

Abstract
Pleiotropic roles are proposed for brain extracellular vesicles (EVs) in the development of Alzheimer's disease (AD). Our previous studies have suggested a beneficial role for EVs in AD, where the endosomal system in vulnerable neurons is compromised, contributing to the removal of accumulated material from neurons. However, the involvement of EVs in propagating AD amyloidosis throughout the brain has been considered because the amyloid‐β precursor protein (APP), APP metabolites, and key APP cleaving enzymes were identified in association with EVs. Here, we undertook to determine whether the secretase machinery is actively processing APP in EVs isolated from the brains of wild‐type and APP overexpressing Tg2576 mice. We found that full‐length APP is cleaved in EVs incubated in the absence of cells. The resulting metabolites, both α‐ and β‐APP carboxyl‐terminal fragments and APP intracellular domain accumulate in EVs over time and amyloid‐β dimerizes. Thus, EVs contribute to the removal from neurons and transport of APP‐derived neurotoxic peptides. While this is potentially a venue for propagation of the pathology throughout the brain, it may contribute to efficient removal of neurotoxic peptides from the brain.
Keywords: Aβ oligomers, Alzheimer's disease, APP‐CTFs, APP metabolism, exosomes
Abbreviations
- Aβ
amyloid‐β
- AD
Alzheimer's disease
- AICD
APP intracellular domain
- APP
Amyloid‐β precursor protein
- APP‐CTFs
APP carboxyl‐terminal fragments
- DMEM
Dulbecco's Modified Eagle Medium
- EVs
Extracellular vesicles
- flAPP
full‐length amyloid‐β precursor protein
- PS1
Presenilin‐1
- PS2
Presenilin‐2
1. INTRODUCTION
Extracellular vesicles (EVs) are highly stable membranous structures that are secreted by most cell types and can be subsequently taken up by recipient cells (reviewed in 1). The various species of EVs have different origins. While microvesicles bud directly from the plasma membrane, exosomes have an endosomal origin because they are formed by invagination of the membrane of late endosomes (also called multivesicular bodies). 2 Given that sorting EVs derived from a particular biogenesis pathway remains difficult, the general term EVs will be used here to refer to small EVs enriched with exosomes, following the International Society for EVs recommendations. 3 EV secretion was originally described as a complementary process to the lysosomal and proteosomal degradative pathways to discard obsolete membrane and cytosolic proteins in differentiating reticulocytes. 4 Accordingly, our previous research has supported a beneficial role for EVs in vulnerable neurons with endosomal pathology, one of the earliest insults observed in Alzheimer's disease (AD), 5 by relieving the cells from toxic waste that was not efficiently degraded intracellularly. 6 , 7 However, the identification of full‐length amyloid‐β precursor protein (flAPP) and amyloid‐β precursor protein (APP) metabolites in EVs 8 , 9 , 10 , 11 has implicated EVs in AD amyloid pathology by carrying and spreading pathology throughout the brain. The accumulation of the amyloid‐β (Aβ) peptide in extracellular senile plaques is one of the pathological hallmarks of AD. 12 Aβ is generated through sequential processing of flAPP, which undergoes proteolytic cleavage by α‐ and β‐secretases to produce α‐ and β‐carboxyl‐terminal fragments (APP‐CTFs), respectively. β‐APP‐CTF (also referred to as C99) is the source of Aβ following cleavage by γ‐secretase, 13 which also results in the generation of the APP intracellular domain (AICD). 14 Processing of flAPP occurs in the plasma membrane and in endosomes 15 and APP metabolites can be incorporated into EVs 16 and released from the cells into the extracellular environment. However, it was suggested that only a minute fraction of total Aβ is associated with EVs, 9 , 16 unlike APP‐CTFs, which are abundantly present within EVs. 8 , 9 , 10 , 11 , 17 Accumulation of β‐APP‐CTFs in specific brain regions is a common early process in different AD mouse models that may contribute to AD pathology (reviewed in 18). Multiple studies have shown that β‐APP‐CTFs are neurotoxic, causing neuronal endosomal‐lysosomal abnormalities 19 , 20 , 21 leading to neuronal cell death and memory loss. 22 , 23 , 24 , 25 , 26 , 27 , 28 Therefore, the transport of β‐APP‐CTF via EVs throughout the brain may result in the uptake of this peptide by neurons with deleterious effects. β‐APP‐CTFs are also a source of Aβ peptides that will assemble into oligomeric forms prior to the formation of senile plaques. 29 These oligomeric forms of Aβ are considered a major culprit in the synaptic dysfunction and neuronal loss that characterize AD. 30 , 31 , 32 The process of oligomerization and subsequent aggregation of Aβ into fibrils can be facilitated by EVs, as it has been shown that they can act as amyloid “seeding” sites. 33 , 34 Further, the identification in EVs of key enzymes involved in APP metabolism 9 , 10 , 35 , 36 has suggested that EVs are a potential site for the de novo generation of APP‐CTFs and Aβ. Because EVs are oriented with the cytoplasmic‐side facing inward, 37 the same orientation as in the plasma membrane, it was hypothesized that cleavage of flAPP would result in capture of APP‐CTFs in the membrane of EVs and in the secretion of EV‐generated Aβ into the extracellular space. Here, we investigated whether flAPP is enzymatically cleaved in EVs in the absence of cells.
2. MATERIALS AND METHODS
2.1. Mice
Brains were derived from wild‐type C57/B6 mice at the age of 12 months and from transgenic mice overexpressing human APP with the K670N/M671L Swedish double mutation (Tg2576) 38 at 4 months of age. Both females and males were used for all analyses. All animal procedures were performed following the National Institutes of Health guidelines with approval from the Institutional Animal Care and Use Committee at the Nathan S. Kline Institute for Psychiatric Research.
2.2. Brain EV isolation
Brain EVs were isolated and purified as we have previously described. 9 Briefly, frozen brain tissues were treated with 20 units/mL of papain (Worthington, Lakewood, NJ, USA) in Hibernate A solution (HA, 3.5 mL/sample; BrainBits, Springfield, IL, USA) for 15 minutes at 37°C. The brain tissue was gently dissociated in 6.5 mL of cold HA supplemented with protease inhibitors, centrifuged at 300 g for 10 minutes at 4°C to discard the cells, and the supernatant was sequentially filtered through a 40 μm mesh filter (BD Biosciences, San Jose, CA, USA) and a 0.2 μm syringe filter (Corning Life Sciences, Teterboro, NJ, USA). The filtrates were sequentially centrifuged at 4°C, at 2000 g for 10 minutes and 10 000 g for 30 minutes to discard membranes and debris, and at 100 000 g for 70 minutes to pellet the EVs. The EV pellet was resuspended in 60 mL of cold PBS (Thermo Fisher Scientific), and centrifuged at 100 000 g for 70 minutes at 4°C. The washed EV pellet was resuspended in 2 mL of 0.95 M sucrose solution and inserted inside a sucrose step gradient column (six 2‐mL steps from 2.0 to 0.25 M sucrose). The sucrose step gradient was centrifuged at 200 000 g for 16 hours and fractions were collected from the top of the gradient. The fractions were diluted in cold PBS and centrifuged at 100 000 g for 70 minutes for pellet collection.
2.3. Incubation of isolated EVs at 37°C
Brain EV pellets from fractions C and D of the sucrose step gradient 9 were resuspended in 30 µL each of Dulbecco's Modified Eagle Medium (DMEM, Thermo Fisher Scientific) and combined. The pooled EVs were divided into experimental groups incubated for the indicated times (0, 1, 4, 24, 48, or 72 hours), in the absence or presence of either the γ‐secretase inhibitor L685,458, or Aβ‐degrading enzyme inhibitors. The same volume of DMEM containing 2X EDTA (except in the experiments using inhibitors of Aβ‐degrading enzymes), without or supplemented with γ‐secretase inhibitor or an Aβ‐degrading enzyme inhibitor, was added to the EV suspensions. The γ‐secretase inhibitor, L685,458 (Tocris Bioscience, Minneapolis, MN, USA) was used at the final concentration of 10 µM. Inhibitors of Aβ‐degrading enzymes (thiorphan [Cayman] and phosphoramidon [Sigma‐Aldrich]) were added to the EV suspensions at the final concentrations of 10 and 100 µM, respectively. Though each can inhibit multiple metalloproteases, at these concentrations thiorphan is selective for neprilysin over endothelin‐converting enzymes (ECEs) and phosphoramidon inhibits neprilysin and ECEs. At time 0 hour EVs were immediately lysed in 2X RIPA buffer (1% Triton‐X, 1% Sodium deoxycholate, 0.1% SDS, 150 mM NaCl, 50 mM Tris‐HCl pH 7.4, and 1 mM EDTA) supplemented with 2X Halt Protease inhibitors (Thermo Fisher Scientific) and 1X EDTA (Thermo Fisher Scientific). At times 1, 4, 24, 48, and 72 hours EVs were placed in a 37°C bath incubator for the time periods indicated and subsequently lysed in 2X RIPA buffer. All lysates were sonicated for 45 seconds, placed on ice for 20 minutes with vortex‐mixing every 5 minutes and kept at −80°C until analysis.
2.4. Preparation of Aβ peptide solution
For preparation of the 10 µM peptide stock, lyophilized Aβ40 peptide (2 µg) was dissolved and equilibrated in dimethyl sulfoxide (Sigma‐Aldrich) for 15 minutes at room temperature with vortex‐mixing every 3 minutes. Subsequently, the 10 µM of Aβ40 stock was diluted in DMEM to make a 300 nM of Aβ40 solution, which was further diluted to the final concentration of 10 nM in the solution used for the Western blot analysis.
2.5. Western blot analysis
The same amount of EV proteins was separated by 4%‐20% Tris‐HCl gel electrophoresis (Criterion precast gel, Bio‐Rad, Hercules, CA, USA) and transferred onto PVDF membranes (Immobilon, Millipore). Membranes were incubated with antibodies to HSC70 (1:1000, Cat# sc‐7298, RRID:AB_62776; Santa Cruz Biotechnology), CD63 (1:1000, Cat# ab217345, RRID:AB_2754982; Abcam), APP and APP‐CTFs (C1/6.1, 39 1:1000), BACE1 (1:1000, Cat# 200‐401‐984, RRID:AB_2243187; Rockland), ADAM10 (1:1000, Cat# AB19026, RRID:AB_2242320; Millipore), Nicastrin (1:1000, Cat# MAB5556, RRID:AB_2235791; Millipore). The antibodies to the subunits of the γ‐secretase complex: PS1 (N‐terminal, 1:1000), PS2 (N‐terminal, 1:50), APH1a (C‐terminal, 1:1000), and APH1b (C‐terminal, 1:1000) were a kind gift from Dr Paul Fraser, University of Toronto. The PEN‐2 antibody (N‐terminal, 1:2500) was a kind gift from Dr Thinakaran, University of Chicago. The secondary antibodies used were HRP‐conjugated anti‐rabbit or anti‐mouse antibodies (Jackson ImmunoResearch, West Grove, PA, USA). The membranes were incubated in chemiluminescent fluid (Pierce, Rockford, IL, USA) and chemiluminescence was visualized on X‐ray films. For identification of Aβ dimers, the same amount of EV proteins was separated by 16.5% tris‐tricine gels, blotted with a 1:1 mixture of the 6E10 (1:1000; Cat# 803001, RRID:AB_2564653; BioLegend) and 4G8 (1:1000; Cat# SIG‐39220, RRID:AB_662812; Covance) antibodies on PVDF. 6E10 and 4G8 antibodies are generated against different epitopes within Aβ. 40 For visualization of Aβ monomers and AICD, EV proteins separated by 16.5% tris‐tricine gels were transferred onto Nitrocellulose (Bio‐Rad), which was subsequently boiled in PBS for 4 minutes, followed by blotting in 6E10 for Aβ monomer or C1/6.1 for AICD detection. Protein bands were quantified using ImageJ (National Institute of Health (NIH), Bethesda, MD, USA).
2.6. ELISA
Aβ peptides were analyzed by sandwich ELISA with specific mouse monoclonal antibodies, as described previously. 41 For human Aβ40 measurement, antibody MM27‐33.1.1 (raised against human Aβ 1‐16) was used for capture and MM32‐13.1.1‐HRP (raised against Aβ35‐40) for detection. For Aβ42, anti‐Aβ42 antibody MM26‐2.1.3 (Aβ35‐42) was used for capture and HRP‐conjugated 4G8 antibody (Aβ17‐24) was used for detection. Aβ antibodies except for 4G8 (Covance, Princeton, NJ, USA) were from Mayo Clinic (Rochester, MN, USA).
2.7. Data analysis and graphing
The statistical and graphing software Prism 5.01 (GraphPad Software, La Jolla, CA, USA) was used for data analysis and graphical representation. For column graphs, column bars represent means ± SEM. To compare the relative changes in the levels of α‐ and β‐APP‐CTFs, Aβ monomers, Aβ dimers and AICD, analysis of variance (one‐way ANOVA) followed by Tukey's post hoc multiple comparison was performed (Figures 2 and 3).
FIGURE 2.
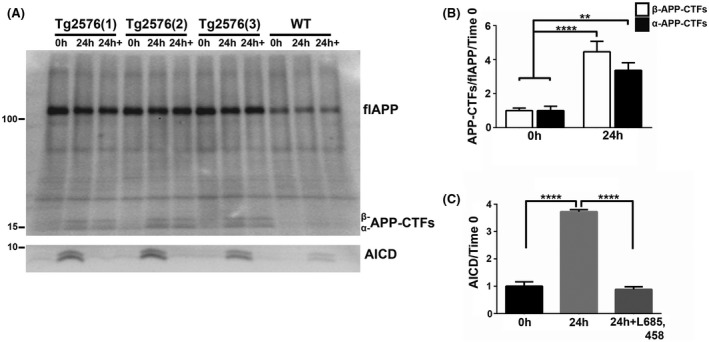
APP‐CTFs in brain EVs are enzymatically cleaved by the γ‐secretase complex, leading to the generation of AICD. A, Western blot analysis with the C1/6.1 antibody of EVs isolated from the brain of 4‐month‐old Tg2576 and wild‐type (WT) mice prior to (0 hour) and after incubation at 37°C in DMEM without (24 hours) or with 10 µM of the γ‐secretase inhibitor L‐685,458 (24 hours+). B, Quantification of α‐ and β‐APP‐CTFs levels in EVs isolated from the brain of Tg2576 mice revealed an increase in the levels of both metabolites. C, The levels of AICD also increased and the increase was blocked by inhibition of the γ‐secretase complex. PVDF membranes were used for APP and APP‐CTFs detection, whereas nitrocellulose was used for detection of AICD. One‐way ANOVA followed by Tukey's post hoc multiple comparison, n = 7 independent experiments (B), n = 3 independent experiments (C). Data are shown as mean ± SEM (**P < .01; ****P < .0001)
FIGURE 3.
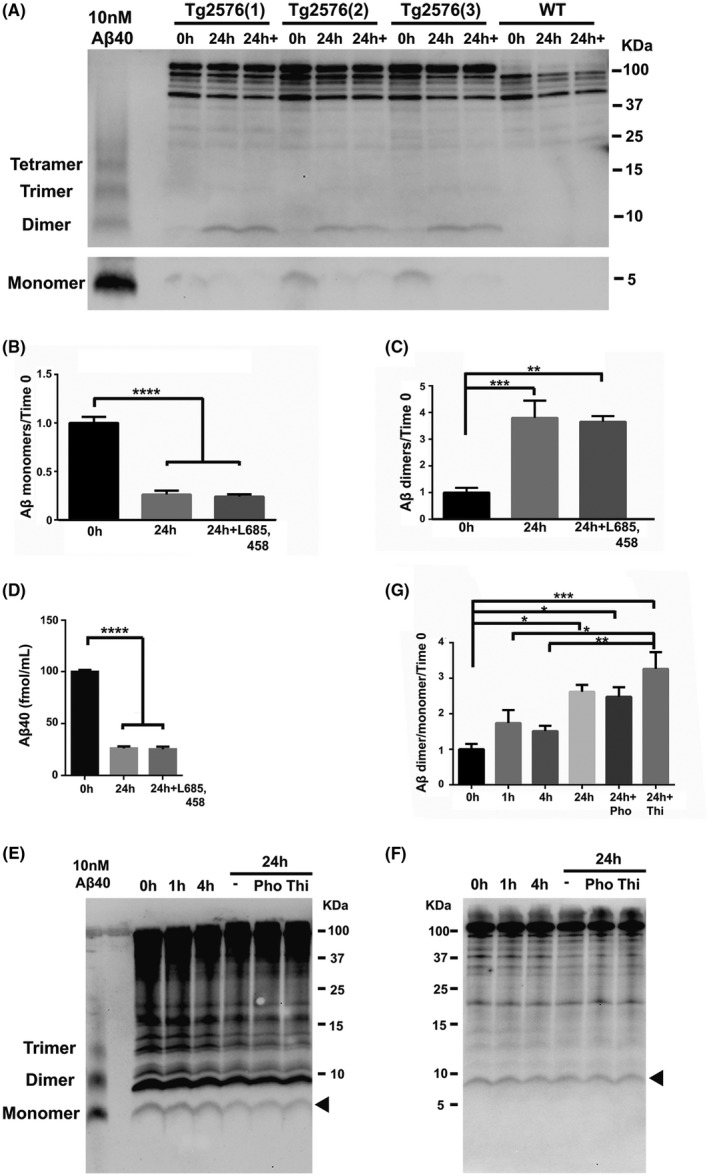
Aβ present at time zero in EVs oligomerizes into dimers. A, Western blot analysis of EVs isolated from the brain of 4‐month‐old Tg2576 and wild‐type (WT) mice prior to (0 hour) and after incubation without (24 hours) or with 10 µM of L‐685,458 (24 hours+) shows the bands for Aβ monomers (lower panel) and dimers. Quantification of the corresponding bands shows a decrease in the level of monomeric Aβ (B) and an increase in the level of the dimers (C) following incubation. DMEM with 10 nM of Aβ40 peptide was used as a positive control of Aβ monomeric and oligomeric forms. D, Aβ40 measurements of brain EVs by ELISA prior to (0 hour) or after a 24‐hour incubation without (24 hours) or with L‐685,458 (24 hours+). E and F, Western blot analysis of brain EVs after incubation for 1, 4, and 24 hours without (−) or with the metalloprotease inhibitors Phosphoramidon (Pho) or Thiorphan (Thi). Aβ monomers (E) and dimers (F) are indicated by arrowheads. G, The protein bands were quantified in all conditions and presented as the ratio between Aβ dimers and Aβ monomers. Nitrocellulose membranes were used for detection of Aβ monomers (E), whereas PVDF was used for detection of Aβ dimers (F). One‐way ANOVA followed by Tukey's post hoc multiple comparison, n = 3 independent experiments (B), n = 7 independent experiments (C), n = 5 independent experiments (D), and n = 4 independent experiments (G). Data are shown as mean ± SEM (*P < .05; **P < .01; ***P < .001; ****P < .0001)
3. RESULTS
3.1. APP present in EVs is enzymatically processed to generate APP‐CTFs
In order to study whether flAPP is cleaved in situ in EVs, we investigated the presence in EVs of the α‐secretase ADAM10, 42 the β‐secretase BACE1, 43 and all the components of the γ‐secretase, a membrane protein complex comprised of Presenilin‐1 (PS1) or Presenilin‐2 (PS2), APH‐1a or APH‐1b, Nicastrin, and PEN‐2. 44 In addition to the confirmation of the presence of previously identified secretases, we found all the γ‐secretase complex subunits in EVs isolated from wild‐type mouse brain (Figure 1A).
FIGURE 1.
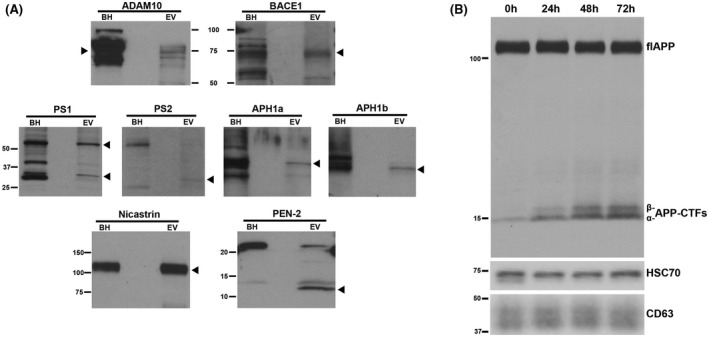
flAPP is enzymatically processed in brain EVs to generate α‐ and β‐APP‐CTFs in a time dependent manner. A, Western blot analyses revealing the presence of the APP processing enzymes ADAM10 (α‐secretase), BACE1 (β‐secretase), and of all the components of the γ‐secretase complex: PS1, PS2 APH1a, APH1b, Nicastrin, and PEN‐2. Wild‐type mouse brain homogenate (BH) was used as a positive control and arrow‐heads indicate the specific protein band for each panel. B, Western blot analysis using an antibody against the carboxyl‐terminus of APP (C1/6.1) showing an increase in the levels of α‐ and β‐APP‐CTFs in wild‐type brain EVs during incubation at 37°C for 24, 48, and 72 hours. Antibodies against the exosomal markers HSC70 and CD63 were used in the lower panels
We then tested whether these secretases are functional in situ, generating APP‐CTFs. EVs isolated from the brain of wild‐type mice were incubated in DMEM in the absence of cells at 37°C for different periods of time. Following incubation, the EVs with the medium in which they were incubated were lysed in RIPA buffer and analyzed by Western blotting. The results showed that flAPP is enzymatically processed to generate α‐ and β‐APP‐CTFs in a time‐dependent manner (Figure 1B), confirming the de novo generation of APP‐CTFs in brain EVs.
3.2. APP‐CTFs are further processed into AICD in EVs and Aβ peptides oligomerize
We next determined whether the γ‐secretase complex in EVs is enzymatically active to generate Aβ and AICD. Given the low Aβ production in the brain of wild‐type mice, we isolated EVs from the brain of APP overexpressing transgenic mice, Tg2576, 38 at an age prior to the initiation of amyloid deposition. We first corroborated that APP processing occurs in EVs isolated from the brain of Tg2576 mice during a 24‐hour incubation, generating both α‐ and β‐APP‐CTFs (Figure 2A,B) at levels higher than in EVs isolated from the brain of wild‐type mice (Figure 2A). Incubation in the presence of the γ‐secretase inhibitor, L685,458 had no significant effect on the levels of the APP‐CTFs (Figure 2A). An indication for the γ‐secretase cleavage of the APP‐CTFs in the incubated EVs is the increase in the level of AICD following 24‐hour incubation and the absence of the increase in AICD level when the EVs were treated with the γ‐secretase inhibitor, L685,458 (Figure 2A,C).
In order to investigate whether Aβ is generated in situ in EVs isolated from the brain of Tg2576 mice, we performed Western blotting experiments. We observed a decrease in the level of monomeric Aβ (Figure 3A,B) and an increase in the level of Aβ dimers (Figure 3A,C) following 24 hours of EV incubation. These findings were not affected by the treatment with the γ‐secretase inhibitor, L685,458 (Figure 3A‐C). While the SDS‐PAGE band of ~8‐9 kDa could derive from N‐terminal extended (NTE) Aβ forms, 45 , 46 the absence of an effect of the γ‐secretase inhibitor on the levels of the ~8‐9 kDa band (Figure 3A), excludes the possibility of NTE Aβ generation during the incubation. These results suggest that EV‐associated monomeric Aβ was oligomerized into Aβ dimers during the incubation. The level of Aβ was also analyzed by sensitive ELISA measurements of Aβ peptides in EVs incubated for 24 hours in DMEM. We found a robust decrease in the level of monomeric Aβ40 (Figure 3D), consistent with the Western blotting results. The level of Aβ42 was below the assay detection level (data not shown). The reduction in Aβ level could be due to the activity of EV‐associated Aβ‐degrading enzymes such as neprilysin, 47 insulin‐degrading enzyme (IDE), 48 or endothelin‐converting enzymes‐1 and −2 (ECE‐1 and ECE‐2), 49 suggested to be active in the extracellular space. 50 Thus, we pretreated the brain EVs with the metalloprotease inhibitors phosphoramidon and thiorphan, in order to inhibit the activities of these enzymes. Treatment with these inhibitors did not affect the monomeric Aβ levels (Figure 3E), suggesting that the decrease in Aβ levels was not due to degradation by the metalloproteases against which inhibitors were used. We also found no effect of the inhibitors on the level of Aβ dimers (Figure 3F). An increase in the ratio of Aβ dimers to Aβ monomers during EV incubation was observed in the absence and presence of the inhibitors (Figure 3G), suggesting a sustained Aβ oligomerization during EVs incubation.
4. DISCUSSION
We have previously reported that EVs are enriched with APP‐CTFs. 8 , 9 Given that an early event that characterizes AD is a dysfunctional endosomal‐lysosomal system (reviewed in 51), we proposed that the release of endosomal materials into the extracellular space via EVs is a beneficial mechanism by which neurons remove accumulated materials, and that failure of efficient exosome production and release can exacerbate 7 and perhaps lead to endosomal‐lysosomal pathway disturbances. 52 Here, we demonstrate that APP‐CTFs are generated by an active secretase machinery and accumulate in EVs over time. Therefore, EVs may contribute to the accumulation in the brain of β‐APP‐CTFs, suggested to be involved in the initiation of neurodegenerative processes associated with endosomal‐lysosomal 21 , 27 , 53 and autophagy dysfunction, 54 , 55 neuroinflammation, 28 , 53 and synaptic alterations leading to cognition and memory deficits in AD. 26 , 28 , 56 Consistently, it was shown that the treatment with γ‐secretase inhibitors leading to APP‐CTFs accumulation impairs cognitive function in transgenic mice 57 and worsens cognitive deficits in AD patients. 58 Interestingly, inhibition of γ‐secretase increased the levels of APP‐CTFs and oligomeric APP‐CTFs in EVs in an AD mouse model. 59 Here, we demonstrate that AICD, previously identified in EVs, 11 , 17 is also generated in brain EVs. Given the orientation of flAPP in the membrane, it is likely that AICD is released into and accumulates within EV lumen. It was demonstrated that in addition to AICD being a regulator of gene expression 60 and cell‐signaling pathways, 61 it has deleterious effects such as causing hyperphosphorylation and age‐dependent aggregation of tau, which recapitulates the features of AD, 62 and impairing hippocampal neurogenesis through the induction of neuroinflammation. 63 While we did not observe significant changes in the levels of APP‐CTF when the EVs were incubated in the presence of a γ‐secretase inhibitor, the decrease in the levels of AICD in the presence of the γ‐secretase inhibitor indicates that γ‐secretase is indeed active in EVs. This is likely due to the continuous cleavage of flAPP by α‐ and β‐secretases leading to the accumulation of α‐ and β‐APP‐CTFs in the EV membrane and a low γ‐secretase activity resulting in only a small part of APP‐CTFs being further processed by γ‐secretase.
Although less abundant than APP‐CTFs, the peptide Aβ has also been identified in association with EVs. 9 , 16 The association between Aβ and EVs is supported by the findings of specific surface molecules in EVs that mediate their interaction with Aβ, such as glycosphingolipids glycans, 64 ceramides, 65 and the GPI‐anchored protein PrPc. 66 The interactions of Aβ with PrP, glycosphingolipids, and ceramide localized in lipid raft domains may also target intracellular amyloids to EVs. 67 Regardless the Aβ origin, it was demonstrated that EVs promote Aβ aggregation on their surface. 33 , 34 , 65 Since APP processing occurs in endosomes, EV‐associated Aβ may have an intracellular origin through its incorporation into intraluminal vesicles that will be released as exosomes upon fusion of the late endosome with the plasma membrane. While we detected an increase in the levels of APP‐CTFs in EVs during ex vivo incubation, the levels of EV‐associated monomeric Aβ decreased dramatically. Although EVs have been implicated in the extracellular enzymatic degradation of Aβ, 48 , 68 the reduction in Aβ levels that we observed was not due to active Aβ‐degrading metalloproteases present in EVs. However, we cannot rule out the possibility that other proteases present in EVs are mildly contributing to the degradation of Aβ. The decrease in the levels of the monomeric Aβ during the incubation was mostly associated with rapid assembly of the existing monomeric Aβ into dimers, which was the most enriched Aβ oligomer species in association with the EVs. Altogether, the data suggest that only negligible de novo generation of Aβ occurs in EVs and that Aβ associated with the EVs has mainly an intracellular origin. During incubation of the EVs, the Aβ dimer/monomer ratio increased in a time‐dependent manner. These results are consistent with previous reports showing that Aβ oligomers are rapidly sequestered on membranes due to their low stability in the aqueous extracellular space. 69 The lower molecular weight Aβ oligomers such as dimers have been suggested to be especially neurotoxic because they can act as seeds for further aggregation. 70 , 71 In addition, a recent study demonstrated that Aβ dimers derived from AD brain mediated neuronal hyperactivity, which characterizes the early stage of AD, through block of synaptic glutamate reuptake. 72 The Aβ dimers‐enriched EVs may thus have a neurotoxic role at the early stages of AD by Aβ oligomerization into dimers and we suggest that EV‐associated Aβ dimers could be used as a diagnostic marker to detect early AD.
More than a decade has passed since EVs were proposed to participate in the spread of Aβ pathology in the AD brain. 16 Accordingly, it has been recently demonstrated that EVs are vehicles for the intercellular transmission of APP and APP metabolites, 17 including oligomeric Aβ. 73 Our results support that brain EVs are not only carriers, but also a site for the generation of APP‐CTFs and AICD, and for the oligomerization of Aβ, all neurotoxic peptides. Thus, EVs may propagate the disease within the brain by delivering toxic metabolites into recipient neurons. Indeed, it was shown that inhibition of EV secretion is protective by decreasing amyloid plaque deposition and ameliorating the cognitive impairment in transgenic APP mice. 65 However, apparently contradictory data suggest that EVs may act as potent scavengers for Aβ in the brain 64 , 74 and contribute to the clearance of toxic proteins by microglia uptake 33 , 75 or by enzymatic degradation of Aβ. 76 Whether EVs drive pathology spread or serve as a pathway for the removal of toxic molecules within the brain, as we have previously suggested, 7 , 8 is dependent on a balance between the release and efficient removal of EVs from the brain extracellular space. Given that the contribution of microglia to Aβ clearance is reduced in advanced AD, 77 this balance is likely to tilt toward the pathological side of EVs as the disease progresses.
To conclude, our data along with a growing body of research suggest a working model where at early stages of AD, EVs have a protective role for cells in the brain. EVs are secreted loaded with APP and neurotoxic APP metabolites (APP‐CTFs, AICD, and Aβ) to relieve neurons from accumulated toxic material. At that stage, EVs transport the material to either microglia for degradation, or out of the brain into the circulation. However, in advanced stages of AD with compromised endosomal‐lysosomal and removal systems, EVs can be sites for the generation and accumulation of APP metabolites, which would potentiate AD‐associated neurodegeneration. Therefore, determining the spatiotemporal functions of brain EVs in AD is crucial to further understand AD pathogenesis.
CONFLICT OF INTERESTS
The authors declare that they have no conflict of interests.
AUTHOR'S CONTRIBUTIONS
E. Levy and R. Pérez‐González conceived the ideas and wrote and revised the manuscript. R. Pérez‐González, Y. Kim, and E. Levy designed research. R. Pérez‐González, Y. Kim, C. Miller, and J. Pacheco‐Quinto performed experiments and analyzed data. R. Pérez‐González, Y. Kim, J. Pacheco‐Quinto, EA Eckman, and E. Levy interpreted results. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
The authors thank Dr Monika Pawlik for managing the mouse colonies. This work was supported by the Alzheimer's Association Grant NIRG‐14‐316622 (to RP‐G.) and the National Institute on Aging (NIA) Grants AG017617 and AG056732 (to EL).
Pérez‐González R, Kim Y, Miller C, Pacheco‐Quinto J, Eckman EA, Levy E. Extracellular vesicles: Where the amyloid precursor protein carboxyl‐terminal fragments accumulate and amyloid‐β oligomerizes. The FASEB Journal. 2020;34:12922–12931. 10.1096/fj.202000823R
Rocío Pérez‐González and Yohan Kim contributed equally to this study.
REFERENCES
- 1. van Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213‐228. [DOI] [PubMed] [Google Scholar]
- 2. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255‐289. [DOI] [PubMed] [Google Scholar]
- 3. Thery C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J Biol Chem. 1987;262:9412‐9420. [PubMed] [Google Scholar]
- 5. Cataldo AM, Mathews PM, Boiteau AB, et al. Down syndrome fibroblast model of Alzheimer‐related endosome pathology: accelerated endocytosis promotes late endocytic defects. Am J Pathol. 2008;173:370‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. D'Acunzo P, Hargash T, Pawlik M, Goulbourne CN, Perez‐Gonzalez R, Levy E. Enhanced generation of intraluminal vesicles in neuronal late endosomes in the brain of a Down syndrome mouse model with endosomal dysfunction. Dev Neurobiol. 2019;79:656‐663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gauthier SA, Perez‐Gonzalez R, Sharma A, et al. Enhanced exosome secretion in Down syndrome brain ‐ a protective mechanism to alleviate neuronal endosomal abnormalities. Acta Neuropathol Commun. 2017;5:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Perez‐Gonzalez R, Gauthier SA, Sharma A, et al. A pleiotropic role for exosomes loaded with the amyloid beta precursor protein carboxyl‐terminal fragments in the brain of Down syndrome patients. Neurobiol Aging. 2019;84:26‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Perez‐Gonzalez R, Gauthier SA, Kumar A, Levy E. The exosome secretory pathway transports amyloid precursor protein carboxyl‐terminal fragments from the cell into the brain extracellular space. J Biol Chem. 2012;287:43108‐43115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sharples RA, Vella LJ, Nisbet RM, et al. Inhibition of gamma‐secretase causes increased secretion of amyloid precursor protein C‐terminal fragments in association with exosomes. FASEB J. 2008;22:1469‐1478. [DOI] [PubMed] [Google Scholar]
- 11. Vingtdeux V, Hamdane M, Loyens A, et al. Alkalizing drugs induce accumulation of amyloid precursor protein by‐products in luminal vesicles of multivesicular bodies. J Biol Chem. 2007;282:18197‐18205. [DOI] [PubMed] [Google Scholar]
- 12. Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885‐890. [DOI] [PubMed] [Google Scholar]
- 13. Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med. 2012;2:a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Passer B, Pellegrini L, Russo C, et al. Generation of an apoptotic intracellular peptide by gamma‐secretase cleavage of Alzheimer's amyloid beta protein precursor. J Alzheimers Dis. 2000;2:289‐301. [DOI] [PubMed] [Google Scholar]
- 15. Nixon RA. Amyloid precursor protein and endosomal‐lysosomal dysfunction in Alzheimer's disease: inseparable partners in a multifactorial disease. FASEB J. 2017;31:2729‐2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rajendran L, Honsho M, Zahn TR, et al. Alzheimer's disease beta‐amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A. 2006;103:11172‐11177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Laulagnier K, Javalet C, Hemming FJ, et al. Amyloid precursor protein products concentrate in a subset of exosomes specifically endocytosed by neurons. Cell Mol Life Sci. 2018;75:757‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lauritzen I, Pardossi‐Piquard R, Bourgeois A, Becot A, Checler F. Does intraneuronal accumulation of carboxyl‐terminal fragments of the amyloid precursor protein trigger early neurotoxicity in Alzheimer's disease? Curr Alzheimer Res. 2019;16:453‐457. [DOI] [PubMed] [Google Scholar]
- 19. Jiang Y, Mullaney KA, Peterhoff CM, et al. Alzheimer's‐related endosome dysfunction in Down syndrome is Abeta‐independent but requires APP and is reversed by BACE‐1 inhibition. Proc Natl Acad Sci U S A. 2010;107:1630‐1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiang Y, Rigoglioso A, Peterhoff CM, et al. Partial BACE1 reduction in a Down syndrome mouse model blocks Alzheimer‐related endosomal anomalies and cholinergic neurodegeneration: role of APP‐CTF. Neurobiol Aging. 2016;39:90‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang Y, Sato Y, Im E, et al. Lysosomal dysfunction in Down Syndrome Is APP‐dependent and mediated by APP‐betaCTF (C99). J Neurosci. 2019;39:5255‐5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oster‐Granite ML, McPhie DL, Greenan J, Neve RL. Age‐dependent neuronal and synaptic degeneration in mice transgenic for the C terminus of the amyloid precursor protein. J Neurosci. 1996;16:6732‐6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Neve RL, Robakis NK. Alzheimer's disease: a re‐examination of the amyloid hypothesis. Trends Neurosci. 1998;21:15‐19. [DOI] [PubMed] [Google Scholar]
- 24. McPhie DL, Golde T, Eckman CB, Yager D, Brant JB, Neve RL. β‐Secretase cleavage of the amyloid precursor protein mediates neuronal apoptosis caused by familial Alzheimer's disease mutations. Brain Res Mol Brain Res. 2001;97:103‐113. [DOI] [PubMed] [Google Scholar]
- 25. Deyts C, Vetrivel KS, Das S, et al. Novel GalphaS‐protein signaling associated with membrane‐tethered amyloid precursor protein intracellular domain. J Neurosci. 2012;32:1714‐1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tamayev R, Matsuda S, Arancio O, D'Adamio L. β‐ but not gamma‐secretase proteolysis of APP causes synaptic and memory deficits in a mouse model of dementia. EMBO Mol Med. 2012;4:171‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lauritzen I, Pardossi‐Piquard R, Bauer C, et al. The beta‐secretase‐derived C‐terminal fragment of betaAPP, C99, but not Abeta, is a key contributor to early intraneuronal lesions in triple‐transgenic mouse hippocampus. J Neurosci. 2012;32:16243‐11655a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lauritzen I, Pardossi‐Piquard R, Bourgeois A, et al. Intraneuronal aggregation of the beta‐CTF fragment of APP (C99) induces Abeta‐independent lysosomal‐autophagic pathology. Acta Neuropathol. 2016;132:257‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid beta‐protein assembly and Alzheimer disease. J Biol Chem. 2009;284:4749‐4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klyubin I, Betts V, Welzel AT, et al. Amyloid beta protein dimer‐containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231‐4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Selkoe DJ. Soluble oligomers of the amyloid beta‐protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shankar GM, Li S, Mehta TH, et al. Amyloid‐beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yuyama K, Sun H, Mitsutake S, Igarashi Y. Sphingolipid‐modulated exosome secretion promotes clearance of amyloid‐beta by microglia. J Biol Chem. 2012;287:10977‐10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yuyama K, Yamamoto N, Yanagisawa K. Accelerated release of exosome‐associated GM1 ganglioside (GM1) by endocytic pathway abnormality: another putative pathway for GM1‐induced amyloid fibril formation. J Neurochem. 2008;105:217‐224. [DOI] [PubMed] [Google Scholar]
- 35. Escrevente C, Morais VA, Keller S, Soares CM, Altevogt P, Costa J. Functional role of N‐glycosylation from ADAM10 in processing, localization and activity of the enzyme. Biochim Biophys Acta. 2008;1780:905‐913. [DOI] [PubMed] [Google Scholar]
- 36. Goetzl EJ, Mustapic M, Kapogiannis D, et al. Cargo proteins of plasma astrocyte‐derived exosomes in Alzheimer's disease. FASEB J. 2016;30:3853‐3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pisitkun T, Shen RF, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci U S A. 2004;101:13368‐13373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hsiao K, Chapman P, Nilsen S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99‐102. [DOI] [PubMed] [Google Scholar]
- 39. Mathews PM, Jiang Y, Schmidt SD, Grbovic OM, Mercken M, Nixon RA. Calpain activity regulates the cell surface distribution of amyloid precursor protein. Inhibition of calpains enhances endosomal generation of beta‐cleaved C‐terminal APP fragments. J Biol Chem. 2002;277:36415‐36424. [DOI] [PubMed] [Google Scholar]
- 40. Baghallab I, Reyes‐Ruiz JM, Abulnaja K, Huwait E, Glabe C. Epitomic characterization of the specificity of the anti‐amyloid abeta monoclonal antibodies 6E10 and 4G8. J Alzheimers Dis. 2018;66:1235‐1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Scheuner D, Eckman C, Jensen M, et al. Secreted amyloid beta‐protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864‐870. [DOI] [PubMed] [Google Scholar]
- 42. Fahrenholz F, Gilbert S, Kojro E, Lammich S, Postina R. Alpha‐secretase activity of the disintegrin metalloprotease ADAM 10. Influences of domain structure. Ann N Y Acad Sci. 2000;920:215‐222. [DOI] [PubMed] [Google Scholar]
- 43. Vassar R, Bennett BD, Babu‐Khan S, et al. Beta‐secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735‐741. [DOI] [PubMed] [Google Scholar]
- 44. Wolfe MS. Gamma‐secretase: structure, function, and modulation for Alzheimer's disease. Curr Top Med Chem. 2008;8:2‐8. [DOI] [PubMed] [Google Scholar]
- 45. Portelius E, Olsson M, Brinkmalm G, et al. Mass spectrometric characterization of amyloid‐beta species in the 7PA2 cell model of Alzheimer's disease. J Alzheimers Dis. 2013;33:85‐93. [DOI] [PubMed] [Google Scholar]
- 46. Welzel AT, Maggio JE, Shankar GM, et al. Secreted amyloid beta‐proteins in a cell culture model include N‐terminally extended peptides that impair synaptic plasticity. Biochemistry. 2014;53:3908‐3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Iwata N, Tsubuki S, Takaki Y, et al. Identification of the major Abeta1‐42‐degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143‐150. [DOI] [PubMed] [Google Scholar]
- 48. Bulloj A, Leal MC, Xu H, Castano EM, Morelli L. Insulin‐degrading enzyme sorting in exosomes: a secretory pathway for a key brain amyloid‐beta degrading protease. J Alzheimers Dis. 2010;19:79‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pacheco‐Quinto J, Clausen D, Perez‐Gonzalez R, et al. Intracellular metalloprotease activity controls intraneuronal Abeta aggregation and limits secretion of Abeta via exosomes. FASEB J. 2018;33:3758‐3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sanderson RD, Bandari SK, Vlodavsky I. Proteases and glycosidases on the surface of exosomes: Newly discovered mechanisms for extracellular remodeling. Matrix Biol. 2019;75‐76:160‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Colacurcio DJ, Pensalfini A, Jiang Y, Nixon RA. Dysfunction of autophagy and endosomal‐lysosomal pathways: roles in pathogenesis of Down syndrome and Alzheimer's disease. Free Radic Biol Med. 2018;114:40‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Peng KY, Perez‐Gonzalez R, Alldred MJ, et al. Apolipoprotein E4 genotype compromises brain exosome production. Brain. 2019;142:163‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kaur G, Pawlik M, Gandy SE, Ehrlich ME, Smiley JF, Levy E. Lysosomal dysfunction in the brain of a mouse model with intraneuronal accumulation of carboxyl terminal fragments of the amyloid precursor protein. Mol Psychiatry. 2017;22:981‐989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid‐beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107‐13120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang DS, Stavrides P, Mohan PS, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pardossi‐Piquard R, Lauritzen I, Bauer C, Sacco G, Robert P, Checler F. Influence of genetic background on apathy‐like behavior in triple transgenic AD mice. Curr Alzheimer Res. 2016;13:942‐949. [DOI] [PubMed] [Google Scholar]
- 57. Tamayev R, D'Adamio L. Inhibition of gamma‐secretase worsens memory deficits in a genetically congruous mouse model of Danish dementia. Mol Neurodegener. 2012;7:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Doody RS, Raman R, Farlow M, et al. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013;369:341‐350. [DOI] [PubMed] [Google Scholar]
- 59. Lauritzen I, Becot A, Bourgeois A, et al. Targeting gamma‐secretase triggers the selective enrichment of oligomeric APP‐CTFs in brain extracellular vesicles from Alzheimer cell and mouse models. Transl Neurodegener. 2019;8:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hebert SS, Serneels L, Tolia A, et al. Regulated intramembrane proteolysis of amyloid precursor protein and regulation of expression of putative target genes. EMBO Rep. 2006;7:739‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Muller T, Meyer HE, Egensperger R, Marcus K. The amyloid precursor protein intracellular domain (AICD) as modulator of gene expression, apoptosis, and cytoskeletal dynamics‐relevance for Alzheimer's disease. Prog Neurogibol. 2008;85:393‐406. [DOI] [PubMed] [Google Scholar]
- 62. Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Pimplikar SW. Alzheimer's disease‐like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci U S A. 2009;106:18367‐18372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ghosal K, Stathopoulos A, Pimplikar SW. APP intracellular domain impairs adult neurogenesis in transgenic mice by inducing neuroinflammation. PLoS One. 2010;5:e11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yuyama K, Sun H, Sakai S, et al. Decreased amyloid‐beta pathologies by intracerebral loading of glycosphingolipid‐enriched exosomes in Alzheimer model mice. J Biol Chem. 2014;289:24488‐24498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dinkins MB, Dasgupta S, Wang G, Zhu G, Bieberich E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer's disease. Neurobiol Aging. 2014;35:1792‐1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid‐beta oligomers. Nature. 2009;457:1128‐1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Joshi P, Turola E, Ruiz A, et al. Microglia convert aggregated amyloid‐beta into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014;21:582‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tamboli IY, Barth E, Christian L, et al. Statins promote the degradation of extracellular amyloid {beta}‐peptide by microglia via stimulation of exosome‐associated insulin‐degrading enzyme (IDE) secretion. J Biol Chem. 2010;285:37405‐37414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hong S, Ostaszewski BL, Yang T, et al. Soluble Abeta oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron. 2014;82:308‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gouras GK, Tampellini D, Takahashi RH, Capetillo‐Zarate E. Intraneuronal beta‐amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol. 2010;119:523‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. 2004;44:181‐193. [DOI] [PubMed] [Google Scholar]
- 72. Zott B, Simon MM, Hong W, et al. A vicious cycle of beta amyloid‐dependent neuronal hyperactivation. Science. 2019;365:559‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sardar Sinha M, Ansell‐Schultz A, Civitelli L, et al. Alzheimer's disease pathology propagation by exosomes containing toxic amyloid‐beta oligomers. Acta Neuropathol. 2018;136:41‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yuyama K, Sun H, Usuki S, et al. A potential function for neuronal exosomes: sequestering intracerebral amyloid‐beta peptide. FEBS Lett. 2015;589:84‐88. [DOI] [PubMed] [Google Scholar]
- 75. Fitzner D, Schnaars M, van Rossum D, et al. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J Cell Sci. 2011;124:447‐458. [DOI] [PubMed] [Google Scholar]
- 76. Katsuda T, Tsuchiya R, Kosaka N, et al. Human adipose tissue‐derived mesenchymal stem cells secrete functional neprilysin‐bound exosomes. Sci Rep. 2013;3:1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta‐amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354‐8360. [DOI] [PMC free article] [PubMed] [Google Scholar]