

We developed a novel vaccine approach based on a new class of vaccine immunogens, called variable epitope libraries (VELs). We showed significant tumor growth inhibition and, most importantly, strong suppression of lung metastasis in mice, challenged with the aggressive and highly metastatic 4T1 cell line, after a single immunization using VEL vaccines. We think that the VELs represent a potent new class of cancer immunotherapy and propose the application of the VEL vaccine concept as a true alternative to currently available vaccine platforms.

Keywords: antigenic variability, cancer epitope vaccine, recombinant M13 phage, variable epitope library
Summary
Immune tolerance is the main challenge in the field of cancer vaccines, so modified peptide sequences or naturally occurring mutated versions of cancer‐related wild‐type (WT) antigens represent a promising pathway. However, the low immunogenicity of mutation‐induced neoantigens and, particularly, their incapacity to activate CD8+ T cells are generating doubts on the success of neoantigen‐based cancer vaccines in clinical trials. We developed a novel vaccine approach based on a new class of vaccine immunogens, called variable epitope libraries (VELs). We used three regions of survivin (SVN), composed of 40, 49 and 51 amino acids, along with the complete SVN protein to generate the VELs as multiepitope vaccines. BALB/c mice, challenged with the aggressive and highly metastatic 4T1 cell line, were vaccinated in a therapeutic setting. We showed significant tumor growth inhibition and, most importantly, strong suppression of lung metastasis after a single immunization using VEL vaccines. We demonstrated vaccine‐induced broad cellular immune responses concomitant with extensive tumor infiltration of T cells, the activation of CD107a+ IFN‐γ + T cells in the spleen and a significant increase in the number of CD3+ CD8+ Ly6C+ effector T cells. In addition, we observed the presence of interferon‐γ‐, granzyme B‐ and perforin‐producing lymphocytes along with modifications in the amount of CD11b+ Ly6Cint/low Ly6G+ granulocytic myeloid‐derived suppressor cells and CD4+ CD25+ FoxP3+ regulatory T cells in the lungs and tumors of mice. In summary, we showed that the VELs represent a potent new class of cancer immunotherapy and propose the application of the VEL vaccine concept as a true alternative to currently available vaccine platforms.
Abbreviations
- anova
analysis of variance
- APC
allophycocyanin
- CFUs
colony forming units
- CTLs
cytotoxic T lymphocytes
- G‐MDSCs
granulocytic myeloid‐derived suppressor cells
- IFN‐γ
interferon‐γ
- ITH
intra/inter‐tumor heterogeneity
- MHC
major histocompatibility complex
- PE
phycoerythrin
- RNA‐seq
RNA sequencing
- SVN
survivin
- TILs
tumor‐infiltrating lymphocytes
- Treg
regulatory T cells
- U.E.
unrelated epitope
- VELs
variable epitope libraries
- WT
wild‐type
Introduction
Recent success of immune checkpoint inhibitor therapy in several cancers has renewed interest in generating cancer vaccines and, most importantly, has revealed that mutated antigens or neoantigens, as well as other cancer‐related antigens, bearing alterations in primary amino acid sequences, are major targets and potential inducers of protective cellular immune responses. 1 , 2 , 3 Immunologically, tumor antigens can be grouped into two broad categories: wild‐type (WT) antigens and their altered/mutated counterparts, which are known as self and non‐self, respectively. 4 Powerful sequencing techniques, such as next‐generation sequencing, including RNA sequencing (RNA‐seq) and whole exome sequencing, enable the identification of mutational landscapes within tumors, in this manner contributing to a better understanding of the degree of intra/inter‐tumor heterogeneity (ITH) and of the evolutionary pathways of cancer development. In addition, the combination of modern sequencing and single‐cell assay techniques demonstrated the molecular composition and spatial heterogeneity of the T‐cell receptor repertoire, reflecting the mutational landscape in lung cancer 5 or ITH of immune response‐related genes, 6 which have a potential impact on the efficacy of immunotherapy. Hence, the presence of multiple private mutations in primary colorectal tumors and in synchronous liver metastases has been shown, supporting the need for such genomic profiling to tailor metastatic colorectal cancer therapies. 7 Importantly, an extensive immunogenomic analysis of thousands of tumors from diverse cancer patients revealed the involvement of multiple modalities of the intracellular and extracellular networks in tumor–immune cell interactions. 8
The concept of neoantigen vaccines is based on the assumption that mutated antigens are not subject to immune tolerance, in contrast to WT antigens; therefore, they can be highly immunogenic, truly cancer‐specific and useful as targets for cellular immune responses. 9 However, the low immunogenicity of neoantigens proved to be a serious challenge, as reported in several human studies, although strong anti‐neoantigen immune responses in animal studies have been frequently demonstrated. 3 Importantly, although currently used neoantigens are those present at the time of tumor sampling, there is clear evidence that the neoantigen landscape is permanently changing (gain/loss of antigen expression) during immune checkpoint inhibitor treatment 10 or adoptive T‐cell therapy of melanoma patients 11 , 12 as well as in untreated cancers, 13 indicating neoantigen editing/depletion as a result of strong selection pressure by the immune microenvironment during tumor evolution.
The response to immunotherapy is largely unpredictable owing to a high level of ITH and the heterogeneous phenotypic profiles of tumor‐infiltrating lymphocytes (TILs), both within individual tumors and among patients. Importantly, experimental testing of in silico predicted neoantigens, using multiplex peptide–major histocompatibility complex (MHC) tetramer staining coupled with mass spectrometry, showed that only 0·18% of neoantigens could be confirmed, which is in line with other studies reporting immune response rates to neoantigens between 0% and 0·5%. 14 Also, in two recent clinical trials, where neoantigens were used to immunize patients with newly diagnosed glioblastoma, only scarce vaccine‐induced CD8+ T‐cell responses against neoepitopes, bearing high‐affinity MHC class I binders were detected while CD4+ T‐cell responses were frequent. 15 , 16 Curiously, the authors concluded that these immunogens are promising vaccine candidates. 15 , 16 In addition, it has been shown that neoantigen‐specific T‐cell lines, generated from neoantigen‐vaccinated melanoma patients, do not recognize autologous tumors from four out of six patients, indicating that this approach may not be beneficial for the majority of patients. 17
The apparent incapacity of these neo‐vaccines to induce strong de novo CD8+ T‐cell responses highlights the observations that such immune responses are scarce or absent in untreated cancer patients. 18 Moreover, Bassani‐Sternberg et al. demonstrated that only four of eleven mutated ligands, identified by mass spectrometry, are immunogenic in melanoma patients. 19 Strikingly, T‐cell immunity was confirmed for only 10% and 2·85% of 70 neoantigen peptides using pre‐vaccine TILs and peripheral blood mononuclear cells, respectively, and these numbers were increased to 70% and 50% when peripheral blood mononuclear cells from two melanoma patients were tested after neoantigen vaccination (vaccines bearing 10 mutated epitopes/peptides) in 10‐day cell‐stimulation assays. 20
Neoantigens bearing somatic point mutations (non‐synonymous mutations), represent only a fraction of the whole landscape of cancer‐related mutated and non‐mutated antigens. Hence, the largest pool of cancer antigens, including neoantigens, arises from diverse cancer‐cell‐specific events, such as the generation of defective ribosomal products, altered transcriptional/translational events, translation of long non‐coding RNAs, insertion‐and‐deletion of DNA mutations and expression of new epitopes encoded by non‐primary open reading frame sequences. 21 Strikingly, this may represent a sustainable source of common neoantigens that, as has been suggested by Koster et al., can be used as ‘personalized’ vaccines in up to 50% of cancer patients. 22
In recent years, we have developed a new class of vaccine immunogens, coined as variable epitope libraries (VELs), based on complex combinatorial libraries carrying from thousands to millions of mutated versions of T‐cell epitopes, in order to address antigenic variability, which is observed as a common critical issue in the field of vaccines against many pathogens and cancer. 23 The feasibility of a VEL‐based vaccine platform has been demonstrated in our previous studies where we have shown that an HIV‐1 epitope‐based VEL induced broadly neutralizing sera in mice that was capable of neutralizing >50% of Tier 2 viruses tested. 24 Furthermore, a VEL vaccine derived from a survivin (SVN) epitope has been successfully tested in the mouse 4T1 breast tumor model leading to significant tumor growth inhibition upon a single immunization during therapeutic treatment. 25 Finally, we have shown recently that VELs expressing epitope libraries derived from MHC class I molecules are able to inhibit lung metastasis in 4T1‐challenged mice. 26 SVN has important functions and is highly expressed in most cancers; therefore, it represents one of the leading targets for the development of therapeutic agents and vaccines. 27 , 28 In the present study, we applied the VEL platform to construct vaccines based on the complete protein and three large antigenic regions covering the entire SVN. We generated immunogens bearing complex libraries and tested their therapeutic efficacy in the 4T1 adenocarcinoma model.
Materials and methods
Generation of vaccine immunogens
As a starting point, two DNA fragments , coding for the mouse SVN protein, were designed and generated synthetically (Blue Heron Biotech, Bothell, WA). The first fragment was the non‐mutated WT version of SVN, and the second one incorporates 20 × 1025 mutated triplets NNK, where N = G, A, T or C and K = G or T, at specific positions (see Supplementary material, Table S1). These fragments, along with three other smaller DNA fragments coding for three regions of 40, 51 and 49 amino acids bearing the N‐terminal, Core and C‐terminal portions of the WT and mutated versions of SVN (VELs), respectively, were obtained by polymerase chain reaction and cloned in the pG8SAET phagemid vector using oligonucleotide pairs carrying NcoI and BamHI restriction sites (underlined in oligonucleotides), as previously described. 25 The immunogens were expressed on the M13 phage surface as peptide fusions with the phage major coat protein (cpVIII) using standard molecular biology protocols. Restriction enzymes, Taq DNA polymerase, DNA isolation/purification kits, T4 DNA ligase and M13KO7 helper phage were obtained from Promega (Madison, WI), Invitrogen (Carlsbad, CA), IDT (San Diego, CA), Sigma‐Aldrich (St Louis, MO) or Qiagen (Valencia, CA). In total, we constructed eight recombinant M13 vaccine immunogens: four bearing WT SVN and multiepitope regions (WT‐Full SVN, WT‐NT, WT‐Core and WT‐CT, respectively) and four carrying corresponding VELs (VEL‐Full SVN, VEL‐NT, VEL‐Core and VEL‐CT, respectively). Computational tools, IEDB (http://tools.iedb.org/mhci/) and SYFPEITHI (http://www.syfpeithi.de/bin/MHCServer.dll/EpitopePrediction.htm) were used to monitor the presence of H‐2D‐restricted 8‐ to 11‐amino‐acid‐long CD8+ T‐cell epitopes. Mutations were introduced into the VELs in determined positions (25, 8, 9 and 8 positions, respectively, where the ‘X’ represents any of 20 amino acids), avoiding alterations in putative MHC I anchor positions. We determined the complexities of the generated VELs by counting colony forming units (CFUs) from samples of the phage libraries (Table 1). To verify for correct DNA synthesis and cloning procedures, we carried out DNA sequencing of several clones representing both WT and VEL‐derived immunogens (Table 2). Phage particles were rescued/amplified by infecting Escherichia coli TG1 cells with M13KO7 helper phage, and then purified by double precipitation with polyethylene glycol (20% PEG/2·5 m NaCl) as described; 25 typical phage yields were 1010–1011 CFU/ml of culture medium. The previously described M13 phage, FB22, expressing an unrelated epitope (U.E.; an HIV‐1 Gag‐derived epitope/peptide ALQRLFETC) was used as a control phage immunogen. 25
Table 1.
Generation of survivin (SVN) ‐derived immunogens
| Immunogens | Amino acid sequences | Library complexity |
|---|---|---|
| WT‐Full SVN | MGAPALPQIWQLYLKNYRIATFKNWPFLEDCACTPERMAEAGFIHCPTENEPDLAQCFFCFKELEGWEPDDNPIEEHRKHSPGCAFLTVKKQMEELTVSEFLKLDRQRAKNKIAKETNNKQKEFEETAKTTRQSIEQLAA | WT1‐140 |
| VEL‐Full SVN | MGAPXLPQIWQLYLXNYXIAXFKNWPFLXDXAXTPXRMAEAGFIHCPTENEPDLAQXFFXFKELEGWXPXDXPIEEXRKHSPGCAFXTXKXQMEXLTXSXFLKLXRQXAKNKXAKETNNKQKXFEETAXTTRQSIEQLAA | 1·5 × 105 |
| WT‐NT | MGAPALPQIWQLYLKNYRIATFKNWPFLEDCACTPERMAE | WT1‐40 |
| VEL‐NT | MGAPXLPQIWQLYLXNYXIAXFKNWPFLXDXAXTPXRMAE | 3·4 × 105 |
| WT‐Core | AGFIHCPTENEPDLAQCFFCFKELEGWEPDDNPIEEHRKHSPGCAFLTVKK | WT41–91 |
| VEL‐Core | AGFIHCPTENEPDLAQXFFXFKELEGWXPXDXPIEEXRKHSPGCAFXTXKX | 2·1 × 105 |
| WT‐CT | QMEELTVSEFLKLDRQRAKNKIAKETNNKQKEFEETAKTTRQSIEQLAA | WT92–140 |
| VEL‐CT | QMEXLTXSXFLKLXRQXAKNKXAKETNNKQKXFEETAXTTRQSIEQLAA | 4·1 × 105 |
X = Any of 20 amino acids. Wild‐type (WT) and variable epitope library (VEL) immunogens were generated by cloning of synthetic DNAs, carrying NNK triplets coding for all 20 amino acids, within phagemid vector as described in the Materials and methods, as well as in the Supplementary material.
Table 2.
Panel of antigens used in cell proliferation assays
| WT‐NT | M | G | A | P | A | L | P | Q | I | W | Q | L | Y | L | K | N | Y | R | I | A | T | F | K | N | W | P | F | L | E | D | C | A | C | T | P | E | R | M | A | E |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VEL‐NT | M | G | A | P | X | L | P | Q | I | W | Q | L | Y | L | X | N | Y | X | I | A | X | F | K | N | W | P | F | L | X | D | X | A | X | T | P | X | R | M | A | E |
| VEL‐NT | ||||||||||||||||||||||||||||||||||||||||
| derived antigen | ||||||||||||||||||||||||||||||||||||||||
| 1 | – | – | – | – | V | – | – | – | – | – | – | – | – | – | N | – | – | M | – | – | Q | – | – | – | – | – | – | – | I | – | N | – | R | – | – | L | – | – | – | – |
| 2 | – | – | – | – | R | – | – | – | – | – | – | – | – | – | T | – | – | P | – | – | R | – | – | – | – | – | – | – | A | – | S | – | M | – | – | S | – | – | – | – |
| 3 | – | – | – | – | Q | – | – | – | – | – | – | – | – | – | Q | – | – | A | – | – | Q | – | – | – | – | – | – | – | T | – | R | – | P | – | – | L | – | – | – | – |
| 4 | – | – | – | – | L | – | – | – | – | – | – | – | – | – | T | – | – | P | – | – | H | – | – | – | – | – | – | – | T | – | R | – | A | – | – | Q | – | – | – | – |
| 5 | – | – | – | – | I | – | – | – | – | – | – | – | – | – | G | – | – | A | – | – | A | – | – | – | – | – | – | – | Y | – | P | – | D | – | – | P | – | – | – | – |
| 6 | – | – | – | – | R | – | – | – | – | – | – | – | – | – | R | – | – | P | – | – | T | – | – | – | – | – | – | – | K | – | S | – | P | – | – | A | – | – | – | – |
| 7 | – | – | – | – | E | – | – | – | – | – | – | – | – | – | S | – | – | S | – | – | L | – | – | – | – | – | – | – | Y | – | P | – | G | – | – | L | – | – | – | – |
| 8 | – | – | – | – | S | – | – | – | – | – | – | – | – | – | Q | – | – | H | – | – | Y | – | – | – | – | – | – | – | T | – | P | – | I | – | – | V | – | – | – | – |
| 9 | – | – | – | – | M | – | – | – | – | – | – | – | – | – | V | – | – | L | – | – | K | – | – | – | – | – | – | – | D | – | H | – | F | – | – | T | – | – | – | – |
| WT‐CT | Q | M | E | E | L | T | V | S | E | F | L | K | L | D | R | Q | R | A | K | N | K | I | A | K | E | T | N | N | K | Q | K | E | F | E | E | T | A | K | T | T | R | Q | S | I | E | Q | L | A | A |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VEL‐CT | Q | M | E | X | L | T | X | S | X | F | L | K | L | X | R | Q | X | A | K | N | K | X | A | K | E | T | N | N | K | Q | K | X | F | E | E | T | A | X | T | T | R | Q | S | I | E | Q | L | A | A |
| VEL‐CT | |||||||||||||||||||||||||||||||||||||||||||||||||
| derived antigen | |||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | – | – | – | Q | – | – | N | – | P | – | – | – | – | P | – | – | P | – | – | – | – | F | – | – | – | – | – | – | – | – | – | A | – | – | – | – | – | G | – | – | – | – | – | – | – | – | – | – | – |
| 2 | – | – | – | R | – | – | H | – | I | – | – | – | – | N | – | – | E | – | – | – | – | Q | – | – | – | – | – | – | – | – | – | G | – | – | – | – | – | W | – | – | – | – | – | – | – | – | – | – | – |
| 3 | – | – | – | Q | – | – | E | – | I | – | – | – | – | P | – | – | T | – | – | – | – | A | – | – | – | – | – | – | – | – | – | A | – | – | – | – | – | R | – | – | – | – | – | – | – | – | – | – | – |
| 4 | – | – | – | I | – | – | R | – | K | – | – | – | – | V | – | – | T | – | – | – | – | H | – | – | – | – | – | – | – | – | – | Q | – | – | – | – | – | G | – | – | – | – | – | – | – | – | – | – | – |
| 5 | – | – | – | Q | – | – | N | – | P | – | – | – | – | P | – | – | P | – | – | – | – | F | – | – | – | – | – | – | – | – | – | A | – | – | – | – | – | G | – | – | – | – | – | – | – | – | – | – | – |
| 6 | – | – | – | Y | – | – | S | – | W | – | – | – | – | H | – | – | S | – | – | – | – | R | – | – | – | – | – | – | – | – | – | P | – | – | – | – | – | G | – | – | – | – | – | – | – | – | – | – | – |
| 7 | – | – | – | A | – | – | R | – | T | – | – | – | – | T | – | – | S | – | – | – | – | V | – | – | – | – | – | – | – | – | – | L | – | – | – | – | – | R | – | – | – | – | – | – | – | – | – | – | – |
| 8 | – | – | – | R | – | – | R | – | V | – | – | – | – | C | – | – | R | – | – | – | – | A | – | – | – | – | – | – | – | – | – | V | – | – | – | – | – | C | – | – | – | – | – | – | – | – | – | – | – |
| 9 | – | – | – | R | – | – | K | – | S | – | – | – | – | K | – | – | S | – | – | – | – | D | – | – | – | – | – | – | – | – | – | K | – | – | – | – | – | L | – | – | – | – | – | – | – | – | – | – | – |
X = Any of 20 amino acids. Amino acid sequences of antigen variants isolated from variable epitope libraries (VELs) at random, as well as wild‐type (WT) antigens, were deduced after DNA sequencing of individual phage clones.
Animal studies
To generate breast tumors, groups of 4‐ to 6‐week‐old female BALB/c mice (n = 5 to n = 7) were transplanted subcutaneously with 104 viable 4T1 mouse mammary carcinoma cells (American Type Culture Collection, Manassas, VA) in 50 µl of phosphate‐buffered saline into the right mammary fat pad, as we previously reported. 25 For the established disease study, mice were immunized once with the above‐mentioned phage particles by intravenous injection (2·5 × 1012 CFU) 5 days after the tumor challenge. Tumor growth was monitored every 3 days and tumor volume (V) was calculated as V = (a*b 2)/2 (largest (a) and smallest (b) superficial diameters of the tumor) using a Vernier caliper. Mice were killed with CO2, 33 days after 4T1 tumor challenge. All animal experiments were conducted under approved protocols from the Animal Care and Use Committee of the Instituto de Investigaciones Biomédicas UNAM (IIB‐UNAM) and in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the NIH.
Cell proliferation assays
Spleen cells were pooled from three animals from each treatment group or from untreated mice 15 days after tumor challenge and were assessed by flow cytometry protocols as described. 25 Briefly, cells were re‐suspended in CFSE (600 nm; Tonbo Biosciences, San Diego, CA) and stimulated by culturing in a 96‐well flat‐bottom plate (2·5 × 105 cells/well) with 1 × 1010 phage particles/well, corresponding to individual epitope variants, during 72 hr in a cell‐culture incubator at 37° in a 5% CO2 atmosphere; Monensin (1×; BioLegend, San Diego, CA) was added during the last 6 hr of incubation. Doublets and dead cells were excluded during gating, and lymphocytes were gated for a CD4+ versus CD8+ dot‐plot graph to measure CD4+ IFN‐γ +, CD8+ IFN‐γ + and proliferation percentages of CD3+ CD4+ and CD3+ CD8+ cells. Cells were analyzed on an Attune Flow Cytometer (ThermoFisher, Waltham, MA) using flowjo free trial (BD, San Jose, CA); at least 100 000 total events were collected (see Supplementary material, Fig. S1).
Immunofluorescence studies
The tumors and lungs were removed at day 33 post‐tumor challenge. The excised tissues were fixed in zinc salts buffer (pH 6·5) for 24 hr at 4° under constant mixing. Excised tissues were processed by incremental sucrose gradients (10%, 20% and 30%) and the samples were frozen at −70° in a mixture of 30% sucrose and 50% OCT (Tissue‐Tek O.C.T. 4583, Sakura Finetek, Torrance, CA). For each sample, 15‐µm tissue sections were obtained at −20° C and placed on glass slides (Corning, Corning, NY). Each slide was submerged in a copper salt solution for 1 hr to minimize autofluorescence. Afterwards, a blocking solution, containing 3% fetal horse serum and 2% fetal bovine serum, was used along with the primary monoclonal antibodies: rat anti‐mouse CD8 (Clone 53‐6.7, 1 : 100 dilution, Santa Cruz Biotechnology, Dallas, TX) and rabbit anti‐mouse CD4 (Clone EPR19514, 1 : 200 dilution, Abcam, Cambridge, UK). Then, secondary polyclonal antibodies were added to the tissue samples [donkey anti‐rat IgG (H + L) Alexa Fluor 488, 1 : 300 dilution and goat anti‐rabbit IgG (H + L) Alexa Fluor 594, 1 : 500 dilution, Jackson ImmunoResearch Laboratories Inc., West Grove, PA]. Finally, samples were mounted using fluorescence medium (Vectashield, Vector Laboratories Inc., Burlingame, CA) and visualized (20×) on an Olympus IX71 inverted fluorescence microscope (Fig. 3c,d).
Cell phenotyping (ex vivo analysis) by multiparametric flow cytometry
Spleen, lung and tumor cells were pooled from three animals from each treatment group or from untreated mice 33 days after tumor challenge and assessed using flow cytometry protocols as described. 25 Briefly, 2·5 × 105 cells were stained with a panel of fluorescence‐labeled monoclonal antibodies to identify cell subsets according to the manufacturer’s instructions: anti‐CD3 allophycocyanin (APC) ‐eFluor780 (clone 145‐2C11); anti‐Ly6c (Gr‐1) phycoerythrin (PE) ‐Cy7 (clone RB6‐8C5); anti‐CD25 APC‐eFluor780 (clone PC61.5); anti‐Ly6G PE (clone RB6‐8C5); anti‐granzyme B fluorescein isothiocyanate (clone NGZB); anti‐FoxP3 PE‐Cy5 (clone FJK‐16s) and anti‐interferon‐γ (IFN‐γ) peridinin chlorophyll protein‐Cy5.5 (clone XMG1.2) (all from eBioscience, San Diego, CA). Also, anti‐CD8α PE (clone 53‐6.7), anti‐perforin APC (clone S16009A), anti‐CD4 AlexaFluor 488 (clone GK1.5); anti‐CD107a (LAMP‐1) APC‐Cy7 (clone 1D4B) (BioLegend); anti‐CD3e PE‐Cy7 (clone 145‐2C11), anti‐FoxP3 PE (clone 3G3), anti‐IFN‐γ PE‐Cy7 (clone XMG1.2), 7‐ aminoactinomycin D (Tonbo Biosciences) and anti‐CD11b AlexaFluor 488 (clone M1/70) from Invitrogen were used. Cells were analyzed on an Attune Flow Cytometer (ThermoFisher) using flowjo free trial (BD); at least 100 000 total events were collected (see Supplementary material, Figs S2, S3 and S4). All phenotyping data were calculated as number of cells/g of tissue, cell numbers in tissues from untreated tumor‐bearing mice were considered as baseline level, and X‐fold changes were calculated from these.
Statistical analysis
All results are expressed as the means ±95% CI. BALB/c group sizes were at least n = 5. All experiments were repeated at least once with comparable results. Before the statistical analysis, tumor volume and metastatic lesion data were converted into logarithmic form to obtain normal distribution (n = 30, *P < 0·033, **P < 0·02, ***P < 0·001). Two‐way analysis of variance (anova) for repeated measurements and Tukey post‐hoc test for multiple comparisons were used, respectively. Cell proliferation data (%) were analyzed with two‐way anova for repeated measurements, and Sidak post‐hoc test for multiple comparisons was applied. All multiparametric flow cytometry analyses are expressed as the X‐fold change as mean ± 95 Cl (n = 5, *P < 0·033, **P < 0·02, ***P < 0·001). All statistical analysis was performed with graphpad prism 8 software version 8.3.0 (GraphPad, San Diego, CA).
Results
Vaccine immunogens and animal studies
To generate immunogens for vaccination experiments, two 517‐bp double‐stranded DNA fragments coding for the complete WT SVN protein, and an extremely large combinatorial library (theoretical complexity of 2025 individual members), carrying SVN‐derived mutated epitopes (VELs), were synthesized. These DNA, along with three smaller DNA fragments (see Supplementary material, Table S1), coding for N‐terminal, Central/Core and C‐terminal regions of WT SVN and VELs (see Supplementary material, Table 1), were amplified by polymerase chain reaction and cloned in the pG8SAET phagemid vector to generate recombinant M13 bacteriophage particles expressing, in total, eight vaccine immunogens: four bearing the complete WT SVN protein and multiepitope regions (WT‐Full SVN, WT‐NT, WT‐Core and WT‐CT, respectively); four carrying corresponding VELs (VEL‐Full SVN, VEL‐NT, VEL‐Core and VEL‐CT, respectively). The theoretical complexities of the libraries are 20 × 1025, 20 × 108, 20 × 109 and 20 × 108 individual members, respectively, but the actual complexity of the generated VELs was 1·5 × 105 to 4·1 × 105 individual members, as shown in Table 1. To verify for correct DNA synthesis and cloning procedures, we carried out DNA sequencing of several clones representing both WT and VEL‐bearing immunogens and determined the amino acid sequences of the corresponding peptides (Table 2).
To assess the anti‐tumor effect of the constructed phage‐displayed immunogens in a therapeutic setting, we administered recombinant phage particles, carrying WT or VELs of SVN‐derived sequences intravenously in mice challenged with the 4T1 adenocarcinoma cell line (Fig. 1a). A single vaccination with the VEL‐Full SVN, VEL‐NT and VEL‐CT resulted in statistically significant inhibition of tumor growth, compared with mice vaccinated with the corresponding WT versions of the immunogens, non‐related epitope (U.E.) and the control group of tumor‐bearing untreated animals; however, this was not seen in mice vaccinated with the VEL bearing the central region (VEL‐Core) of SVN (Fig. 1b–e). We also analyzed whether the VEL vaccines have an effect on the development of lung metastasis (Fig. 1j–m), wherein we observed a significant decrease in the number of metastatic lesions (macrometastases) in the lungs of VEL‐vaccinated mice compared with the control group of tumor‐bearing untreated animals (Fig. 1f–i). However, when we compared the reduction of lung macrometastases between WT SVN and the corresponding VEL, a significant decrease was only observed in the case of VEL‐NT. Interestingly, although immunization with the complete SVN VEL resulted in significantly reduced metastases compared with mice from the control group, this inhibition was not significant when compared with the complete WT SVN or U.E. vaccinated animals. Strikingly, we observed few metastatic lung lesions in several animals treated with either the VEL‐NT or VEL‐CT immunogens. Taken together, these data show the superior vaccine efficacy of immunogens derived from the N‐ and C‐terminal portions of SVN, compared with VEL vaccines derived from the core region or from the complete SVN protein. Superior anti‐tumor effects induced by VEL‐NT and VEL‐CT compared with full‐length SVN‐derived VEL (VEL‐Full SVN) or VEL‐Core may be explained by differential dynamics of antigen processing, epitope presentation and immunodominance events intrinsically related to these antigenic regions during tumor growth.
Figure 1.
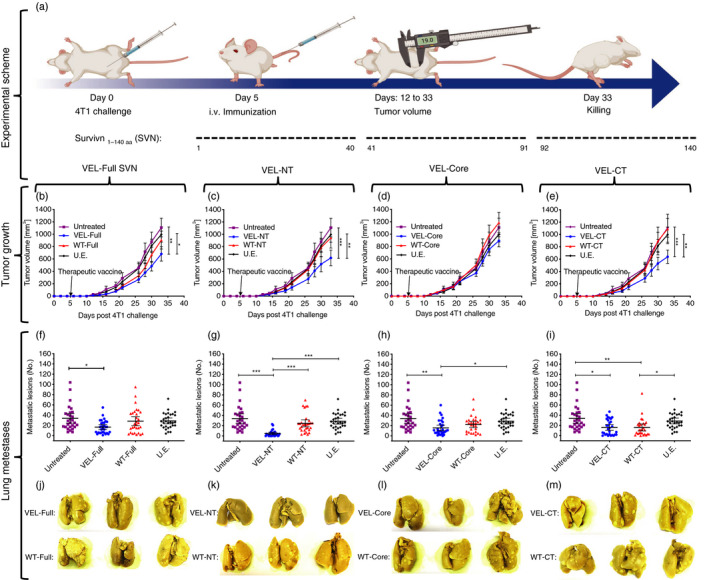
Therapeutic vaccination with variable epitope libraries (VELs) inhibits 4T1 tumor growth and the development of spontaneous metastases. (a) Groups of 4T1 tumor‐bearing BALB/c mice were immunized (at day 5) intravenously (i.v.) with 2·5 × 1012 recombinant M13 phage particles, expressing four vaccines bearing combinatorial VELs, based on full‐length (VEL‐Full SVN), N‐terminal (VEL‐NT), core (VEL‐Core) and C‐terminal (VEL‐CT) sequences of surviving (SVN), respectively (figure created with BioRender.com). (b–e) Tumor growth was monitored by measuring tumor volumes. (f–i) Lung macrometastases were counted at day 33. Mice were also vaccinated with the corresponding wild‐type (WT) counterparts of the four VELs. Tumor‐bearing untreated mice and mice vaccinated with an unrelated phage (U.E.) were used as controls. (j–m) Lungs from each group were fixed in Bouin’s solution, representative images are shown. (b–e) Tumor volumes are presented as mean ± 95% CI mm3; n = 30, *P < 0·033, **P < 0·02, ***P < 0·001. Two‐way analysis of variance for repeated measurements and Tukey post‐hoc test for multiple comparisons were used. (f–i) Lung macrometastases are presented as mean ± 95 CI; n = 30; *P < 0·033, **P < 0·02, ***P < 0·001. One‐way analysis of variance with Tukey post‐hoc test for multiple comparisons was used.
Cellular immune responses induced by VEL vaccines
We further analyzed the induced immune responses in control groups and in mice vaccinated with VEL‐NT and VEL‐CT, because in these two groups we observed the strongest inhibition of tumor growth and lung metastasis development. First, to evaluate the immunogenicity of our vaccines, we immunized both 4T1 tumor‐bearing and non‐tumor‐bearing mice with two VELs along with their WT counterparts. Afterwards, spleen cells from corresponding mice were stimulated with a panel of nine variant antigens from each VEL immunogen bearing mutated versions of their respective WT antigen (Table 2). As shown in Figs 2(a) and 3(a), several variant epitopes from the panel of mutated antigens induced higher proliferation levels of CD8+ and CD4+ spleen cells from VEL‐NT and VEL‐CT immunized or vaccinated mice, compared with cells from mice immunized or vaccinated with the corresponding WT counterparts. However, it is important to mention that stimulation with three antigen variants (antigens 6–8), derived from VEL‐NT, induced stronger proliferation of cells from WT‐NT‐vaccinated mice than those from the VEL‐NT‐vaccinated group, indicating that these variant antigens are cross‐stimulating T cells induced by both immunogens (Fig. 2a). In all experiments, cells obtained from mice immunized with U.E. phage, carrying an HIV‐1‐derived epitope, showed low levels of proliferation upon stimulation with the panel of antigens (shown as dotted line in Figs 2 and 3), indicating that vaccine‐induced immune responses are antigen‐specific and are not directed against the vaccine carrier, i.e. M13 phage.
Figure 2.
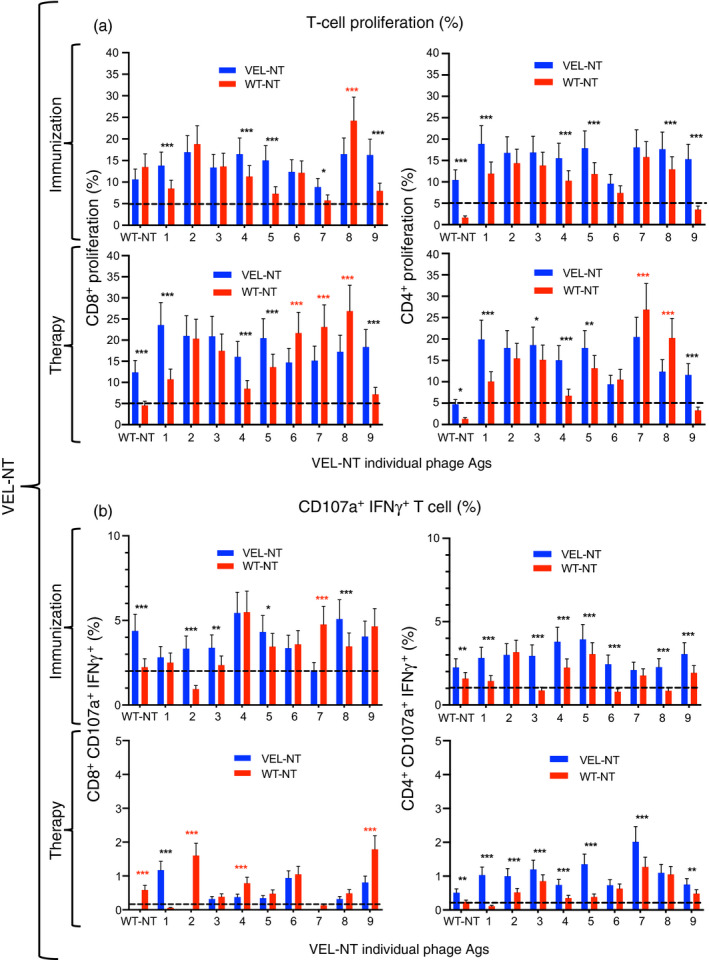
Immune responses of spleen cells upon immunization and therapeutic vaccination with the N‐terminal variable epitope library (VEL‐NT). (a, b) Mouse splenocyte pools (n = 3, each point represents cells from three independent experiments) were generated 15 days after vaccination with VEL‐NT and wild‐type (WT) ‐NT and analyzed by multiparametric flow cytometry. (b) CD8+ CD107a+ IFN‐γ + and CD4+ CD107a+ IFN‐γ + cell subsets were determined within proliferating cells. Spleen cells were also obtained from non‐tumor‐bearing mice, immunized with the same immunogens. Cell proliferation data (%) are presented as mean ± 95 CI and were analyzed with two‐way analysis of variance for repeated measurements and Sidak post‐hoc test for multiple comparisons. *P < 0·033, **P < 0·02, ***P < 0·001.
Figure 3.
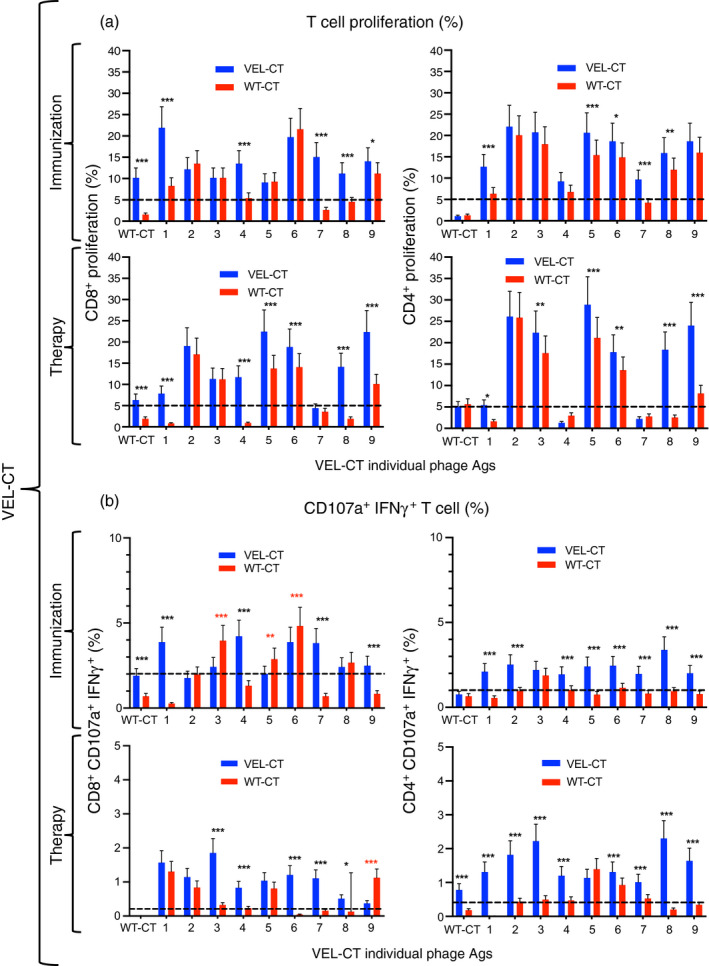
Immune responses of spleen cells upon immunization and therapeutic vaccination with the C‐terminal variable epitope library (VEL‐CT). (a, b) Mouse splenocyte pools (n = 3, each point represents cells from three independent experiments) were generated 15 days after vaccination with VEL‐CT and wild‐type (WT) ‐CT and analyzed by multiparametric flow cytometry. (b) CD8+ CD107a+ IFN‐γ + and CD4+ CD107a+ IFN‐γ + cell subsets were determined within proliferating cells. Spleen cells were also obtained from non‐tumor‐bearing mice, immunized with the same immunogens. Cell proliferation data (%) are presented as mean ± 95 CI and were analyzed with two‐way analysis of variance for repeated measurements and Sidak post‐hoc test for multiple comparisons. *P < 0·033, **P < 0·02, ***P < 0·001.
We further determined the phenotypes of splenocytes by measuring additional activation markers, such as the degranulation marker CD107a and IFN‐γ, showing a generalized higher proliferation levels of CD4+ CD107a+ IFN‐γ + cells, and in a lesser extent of CD8+ CD107a+ IFN‐γ + cells, from non‐tumor‐bearing mice immunized with the VELs compared with cells from mice immunized with the respective WT counterparts (Figs 2b and 3b). The above‐mentioned immune responses were inhibited in similarly vaccinated tumor‐bearing mice, particularly in animals vaccinated with VEL‐NT, indicating tumor‐induced immune suppression; although splenocytes from VEL‐vaccinated mice still showed relatively higher levels of proliferation compared with cells derived from WT immunogen‐vaccinated animals, with the exception of CD8+ CD107a+ IFN‐γ + cells from VEL‐NT‐vaccinated mice (Figs 2b and 3b). It is important to mention that CD4+, CD8+ CD107a+ IFN‐γ + and CD4+ CD107a+ IFN‐γ + cells from mice immunized with VEL‐NT showed higher proliferation levels upon stimulation with WT‐NT antigen compared with the cells from WT‐NT‐immunized animals (Fig. 2a,b). Also, WT‐NT antigen induced higher levels of proliferation of CD8+ cells from mice therapeutically vaccinated with VEL‐NT (Fig. 2a). By contrast, WT‐CT antigen was able to stimulate the proliferation of CD8+ cells from mice immunized with VEL‐CT, and also, some increase in proliferation of CD4+ CD107a+ IFN‐γ + cells from mice therapeutically vaccinated with VEL‐CT (Fig. 3a,b).
In addition, we measured both central and effector memory T‐cell responses. Mice were immunized with the above‐mentioned immunogens and 90 days later, spleen cells were analyzed by flow cytometry. As shown in the Supplementary material (Fig. S5), a significant increase of both CD8+ and CD4+ effector memory (CD44+ CD62L−) cells in relation to central memory CD44+ CD62L+ cells was observed in mice immunized with the VELs and WTs as compared with naive mice or animals immunized with U.E. Interestingly, the increase in the number of effector memory cells was significantly higher in the spleen of VEL‐NT‐immunized animals compared with WT‐NT‐immunized mice. Antibody responses measured by ELISA were not detected in any group of mice (data not shown).
Immune response in lungs and TILs
Next, we performed multiparametric flow cytometry and immunofluorescence analysis of tumor and lung‐derived TILs from vaccinated mice at day 33 after tumor challenge, to test whether there is a correlation between data from T‐cell assays and effector tissue‐penetrating cells. Strikingly, we observed a 5·5‐fold and 7·8‐fold increase in the number of CD8+ T cells in the lungs of mice vaccinated with VEL‐NT and VEL‐CT, respectively, compared with untreated mice (Figs 4a and 5a). Immunization with the WT immunogens, WT‐NT and WT‐CT, resulted in 3·4‐fold and 2·7‐fold increase in CD8+ cells in the lungs, respectively. Regarding TILs, the number of CD8+ cells increased about 2·5‐fold in tumors from mice vaccinated with the VELs and, to a lesser extent, in tumors from WT‐vaccinated mice. These data, as per immunofluorescence studies, were reflected as extensive CD8+ cell infiltration in the lungs and tumors from these mice (Figs 4b and 5b). Similar analysis of CD4+ cells showed a 1·6‐ to 2·2‐fold rise in their numbers, in the lungs and tumors of both WT‐CT‐, WT‐NT‐ and VEL‐CT‐vaccinated mice, and a 4·6‐fold increase in the tumors from VEL‐NT‐vaccinated animals (Figs 4a and 5a). Again, these data correlated with the analysis of lung infiltrating CD4+ cells and TILs (Figs 4b and 5b).
Figure 4.
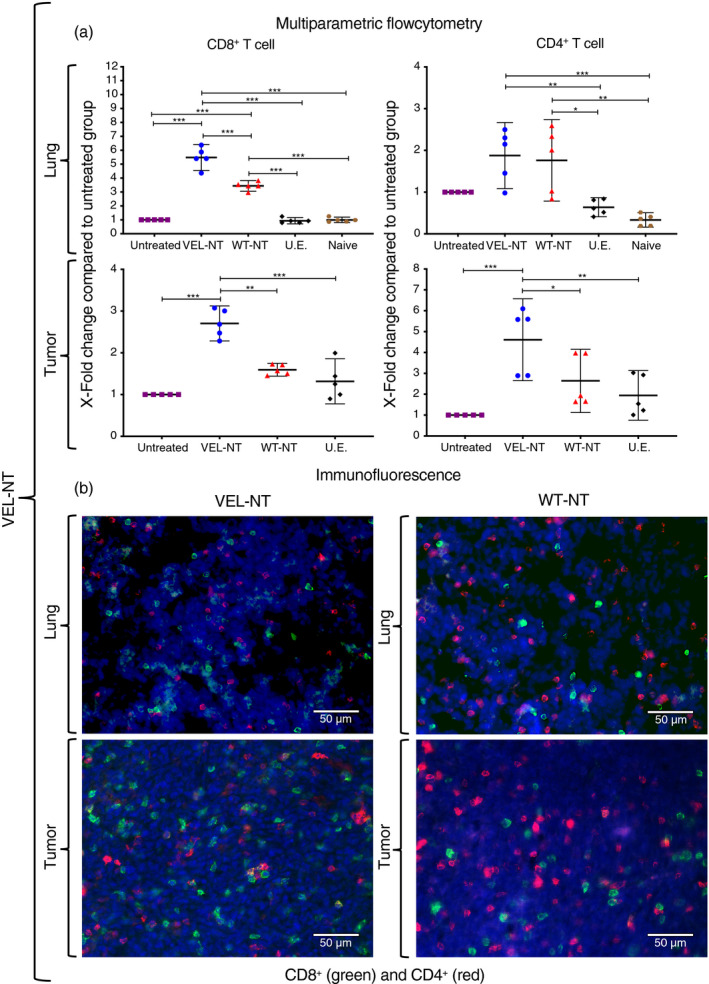
TILs and pulmonary T‐cell infiltration in the N‐terminal variable epitope library (VEL‐NT vaccinated mice. (a) Multiparametric flow cytometry results showing X‐fold change in the numbers of CD8+and CD4+ T cells within lungs and tumors compared to tissues from the untreated group. X‐fold change is presented as mean ± 95 CI (n = 5, each point represents a pool of three distinct tissues from different experiments). *P < 0·033, **P < 0·02, ***P < 0·001. One‐way anova with Tukey post‐hoc test for multiple comparisons was used. (b) Representative immunofluorescence staining of tumor and lung‐derived CD8+ and CD4+ T cells in VEL‐NT and WT‐NT treatments. DAPI was used to stain nuclei. (Scale bars, 50 µm).
Figure 5.
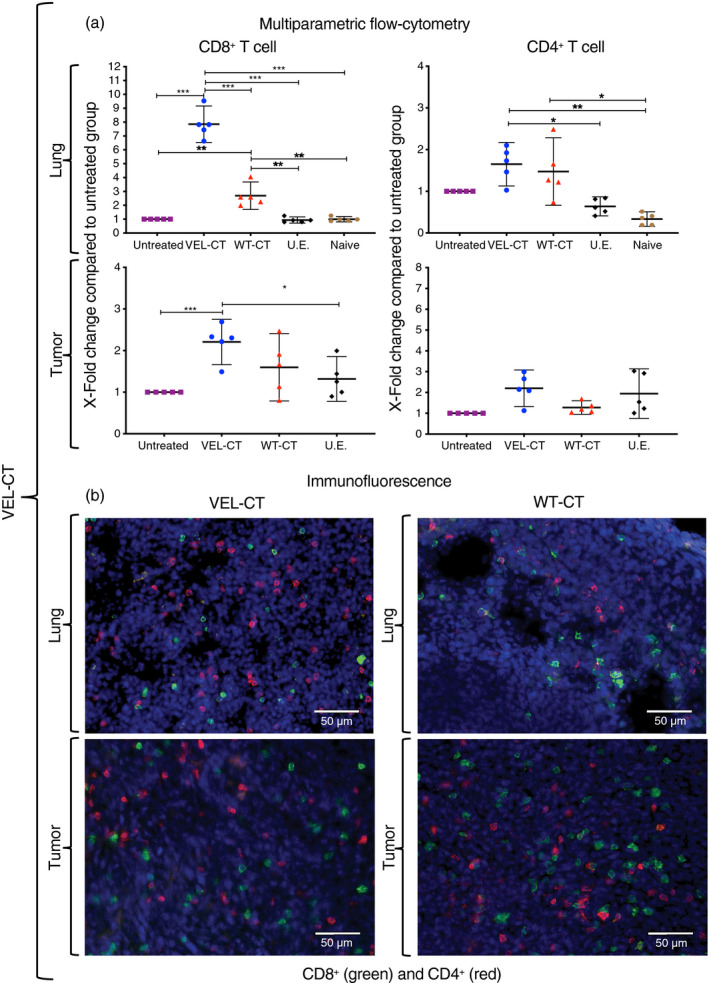
Tumor‐infiltrating lymphocytes (TILs) and pulmonary T‐cell infiltration in the C‐terminal variable epitope library (VEL‐CT) ‐vaccinated mice. (a) Multiparametric flow cytometry results showing X‐fold change in the numbers of CD8+ and CD4+ T cells within lungs and tumors compared with tissues from the untreated group. X‐fold change is presented as mean ± 95 CI (n = 5, each point represents a pool of three distinct tissues from different experiments). *P < 0·033, **P < 0·02, ***P < 0·001. One‐way analysis of variance with Tukey post‐hoc test for multiple comparisons was used. (b) Representative immunofluorescence staining of tumor‐ and lung‐derived CD8+ and CD4+ T cells in VEL‐CT and wild‐type (WT) ‐CT treatments. DAPI was used to stain nuclei. (Scale bars, 50 µm).
Also, we measured ex vivo the presence of IFN‐γ‐, granzyme B‐ and perforin‐producing CD8+ cells, and CD4+ IFN‐γ + lymphocytes within the lungs, showing a significantly higher level of infiltration in mice vaccinated with VEL‐NT and VEL‐CT, compared with the lungs from untreated or U.E.‐vaccinated mice (Fig. 6a–d). Importantly, when compared with the WT counterparts, significantly higher levels of IFN‐γ‐, granzyme B‐ and perforin‐producing CD8+ cells were observed in the lungs of VEL‐NT‐vaccinated mice but not in VEL‐CT‐vaccinated animals (Fig. 6a–d). CD4+ IFN‐γ + lymphocytes were increased in the lungs of both VEL‐vaccinated groups compared with the WT counterparts.
Figure 6.
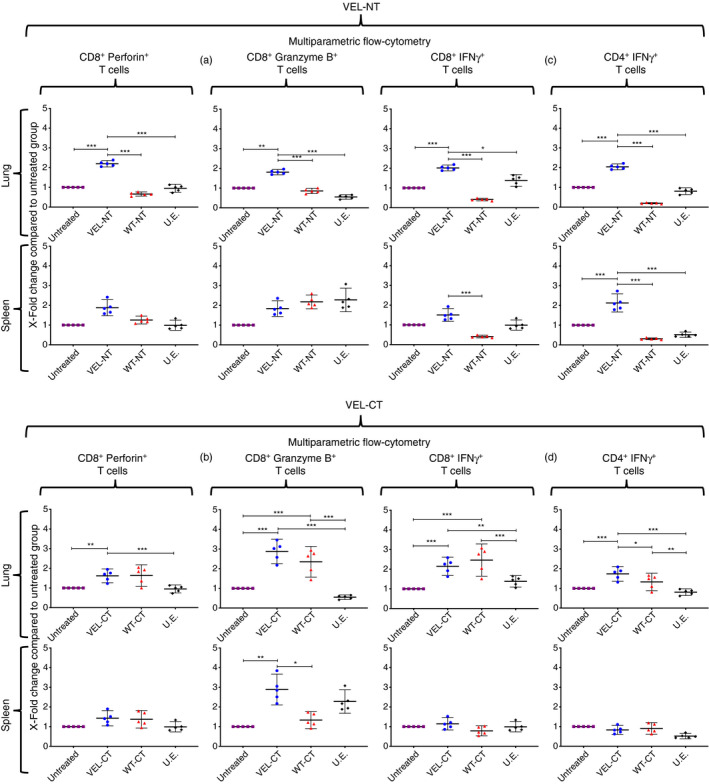
Effector T‐cell phenotyping. Multiparametric flow cytometry measurement of infiltrating interferon‐γ (IFN‐γ‐), granzyme B‐ and perforin‐producing CD8+ cells and CD4+ IFN‐γ + lymphocytes within lungs and tumors. (a, c) Mice vaccinated with the N‐terminal variable epitope library (VEL‐NT); (b, d): mice vaccinated with the C‐terminal variable epitope library (VEL‐CT). The numbers of lymphocytes in tissues from untreated tumor‐bearing mice were considered as baseline level and X‐fold changes were calculated from them. X‐fold change is presented as mean ± 95 CI (n = 5, each point represents a pool of three distinct tissues from different experiments). *P < 0·033, **P < 0·02, ***P < 0·001. One‐way analysis of variance with Tukey post‐hoc test for multiple comparisons was used.
Similar analysis of splenocytes revealed only a significant increase in the number of CD4+ IFN‐γ + cells in VEL‐NT‐vaccinated and CD8+ granzyme B+ cells in VEL‐CT‐vaccinated mice, respectively (Fig. 6b,c). Measurement of TILs revealed only minor differences between these cell subpopulations within the tumors (data not shown). These data indicate that the therapeutic efficacy of our vaccines might be mediated by diverse subpopulations of effector T cells.
Finally, to further characterize effector cells and to know whether a T‐cell regulatory network is involved in the observed anti‐tumor effect, we performed immune‐phenotyping of the cells obtained from lungs and tumors of vaccinated mice. We observed a significant increase in the number of CD3+ CD8+ Ly6C+ effector T cells in the lungs of mice vaccinated with VEL‐NT (Fig. 7a) compared with the lungs from animals immunized with WT‐NT, U.E. and tumor‐bearing untreated mice; in contrast, although the number of these cells significantly increased in the lungs of VEL‐CT‐vaccinated mice compared with U.E.‐vaccinated and tumor‐bearing untreated mice, no differences were observed when compared with WT‐CT‐vaccinated animals (Fig. 7b). In addition, we observed a significant increase in the number of CD3+ CD8+ Ly6C+ effector T cells in the tumors from VEL‐NT‐vaccinated mice but not in VEL‐CT‐vaccinated animals (Fig. 7a,b). Interestingly, the significant increase in the number of CD11b+ Ly6Cint/low Ly6G+ granulocytic myeloid‐derived suppressor cells (G‐MDSCs) was observed in the tumors but not in the lungs of mice vaccinated with VEL‐NT (Fig. 7a). In contrast, the number of G‐MDSCs was increased in the lungs but not in the tumors derived from VEL‐CT‐vaccinated mice (Fig. 7b). These data, along with the observation that the ratio of effector/suppressor cells was significantly higher in the lungs of mice vaccinated with VEL‐NT (Fig. 7c,d), may explain the stronger inhibition of lung metastases in these mice compared with those vaccinated with VEL‐CT. Interestingly, we found a significant increase in the number of CD4+ CD25+ FoxP3+ regulatory T cells in the lungs of mice vaccinated with VEL‐CT compared with the control groups, whereas in the VEL‐NT‐vaccinated group the number of these cells was unchanged, which may also contribute to the stronger inhibition of lung metastases in mice vaccinated with the VEL‐NT compared with VEL‐CT‐vaccinated animals. However, the opposite effect was observed within the tumors: regulatory T cells infiltrated primary tumors in significantly higher numbers in VEL‐NT vaccination settings versus VEL‐CT (Fig. 7c,d). In summary, although the observed anti‐tumor effects are mediated mainly by effector T cells, suppressor/regulatory cells may also contribute to the induction and maintenance of protective cellular immune responses.
Figure 7.
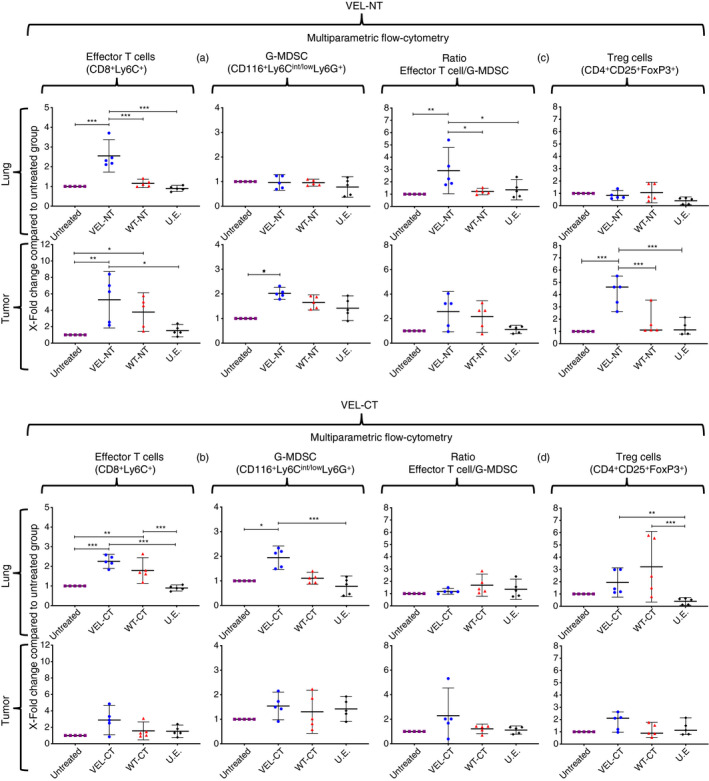
Analysis of variable epitope library (VEL) vaccine‐induced effector/suppressor/regulatory cells. Multiparametric flow cytometry measurement of CD3+ CD8+ Ly6C+ effector T cells, CD11b+ Ly6Cint/low Ly6G+ granulocytic myeloid‐derived suppressor cells (G‐MDSCs) and CD4+ CD25+ FoxP3+ regulatory T (Treg) cells in the lungs and tumors of mice vaccinated with N‐terminal VEL (VEL‐NT) (a, c) and C‐terminal VEL (VEL‐CT) (b, d). Ratios of effector/suppressor cells (CD3+ CD8+ Ly6C+/G‐MDSC) were calculated (c, d). X‐fold changes are presented as mean ± 95 CI (n = 5, each point represents a pool of three distinct tissues of different experiments). *P < 0·033, **P < 0·02, ***P < 0·001. One‐way analysis of variance with Tukey post‐hoc test for multiple comparisons was used.
Discussion
In this study, we demonstrated that large combinatorial libraries, carrying thousands of SVN‐based mutated epitopes, are efficient vaccine immunogens capable of inducing a protective immune response in the highly malignant, metastatic 4T1 mouse mammary adenocarcinoma model. The efficacy of any vaccine depends not only on its strong immunogenicity, but on the presence of epitopes on the target cells and, in the case of therapeutic cancer vaccines, a desirable scenario is the capacity of a vaccine to induce both tumor‐specific and vaccine‐specific immune responses. In our previous study, we showed that about 70% of 87 variant epitopes, isolated at random from the VEL, based on immunodominant SVN‐derived CTL epitope, were stimulating spleen cells from mice challenged with 4T1, although a minority of variants were able to stimulate cells obtained from naive mice. 25 These data indicated that the majority of these mutant variants and, consequently, the whole VEL, are able to specifically stimulate T cells from tumor‐bearing mice. 25 We obtained similar results in the current study demonstrating preferential stimulation of splenocytes from tumor‐bearing mice by the majority of mutated antigen variants, whereas only a minority of variant antigens were stimulating cells obtained from non‐immunized intact mice (data not shown). Furthermore, a clear indication of tumor specificity is a severalfold higher presence of T cells, particularly CD8+ cells, within the lungs and tumors of mice vaccinated with VELs compared with non‐related immunogen‐treated, tumor‐bearing untreated and naive animals (Figs 4 and 5). Also, we have shown the tumor‐specific nature of SVN‐derived VEL by including a non‐related control VEL immunogen in our previous study. 25 Importantly, in the current study we showed the reduction of lung metastases that was not achieved in our previous study, indicating that large multiepitope VELs are more potent vaccine immunogens. Equally significant is the demonstration that the VELs are superior vaccine immunogens compared with their WT counterparts. Importantly, cells from VEL‐NT‐immunized animals were proliferating upon stimulation with WT‐NT antigen, indicating that VEL‐induced effector T cells may also target non‐mutated tumor antigen. Furthermore, considering that variant epitopes within the VELs are not the subjects of negative selection under host immune pressure, which is the case with neoantigens, we suggest that the VELs may have an advantage over traditional cancer vaccine approaches based on the application of cancer‐related WT or mutated defined antigen sequences.
Our aim was not the construction of the largest possible VELs (e.g. the theoretical complexity of the full‐length SVN VEL is of an astronomical order of 1025 individual members), instead, in the VELs used in this study, 25 out of 140 amino acids from the SVN sequence were mutated. There are two reasons for such a decision. First, we wanted to achieve structural diversity in the immunogen where the majority of amino acids are of WT origin. It is known that WT antigens along with neoantigens are important targets for T‐cell responses and it has been shown recently that, although both WT germline and mutated epitopes were capable of inducing protection in a melanoma mouse model, the best therapeutic effect was achieved when these multi‐target nano‐vaccines were applied in combination. 29 Second, we have previously shown that these low‐to‐moderate levels of mutations (15%–30% of amino acid positions mutated) are sufficient to induce broad T‐cell responses. 25
According to the cancer immune surveillance theory, opposing forces act to maintain the equilibrium between the elimination of tumor cells and immune escape, mediated by repeated cycles of immune pressure and the selection of cancer cells carrying certain mutation(s). 13 , 30 Extensive data are underpinning the striking similarities between cancer and antigenically variable pathogens, such as viruses, in terms of induced immune responses, 23 which in turn, allow us to conclude that the successful vaccine against antigenically variable pathogens should share critical features of the vaccine immunogen intended to treat cancer. Indeed, a recent study applying a neoantigen fitness model to predict the survival of patients treated with immune checkpoint inhibitor therapy revealed broad similarities between the evolution of tumors and rapidly evolving pathogens. 31 The phenomenon of immune interference, also known as ‘original antigenic sin’ described in many antigenically variable pathogens, 32 might be the major reason and a critical separate phenomenon responsible for the low immunogenicity of cancer neoepitopes; but, surprisingly, it has not been properly addressed in cancer research. In evaluating the chance of success of neoantigen vaccines, we first should try to understand the reasons for the failure of the only phase 3 vaccine efficacy trial, where vaccination with a neoantigen bearing a peptide from EGFRvIII‐positive glioblastoma patients did not increase the patient’s survival. 33 Similarly, it will be useful to compare clinical data from whole‐cell and dendritic cell vaccines 34 , 35 with neoantigen vaccine studies, to evaluate whether the latter have advantages that are currently not obvious in terms of immunogenicity or expected vaccine efficacy.
In other words, the immune response will always stay behind (i.e. lag) mutational events and will prove incapable of mounting efficient anti‐tumor pressure. Unfortunately, the above‐mentioned issues clearly demonstrate that cancer is a moving target and as such, is not considered adequately in current vaccine efforts. We think that the VEL vaccines, most probably, act through attenuation or a decrease in the ‘original antigenic sin’‐related events in cancer. Importantly, the mutational landscape of cancer cells, determined by powerful next‐generation sequencing techniques (see Introduction) at the specific time of sampling, will lead to the identification of neoantigens that have already passed through multiple rounds of elimination/selection/evolution events during long periods of time by both negative and positive selection events 13 , 30 , 36 and therefore, can hardly be considered bona fide vaccine targets. Furthermore, intra‐tumoral and circulating T cells, reactive to neoantigens that bear homology to infectious disease‐derived peptides, were detected in long‐term survivors of pancreatic cancer consistent with neoantigen molecular mimicry and, importantly, selective loss of high‐quality neoantigen clones on metastatic progression was observed, suggesting neoantigen immunoediting. 37
All of these observations may explain, in part, why immunization with highly immunogenic neoantigens is not sufficient to achieve anti‐tumor effects, as was shown in a recent study, where vaccination with mutated p53 and KRAS peptides resulted in a strong increase in regulatory T cells and accelerated tumor growth in a syngeneic sarcoma model. 38 In our study, the differential changes in the amount of CD11b+ Ly6Cint/low Ly6G+ suppressor G‐MDSCs in the lungs and tumors of vaccinated mice may explain the stronger inhibition of lung metastasis by VEL‐NT compared with VEL‐CT (Fig. 1). Indeed, as was shown recently, adoptive transfer of these cells, isolated from 4T1 tumor‐bearing mice, induces metastases by suppressing CD8+ T cells in EMT6‐primed mice. 39
The logic behind the VEL concept is that instead of using defined neoantigens or WT antigens as immunogens, we should use libraries of artificial epitopes that were not subject to the above‐mentioned selection phenomenon and therefore, may effectively target not only the epitopes present at a certain time‐point within the tumor, but, more importantly, may prevent immune escape by generating memory T cells against neoantigens that may appear later. Furthermore, the VELs may also activate T cells from the memory pool that reflect the immunological history of an individual, thus ‘compacting’ the time required for epitope evolution, which may exert additional anti‐tumor pressure. Therefore, most probably, the VEL vaccines are capable of inducing the ‘generalized’ activation of a large repertoire of T‐cell receptors, which may permit us to target not only neoantigens defined by next‐generation sequencing techniques, but also as yet unknown and difficult to determine ‘cancer‐specific’ antigens, generated by other mechanisms.
Collectively, our study showed that large combinatorial epitope libraries are efficient and potent vaccine immunogens and should be considered by vaccine researchers when designing clinical trials in cancer patients. These libraries represent true alternatives to the existing vaccine platforms, both as stand‐alone treatment or in combination immunotherapy, and, hopefully, the VEL concept may change the way we approach cancer vaccines.
Author contributions
KM designed the study, performed data analysis and wrote the paper; ANDR performed all experiments and data analysis; ANDR also prepared the tables and figures; FMC and AJOP performed the animal studies; MEM performed the immunofluorescence studies; GG was involved in the experimental design and wrote the paper.
Disclosures
The authors state no conflict of interest.
Supporting information
Table S1. Synthetic 517‐bp DNAs and primers used to generate survivin (SVN) ‐derived immunogens.
Figure S1. Representative flow cytometric analysis of CD4+ and CD8+ T‐cell proliferation, derived from spleen cells using CFSE.
Figure S2. Representative flow cytometric analysis of CD8+ Ly6C+ effector T‐cell phenotyping from lung tissue samples.
Figure S3. Representative flow cytometric analysis of CD11b+ Ly6Cint/low Ly6G+ granulocytic myeloid‐derived suppressor cells (G‐MDSCs) phenotyping from tumor tissue sample.
Figure S4. Representative flow cytometric analysis of CD4+ CD25+ FoxP3+ regulatory T‐cell phenotyping from lung tissue sample.
Figure S5. The phenotyping of memory T cells.
Acknowledgements
Funding was provided by PAPIIT‐UNAM (IN205216) and CONACyT (N283036), MEXICO, to KM. ANDR is a recipient of a scholarship from CONACyT and Posgrado en Ciencias Biológicas, UNAM. The authors thank: J. Guzman Valle for editing and proofreading; Patricia de la Torre for DNA sequencing; Patricia Espinosa‐Cueto for technical assistance; LabNalCit‐UNAM (CONACYT) for the technical support for the acquisition (and/or sorting) of flow cytometry samples and Unidad de Modelos Biológicos (Instituto de Investigaciones Biomédicas, UNAM) for animal care.
References
- 1. Yarchoan M, Johnson BA, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer 2017; 17:209–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhuting H, Ott PA, Wu CJ. Towards personalized, tumour‐specific, therapeutic vaccines for cancer. Nat Rev Immunol 2017; 18:168–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fennemann FL, de Vries IJM, Figdor CG, Verdoes M. Attacking tumors from all sides: personalized multiplex vaccines to tackle intratumor heterogeneity. Front Immunol 2019; 10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Capietto A‐H, Jhunjhunwala S, Delamarre L. ScienceDirect Characterizing neoantigens for personalized cancer immunotherapy. Curr Opin Immunol 2017; 46:58–65. [DOI] [PubMed] [Google Scholar]
- 5. Joshi K, de Massy MR, Ismail M, Reading JL, Uddin I, Woolston A et al Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer. Nat Med 2019; 25:1549–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma K‐Y, Schonnesen AA, Brock A, Van Den Berg C, Eckhardt SG, Liu Z et al Single‐cell RNA sequencing of lung adenocarcinoma reveals heterogeneity of immune response‐related genes. JCI Insight 2019; 4:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mogensen MB, Rossing M, Østrup O, Larsen PN, Heiberg Engel PJ, Jørgensen LN et al Genomic alterations accompanying tumour evolution in colorectal cancer: tracking the differences between primary tumours and synchronous liver metastases by whole‐exome sequencing. BMC Cancer 2018; 18:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Yang T‐HO et al The immune landscape of cancer. Immunity 2018; 48:812–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019; 7:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Verdegaal EME, de Miranda NFCC, Visser M, Harryvan T, van Buuren MM, Andersen RS et al Neoantigen landscape dynamics during human melanoma–T‐cell interactions. Nature 2016; 536:91–5. [DOI] [PubMed] [Google Scholar]
- 11. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J et al Evolution of neoantigen landscape during immune checkpoint blockade in non‐small cell lung cancer. Cancer Discov 2017; 7:264–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS et al Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017; 171:934–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rosenthal R, Cadieux EL, Salgado R, Bakir Al M, Moore DA, Hiley CT et al Neoantigen‐directed immune escape in lung cancer evolution. Nature 2019; 567:479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Simoni Y, Becht E, Fehlings M, Loh CY, Koo S‐L, Teng KWW et al Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018; 557:575–9. [DOI] [PubMed] [Google Scholar]
- 15. Hilf N, Kuttruff‐Coqui S, Frenzel K, Bukur V, Stevanović S, Gouttefangeas C et al Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019; 565:240–5. [DOI] [PubMed] [Google Scholar]
- 16. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S et al Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2018; 565:234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ott PA, Zhuting H, Keskin DB, Shukla SA, Sun J, Bozym DJ et al An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017; 547:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bjerregaard A‐M, Nielsen M, Jurtz V, Barra CM, Hadrup SR, Szallasi Z et al An analysis of natural T cell responses to predicted tumor neoepitopes. Front Immunol 2017; 8:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bassani‐Sternberg M, Bräunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S et al Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun 2016; 7:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Linette GP, Becker‐Hapak M, Skidmore ZL, Baroja ML, Xu C, Hundal J et al Immunological ignorance is an enabling feature of the oligo‐clonal T cell response to melanoma neoantigens. Proc Natl Acad Sci USA 2019; 116:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ruiz‐Orera J, Messeguer X, Subirana JA, Alba MM. Long non‐coding RNAs as a source of new peptides. eLife 2014; 3:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koster J, Plasterk RHA. A library of Neo Open Reading Frame peptides (NOPs) as a sustainable resource of common neoantigens in up to 50% of cancer patients. Sci Rep 2019; 9:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Servín‐Blanco R, Zamora‐Alvarado R, Gevorkian G, Manoutcharian K. Antigenic variability: obstacles on the road to vaccines against traditionally difficult targets. Hum Vaccin Immunother 2016; 12:2640–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Charles‐Niño C, Pedroza‐Roldan C, Viveros M, Gevorkian G, Manoutcharian K. Variable epitope libraries: new vaccine immunogens capable of inducing broad human immunodeficiency virus type 1‐neutralizing antibody response. Vaccine 2011; 29:5313–21. [DOI] [PubMed] [Google Scholar]
- 25. NoeDominguez‐Romero A, Zamora‐Alvarado R, Servín‐Blanco R, Pérez‐Hernández EG, Castrillon‐Rivera LE, Munguia ME et al Variable epitope library carrying heavily mutated survivin‐derived CTL epitope variants as a new class of efficient vaccine immunogen tested in a mouse model of breast cancer. Hum Vaccin Immunother 2014; 10:3201–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Servín‐Blanco R, Chávaro‐Ortiz RM, Zamora‐Alvarado R, Martínez‐Cortes F, Gevorkian G, Manoutcharian K. Generation of cancer vaccine immunogens derived from major histocompatibility complex (MHC) class I molecules using variable epitope libraries. Immunol Lett 2018; 204:47–54. [DOI] [PubMed] [Google Scholar]
- 27. Onodi F, Maherzi‐Mechalikh C, Mougel A, Hamouda NB, Taboas C, Gueugnon F et al High therapeutic efficacy of a new survivin LSP‐cancer vaccine containing CD4+ and CD8+ T‐cell epitopes. Front Oncol 2018; 8:15–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fenstermaker RA, Figel SA, Barone TA, Dharma SS, Winograd EK, Galbo PM et al Survivin monoclonal antibodies detect survivin cell surface expression and inhibit tumor growth in vivo . Clin Cancer Res 2018; 24:2642–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mohsen MO, Vogel M, Riether C, Muller J, Salatino S, Ternette N et al Targeting mutated plus germline epitopes confers pre‐clinical efficacy of an instantly formulated cancer nano‐vaccine. Front Immunol 2019; 10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zapata L, Pich O, Serrano L, Kondrashov FA, Ossowski S, Schaefer MH. Negative selection in tumor genome evolution acts on essential cellular functions and the immunopeptidome. Genome Biol 2018; 31:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Łuksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, Solovyov A et al A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017; 551:517–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Oberle SG, Hanna‐El‐Daher L, Chennupati V, Enouz S, Scherer S, Prlic M et al A minimum epitope overlap between infections strongly narrows the emerging T cell repertoire. Cell Rep 2016; 17:627–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H et al Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII‐expressing glioblastoma (ACT IV): a randomised, double‐blind, international phase 3 trial. Lancet Oncol 2017; 18:1373–85. [DOI] [PubMed] [Google Scholar]
- 34. Tanyi JL, Bobisse S, Ophir E, Tuyaerts S, Roberti A, Genolet R et al Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci Transl Med 2018; 10:1–14. [DOI] [PubMed] [Google Scholar]
- 35. Dillman RO, Cornforth AN, Nistor GI, McClay EF, Amatruda TT, Depriest C. Randomized phase II trial of autologous dendritic cell vaccines versus autologous tumor cell vaccines in metastatic melanoma: 5‐year follow up and additional analyses. J Immunother Cancer 2018; 6:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang W, Lee K‐W, Srivastava RM, Kuo F, Krishna C, Chowell D et al Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med 2019; 25:767–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R et al Identification of unique neoantigen qualities in long‐term survivors of pancreatic cancer. Nature 2017; 551:512–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Quandt J, Schlude C, Bartoschek M, Will R, Cid‐Arregui A, Schölch S et al Long‐peptide vaccination with driver gene mutations in p53 and Kras induces cancer mutation‐specific effector as well as regulatory T cell responses. OncoImmunology 2018; 7:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Piranlioglu R, Lee E, Ouzounova M, Bollag RJ, Vinyard AH, Arbab AS et al Primary tumor‐induced immunity eradicates disseminated tumor cells in syngeneic mouse model. Nat Commun 2019; 10:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Synthetic 517‐bp DNAs and primers used to generate survivin (SVN) ‐derived immunogens.
Figure S1. Representative flow cytometric analysis of CD4+ and CD8+ T‐cell proliferation, derived from spleen cells using CFSE.
Figure S2. Representative flow cytometric analysis of CD8+ Ly6C+ effector T‐cell phenotyping from lung tissue samples.
Figure S3. Representative flow cytometric analysis of CD11b+ Ly6Cint/low Ly6G+ granulocytic myeloid‐derived suppressor cells (G‐MDSCs) phenotyping from tumor tissue sample.
Figure S4. Representative flow cytometric analysis of CD4+ CD25+ FoxP3+ regulatory T‐cell phenotyping from lung tissue sample.
Figure S5. The phenotyping of memory T cells.