunction, and arterial hypertension in psoriasis‐like skin disease. Arterioscler Thromb Vasc Biol 2014; 34: 2658–2668. [DOI] [PubMed] [Google Scholar]
- 62. Schüler R, Brand A, Klebow S et al Antagonization of IL‐17A attenuates skin inflammation and vascular dysfunction in mouse models of psoriasis. J Invest Dermatol 2019; 139: 638–647. 10.1016/j.jid.2018.09.021 [DOI] [PubMed] [Google Scholar]
- 63. Subramanian M, Thorp E, Tabas I. Identification of a non‐growth factor role for GM‐CSF in advanced atherosclerosis: promotion of macrophage apoptosis and plaque necrosis through IL‐23 signaling. Circ Res 2015; 116: e13–e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Abbas A, Gregersen I, Holm S et al Interleukin 23 levels are increased in carotid atherosclerosis: possible role for the interleukin 23/interleukin 17 axis. Stroke 2015; 46: 793–799. [DOI] [PubMed] [Google Scholar]
- 65. Hauer AD, Uyttenhove C, de Vos P et al Blockade of interleukin‐12 function by protein vaccination attenuates atherosclerosis. Circulation 2005; 112: 1054–1062. [DOI] [PubMed] [Google Scholar]
- 66. Li Y, Golden JB, Camhi MI et al Protection from psoriasis‐related thrombosis after inhibition of IL‐23 or IL‐17A. J Invest Dermatol 2018; 138: 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Furue M, Tsuji G, Chiba T, Kadono T. Cardiovascular and metabolic diseases comorbid with psoriasis: beyond the skin. Intern Med Tokyo Jpn 2017; 56: 1613–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hjuler KF, Böttcher M, Vestergaard C et al Increased prevalence of coronary artery disease in severe psoriasis and severe atopic dermatitis. Am J Med 2015; 128: 1325–1334.e2. [DOI] [PubMed] [Google Scholar]
- 69. Mahiques‐Santos L, Soriano‐Navarro CJ, Perez‐Pastor G, Tomas‐Cabedo G, Pitarch‐Bort G, Valcuende‐Cavero F. Psoriasis and ischemic coronary artery disease. Actas Dermosifiliogr 2015; 106: 112–116. [DOI] [PubMed] [Google Scholar]
- 70. Picard D, Bénichou J, Sin C et al Increased prevalence of psoriasis in patients with coronary artery disease: results from a case‐control study. Br J Dermatol 2014; 171: 580–587. [DOI] [PubMed] [Google Scholar]
- 71. Zhang M, Cai Z‐R, Zhang B et al Functional polymorphisms in interleukin‐23 receptor and susceptibility to coronary artery disease. DNA Cell Biol 2014; 33: 891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kave M, Shadman M, Alizadeh A, Samadi M. Analysis of the association between IL‐23R rs11209026 polymorphism and incidence of atherosclerosis. Int J Immunogenet 2015; 42: 341–345. [DOI] [PubMed] [Google Scholar]
- 73. Ávalos AM, Apablaza FA, Quiroz M et al IL‐17A levels increase in the infarcted region of the left ventricle in a rat model of myocardial infarction. Biol Res 2012; 45: 193–200. [DOI] [PubMed] [Google Scholar]
- 74. Arslan F, de Kleijn DP, Pasterkamp G. Innate immune signaling in cardiac ischemia. Nat Rev Cardiol 2011; 8: 292–300. [DOI] [PubMed] [Google Scholar]
- 75. Yan X, Shichita T, Katsumata Y et al Deleterious effect of the IL‐23/IL‐17A axis and γδT cells on left ventricular remodeling after myocardial infarction. J Am Heart Assoc 2012; 1: e004408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Thygesen K, Alpert JS, Jaffe AS et al Third universal definition of myocardial infarction. J Am Coll Cardiol 2012; 60: 1581–1598. [DOI] [PubMed] [Google Scholar]
- 77. Hu X, Ma R, Lu J et al IL‐23 promotes myocardial I/R injury by increasing the inflammatory responses and oxidative stress reactions. Cell Physiol Biochem 2016; 38: 2163–2172. [DOI] [PubMed] [Google Scholar]
- 78. Liao Y, Hu X, Guo X, Zhang B, Xu W, Jiang H. Promoting effects of IL‐23 on myocardial ischemia and reperfusion are associated with increased expression of IL‐17A and upregulation of the JAK2‐STAT3 signaling pathway. Mol Med Rep 2017; 16: 9309–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhu H, Li J, Wang S, Liu K, Wang L, Huang L. Hmgb1‐TLR4‐IL‐23‐IL‐17A axis promote ischemia‐reperfusion injury in a cardiac transplantation model. Transplantation 2013; 95: 1448–1454. [DOI] [PubMed] [Google Scholar]
- 80. Wu JJ, Poon K‐YT, Channual JC, Shen AY‐J. Association between tumor necrosis factor inhibitor therapy and myocardial infarction risk in patients with psoriasis. Arch Dermatol 2012; 148: 1244–1250. [DOI] [PubMed] [Google Scholar]
- 81. Ahlehoff O, Skov L, Gislason G et al Cardiovascular disease event rates in patients with severe psoriasis treated with systemic anti‐inflammatory drugs: a Danish real‐world cohort study. J Intern Med 2013; 273: 197–204. [DOI] [PubMed] [Google Scholar]
- 82. Gulliver WP, Randell S, Gulliver S et al Do biologics protect patients with psoriasis from myocardial infarction? a retrospective cohort. J Cutan Med Surg 2016; 20: 536–541. [DOI] [PubMed] [Google Scholar]
- 83. Ridker PM, Everett BM, Thuren T et al Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017; 377: 1119–1131. [DOI] [PubMed] [Google Scholar]
- 84. Ridker PM, Everett BM, Pradhan A et al Low‐dose methotrexate for the prevention of atherosclerotic events. N Engl J Med 2019; 380: 752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zhao S‐C, Ma L‐S, Chu Z‐H, Xu H, Wu W‐Q, Liu F. Regulation of microglial activation in stroke. Acta Pharmacol Sin 2017; 38: 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ma S, Zhong D, Chen H et al The immunomodulatory effect of bone marrow stromal cells (BMSCs) on interleukin (IL)‐23/IL‐17‐mediated ischemic stroke in mice. J Neuroimmunol 2013; 257: 28–35. [DOI] [PubMed] [Google Scholar]
- 87. Wang M, Zhong D, Zheng Y et al Damage effect of interleukin (IL)‐23 on oxygen‐glucose‐deprived cells of the neurovascular unit via IL‐23 receptor. Neuroscience 2015; 289: 406–416. [DOI] [PubMed] [Google Scholar]
- 88. Zheng Y, Zhong D, Chen H et al Pivotal role of cerebral interleukin‐23 during immunologic injury in delayed cerebral ischemia in mice. Neuroscience 2015; 290: 321–331. [DOI] [PubMed] [Google Scholar]
- 89. Brait VH, Arumugam TV, Drummond GR, Sobey CG. Importance of T lymphocytes in brain injury, immunodeficiency, and recovery after cerebral ischemia. J Cereb Blood Flow Metab 2012; 32: 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gelderblom M, Gallizioli M, Ludewig P et al IL‐23 (Interleukin‐23)‐producing conventional dendritic cells control the detrimental IL‐17 (interleukin‐17) response in stroke. Stroke 2018; 49: 155–164. [DOI] [PubMed] [Google Scholar]
- 91. Hu Y, Zheng Y, Wu Y, Ni B, Shi S. Imbalance between IL‐17A‐producing cells and regulatory T cells during ischemic stroke. Mediators Inflamm 2014; 2014: 813045 10.1155/2014/813045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jiang C, Kong W, Wang Y et al Changes in the cellular immune system and circulating inflammatory markers of stroke patients. Oncotarget 2016; 8: 3553–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Konoeda F, Shichita T, Yoshida H et al Therapeutic effect of IL‐12/23 and their signaling pathway blockade on brain ischemia model. Biochem Biophys Res Commun 2010; 402: 500–506. [DOI] [PubMed] [Google Scholar]
- 94. Prodanovich S, Kirsner RS, Kravetz JD, Ma F, Martinez L, Federman DG. Association of psoriasis with coronary artery, cerebrovascular, and peripheral vascular diseases and mortality. Arch Dermatol 2009; 145: 700–703. [DOI] [PubMed] [Google Scholar]
- 95. David A, Saitta S, De Caridi G et al Interleukin‐23 serum levels in patients affected by peripheral arterial disease. Clin Biochem 2012; 45: 275–278. [DOI] [PubMed] [Google Scholar]
- 96. Armstrong AW, Lin SW, Chambers CJ, Sockolov ME, Chin DL. Psoriasis and hypertension severity: results from a case‐control study. PLoS ONE 2011; 6: e18227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med 2011; 17: 1402–1409. [DOI] [PubMed] [Google Scholar]
- 98. Kirabo A, Fontana V, de Faria APC et al DC isoketal‐modified proteins activate T cells and promote hypertension. J Clin Invest 2014; 124: 4642–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dixon KB, Davies SS, Kirabo A. Dendritic cells and isolevuglandins in immunity, inflammation, and hypertension. Am J Physiol Heart Circ Physiol 2017; 312: H368–H374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Loperena R, Van Beusecum JP, Itani HA et al Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: roles of STAT3, interleukin 6 and hydrogen peroxide. Cardiovasc Res 2018; 114: 1547–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Madhur MS, Lob HE, McCann LA et al Interleukin 17 promotes angiotensin II‐induced hypertension and vascular dysfunction. Hypertens Dallas Tex 1979 2010; 55: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Liu Z, Zhao Y, Wei F et al Treatment with telmisartan/rosuvastatin combination has a beneficial synergistic effect on ameliorating Th17/Treg functional imbalance in hypertensive patients with carotid atherosclerosis. Atherosclerosis 2014; 233: 291–299. [DOI] [PubMed] [Google Scholar]
- 103. Langan SM, Seminara NM, Shin DB et al Prevalence of metabolic syndrome in patients with psoriasis: a population‐based study in the United Kingdom. J Invest Dermatol 2012; 132: 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Singh S, Young P, Armstrong AW. Relationship between psoriasis and metabolic syndrome: a systematic review. G Ital Dermatol E Venereol 2016; 151: 663–677. [PubMed] [Google Scholar]
- 105. Carrascosa JM, Bonanad C, Dauden E, Botella R, Olveira‐Martín A; en nombre del Grupo de Trabajo en Inflamación Sistémica en Psoriasis . Psoriasis and nonalcoholic fatty liver disease. Actas Dermosifiliogr 2017; 108: 506–514. [DOI] [PubMed] [Google Scholar]
- 106. Jung UJ, Choi M‐S. Obesity and its metabolic complications: the role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int J Mol Sci 2014; 15: 6184–6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Upadhyay J, Farr O, Perakakis N, Ghaly W, Mantzoros C. Obesity as a disease. Med Clin North Am 2018; 102: 13–33. [DOI] [PubMed] [Google Scholar]
- 108. Gisondi P, Del Giglio M, Girolomoni G. Considerations for systemic treatment of psoriasis in obese patients. Am J Clin Dermatol 2016; 17: 609–615. [DOI] [PubMed] [Google Scholar]
- 109. Gisondi P, Del Giglio M, Di Francesco V, Zamboni M, Girolomoni G. Weight loss improves the response of obese patients with moderate‐to‐severe chronic plaque psoriasis to low‐dose cyclosporine therapy: a randomized, controlled, investigator‐blinded clinical trial. Am J Clin Nutr 2008; 88: 1242–1247. [DOI] [PubMed] [Google Scholar]
- 110. Carrascosa JM, Vilavella M, Garcia‐Doval I et al Body mass index in patients with moderate‐to‐severe psoriasis in Spain and its impact as an independent risk factor for therapy withdrawal: results of the Biobadaderm Registry. J Eur Acad Dermatol Venereol 2014; 28: 907–914. [DOI] [PubMed] [Google Scholar]
- 111. Monteiro‐Sepulveda M, Touch S, Mendes‐Sá C et al Jejunal T cell inflammation in human obesity correlates with decreased enterocyte insulin signaling. Cell Metab 2015; 22: 113–124. [DOI] [PubMed] [Google Scholar]
- 112. Chen Y, Tian J, Tian X et al Adipose tissue dendritic cells enhances inflammation by prompting the generation of Th17 cells. PLoS ONE 2014; 9: e92450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lynch M, Ahern T, Sweeney CM et al Adipokines, psoriasis, systemic inflammation, and endothelial dysfunction. Int J Dermatol 2017; 56: 1103–1118. [DOI] [PubMed] [Google Scholar]
- 114. Sumarac‐Dumanovic M, Stevanovic D, Ljubic A et al Increased activity of interleukin‐23/interleukin‐17 proinflammatory axis in obese women. Int J Obes 2005 2009; 33: 151–156. [DOI] [PubMed] [Google Scholar]
- 115. Gisondi P, Targher G, Zoppini G, Girolomoni G. Non‐alcoholic fatty liver disease in patients with chronic plaque psoriasis. J Hepatol 2009; 51: 758–764. [DOI] [PubMed] [Google Scholar]
- 116. Wong RJ, Aguilar M, Cheung R et al Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015; 148: 547–555. [DOI] [PubMed] [Google Scholar]
- 117. Gisondi P, Barba E, Girolomoni G. Non‐alcoholic fatty liver disease fibrosis score in patients with psoriasis. J Eur Acad Dermatol Venereol 2016; 30: 282–287. [DOI] [PubMed] [Google Scholar]
- 118. He B, Wu L, Xie W et al The imbalance of Th17/Treg cells is involved in the progression of nonalcoholic fatty liver disease in mice. BMC Immunol 2017; 18: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Paquissi FC. Immune imbalances in non‐alcoholic fatty liver disease: from general biomarkers and neutrophils to interleukin‐17 axis activation and new therapeutic targets. Front Immunol 2016; 7: 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Tang Y, Bian Z, Zhao L et al Interleukin‐17 exacerbates hepatic steatosis and inflammation in non‐alcoholic fatty liver disease. Clin Exp Immunol 2011; 166: 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Skov L, Thomsen SF, Kristensen LE, Dodge R, Hedegaard MS, Kjellberg J. Cause‐specific mortality in patients with psoriasis and psoriatic arthritis. Br J Dermatol 2018; 180: 100–107. [DOI] [PubMed] [Google Scholar]
- 122. Eirís N, González‐Lara L, Santos‐Juanes J, Queiro R, Coto E, Coto‐Segura P. Genetic variation at IL12B, IL23R and IL23A is associated with psoriasis severity, psoriatic arthritis and type 2 diabetes mellitus. J Dermatol Sci 2014; 75: 167–172. [DOI] [PubMed] [Google Scholar]
- 123. Lehuen A, Diana J, Zaccone P, Cooke A. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol 2010; 10: 501–513. [DOI] [PubMed] [Google Scholar]
- 124. Bellemore SM, Nikoopour E, Schwartz JA, Krougly O, Lee‐Chan E, Singh B. Preventative role of interleukin‐17 producing regulatory T helper type 17 (Treg 17) cells in type 1 diabetes in non‐obese diabetic mice. Clin Exp Immunol 2015; 182: 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Fatima N, Faisal SM, Zubair S et al Role of Pro‐inflammatory cytokines and biochemical markers in the pathogenesis of Type 1 diabetes: correlation with age and glycemic condition in diabetic human subjects. PLoS ONE 2016; 11: e0161548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Fatima N, Faisal SM, Zubair S, Siddiqui SS, Moin S, Owais M. Emerging role of interleukins IL‐23/IL‐17 axis and biochemical markers in the pathogenesis of Type 2 diabetes: association with age and gender in human subjects. Int J Biol Macromol 2017; 105: 1279–1288. [DOI] [PubMed] [Google Scholar]