

Abstract
In the last decade, screening compound libraries on live cells has become an important step in drug discovery. The abundance of compounds in these libraries requires effective high‐throughput (HT) analyzing methods. Although current cell‐based assay protocols are suitable for HT analyses, the analysis itself is often restrained to simple, singular outcomes. Incorporation of HT samplers on flow cytometers has provided an interesting approach to increase the number of measurable parameters and increase the sensitivity and specificity of analyses. Nonetheless, to date, the labor intensive and time‐consuming strategies to detach and stain adherent cells before flow cytometric analysis has restricted use of HT flow cytometry (HTFC) to suspension cells. We have developed a universal “no‐touch” HTFC antibody staining protocol in 384‐well microplates to bypass washing and centrifuging steps of conventional flow cytometry protocols. Optimizing culture conditions, cell‐detachment and staining strategies in 384‐well microplates resulted in an HTFC protocol with an optimal stain index with minimal background staining. The method has been validated using six adherent cell lines and simultaneous staining of four parameters. This HT screening protocol allows for effective monitoring of multiple cellular markers simultaneously, thereby increasing informativity and cost‐effectiveness of drug screening. © 2019 The Authors. Cytometry Part A published by Wiley Periodicals LLC. on behalf of International Society for Advancement of Cytometry.
Keywords: compound library screening, high‐throughput screening, flow Cytometry, adherent Cells
The new era of biologicals used in anticancer therapy shifts the focus of therapeutic intervention from achieving cell death to (immune) cell modulation, thereby making cells more susceptible to other compounds or immune clearance. Cell‐based screening of drug libraries has become an important approach in discovering new drugs against these targets of interest. Biochemical assays are more and more replaced by cell‐based assays, as they enable studying underlying cellular mechanisms 1. Many cell‐based assays have been developed in recent years, including functional, reporter, and phenotypic assays. Nonetheless, high‐throughput (HT) cell‐based screening is still limited by labor‐intensive, expensive, and throughput limiting analyzing methods.
Equipment of flow cytometers with HT samplers (both 96‐ and 384‐well format) has made otherwise low‐throughput flow cytometry more suitable as a HT analyzing strategy 2. In this way, whole 96‐well microplates can be analyzed in as little as 15 min. One of the most favorable aspects of flow cytometry is the opportunity to multiplex the measurement of protein expression on the surface or in the cytosol of individual cells. In this way, multiple effects or the underlying mechanisms of drugs can be studied in more detail, contributing to the informativity of drug screens.
The application of HT flow cytometry (HTFC) multiparameter analysis may, however, still be compromised due to labor intensive and time‐consuming staining protocols, especially when analyzing adherent cell types. As a result, HT microscopy (HTM) is currently recommended as the method of choice when analyzing adherent cells 3. It is important to note that different research questions can be answered with HTM compared to HTFC. A major advantage of HTM analysis is that cells can be analyzed in their natural shape and effects of compounds on morphology can be assessed. Disadvantages of HTM are the requisite of multiple wash steps, causing cell loss, the fact that HTM output files are typically 10–100 times bigger and less straightforward to analyze, as well as the restricted potential to multiplex protein expression measurements when compared to HTFC 3. This clearly indicates the potential of a “no‐touch” antibody staining protocol to efficiently use HTFC to analyze protein expression on adherent cells.
A limited number of studies reports the preparation of adherent cells for HTFC 4, 5, 6, 7, 8, 9, 10. The published protocols either involve one or more washing steps or use trypsin/EDTA to generate single‐cell suspensions 4, 5, 6, 7. Washing steps may cause loss of cells, thereby hampering HT screening protocols, whereas trypsin is reported to potentially cause loss of surface antigen expression 8, 9. A recent article by Kaur and Esau 10 describes a two‐step protocol to prepare adherent cells for HTFC. They show that the use of EDTA as a cell detachment reagent bypasses the need of washing, enzymatic inactivation, centrifugation, and transfer between plates, reducing cell loss and labor‐intensity of the protocol 10. Even though the report shows the 384‐well protocol is compatible with several commercially available dyes to measure, for instance, apoptosis and production of reactive oxygen species, there is no data on the use of antibody staining to detect the dynamics of protein expression within or on the surface of the 384‐well microplate seeded cells. The described universal protocol allows for antibody staining in 384‐well format and is validated with six adherent cell lines, including neuroblastoma, cervical, hepatocellular, and breast cancer lines. We demonstrate that there is no difference in staining effectiveness between single and multiple stained samples, showing the potential of adherent‐cell HTFC to increase the informativity of HT screening protocols.
Material and Methods
Cell Lines and Reagents
MCF‐7 (human breast adenocarcinoma; ATCC HTB‐22), SKBR3 (human breast adenocarcinoma; ATCC HTB30), HepG2 (human hepatocellular carcinoma; ATCC HB‐8065), HeLa (human cervical adenocarcinoma; ATCC CCL‐2), and HEK‐293 T (human embryonic kidney; ATCC CRL‐3216) cells were obtained from ATCC (Manassas, VA). The GIMEN neuroblastoma cell line was obtained from the Academic Medical Center of Amsterdam. GIMEN NFkB reporter cells were generated as previously described 11. SKBR3 cells were maintained in RPMI 1640 GlutaMAX supplement medium (Life Technologies, Carlsbad, CA), supplemented with 10% FCS (Sigma‐Aldrich, Steinheim, Germany) and 1% penicillin/streptomycin (50 U/ml, Life Technologies). GIMEN, HeLa, and HEK‐293 T were maintained in Dulbecco's Modified eagle medium (DMEM) GlutaMAX supplement medium (Life Technologies), supplemented with 10% FCS (Sigma‐Aldrich) and 1% penicillin/streptomycin (50 U/ml, Life Technologies).
Cell Plating and Compound Addition
Cells were cultured in T75 flasks until 80% confluency, detached with 0.05% Trypsin/EDTA (Life Technologies), and counted using the Countess automated cell counter (Life Technologies). Optimal seeding density was determined. Cells were plated in a culture volume of 15 μl in a low flange, polystyrene, tissue culture treated, flat bottom 384‐well tissue‐culture treated microplate (stock number: 3764, lot: 22017037, Corning, NY) using a multidrop combi reagent dispenser (Life Technologies). Cells were cultured for 16–24 h under standard culturing conditions (5% CO2, 37°C), after which 5 μl of compound library would normally have been added to the cells using a liquid handling system, for example, the Sciclone G3 liquid handling system (PerkinElmer, Waltham, MA). As a proof of principle for this article, we added TNF‐α and IFN‐γ as a positive control for upregulation of the markers of interest 11. TNF‐α was added at a final concentration of 50 ng/μL (Miltenyi Biotec, Bergisch Gladbach, Germany), IFN‐γ at a final concentration of 1,000 U/ml (R&D, Abingdon, UK). Cells were incubated in the presence of compounds for 16–24 h (5% CO2, 37°C) after which the effect on the protein(s) of interest was measured with HTFC.
Sample Preparation for HTFC
EDTA (Life Technologies) was diluted with deionized H2O (pH = 6.14), after which the optimal EDTA concentration was determined. Addition of 5 μl 15 mM EDTA per well resulted in an optimal EDTA concentration of 3 mM per well. EDTA was added to the plate and after shaking at 1000 RPM with an orbital shaker (Heidolph Titramax 1,000; Schabwach, Germany) for 30 s, incubated at 37°C for 45 min to allow detachment of the cells. Plates were shaken again at 1000 RPM for 30 s, after which 5 μl antibody, diluted in serum‐free medium, was added in the concentration established with titration. The following monoclonal antibodies have been used: AlexaFluor‐647‐labeled mouse‐anti‐human HLA‐ABC (W6/32; Biolegend, London, UK), FITC‐labeled mouse‐anti‐human HLA‐ABC (W6/32, Sony Biotechnology, Weybridge, UK), APC‐labeled mouse‐anti‐human CD274 (PD‐L1) (Clone MIH1, Life Technologies), and PE‐labeled mouse‐anti‐human CD54 (ICAM‐1) (MEM‐111, Exbio, London, UK). The nucleic acid dye 7‐AAD (BD Biosciences, Eysins, Switzerland) was used for exclusion of nonviable cells. Gating was based on unstained samples and verified using conventional flow cytometry. Cell viability has been validated with the mitochondrial membrane potential dye tetramethylrhodamine (TMRM) at a concentration of 50 nM (Sigma‐Aldrich). Addition of 50 uM carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone (FCCP), a mitochondrial oxidative phosphorylation uncoupler (Sigma‐Aldrich), was used as a negative control for TMRM staining. Cells were incubated for 20 min on an orbital shaker at 4°C to allow for antibody staining (300 RPM). Subsequently, 50 μl of PBS supplemented with 2% FCS and 2 μM EDTA was added per well using the multidrop combi reagent dispenser to dilute the samples. Plates are kept on ice until analysis.
Flow Cytometry and Analysis
Cells were acquired on the FACSCanto II HT sampler (BD Biosciences), by measuring a fixed volume of 50 μl sample per well at a flow rate of 3 μl/s. Analysis of a full 384‐well plate will take about 100 min. Fluorescent‐labeled beads (CS&T beads, Becton Dickinson) were used to check the performance and verify the optical path and stream flow of the flow cytometer. This procedure enables controlled standardized results and allows the determination of long‐term drifts and incidental changes within the flow cytometer. No changes were observed which could affect the results. The data shown is a representation of at least six independent experiments. The Stain Index (SI) was used to reflect the ratio of separation between the positive and the negative population divided by two times the standard deviation (SD) of the negative population 12. Data were analyzed using FACS Diva Version 8.0.1 (BD Bioscience), FlowJo version 10.1, and Graphpad Prism version 7.
Statistical Analysis
The nonparametric Mann–Whitney U‐test was performed for statistical testing between single‐cell counts before and after optimization. P‐values <0.05 are considered significant. A Z‐score was calculated to define the difference in fluorescent intensity between medium‐ and cytokine‐treated samples (n = 8 per group) using the following equation:
In which X is the mean fluorescent intensity (MFI) of the cytokine treated group, μ is the mean MFI of the medium control group, and σ is the standard deviation of the medium control group. All data shown ±SD.
Results
Optimization of Cell Seeding Density, EDTA Concentration, and Cell Density during Analysis Results in a 12‐Fold Increase in Single‐Cell Retrieval
The first goal in the development of this HTFC protocol was to find a strategy to optimize reproducible cell retrieval, using the adherent GIMEN neuroblastoma cell line. Initially, we adapted the cell detachment protocol of Kaur and Esau to a 384‐well format 10 but were unable to achieve sufficient and reproducible cell retrieval (Fig. 1A, before optimization).
Figure 1.
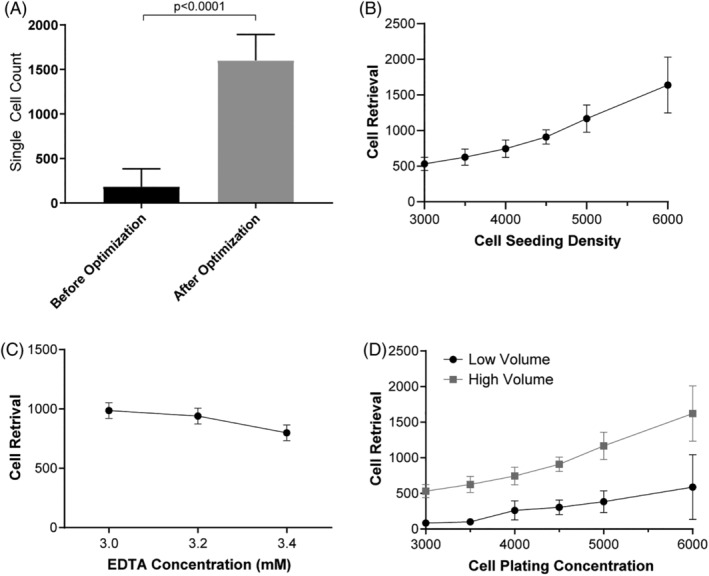
Optimization of flow cytometric cell retrieval using GIMEN cells. An over 12‐fold increase in single‐cell retrieval is observed upon sample preparation optimization. (A) Bar graph representing average single‐cell retrieval prior to and after optimization. Before optimization: n = 60, after optimization: n = 7,153. (B) Graphical display of flow cytometric cell retrieval when increasing cell‐seeding density. (C) Graphical display of cell retrieval after incubation with increasing EDTA concentrations at a seeding density of 4,500 cells/well, n = 2 per group. (D) Cell retrieval when well volume is 30 μL (low volume) or 80 μl (high volume). Graphs: Dots reflect mean, error bars reflect SD between samples, n = 6 per group unless otherwise indicated. Mann–Whitney U‐test was performed, p < 0.05 was considered significant. SD = standard deviation.
First, cell seeding density was evaluated by seeding increasing numbers of cells per well. As expected, cell retrieval markedly improved when more cells were plated (Fig. 1B). However, reproducibility of cell retrieval decreased when seeding density exceeded 5,000 cells/well, as observed by an increase in SD. Based on these data, it was concluded that a cell seeding density of 4,500 cells/well was optimal. Second, microscopic evaluation of the cell suspensions after different lengths of incubation periods with increasing EDTA concentrations revealed a minimum incubation time of 45 min and a minimum EDTA concentration of 3 mM (data not shown). Further increase in the EDTA concentration to 3.2 and 3.4 mM did not result in further improvement of cell retrieval (Fig. 1C). Finally, we assessed the effect of sample dilution prior to flow cytometric analysis. Dilution of the samples with 50 μL PBS supplemented with 2% FCS + 2 μM EDTA resulted in a 3.8‐fold increase in cell retrieval (Fig. 1D). Optimal cell seeding density, EDTA cell detachment concentration and incubation times, and final cell suspension density of the flow cytometry sample per well resulted in an over 12‐fold increase in single‐cell retrieval (Fig. 1A). More than 90% of the nondebris cell population were single cells and flow rate was constant. 7‐AAD and TMRM staining confirmed the cells were alive (Supporting Information S1).
The reproducibility of the cell numbers retrieved with the protocol can be concluded from a HTFC compound screen we have performed utilizing this protocol, in which over 10,000 wells were analyzed with an average retrieval of 1,600 (±SD 294) alive single cells/well. This corresponds to 74% alive single cell retrieval when corrected for sampled volume.
Optimized Antibody Staining Allows for (Multiplexed) Staining with Minimal Nonspecific Background Signal
Elimination of all washing steps from the HTFC protocol contributes to the HT nature of the protocol by decreasing the labor intensity, while minimizing cell loss inherent to washing. On the other hand, elimination of these steps also clearly indicates the need for antibody concentration titration. TNF‐α and IFN‐γ are involved in the upregulation of cell surface protein expression of MHC‐I 11, CD54 (ICAM‐1) (13, 14), and CD274 (PD‐L1) 15 in several tumor cell types. Effects of these cytokines on protein expression have been validated for all utilized cell lines using conventional flow cytometry (shown for GIMEN; Fig. 2A and Supporting Information S2).
Figure 2.
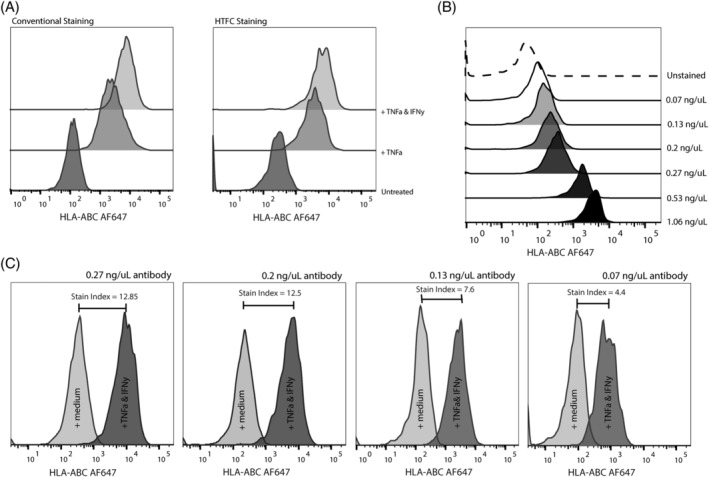
Antibody staining optimization in GIMEN cells. (A) The optimized HTFC staining protocol (right) shows similar expression patterns to a typical conventional staining protocol (left). Z‐score of expression in untreated versus TNF‐α‐treated cells is 49 (X = 1911, μ = 217, σ = 35), and 94 (X = 3,466, μ = 217, σ = 35) in TNF‐α + IFN‐γ treated cells (n = 8 per group). (B) HLA‐ABC background staining decreases when antibody concentration (ng/μL) decreases. The dashed line represents the unstained control. (C) Histograms depicting HLA‐ABC MFIs of untreated and TNF‐α + IFN‐γ‐treated samples with diluting antibody concentrations. The stain index decreases when antibody concentration is reduced. Data shown are from a representative experiment using the HTFC protocol on GIMEN neuroblastoma cells. MFI = mean fluorescent intensity.
Decreasing the antibody concentration caused a clear decrease in background staining in medium‐treated HLA‐ABC stained MHC‐I lacking GIMEN cells (Fig. 2B). However, a marked decrease in the stain index between untreated and TNF‐α + IFN‐γ treated samples was observed when decreasing the antibody concentration (Fig. 2C). This indicated a delicate balance between nonspecific background staining and discriminative ability of the antibody staining. Titration showed that HLA‐ABC antibody concentration was optimal at a concentration of 8 ng/well (final concentration of 0.27 ng/μL). The HTFC staining protocol allows for distinct discrimination between HLA‐ABC expression in untreated versus TNF‐α‐treated (Z = 49) and versus TNF‐α + IFN‐γ treated cells (Z = 94) (n = 8 per group), comparable with results from a typical conventional staining protocol (Fig. 2A). The same effect is observed for the other utilized antibodies (Supporting Information S2).
A unique aspect of flow cytometry is the opportunity to multiplex expression analysis of proteins on/in the same cell. Combining this aspect with HTS allows for an opportunity to increase the possibilities and informativity of HTS analysis. Combining antibody staining of HLA‐ABC with the nucleic acid dye 7‐AAD and antibody staining against PD‐L1 and ICAM‐1 revealed no noticeable differences in individual staining efficacy of the HTFC protocol, as shown for HLA‐ABC antibody staining (Fig. 3A).
Figure 3.
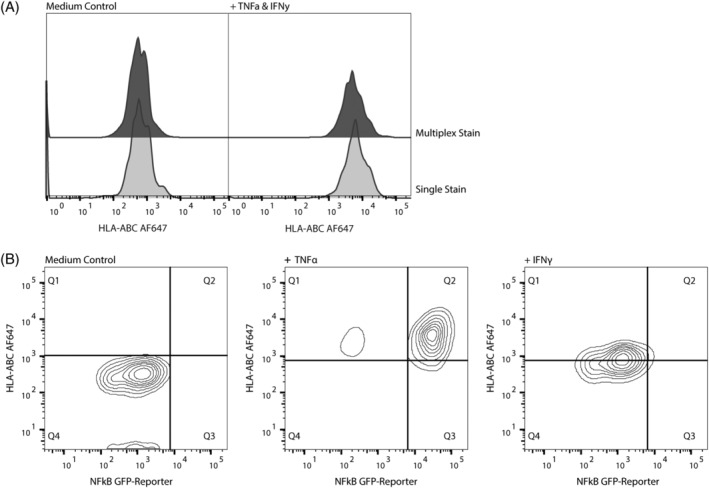
Multiplexed antibody staining in GIMEN cells. Multiplexing antibody staining using the HTFC protocol is technically feasible and as effective as singular staining. (A) HLA‐ABC MFI in untreated controls (left) and TNF‐α + IFN‐γ‐treated cells (right). Top graphs show the MFI in multiplexed stained samples, lower graphs show MFIs in single stained samples. (B) NFkB‐GFP‐reporter × HLA‐ABC. Treatment of GIMEN NFkB GFP reporter cells with TNF‐α or IFN‐γ shows dependence of TNF‐α induced upregulation of MHC‐I, and independence of IFN‐γ induced upregulation. Left graph: untreated control, middle graph: TNF‐α‐treated cells, right graph: IFN‐γ‐treated cells. Data shown are from a representative experiment using the HTFC protocol on GIMEN neuroblastoma cells. MFI = mean fluorescent intensity.
We previously generated a GIMEN NFkB reporter cell line and found that TNF‐α upregulates MHC‐I expression in an NFkB‐dependent manner, whereas IFN‐γ‐induced MHC‐I upregulation is independent of NFkB 11. Utilizing our HTFC protocol combining HLA‐ABC antibody staining with evaluation of the intrinsic NFkB reporter expression confirmed NFkB (in)dependency of the observed MHC‐I upregulations (Fig. 3B). This shows that the protocol is also suitable to study the effects of compounds on intracellular transcription factors using reporter cell lines.
The Staining Protocol is Translatable to Multiple Cell Lines without any Modifications
The optimized HTFC protocol was subsequently performed using five additional cell lines, including breast cancer, cervical cancer, hepatocellular cancer, and human embryonal kidney cells. Retrieved single cell counts were lower (HepG2), comparable (MCF‐7, SKBR3), or superior (HeLa, HEK293T) to the counts obtained in GIMEN neuroblastoma cells (Supporting Information S3). Individual and multiplex HTFC staining was performed and validated with conventional flow cytometry staining (data not shown). The HepG2 and MCF‐7 cell lines were selected based on their trypsinization resistant nature. The MCF‐7 line shows sufficiently high and reproducible cell retrieval, even though we do observe more doublets, which were excluded from analysis (Supporting Information S3A). In contrast, the slower growing, clumping HepG2 cells showed decreased cell counts, with an average single cell retrieval of 814 (n = 44, two individual experiments), but the counts were still sufficiently high and reproducible for reliable results (Supporting Information S3E). This indicates the potential to translate the HTFC protocol universally to other adherent cell lines of interest, contributing to the versatility of the protocol.
Discussion and Conclusion
HT cell‐based screening of drug libraries is currently still limited by labor‐intensive, time‐consuming, expensive, and throughput limiting analyzing methods. The new era of biologicals used in anticancer therapy, including (immune) cell modulating biologicals, asks for novel, more delicate HT analyzing protocols to screen for effects beyond cell death. Cell surface expression of proteins is often key in treatment response to (immuno)therapies in cancer (11, 16, 17, 18, 19, 20, 21). Screening for compounds affecting expression of these proteins may contribute to therapy efficacy in the future. For example, neuroblastoma immunotherapy efficacy is hampered by low MHC‐I expression and requires upregulation 11, for which potential compounds can be selected by a compound screen. Similarly, novel compounds may affect immune checkpoint regulator expression as expression is correlated with poor survival in multiple cancers 16, 17, 18, 19, 20, 21. Here, we report the development of a universal “no‐touch” HTFC antibody staining protocol in 384‐well microplate format for adherent cells in which we are able to bypass washing and centrifuging steps of conventional flow cytometry protocols.
We have adapted a protocol from Kaur and Esau, in which the potential of EDTA as a nonenzymatic cell detachment agent in “no‐touch” HTFC was demonstrated 10. Using EDTA instead of enzymatic detachment agents bypasses the need of indispensible wash steps to wash away the fetal calf serum and to neutralize the enzymatic activity to decrease cell toxicity and potential antigen loss 4, 5, 6, 7, 8, 9. Bypassing washing steps not only contributes to the HT format of the protocol, but it also contributes to the cell retrieval by preventing cell loss, which is especially a problem when working with small cell numbers. Even though Kaur and Esau show their 384‐well protocol is compatible with several commercially available dyes, they have not optimized the 384‐well format protocol in combination with (multiplexed) antibody staining 10. Furthermore, they show a clear need for cell line specific optimization of their protocol, limiting the throughput potential of the protocol.
Based on our data, decreasing the cell density prior to flow cytometric analysis markedly increased cell retrieval (3.8‐fold). This indicates that cell density during analysis is a crucial factor in this protocol: when cell density is too high, the EDTA cannot avoid the tendency of cells to clump, thereby affecting (reproducibility of) cell retrieval. This also explains the reduction in reproducibility of the protocol when exceeding a seeding density of 5,000 cells/well. The fact that cell density rather than other cellular parameters is so critical in this protocol also emphasizes the universal potential of this protocol.
Analysis of the very slow growing and clumping HepG2 cell line with our protocol showed decreased cell retrieval when compared to the other cell lines. We believe this can be explained by the extreme adherent nature of the cells, as well as by the slow growth rate. This was further confirmed by an ~1.5‐fold increase in average cell retrieval (average single‐cell count = 1,115 [± SD = 236] [n = 9]), without a marked increase in variability between wells when increasing the plating density to 5,000 cells/well. Even though the unmodified protocol still gave sufficient cell retrieval, these results indicate that the protocol might benefit from cell plating density titration when analyzing slow‐growing cell lines. However, the extreme clumping nature of the HepG2 line makes its suitability questionable for flow cytometry analysis in general, and alternative hepatocyte cell lines may be preferred.
It has been reported that EDTA could have a significant impact on antibody binding capacity 22, especially when the structure of the epitope depends on ions, such as calcium. It is therefore of importance to compare conventional and high‐throughput protocol staining for every newly utilized antibody. Furthermore, more general, we recommend to include appropriate controls for every utilized antibody on every HTS plate to be able to monitor basal protein expression and antibody staining efficiency.
To the best of our knowledge, we are the first to report a universal “no‐touch” HTFC antibody staining protocol for adherent cell lines in 384‐well microplate format. We show that our protocol allows for multiplexing antibody staining and addition of reporter gene analysis, thereby improving the output of cell‐based screening of drug libraries in a cost‐efficient manner.
Author Contributions
C.S., S.N., and A.C. designed the study, and A.C. wrote the manuscript. C.S. and A.C. performed the experiments and A.C. analyzed the data with critical comments from C.S,. J.V., N.T., S.N., and J.J.B. All authors read and approved the manuscript.
Conflict of Interest
The authors declare no competing financial interest.
Supporting information
MIFlowCyt MIFlowCyt‐Compliant Items
Supplementary1 – Flow cytometric characteristics of GIMEN cells after protocol optimization.
(A) FSC/SSC after optimization, gate reflects the non‐debris population (total cell retrieval = 1,584, single cell retrieval = 1,512); (B) FSC‐W/FSC‐A graph of the non‐debris population. Gate contains the single cell population (95.5% of non‐debris population is single cell). (C) FSC‐H/time were plotted against each other to detect potential drifts. Flow rate is constant. (D) 7‐AAD staining of single cell population. The gate contains the alive cell population (98.2% of the single cells are alive). (E) TMRM staining of single cell population. TMRM staining intensity in unstained cells (bottom), TMRM‐stained cells (middle), and TMRM stained cells to which the mitochondrial oxidative phosphorylation uncoupler FCCP is added (top). Data shown are from a typical experiment using the HTFC protocol on GIMEN neuroblastoma cells and is representative of at least six independent experiments.
Supplementary2 – PD‐L1 and CD54 staining using the conventional and HTFC protocol show similar results. The optimized HTFC staining protocol shows similar PD‐L1 (A) and CD54 (B) expression patterns (right) to a typical conventional staining protocol (left). Z‐score of PD‐L1 expression in untreated versus TNF‐α treated cells is 14 (X = 3,453, μ = 978, σ = 175), and 23 (X = 5,081, μ = 978, σ = 175) in TNF‐α + IFN‐γ treated cells (n = 3 per group). Z‐score of CD54 expression between untreated versus TNF‐α treated cells is 151 (X = 2,511, μ = 205, σ = 15), and 236 (X = 3,817, μ = 205, σ = 15) between TNF‐α + IFN‐γ treated cells (n = 3 per group).Data shown are from a representative experiment using the HTFC protocol on GIMEN neuroblastoma cells.
Supplementary 3– Cell retrieval and HLA‐ABC antibody staining of additional analyzed cell lines analyzed with the unmodified HTFC staining protocol. Left: FSC/SSC of MCF‐7 (A), SKBR3 (B), HEK‐293 T (C), HeLa (D), and HepG2 (E) cell lines, gate reflects the non‐debris population. Single cell retrieval is based on exclusion via FSC‐W/FSC‐A characteristics (data not shown). Cells outside the non‐debris gate are confirmed to be doublets. Middle: Viability of MCF‐7 (A), SKBR3 (B), HEK‐293 T (C), and HeLa (D), and HepG2 (E) cell lines. Gating is based on unstained controls of the respective cell lines. Right: HLA‐ABC staining intensity in untreated controls (bottom), TNF‐α (middle) or TNF‐α + IFN‐γ (top) treated MCF‐7(A), SKBR3 (B), HEK‐293 T (C), HeLa (D), and HepG2 (E) cell lines. Data shown are from a representative experiment using the HTFC protocol on the respective cell line.
Acknowledgment
This work was supported by the Villa Joep Foundation [IWOV‐Actief.51391.180034].
Literature Cited
- 1. An WF, Tolliday N. Cell‐based assays for high‐throughput screening. Mol Biol 2010;45:180–186. 10.1007/s12033-010-9251-z. [DOI] [PubMed] [Google Scholar]
- 2. Picot J, Guerin CL, Kim CLV, Boulanger CM. Flow cytometry: Retrospective, fundamentals and recent instrumentation. Cytotechnology 2012;64(2):109–130. 10.1007/s10616-011-9415-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Black CB, Duensing TD, Trinkle LS, Dunlay RT. Cell‐based screening using high‐throughput flow cytometry. Technol Rev 2011;9(1):13–20. 10.1089/adt.2010.0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haynes MK, Strouse JJ, Waller A, Leitao A, Curpan RF, Bologa C, Oprea TI, Prossnitz ER, Edwards BS, Sklar LA, et al. Detection of intracellular granularity induction in prostate cancer cell lines by small molecules using the HyperCyt® high‐throughput flow cytometry system. J Biomed Screen 2009;14:596–609. 10.1177/1087057109335671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Young SM, Bologa CM, Fara D, Bryant BK, Strouse JJ, Arterburn JB, Ye RD, Oprea TI, Prossnitz ER, Sklar LA, et al. Duplex high throughput flow cytometry screen identifies two novel formylpeptide receptor family probes. Cytometry 2009;75A:253–263. 10.1002/cyto.a.20645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Glazer ES, Massey KL, Curley SA. A protocol to effectively create single cell suspensions of adherent cells for multiparameter high‐throughput flow cytometry. In Vitro Cell Dev Biol 2010;46(2):97–101. 10.1007/s11626-009-9256-8. [DOI] [PubMed] [Google Scholar]
- 7. Martinez EM, Klebanoff SD, Secrest S, Romain G, Haile ST, Emtage PCR, Gilbert AE. High‐throughput flow cytometric method for the simultaneous measurement of CAR‐T cell characterization and cytotoxicity against solid tumor cell lines. SLAS Discov 2018;23(7):603–612. 10.1177/2472555218768745. [DOI] [PubMed] [Google Scholar]
- 8. Tsuji K, Ojima M, Otabe K, Ho rie M, Koga H, Sekiya I, Menuta T. Effects of different cell‐detaching methods on the viability and cell surface antigen expression of synovial mesenchymal stem cells. Cell Transplant 2017;26(6):1089–1102. 10.3727/096368917X694831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang HL, Hsing HW, Lai TC, Chen YW, Lee TR, Chan HT, Lyu PC, Wu CL, Lu YC, Lin ST, et al. Trypsin‐induced proteome alteration during cell subculture in mammalian cells. J Biomed Sci 2010;17(1):36 10.1186/1423-0127-17-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kaur M, Esau L. Two‐step protocol for preparing adherent cells for high‐throughput flow cytometry. Biotechniques 2015;59:119–126. 10.2144/000114325. [DOI] [PubMed] [Google Scholar]
- 11. Spel L, Nieuwenhuis J, Haarsma R, Stickel E, Bleijerveld OB, Altelaar M, Boelens JJ, Brummelkamp TR, Nierkens S, Boes M. Nedd4 binding protein 1 (N4BP1) and TNFAIP3 interacting protein 1 (TNIP1) control MHC‐1 display in neuroblastoma. Cancer Res 2018;78:6621–6631. 10.1158/0008-5472.CAN-18-0545. [DOI] [PubMed] [Google Scholar]
- 12. Maecker HT, Frey T, Nomura LE, Trotter J. Selecting fluorochrome conjugates for maximum sensitivity. Cytometry A 2004;62A(2):169–173. 10.1002/cyto.a.20092. [DOI] [PubMed] [Google Scholar]
- 13. Chang YJ, Holtzman MJ, Chen CC. Interferon‐γ‐induced epithelial ICAM‐1 expression and monocyte adhesion. J Biol Chem 2002;277(9):7118–7126. 10.1074/jbc.M109924200. [DOI] [PubMed] [Google Scholar]
- 14. Ren G, Zhao X, Zhang L, Zhang J, L'Huillier A, Ling W, Roberts AI, Le AD, Shi S, Shao C, et al. Inflammatory cytokine‐induced intercellular adhesion molecule‐1 and vascular cell adhesion molecule‐1 in mesenchymal stem cells are critical for immunosuppression. J Immunol 2010;184:2321–2328. 10.4049/jimmunol.0902023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dondero A, Pastorino F, Della Chiesa M, Corrias MV, Morandi F, Pistoia V, Olive D, Bellora F, Locatelli F, Castellano A, et al. PD‐L1 expression in metastatic neuroblastoma as an additional mechanism for limiting immune surveillance. Oncoimmunology 2016;5(1):e1064578 10.1080/2162402X.2015.1064578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hino R, Kabashima K, Kato Y, Yagi H, Nakamura M, Honjo T, Okazaki T, Tokura Y. Tumor cell expression of programmed cell death‐1 ligand 1 is a prognostic factor for malignant melanoma. Cancer 2010;116(7):1757–1766. 10.1002/cncr.24899. [DOI] [PubMed] [Google Scholar]
- 17. Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L. Association of PD‐1, PD‐1 ligands, and other features of the tumor immune microenvironment with response to anti‐PD‐1 therapy. Clin Cancer Res 2014;20(19):5064–5074. 10.1158/1078-0432.CCR-13-3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pistillo MP, Tazzari PL, Palmisano GL, Pierri I, Bolognesi A, Ferlito F, Capanni P, Polito L, Ratta M, Pileri S, et al. CTLA‐4 is not restricted to the lymphoid cell lineage and can function as a target molecule for apoptosis induction of leukemic cells. Blood 2003;101(1):202–209. 10.1182/blood-2002-06-1668. [DOI] [PubMed] [Google Scholar]
- 19. Paulsen EE, Thomas KK, Rakaee M, Richardsen E, Hald SM, Andersen S, Busund LT, et al. CTLA‐4 expression in the non‐small cell lung cancer patient tumor microenvironment: Diverging prognostic impact in primary tumors and lymph node metastases. Cancer Immunol Immunother 2017;66(11):1449–1461. 10.1007/s00262-017-2039-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zaretsky JM, Garcia‐Diaz A, Shin DS, Escuin‐Ordinas H, Hugo W, Hu‐Lieskovan S, Torrejon DY, Abril‐Rodriguez G, Sandoval S, Barthly L, et al. Mutations associated with acquired resistance to PD‐1 blockade in melanoma. N Engl J Med 2016;375(9):819–829. 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pfirschke C, Engblom C, Rickelt S, Cortez‐Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier‐Colame V, Newton A, Redouane Y, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 2016;44(2):343–354. 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Giroux M, Denis F. Influence of calcium ions in the flow cytometric analysis of human CD8‐positive cells. Cytometry 2004;62A:61–64. 10.1002/cyto.a.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MIFlowCyt MIFlowCyt‐Compliant Items
Supplementary1 – Flow cytometric characteristics of GIMEN cells after protocol optimization.
(A) FSC/SSC after optimization, gate reflects the non‐debris population (total cell retrieval = 1,584, single cell retrieval = 1,512); (B) FSC‐W/FSC‐A graph of the non‐debris population. Gate contains the single cell population (95.5% of non‐debris population is single cell). (C) FSC‐H/time were plotted against each other to detect potential drifts. Flow rate is constant. (D) 7‐AAD staining of single cell population. The gate contains the alive cell population (98.2% of the single cells are alive). (E) TMRM staining of single cell population. TMRM staining intensity in unstained cells (bottom), TMRM‐stained cells (middle), and TMRM stained cells to which the mitochondrial oxidative phosphorylation uncoupler FCCP is added (top). Data shown are from a typical experiment using the HTFC protocol on GIMEN neuroblastoma cells and is representative of at least six independent experiments.
Supplementary2 – PD‐L1 and CD54 staining using the conventional and HTFC protocol show similar results. The optimized HTFC staining protocol shows similar PD‐L1 (A) and CD54 (B) expression patterns (right) to a typical conventional staining protocol (left). Z‐score of PD‐L1 expression in untreated versus TNF‐α treated cells is 14 (X = 3,453, μ = 978, σ = 175), and 23 (X = 5,081, μ = 978, σ = 175) in TNF‐α + IFN‐γ treated cells (n = 3 per group). Z‐score of CD54 expression between untreated versus TNF‐α treated cells is 151 (X = 2,511, μ = 205, σ = 15), and 236 (X = 3,817, μ = 205, σ = 15) between TNF‐α + IFN‐γ treated cells (n = 3 per group).Data shown are from a representative experiment using the HTFC protocol on GIMEN neuroblastoma cells.
Supplementary 3– Cell retrieval and HLA‐ABC antibody staining of additional analyzed cell lines analyzed with the unmodified HTFC staining protocol. Left: FSC/SSC of MCF‐7 (A), SKBR3 (B), HEK‐293 T (C), HeLa (D), and HepG2 (E) cell lines, gate reflects the non‐debris population. Single cell retrieval is based on exclusion via FSC‐W/FSC‐A characteristics (data not shown). Cells outside the non‐debris gate are confirmed to be doublets. Middle: Viability of MCF‐7 (A), SKBR3 (B), HEK‐293 T (C), and HeLa (D), and HepG2 (E) cell lines. Gating is based on unstained controls of the respective cell lines. Right: HLA‐ABC staining intensity in untreated controls (bottom), TNF‐α (middle) or TNF‐α + IFN‐γ (top) treated MCF‐7(A), SKBR3 (B), HEK‐293 T (C), HeLa (D), and HepG2 (E) cell lines. Data shown are from a representative experiment using the HTFC protocol on the respective cell line.