

Abstract
Photodynamic therapy (PDT) leads to cancer remission via the production of cytotoxic species under photosensitizer (PS) irradiation. However, concomitant damage and dark toxicity can both hinder its use. With this in mind, we have implemented a versatile peptide‐based platform of bioorthogonally activatable BODIPY‐tetrazine PSs. Confocal microscopy and phototoxicity studies demonstrated that the incorporation of the PS, as a bifunctional module, into a peptide enabled spatial and conditional control of singlet oxygen (1O2) generation. Comparing subcellular distribution, PS confined in the cytoplasmic membrane achieved the highest toxicities (IC50=0.096±0.003 μm) after activation and without apparent dark toxicity. Our tunable approach will inspire novel probes towards smart PDT.
Keywords: bioorthogonal reactions, BODIPY, peptides, photodynamic therapy, tetrazine
Modular peptide‐based platform for organelle‐confinement of the photodynamic effect. We report a general approach capable of directing the bioorthogonal activation of tetrazine‐BODIPY photosensitizers to specific subcellular organelles.
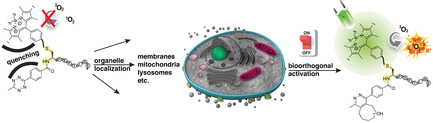
Introduction
Recent years have witnessed an immense growth in interest in the photoregulation of biological events1 because light offers the possibility of exerting remote control of cellular functions and, consequently, cures for a variety of diseases too. A traditional clinical treatment based on light exposure is photodynamic therapy (PDT),2 which is nowadays the standard treatment for several illnesses,3 including some cancers.4 PDT requires only three primary components to trigger cell death: an efficient photosensitizer (PS), light, and in situ production of radicals5 and/or highly reactive oxygen species (ROS)6 as cytotoxic agents. During the last decade, we have moved from constitutively active PSs to smart compounds,7 whose toxicity is triggered by controlled stimuli such as environmental conditions (pH,8 glutathione concentration,9 enzymes,9b, 10 and cations11), electrostatic assemblies,12 oligonucleotide displacement,13 aptamer recognition,14 as well as photochromic switching.15 These activatable photosensitizers (aPSs) overcome one of the biggest limitations of PDT: dark toxicity. Last year, we first introduced a novel type of aPSs based on halogenated BODIPY‐tetrazine (mTz‐2I‐BODIPY, 1OFF) probes (Figure 1).16 We demonstrated that the direct incorporation of the tetrazine moiety into the core of the 2I‐BODIPY PS results in an efficient FRET deactivation of the 1O2 production. Importantly, the phototoxic effect can be restored via the bioorthogonal inverse‐electron‐demand Diels–Alder (iEDDA) reaction in presence of a dienophile such as trans‐cyclooctenol (TCO) (Figure 1).
Figure 1.
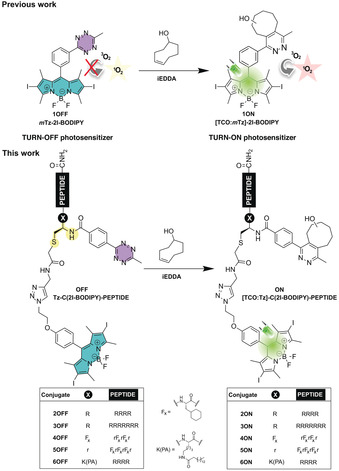
Structures of our precedent work on the unimodular turn‐off mTz‐2I‐BODIPY (1OFF) photosensitizer16 (PS) and the turn‐on [TCO:mTz]‐2I‐BODIPY (1ON) PS, as well as the novel modular peptide‐based turn‐off Tz‐C(2I‐BODIPY)‐PEPTIDE (2‐6OFF) PSs and turn‐on [TCO:Tz]‐C(2I‐BODIPY)‐PEPTIDE (2‐6ON) analogues from this project.
Now, we further expand this concept by exploring a modular platform, in which the halogenated BODIPY and the quencher tetrazine are separately integrated into a peptide scaffold via a cysteine residue (Figure 1, Tz‐C(2I‐BODIPY)‐PEPTIDE, 2‐6OFF). As yet, there are only few examples of BODIPY‐peptide conjugates in PDT;17 they have been mostly used as fluorescent probes.18 However, if the new molecular arrangement maintains the precise control of the FRET process, peptide bioconjugation may not only improve the properties of our aPS in line with other hydrophobic PSs,19 but since peptide signalling sequences are powerful delivery vehicles,20 may also direct photodynamic action to specific subcellular locations. Notably, the efficiency of cell photodamage is strongly determined by the PS biodistribution.21 To investigate our hypothesis, we aimed for the synthesis and study of five bioorthogonally aPS peptide conjugates (Figure 1, Tz‐C(2I‐BODIPY)‐PEPTIDE, 2‐6OFF) and the corresponding active PS analogues: [TCO:Tz]‐C(2I‐BODIPY)‐PEPTIDE, 2‐6ON. We selected the conventional polyarginines (Rn) composed of five (R5) (Tz‐C(2I‐BODIPY)‐R5, 2OFF and [TCO:Tz]‐C(2I‐BODIPY)‐R5, 2ON) or eight (R8) (Tz‐C(2I‐BODIPY)‐R8, 3OFF and [TCO:Tz]‐C(2I‐BODIPY)‐R8, 3ON) consecutive arginine residues capable of delivering cargos to the cytoplasm and/or nucleus,22 as well as two mitochondrial‐penetrating peptides (MPPs) from Kelley's lab (Tz‐C(2I‐BODIPY)‐MPP1, 4OFF and [TCO:Tz]‐C(2I‐BODIPY)‐MPP1, 4ON; Tz‐C(2I‐BODIPY)‐MPP2, 5OFF and [TCO:Tz]‐C(2I‐BODIPY)‐MPP2, 5ON), as our targeting vectors. The latter peptides, MPP123 and MPP2,24 are based on the combination of two unnatural amino acids: cyclohexylalanine (Fx) and d‐arginine (r), the difference between them is only a single extra d‐arginine residue in MPP2. We also pursued the preparation of a conjugate pair embedded in the cell membrane. To achieve the appropriate lipophilicity,25 we planned to combine the hydrophilic tetra‐arginine (R4) moiety with the palmitic acid (PA) to form the corresponding pair Tz‐C(2I‐BODIPY)‐K(PA)R4 (6OFF) and [TCO:Tz]‐C(2I‐BODIPY)‐ K(PA)R4 (6ON).
Results and Discussion
For the assembly of the hybrids, we followed a convergent strategy that involves the synthesis of the 2‐bromoacetamide BODIPY 9 and the corresponding cysteine‐containing peptides (11–14) (Scheme 1). The tetrazine could successfully be introduced in solution via standard N‐hydroxysuccinimide (NHS) conjugation26 to yield the expected turn‐off BODIPY‐peptide PSs (Tz‐C(2I‐BODIPY)‐PEPTIDE, 2‐6OFF). These could be activated, in the presence of TCO, to afford the pyridazine analogues ([TCO:Tz]‐C(2I‐BODIPY)‐PEPTIDE, 2‐6ON) with the intention of generating the 1O2 upon irradiation.
Scheme 1.
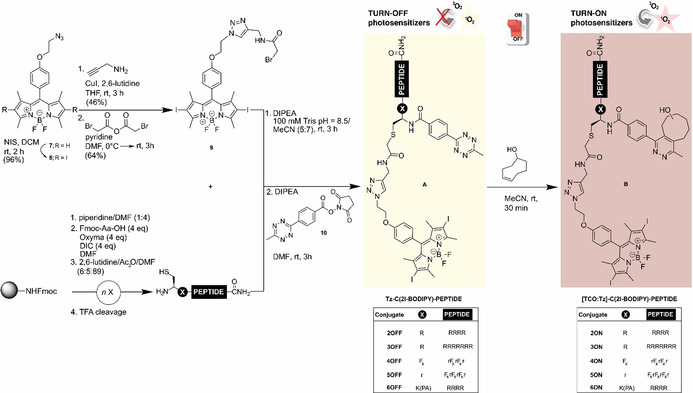
Convergent synthetic approach for the preparation of the turn‐off Tz‐C(2I‐BODIPY)‐PEPTIDE conjugates (2‐6OFF), and the corresponding turn‐on [TCO:Tz]‐C(2I‐BODIPY)‐PEPTIDE analogues (2‐6ON).
The synthetic route to these bioconjugates is shown in Scheme 1. Thus, the azide‐functionalized BODIPY 7, which had previously been described as an accessible scaffold for the generation of various BODIPY dyads,27 was first iodinated with N‐iodosuccinimide (NIS) in DCM to maximize the intersystem crossing process, and hence 1O2 production.28 The resulting 2I‐BODIPY compound 8 was then converted to bromoacetamide 9 via a sequence of a 1,3‐dipolar cycloaddition followed by substitution. For the modular attachment of our PS/quencher pair, all peptides included an N‐terminal cysteine. The synthesis of these cysteinyl fragments (11–14) was performed using the classical Fmoc solid‐phase methodology. After cleavage from the solid support and reversed‐phase (RP) HPLC purification, the single cysteine residue enabled the efficient alkylation with the thiol‐reactive 2I‐BODIPY 9. Taking advantage of the preference of the NHS active esters for primary amines, the peptides were tetrazine derivatized at the N‐terminus in the final step. The structures of the activatable BODIPY‐peptide PSs (Tz‐C(2I‐BODIPY)‐PEPTIDE, 2‐5OFF) were verified by high‐resolution mass spectrometry. For the Tz‐C(2I‐BODIPY)‐K(PA)R4 (6OFF), the palmitic acid (PA) was on‐resin coupled to the lysine Nϵ‐amino group, which was previously protected as allyloxycarbonyl.
The remainder of the synthesis was then analogous to those described above. Lastly, all of the obtained Tz‐C(2I‐BODIPY)‐PEPTIDE conjugates (2‐6OFF) reacted successfully with the strained dienophile TCO to form the desired active peptide‐based PSs ([TCO:Tz]‐C(2I‐BODIPY)‐PEPTIDE, 2‐6ON).
Next, we explored the photodynamic activity and subcellular localization of all BODIPY peptide‐based conjugates. First, we analysed their ability to produce 1O2 under irradiation. We used 1,3‐diphenylisobenzofuran (DPBF) as a trap compound.29 In the presence of 1O2, DPBF decomposes in a 1:1 stoichiometry via a [4+2] cycloaddition to give 1,2‐dibenzoylbenzene, resulting in reduced absorbance at 415 nm, and thereby, allowing the measurement of 1O2 production. Our kinetics (Figure 2, Figure S43–S45 and Table 1) together with the determined 1O2 quantum yields (Table 1) showed that bioconjugation, that is, conjugation to the peptide sequences, does not have any impact on the in vitro production of 1O2. The PS capacity of the turn‐on [TCO:Tz]‐C(2I‐BODIPY)‐PEPTIDE conjugates (2‐6ON) is similar (Φ Δ∼0.60) to the one of standard PSs.30 Under 525 nm irradiation, all pyridazine BODIPY [TCO:Tz]‐C(2I‐BODIPY)‐PEPTIDE analogues (2‐6ON) retained the same PS capacity as the halogenated azide‐functionalized BODIPY 8 (k∼4.52 s−1 10−3 versus k=4.97 s−1 10−3; Φ Δ∼0.59 versus Φ Δ=0.61, respectively) while, gratifyingly, there was a slower decrease in the 1O2 generation of all Tz‐C(2I‐BODIPY)‐PEPTIDE conjugates (2‐6OFF) (k∼2.61 s−1 10−3; Φ Δ∼0.55). Despite the efficient 1O2 quenching observed, as expected from a FRET‐quenching mechanism,16, 31 this effect is higher when the tetrazine is directly conjugated with the 2I‐BODIPY core, as in PSs 1 16 than in the case of the Tz‐C(2I‐BODIPY)‐PEPTIDE compounds (2‐6OFF).
Figure 2.
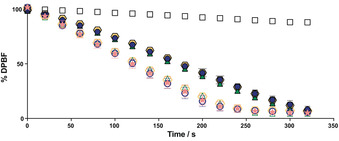
Consumption of DPBF in MeCN (200 μm) over time due to 1O2 generation in the presence of 0.1 μm of: 3OFF (solid dark blue circle); 3ON (white circle with dark blue border); 5OFF (solid dark green triangle); 5ON (white triangle with dark green border); 6OFF (solid yellow hexagon); 6ON (white hexagon with yellow border); 8 (pink star) under irradiation at 525 nm with a custom‐made 96‐well plate LED array (69.4±0.6 W m−2). Control: solution of DPBF in MeCN (200 μm, white squares). Mean data‐points and standard deviations are derived from at least two independent experiments, each concentration in triplicate. For better comprehension only the turn‐off/turn‐on photosensitizer (PS) pairs with the highest difference between off and on states for each vector type are shown. The remaining pairs: 2OFF/ON and 4OFF/ON are analysed in Figure S43.
Table 1.
Photophysical properties and photodynamic activity of the 2I‐BODIPY‐peptide conjugates in HeLa cells.
|
Compound |
k [s−1×10−3][a] |
Φ Δ [b] |
IC50 [μm][c] |
|---|---|---|---|
|
2OFF |
2.62 |
0.55 |
0.617±0.024 |
|
3OFF |
2.55 |
0.55 |
0.716±0.078 |
|
4OFF |
2.82 |
0.56 |
0.599±0.043 |
|
5OFF |
2.70 |
0.56 |
0.598±0.037 |
|
6OFF |
2.40 |
0.55 |
0.369±0.030 |
|
2ON |
4.50 |
0.59 |
0.201±0.012 |
|
3ON |
4.96 |
0.60 |
0.196±0.017 |
|
4ON |
4.14 |
0.59 |
0.228±0.008 |
|
5ON |
4.32 |
0.59 |
0.225±0.011 |
|
6ON |
4.68 |
0.59 |
0.096±0.003 |
[a] Rate constant of the DPBF consumption in MeCN; [b] 1O2 quantum yields calculated relative to: rose bengal (Φ Δ=0.76) and erythrosine B (Φ Δ=0.60) in MeOH;30 [c] half maximal inhibitory concentration obtained from resazurin cell viability assays (irradiation dose: 69.4±0.6 W m−2 for 160 s). Mean values derived from two independent experiments with errors ±6 % for k, ±7 % for Φ Δ.
Afterwards, we evaluated the cellular uptake and subcellular localization of these derivatives in HeLa cells by flow cytometry and live‐cell confocal microscopy. For this purpose, we prepared the fluorescent non‐halogenated H2N‐C(BODIPY)‐PEPTIDE analogues (2‐6FL) lacking the tetrazine group. (Figure 3 A). Flow cytometry enables cellular‐uptake quantification by measuring fluorescence intensity. Thus, HeLa cells were incubated with the BODIPY‐labelled peptides (H2N‐C(BODIPY)‐PEPTIDE 2‐6FL) at different concentrations for two hours, followed by a washing protocol including a trypsin treatment before the cytometry trials. At 2 μm concentration, all peptides displayed a pronounced fluorescence shift, which is clearly different to that of the untreated cells. This signal corresponds to quantitative labelling (Figure 3 B–D). However, if the concentration were decreased 100 times (0.02 μm), cells were hardy labelled, except when using the lipopeptide H2N‐C(BODIPY)‐K(PA)R4 (6FL) (28 % labelled cells). Interestingly, this effect was more dramatic for the oligoarginine conjugates, where low labelling rates (∼10 %) were already detected at 0.2 μm (Figure S67 and S68). The fluorescence intensity was directly proportional to the concentration and dependent on the type of peptide (Figure 3 D). The lipopeptide H2N‐C(BODIPY)‐K(PA)R4 (6FL) displayed at least an ∼three‐fold signal increase in comparison to 2FL, 3FL, 4FL and 5FL at all concentrations (Table S4), with a maximum of 15‐fold signal intensity increase for the 2 μm concentration of H2N‐C(BODIPY)‐K(PA)R4 (6FL) compared to H2N‐C(BODIPY)‐R5 (2FL).
Figure 3.
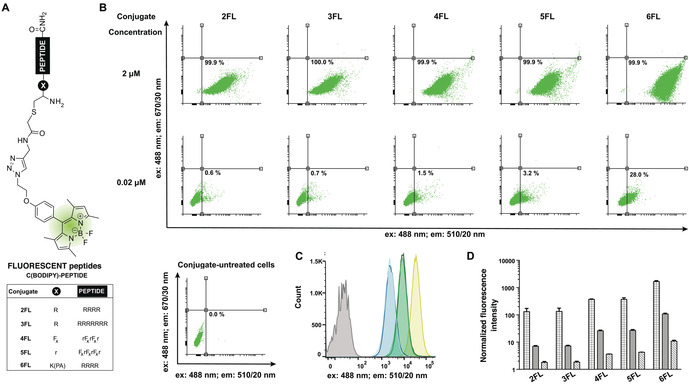
Evaluation of cellular uptake via flow cytometry. A) Structure of the fluorescent H2N‐C(BODIPY)‐PEPTIDE conjugates (2‐6FL). B) Representative dot plots of the flow cytometry analysis of conjugate‐untreated and ‐treated HeLa cells with the conjugates 2‐6FL, including quantification at 2 μm (top) and 0.02 μm (bottom). C) Overlayed histogram profiles of representative flow cytometry of conjugate‐untreated (grey) and 2FL (light blue), 3FL (dark blue), 4FL (light green), 5FL (dark green) and 6FL (yellow) treated cells showing the shift of flow cytometry fluorescence intensity signal at 2 μm conjugate concentration. D) Quantified fluorescent signatures of the HeLa cells treated with the conjugates 2‐6FL at concentrations: 2 μm (grid pattern), 0.2 μm (solid pattern) and 0.02 μm (diagonal line pattern). Data are normalized to the vehicle‐treated (ultrapure H2O, 1.25 % MeCN) treated cells, which are assigned a value of 1.
Since standard flow cytometry cannot discriminate between fluorescence from internalized fluorophores and that resulting from membrane‐bound ones, we employed live‐cell confocal microscopy (Figure 4). Images recording across consecutive Z‐stacks and the use of commercial fluorescent markers specific for lysosomes (LysoTracker® Red DND‐99),32 mitochondria (tetramethylrhodamine ethyl ester perchlorate: TMRE )33 and cytoplasmic membranes (1,1′‐dioctadecyl‐3,3,3′‐tetramethyl‐indocarbocyanine perchlorate: DiI)34 allowed us to decipher the intracellular distribution of our fluorescent conjugates. For comparison, we used the same parameters for all BODIPY‐peptide derivatives (H2N‐C(BODIPY)‐PEPTIDEs, 2‐6FL), that is, 2 μm concentration and two hours of incubation. The analysis of the results revealed that the lipopeptide H2N‐C(BODIPY)‐K(PA)R4 (6FL) was unable to penetrate the cells, despite having an oligoargenine sequence. Indeed, the incapacity of other R4 peptide constructs to efficiently translocate the cell membrane has previously been reported.25, 35 In this case, we observed a very intense fluorescence signal around the cytoplasmatic membrane.
Figure 4.
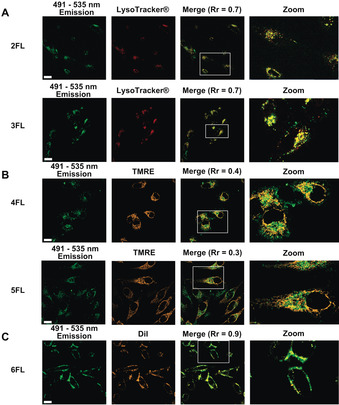
Localization of the fluorescent BODIPY‐peptides 2‐6FL in live HeLa cells. Images displayed in green correspond to these peptides at 488 nm excitation/491–535 nm emission. Before confocal imagining, cells were incubated for 2 h (in FluoroBrite media, 2.5 % FBS, 1.25 % MeCN, 8.75 % ultrapure H2O). Concentrations of 2 μm BODIPY conjugates were used. The conditions were: A) BODIPY‐oligoarginine (Rn) peptides 2FL or 3FL: following a DPBS washing step, 50 nm of LysoTracker® Red DND‐99 in FluoroBrite media (2.5 % FBS, 0.5 % DMSO) was added and left 2 h; cells were DPBS washed again before image acquisition; images displayed in red correspond to LysoTracker® Red DND‐99 fluorescence (633 nm excitation/635–700 nm emission); or B) BODIPY‐mitochondria penetrating peptides (MPPs) 4FL or 5FL: following a DPBS washing step, 25 nm of TMRE in FluoroBrite media (2.5 % FBS, 0.5 % DMSO) was added and left for 30 min; cells were DPBS washed again before image acquisition; images displayed in orange correspond to TMRE fluorescence (543 nm excitation/545–589 nm emission); or C) BODIPY‐lipopeptide 6FL: following a DPBS washing step, 10 μm of DiI in FluoroBrite media (2.5 % FBS, 0.5 % DMSO) was added and left for 15 min; cells were DPBS washed again before image acquisition; images displayed in orange correspond to DiI fluorescence parameters (543 nm excitation/545–589 nm emission). All images collected were analysed by Zeiss ZEN; ImageJ software was used for the calculation of the Rr to determine the extent of overlay.37 Values reported were calculated by linear regression,38 using more than 10 cells analysed in two independent experiments. Scale bar: 20 μm; TMRE: tetramethylrhodamine ethyl ester perchlorate; DiI: 1,1′‐dioctadecyl‐3,3,3′‐tetramethyl‐indocarbocyanine perchlorate.
Co‐localization experiments with H2N‐C(BODIPY)‐K(PA)R4 conjugate (6FL) and the membrane stain DiI demonstrated a close‐to‐perfect overlay with a Pearson's correlation coefficient (Rr) of 0.907. However, the conjugates containing either at least five arginine residues (R5 and R8) or either the MPP sequences were efficiently taken up by the HeLa cells under the same conditions (Figure 4). For the polyarginine vectors, in general, it is assumed that a minimum of six arginine residues (R6) are needed to induce intracellular uptake.22a, 36 However, we observed that five consecutive arginines (R5) in our BODIPY‐peptide H2N‐C(BODIPY)‐R5 (2FL) are enough to achieve almost the same cell‐penetration as the R8 derivatives (H2N‐C(BODIPY)‐R8, 3FL). Regarding the subcellular localization, significant differences between the Rn and MPP hybrids were observed. Thus, LysoTracker® colocalization experiments verified that the punctate BODIPY‐Rn distribution—hybrids H2N‐C(BODIPY)‐R5 (2FL) and H2N‐C(BODIPY)‐R8 (3FL)—is predominantly within the lysosomes (Rr∼0.7). Unexpectedly, the compounds designed specifically to target mitochondria (H2N‐C(BODIPY)‐MPP1, 4FL and H2N‐C(BODIPY)‐MPP2, 5FL) showed poor colocalization with TMRE (Rr∼0.34) even when the incubation time was reduced to 30 min (Rr∼0.42, Figure S65). Further colocalization studies with LysoTracker® (Figure S64) suggested that the MPP derivatives were located within the lysosomes (Rr∼0.6 and Rr∼0.65 for 2 h and 30 min incubation, respectively).
Long‐distance diffusion of 1O2 is impossible due to its brief lifetime.39 Consequently, PS distribution may determine the efficiency of photoinduced cell death.40 Once we had achieved different subcellular localization by using peptides capable of targeting specific cellular compartments, we next studied this effect on both phototoxicity and in situ intracellular turn‐on activation of our bioorthogonally activatable PS. To this end, the different OFF/ON BODIPY‐peptide PSs were incubated at different concentrations, and irradiated at 525 nm with a custom‐made 96‐well plate LED array (69.4±0.6 W m−2 for 160 s) after two hours of incubation. Before the viability assays, the total incubation time was 20 h. The obtained IC50 values (Table 1; Figure S47–S49) displayed a clear difference between the tetrazine Tz‐C(2I‐BODIPY)‐PEPTIDE derivatives (2‐6OFF) and the corresponding pyridazine [TCO:Tz]‐C(2I‐BODIPY)‐PEPTIDE ones (2‐6ON) in the intracellular environment. Importantly, without the PS motif, the peptidic vectors for cellular‐compartment targeting displayed a total absence of toxicity at the studied concentration range, up to 4 μm (Figure S51).
In concordance with the in vitro 1O2 quenching experiments (Figure 2), the phototoxicity window increases when the tetrazine is directly conjugated with the 2I‐BODIPY core, for example PS 1 (IC50=1.92±0.27 μm for the tetrazine derivative 1OFF versus IC50=0.354±0.017 μm for its pyridazine analogue 1ON after iEDDA with TCO).16 However, divergence remains evident in the new bioorthogonally activatable BODIPY‐peptide conjugates. Furthermore, all the turn‐on [TCO:Tz]‐C(2I‐BODIPY)‐PEPTIDE compounds (2‐6ON) showed higher cytotoxic potency than the pyridazine analogue of the unimodular PS [TCO:mTz]‐2I‐BODIPY (1ON), which must be due to the advantages of peptide conjugation. It is known than peptides increase aqueous solubility and reduce the general aggregation tendency observed in hydrophobic photosensitizers such as BODIPYs.19 As expected, there was almost no difference in the intracellular phototoxicity of the turn‐off/turn‐on pairs bearing oligoarginines (Rn) or mitochondria penetrating peptides (MPPs), which were almost identical in both vitro reactivity (Figure 2 and Table 1) and cellular localization (Figure 4). It is noteworthy to highlight the BODIPY‐lipopeptide conjugates 6 because, among our BODIPY‐peptide PSs, the membrane‐tagging turn‐on [TCO:Tz]‐C(2I‐BODIPY)‐K(PA)R4 (6ON) was the most cytotoxic, and, simultaneously, this construct displayed the highest phototoxicity variation between turn‐off Tz‐C(2I‐BODIPY)‐K(PA)R4 (6OFF)/turn‐on [TCO:Tz]‐C(2I‐BODIPY)‐K(PA)R4 (6ON) (3.8‐fold). Its cellular phototoxicity is higher than that of the other constructs (2‐5ON) yet DPBF behaviour (Figure 2) is comparable. The superior phototoxicity of 6ON was verified in other cell types too (Figure S52–S54, Table S3). Importantly, in the highly metastatic PC‐3 cells, the conjugates 6 displayed similarly the highest toxicity as well as the best OFF/ON ratio (3.3‐fold; IC50=0.392±0.028 μm for 6OFF versus IC50=0.119±0.012 μm for 6ON) among the pairs. In addition, we demonstrated that the acidic pH of the lysosomes does not affect the 1O2 generation by our PSs (Figure S44), unlike in other lysosome targeted BODIPYs.41 Consequently, the highest phototoxicity of [TCO:Tz]‐C(2I‐BODIPY)‐K(PA)R4 (6ON) must be due to the specific localisation within the cell. These factors jointly reveal the importance of photoinduced membrane damage. This observation is in agreement with the general theory that cell membranes are essential sites for the photosensitized cell damage,42 as reported for other PSs like rose bengal,43 or KillerRed.44 Of note, no dark toxicity was detected for any compound up to a maximum of 4 μm concentration (Figure S50). Similarly, conjugates of the well‐validated PS tetraphenyl‐porphyrin (TPP) with our peptides 12, 14 and 15 displayed an increased phototoxicity for the H2N‐C(TPP)‐K(PA)R4 conjugate (31, IC50=2.310±0.301 μm) compared to the H2N‐C(TPP)‐R8 (29, IC50=3.894±0.453 μm) and H2N‐C(TPP)‐MPP2 (30, IC50=3.923±0.469 μm) conjugates (Figure S49). These results suggest a general behaviour of PA‐R4 conjugates, which is independent of the cargo, and therefore, applicable to other PSs.
Finally, we investigated whether we could achieve activation of our turn‐off BODIPY‐peptide PSs via an in situ bioorthogonal iEDDA, and whether there is an influence of PS localization. For this purpose, HeLa cells were treated with the corresponding turn‐off BODIPY‐peptide PSs (2‐6OFF) in varied concentrations from 0.13 μm to 1 μm for 90 min, followed by a washing step to remove any traces of extracellular derivatives. Afterwards, the dienophile TCO was added in excess (12.5 equiv). After 30 min, the plates were irradiated with 69.4±0.6 W m−2 for 160 s and incubated for another 18 h, before determining cell viability via a resazurin‐based assay.42 We observed that the addition of TCO to the turn‐off Tz‐C(2I‐BODIPY)‐PEPTIDE probes (2‐6OFF) caused more cellular death than either compound alone, across the entire range of concentrations studied (Figure S56–S58). Importantly, such toxicity perfectly aligns to that measured for the synthesized turn‐on [TCO:Tz]‐C(2I‐BODIPY)‐PEPTIDE PSs, (2‐6ON) (Figure S47–S49). Altogether, these results demonstrate that regardless of the cellular localization of the PSs, all conjugates are assembled inside the cells, and upon irradiation, the newly synthesized [TCO:Tz]‐C(2I‐BODIPY)‐PEPTIDE cycloadducts (2‐6ON) display enhanced phototoxicity. The Figure 5 compares the in situ activation processes for the different peptide‐based PSs at concentrations, where the cytotoxic difference between the turn‐off and in situ activated analogues is the highest. Despite similar, satisfactory tendencies across conjugates in the in situ activation process, the membrane‐embedded conjugates 6OFF/ON again surpassed the rest of the conjugates. Indeed, its effectiveness in triggering cellular mortality due to its specific membrane localization enabled reduction of the concentration of the turn‐off Tz‐C(2I‐BODIPY)‐K(PA)R4 PS (6OFF) by half, which minimized background toxicity and improved its applicability as an aPS.
Figure 5.
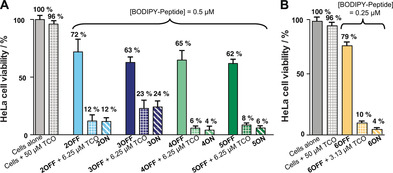
Intracellular activation and phototoxicity effects on HeLa cells of the turn‐off 2I‐BODIPY‐peptides 2‐6OFF at concentrations A) 0.5 μm of 2 (light blue), 3 (dark blue), 4 (light green) or 5 (dark green), and B) 0.25 μm for 6 (yellow) in the presence (grid pattern) and absence (solid pattern) of the dienophile trans‐cyclooctenol (TCO), and the corresponding turn‐on 2I‐BODIPY‐peptides 2‐6ON as controls (diagonal line pattern; same colours). After standing for 30 min, cells were irradiated (69.4±0.6 W m−2 for 160 s) at 525 nm with a custom made 96‐well plate LED array, followed by 18 h incubation in the dark. Mean data‐points and standard deviations are derived from two independent experiments.
Conclusions
Most small‐molecules fail to target cellular compartments specifically: instead, they distribute either between several areas simultaneously, or at random without reaching the desired localization. In this study, we successfully provided access to a modular peptide‐based platform, in which the halogenated BODIPY photosensitizer (PS) and the quencher tetrazine are separated. Peptide conjugation not only represents a straightforward approach to improve the photochemical properties of PSs but also enables spatial activation due to the accumulation of the PS in specific organelles. We reported a range of dual‐labelled peptides that only differ in their peptidic vectors, sharing the same PS/quencher pair. This type of bioorthogonally activatable PSs achieved control of the confined photodynamic effect. Thus, the bioorthogonal activation in specific subcellular organelles was successful. Importantly, PS distribution determined the cellular response to photodamage in multiple cell lines. Among our conjugates, the example capable of being activated specifically in the cytoplasmic membrane, Tz‐C(2I‐BODIPY)‐K(PA)R4, (6OFF) via an bioorthogonal iEDDA surpassed the rest. Once active, the in situ formed, [TCO:Tz]‐C(2I‐BODIPY)‐K(PA)R4 PS, (6ON) attained superior PDT performance, that is, enhancement of the photodynamic effect at lower concentrations without dark toxicity. Importantly, since our approach used a cysteine residue as workhorse, this methodology is compatible with the common bioconjugation strategies, making it highly versatile and tunable for the incorporation of other PS as well as other biopolymers. Thus, we believe that our modular approach will assist the exploration of further subcellular compartments and bioconjugation possibilities, as well as additional peptidic functionalities such as tumour‐targeting peptides and antibodies to tackle the selectivity issues in PDT. Therefore, this modular peptide‐based platform will further advance the applicability of the bioorthogonally activatable PSs.
Experimental Section
Materials
All commercially available compounds were used without further purification as delivered from the corresponding companies, 3‐diphenylisobenzofuran from Alfa Aesar (USA); propagylamine from Acros Organics (Belgium); TMRE from Cayman Chemical (USA); DiI from MedChemExpress (USA); LysoTracker® Red DND‐99 from Thermo Fisher (USA); bromoacetic anhydride, palmitic acid, trypsin‐EDTA and penicillin‐streptomycin from Sigma Aldrich (USA); Oxyma, Fmoc‐Lys(Alloc)‐OH and Fmoc‐D‐Arg(Pbf)‐OH from Carbolution (Germany); Fmoc‐Fx‐OH from Fluorochem; DMF (peptide grade), Fmoc‐protected amino acids and TentaGel S RAM resin from Iris Biotech (Germany); DPBS, DMEM and FluoroBrite from Gibco Thermo Fisher Scientific (USA); FBS from PAN Biotech (Germany); MeCN (HPLC grade) from VWR (France). TEA, THF and DCM were dried using standard procedures.45 Ultrapure H2O of type 1 was obtained with a MicroPure Water Purification System (TKA, Germany).
Characterization
NMR spectra were recorded at 300 K on Bruker AV III HD 300 and 600 MHz spectrometers, whereas HRMS (ESI) results were acquired with a LTQ‐FT Finnigan Ultra mass spectrometer (Thermo Fischer Scientific), the resolution was set to 100 000. Peptide and conjugate characterization was performed via RP‐HPLC‐MS on an Agilent 1260 Infinity II HPLC‐System (Agilent Technologies) with an UV detector (220 nm). The analytical column was an Agilent eclipse XDB‐C18 column (5 μm, 4.6×150 mm) or Macherey–Nagel Nucleodur 100‐C18 ec column (3 μm, 2×125 mm). The following two eluents were used: A) ultrapure H2O with addition of 0.05 % TFA, and B) MeCN with addition of 0.03 % TFA.
Singlet oxygen measurements
Singlet oxygen production: This was determined in black μclear 96 well plates (Greiner Bio‐One, Item: 655096) by recording endpoint absorbance at 415 nm with a Tecan Spark 20 m plate reader at 25 °C. Compound concentrations were determined from stocks using the molar extinction coefficients (Table S1). 50 μL of 800 μm DPBF solution (200 μm as final concentration per well) and 20 μL of 1 μm PS (100 nm as final concentration per well) were added in MeCN (200 μL as final volume per well). As controls the background (MeCN), and a sample containing 200 μm DPBF were used. Irradiation was performed in 20 s intervals with an irradiation of 69.4±0.6 W m−2 until 320 s by a custom‐made LED‐array bearing 96 LEDs (Broadcom Limited, 525 nm, 16 000–27 000 mcd, viewing angle: 23°, AVAGO HLMP‐CM2B‐120DD). For all measurements, before the first irradiation cycle, an initial measurement was performed as time zero without irradiation. Results were given as mean of the individual values obtained, in which the background was previously subtracted, displayed as percentage of DPBF present in each sample in triplicate. Measurements were performed at least in two independent experiments, that is, from two different stocks.
Singlet oxygen quantum yields ( Φ Δ ): They were determined as we described before.16 The procedure was alike the determination of 1O2 described above, but using 1 μm as final PS concentration. Erythrosine B (EB) and rose bengal (RB) in MeOH were used as reference (Φ Δ=0.76 for RB and 0.6 for EB).30 Irradiation was performed in 10 s intervals with irradiation of 69.4±0.6 Wm−2 for 160 s.
Peptide synthesis
Peptides 11–14 were synthesized in a 20 μmol scale according to the standard SPPS methodology, using Fmoc‐amino acids and Oxyma/DIC as coupling agent. Obtained peptides were purified by semipreparative RP‐HPLC on a Varian (USA) ProStar Preparative HPLC system with a semi‐preparative Phenomenex Juptier 10 u C18 300 Å column. The eluents were ultrapure H2O (A) and MeCN (B) with addition of 0.1 % TFA. Analytical RP‐HPLC chromatograms and used gradients are in Figure S1–S4.
H2N‐CRRRRR‐CONH2 (11): White solid (13.3 mg, 8.4 μmol, 42 %). tR=13.16 min (Figure S1). Chemical formula: C33H68N22O6S. HRMS‐ESI+ (m/z): [M+2H]2+calcd: 451.2779; found: 451.2778.
H2N‐CRRRRRRRR‐CONH2 (12): White solid (15.4 mg, 6.4 μmol, 32 %), tR=16.48 min (Figure S2). Chemical formula: C51H104N34O9S. HRMS‐ESI+ (m/z): [M+3H]3+ calcd: 457.2888; found: 457.2887.
H2N‐CFxrFxrFxr‐CONH2 (13): White solid (18.8 mg, 12.5 μmol, 62 %). tR=22.15 min (Figure S3). Chemical formula: C48H89N17O7S. HRMS‐ESI+ (m/z): [M+2H]2+ calcd: 524.8499; found: 524.8516.
H2N‐CrFxrFxrFxr‐CONH2 (14): White solid (16.4 mg, 9.2 μmol, 46 %). tR=25.05 min (Figure S4). Chemical formula: C54H101N21O8S. HRMS‐ESI+ (m/z): [M+2H]2+ calcd: 602.9004; found: 602.9022.
Organic synthesis
NMR spectra for compounds 19 and 9 are in Figure S12–S17.
Compound 9 : This was obtained in a two‐step synthesis from 8. 8 (165 mg, 250 μmol, 1.00 equiv) and CuI (7.6 mg, 40.0 μmol, 0.16 equiv) were dissolved in dry THF (3.00 mL) under inert atmosphere. Propargylamine (240 μL, 3.75 μmol, 15.0 equiv) was added last, and the solution was stirred for three hours. The reaction mixture was concentrated under reduced pressure and purified by column flash chromatography (DCM to DCM/MeOH/TEA; 94:5:1, v/v/v). The intermediate 19 was obtained as red solid (82.9 mg, 116 μmol, 46 %). TLC: R f=0.30 (DCM/MeOH/TEA, 94:5:1, v/v/v). 1H NMR (300 MHz, 300 K, CDCl3): δ=8.68 (brs, 3 H, NH 3), 8.09 (s, 1 H,H triazole), 7.13 (d, 3 J=8.5 Hz, 2 H, 2×H arom), 7.01 (d, 3 J=8.6 Hz, 2 H, 2×H arom), 4.81 (t, 3 J=4.6 Hz, 2 H, CH 2), 4.44–4.41 (m, 4 H, 2×CH 2), 2.61 (s, 6 H, 2×CH 3), 1.38 ppm (s, 6 H, 2×CH 3). 13C NMR (75 MHz, 300 K, CDCl3): δ=158.8 (1 C, C q.), 157.0 (2 C, 2×C q.), 145.3 (2 C, 2×C q.), 141.0 (1 C, C q.), 140.3 (1 C, C q), 131.7 (2 C, 2×C q.), 129.5 (2 C, 2×C arom), 128.1 (1 C, C q.triazole) 125.5 (1 C, HC trialzole), 115.6 (2 C, 2×C arom), 85.9 (2 C, 2×CI), 66.2 (1 C, CH2), 50.1 (1 C, CH2), 34.9 (1 C, CH2‐NH3), 17.4 (2 C, 2×CH3), 16.2 ppm (2 C, 2×CH3). 19F NMR (470 MHz, 300 K, CDCl3): δ=−146.2 ppm (q, 2 F, 1 J=32.4 Hz, BF2). 11B NMR (160 MHz, 300 K, CDCl3): δ=0.28 ppm (t, 1 B, 1 J=32.4 Hz, BF2). HRMS‐ESI+ (m/z): calcd for C24H26BF2I2N6O [M+Na]+: 740.0216; found: 740.0212. The intermediate 19 (50.0 mg, 69.8 μmol, 1.00 equiv), and pyridine (5.6 μL, 69.8 μmol, 1.00 equiv) were dissolved in degassed DMF (2.50 mL) under inert atmosphere and placed in an ice bath. Bromoacetic anhydride (54.4 mg, 209 μmol, 3.00 equiv) dissolved in degassed DMF (1.50 mL) was added dropwise. After 15 min the ice bath was removed, and the red solution was stirred for three hours at room temperature. The mixture was then concentrated under reduced pressure, diluted with EtOAc (10 mL), washed with distilled H2O (2×25 mL) and brine (1×25 mL). The organic phase was dried over anhydrous MgSO4, filtered, concentrated under reduced pressure and purified by column flash chromatography (EtOAc) to obtain 9 as dark red solid (37.5 mg, 44.8 μmol, 64 %). TLC: R f=0.36 (EtOAc). 1H NMR (300 MHz, 300 K, CDCl3): δ=7.79 (s, 1 H, CH triazol), 7.15 (d, 3 J=8.7 Hz, 2 H, 2×H arom), 7.01 (d, 3 J=8.7 Hz, 2 H, 2×H arom), 4.81 (t, 3 J=5.1 Hz, 2 H, CH 2), 4.59 (d, 3 J=5.9 Hz, 2 H, CH 2), 4.44 (t, 3 J=5.1 Hz, 2 H, CH 2), 3.88 (s, 2 H, CH 2), 2.63 (s, 6 H, 2× CH 3), 1.41 ppm (s, 6 H, 2×CH 3). 13C NMR (75 MHz, 300 K, CDCl3): δ=165.9 (1 C, CO), 158.9 (1 C, C q.), 157.0 (2 C, 2×C q.), 145.4 (2 C, 2×C q.), 144.2 (1 C, C q), 141.0 (1 C, C q.), 131.8 (2 C, 2×C q.), 129.6 (2 C, 2×C arom), 128.1 (1 C, C q.triazole), 123.8 (1 C, HC triazole), 115.6 (2 C, 2×C arom), 85.8 (2 C, 2×CI), 66.5 (1 C, CH2), 49.9 (1 C, CH2), 35.7 (1 C, CH2‐NH), 28.9 (1 C, CH2‐Br), 17.4 (2 C, 2×CH3), 16.2 ppm (2 C, 2×CH3). 19F NMR (470 MHz, 300 K, CDCl3): δ=−145.8 ppm (q, 2 F, 1 J=32.4 Hz, BF 2). 11B NMR (160 MHz, 300 K, CDCl3): δ=0.27 ppm (t, 1 B, 1 J=32.4 Hz, BF2). HRMS‐ESI+ (m/z): calcd for C26H26BBrF2I2N6O2 [M+Na]+: 858.9348; found: 858.9346.
Conjugate synthesis
Compounds 2 – 5A: They were obtained in a two‐step synthesis. Initially, in a microcentrifuge tube DIPEA (2.20 equiv) was added to a 0.12 m solution of the corresponding peptide (1.20 equiv) in degassed Tris buffer (0.10 m, pH 8.5). Afterwards, the 2‐bromoacyl photosensitizer 9 (1.00 equiv, 0.10 m in MeCN) was added. The reaction was mixed at room temperature on an Eppendorf Thermoshaker, and followed by analytical RP‐HPLC‐MS until the complete conversion of the starting material 9 was observed, and the desired 2I‐BODIPY‐peptides 24–27 were observed (Figure S23–S26). These compounds were purified by semipreparative RP‐HPLC using a linear gradient over 30 minutes from 5 % to 75 % of solvent B. Detection of the signals was achieved with a dual wavelength UV detector at 220 nm and 260 nm. The 2I‐BODIPY‐peptides were characterized as explained before (characterization section above). Analytical RP‐HPLC chromatograms and used gradients are in Figures S23–S26. Afterwards, to a 0.07 m solution of the corresponding 2I‐BODIPY‐peptide (1.00 equiv) dissolved in DMF, it was added first DIPEA (2.00 equiv), and subsequently the NHS‐active ester 10 (2.00 equiv, 0.20 m in DMF). The reaction was mixed at room temperature on an Eppendorf Thermoshaker, and followed by analytical RP‐HPLC‐MS until complete conversion was observed. 2‐5A conjugates were purified by semi‐preparative RP‐HPLC. A linear gradient over 30 minutes from 5 % to 75 % of solvent B was applied for compounds 2‐5A, Detection of the signals was achieved with a dual wavelength UV detector at 220 nm and 260 nm. Analytical RP‐HPLC chromatograms and used gradients are in Figures S28–S31.
Tz‐C(2I‐BODIPY)‐RRRRR‐CONH2 (2OFF): obtained from peptide 24 and compound 10; reaction time: 3 h; red solid; yield: 46 %; tR=20.81 min (Figure S28). Chemical formula: C69H99BF2I2N32O9S. HRMS‐ESI+ (m/z): [M+2H]2+calcd: 928.3150; found:928.3153.
Tz‐C(2I‐BODIPY)‐RRRRRRRR‐CONH2 (3OFF): obtained from peptide 25 and compound 10; reaction time: 3 h; red solid; yield: 61 %; tR=23.00 min (Figure S29). Chemical formula: C87H135BF2I2N44O12S. HRMS‐ESI+ (m/z): [M+2H]2+ calcd: 1162.9677; found: 1162.9682.
Tz‐C(2I‐BODIPY)‐FxrFxrFxr‐CONH2 (4OFF): obtained from peptide 26 and compound 10; reaction time: 3 h; pink solid; yield: 53 %; tR=26.32 min (Figure S30). Chemical formula: C84H120BF2I2N27O10S. HRMS‐ESI+ (m/z): [M+2H]2+ calcd: 1001.8871; found: 1001.8899.
Tz‐C(2I‐BODIPY)‐rFxrFxrFxr‐CONH2 (5OFF): obtained from peptide 27 and compound 10; reaction time: 3 h; pink solid; yield: 57 %; tR=23.92 min (Figure S31). Chemical formula: C90H132BF2I2N31O11S. HRMS‐ESI+ (m/z): [M+2H]2+ calcd: 1079.9375; found: 1079.9415.
Compound 6OFF : Following the explained SPPS methodology, •‐RRRRK(Alloc)C‐Fmoc was synthesised. The allyloxycarbonyl group was removed on‐resin. Thus, under nitrogen, the resin was washed with DCM, and then Pd(OAc)2 (0.05 equiv) and triphenylphosphine (1.50 equiv) were added to the reactor together with a solution of phenylsilane (20.0 equiv) and NMM (20.0 equiv) in 1 mL DCM (0.02 m). The resin was shaken for 90 min, and afterwards, washed with sodium diethyldithiocarbamate (5 mg mL−1 in DMF, 5×1.5 mL), DCM (5×1.5 mL) and DMF (5×1.5 mL). Then, palmitic acid (4.00 equiv), Oxyma (4.00 equiv) and DIC (4.00 equiv) were dissolved in 400 μL of DMF and pre‐activated for 3 min. The mixture was added to the resin. After 90 min, the coupling was repeated to ensure quantitative conversion. Test cleavage and MS (Figure S5) confirmed the identity of the Fmoc‐CK(PA)RRRR‐CONH2 (15) intermediate. Afterwards, the N‐terminus was deprotected under standard SPPS conditions, this lipopeptide was cleaved from the solid support. The obtained compound underwent the same bioconjugation conditions as 24–27 but using DMSO as solvent to avoid the micelle formation of the starting material lipopeptide, which was observed in the Tris buffer/MeCN solvent mixture. The reaction mixture was further diluted with ultrapure H2O/MeCN (7:3) to have a DMSO concentration ≤5 % before semipreparative RP‐HPLC purification at a linear gradient over 30 min from 15 % to 95 % of solvent B. This procedure yielded the H2N‐C(2I‐BODIPY)‐K(PA)RRRR‐CONH2 (28) (Figure S27). Finally the tetrazine conjugation proceed as for 2‐5OFF using a purification gradient from 15 % to 95 % of solvent B, which afforded the compound 6OFF. Analytical RP‐HPLC chromatogram and used gradients are in Figure S32.
Tz‐C(2I‐BODIPY)‐K(PA)RRRR‐CONH2 (6OFF): obtained from peptide 28 and compound 10; reaction time: 4 h; pink solid; yield: 42 %; tR=31.16 min (Figure S32). Chemical formula: C85H129BF2I2N30O10S. HRMS‐ESI+ (m/z): [M+2H]2+ calcd: 1033.4269; found: 1033,4309.
Compounds 2 – 6ON : In a microcentrifuge tube, TCO (10.0 equiv) was added to the corresponding 2I‐BODIPY peptide OFF (1.00 equiv, 50 mm) dissolved in MeCN. The reaction was mixed at room temperature on an Eppendorf Thermoshaker for 30 min. The crudes were purified by semipreparative RP‐HPLC purification to obtain the compounds 2‐6ON. A linear gradient over 30 min from 5 % to 75 % of solvent B was applied for compounds 2‐5ON, and from 15 % to 95 % of solvent B for compound 6ON. Detection of the signals was achieved with a dual wavelength UV detector at 220 nm and 260 nm. Analytical RP‐HPLC chromatograms and used gradients are in Figures S33–S37.
[TCO:Tz]‐C(2I‐BODIPY)‐RRRRR‐CONH2 (2ON): Pink solid; yield: 54 %; (Figure S33). Chemical formula: C77H111BF2I2N30O10S. HRMS‐ESI+ (m/z): [M+2H]2+ calcd: 976.3564; found: 976.3571.
[TCO:Tz]‐C(2I‐BODIPY)‐RRRRRRRR‐CONH2 (3ON): Pink solid; yield: 47 %; (Figure S34). Chemical formula: C95H147BF2I2N42O13S. HRMS‐ESI+ (m/z): [M+2H]2+ calcd: 1211.0092; found: 1211.0106.
[TCO:Tz]‐C(2I‐BODIPY)‐FxrFxrFxr‐CONH2 (4ON): Pink solid; yield: 61 %; (Figure S35). Chemical formula: C92H132BF2I2N25O11S. HRMS‐ESI+ (m/z): [M+2H]2+ calcd: 1049.9283; found: 1049.9305.
[TCO:Tz]‐C(2I‐BODIPY)‐rFxrFxrFxr‐CONH2 (5ON): Pink solid; yield: 57 %; (Figure S36). Chemical formula: C98H144BF2I2N29O12S. HRMS‐ESI+ (m/z): [M+2H]2+ calcd: 1127.9788; found: 1127.9813.
[TCO:Tz]‐C(2I‐BODIPY)‐K(PA)RRRR‐CONH2 (6ON): Pink solid; yield: 52 %; (Figure S37). Chemical formula: C93H141BF2I2N28O11S. HRMS‐ESI+ (m/z): [M+2H]2+ calcd: 1081.4683; found: 1081.4720.
Cell‐bassed assays
Cell culture: Most of the studies were performed in HeLa cells (details on PC‐3 cells see cell culture in SI). HeLa cells were grown in flat‐bottomed culture flasks (T75, Sarstedt, Germany) in a fully humidified cell‐culture incubator (Galaxy CO‐170 S incubator, New Brunswick Scientific, USA) at 37 °C under CO2 (g) (5 % v/v). Growth medium was DMEM supplemented with FBS (10 % v/v), penicillin (100 units mL−1), and streptomycin (100 μg mL−1). DMEM and DPBS were stored at 6 °C, additives and trypsin at −20 °C, solutions were tempered to 37 °C prior usage by a water bath. Cells were grown to confluency and passaged every 2 to 4 days using trypsin‐EDTA solution till passage 17. Cells were counted using a Neubauer improved cell counter (Laboroptik Ltd., Germany) for the determination of seeding densities. All cell‐based assays were performed under reduced media conditions (2.5 % FBS). A detailed step‐wise protocol of the cell‐bassed assays is in the supporting information (Cell Culture).
IC 50 determinations: HeLa cells (2×104) were seeded into black μclear 96‐well microtiter plates (Greiner Bio‐One, Item: 655090) in DMEM (200 μL, 2.5 % FBS). After 21 h, 110 μL media were removed from each well, and cells were treated with 10 μL of the conjugates 2‐6OFF, 2‐6ON and 29–31 in ultrapure H2O with 12.5 % MeCN at different concentrations (4.00–0.03 μm as final conjugate concentrations), each concentration in triplicate. Compounds were incubated for 2 h. Then, the plate was irradiated 69.4±0.6 Wm−2 for 160 s at 525 nm and further incubated for 18 h. Resazurin fluorescence‐based cell viability assay was performed to evaluate cell viability. IC50 values were determined using GraphPad Prism version 6 (GraphPad Software, USA) applying the log(inhibitor) vs. response—variable slope (four parameters) fit. Untreated cells and cells treated only with the vehicle solvent (12.5 % MeCN in ultrapure H2O), were considered as negative controls. All conditions were assessed in triplicates per sample/control. The IC50 derived from, at least, two independent experiments.
Intracellular iEDDA cell viability assays: HeLa cells were seeded as for the IC50 determination experiments. 21 h after seeding, 110 μL media per well were removed, and cells were treated with 10 μL of the conjugates 2‐6OFF in ultrapure H2O with 12.5 % MeCN at different concentrations (1.00–0.13 μm as final conjugate concentration), each concentration in triplicate. Compounds were incubated for 90 min. Then, the medium was carefully aspirated, and the cells were washed twice with DPBS, and once with media (DMEM, 2.5 % FBS). Afterwards, fresh reduced serum culture media (100 μL DMEM, 2.5 % FBS) was added to the wells as well as 12.5 equiv (according to the peptide concentration) of TCO. After 30 min of incubation, the plate was irradiated 69.4±0.6 W m−2 for 160 s at 525 nm and incubated for 18 h. Untreated cells, cells treated only with the vehicle solvent (12.5 % MeCN in ultrapure H2O) and only TCO were controls. All conditions were assessed in triplicates per sample/control. The results derived from, at least, two independent experiments.
Flow cytometry measurements
HeLa cells (1×106) were seeded in 3.0 mL DMEM (2.5 % FBS) into 6‐well plates (Sartetedt, Item: 83.3920), and incubated for 18 h. The cells were treated with three different concentrations: 2.00 μm, 0.20 μm and 0.02 μm in 1.25 % MeCN in ultrapure H2O of the fluorescent peptides 2‐6FL for 2 h. Afterwards they were washed with DPBS (2×2.0 mL), and treated with a trypsin‐EDTA solution (500 μL) for 3 min at 37 °C. After that time, DMEM (1000 μL, 2.5 % FBS) was added, and the cell suspensions were transferred to microcentrifuge tubes and centrifuged (6000 rpm, 1 min) in a Biofuge pico (Heraeus, Germany), the supernatant was discarded. This was repeated one more time using DPBS. Finally, the cells were suspended in DPBS containing 0.5 % FBS for measurement. Fluorescence analysis was performed immediately with a LSR Fortessa cell analyser (BD Biosciences, USA). A minimum of 50 000 events per sample were analysed. The experiment was performed in two independent experiments.
Live‐cell microscopy measurements
HeLa cells (2.5×104) were seeded in 8‐well on cover glass cell culture chambers (Sarstedt, Item: 94.6190.802) in 500 μL FluoroBrite DMEM (2.5 % FBS) and incubated for 18 h. Then, fluorescent peptides 2‐6FL were added from freshly prepared stocks (20 μm in 12.5 % MeCN ultrapure H2O) resulting into a final 2 μm concentration and incubated for 2 h. After that, cells were rinsed with DPBS (3×500 μL). Cells were then treated with commercial dyes for labelling cell compartments. For lysosome labelling: cells were treated with 100 nm LysoTracker® Red DND‐99 in FluoroBrite media (2.5 % FBS, 0.5 % DMSO) for 2 h. For mitochondria labelling, cells were treated with 25 nm TMRE in FluoroBrite media (2.5 % FBS, 0.5 % DMSO) for 30 min. For cytoplasmic membrane labelling, cells were treated with 10 μm DiI in FluoroBrite media (2.5 % FBS, 0.5 % DMSO) for 15 min. Afterwards, cells were rinsed with DPBS (2×500 μL), then, 500 μL FluoroBrite DMEM with 2.5 % FBS was added to each well. Images were acquired on a LSM880 Laser Scanning Confocal Microscope (Zeiss, Germany) with a 63×1.4 oil immersion objective at 37 °C. Z‐stack images (1 μm steps) corresponding to images in Figure 4 are depicted in Figures S59–S63.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We gratefully thank Prof. Dr. E. Meggers (Philipps‐Universität Marburg) for accessibility to the S1 laboratory; Dr. G. Malengo (Max Plack Institute for Terrestrial Microbiology) for microscopy image acquisition; Nina Büttner (Max Plack Institute for Terrestrial Microbiology) for flow cytometry acquisition; Jun.‐Prof. Dr. L. Schulte for initial assistance in flow cytometry analysis. Prof. Dr. U. Bakowsky (Philipps‐Universität Marburg) for providing LysoTracker® Red DND‐99; N. Frommknecht (Philipps‐Universität Marburg) for design assistance and construction of custom‐made 96‐well plate LED arrays, Dr. K. Hoogewijs (Ghent University) for his insight into mitochondria‐tagging and T. Roider (Philipps‐Universität Marburg) for LED irradiance measurements. Open access funding enabled and organized by Projekt DEAL.
G. Linden, O. Vázquez, Chem. Eur. J. 2020, 26, 10014.
Dedicated to Professor Jesús Jiménez–Barbero on the occasion of his 60th birthday
References
- 1.
- 1a. Ahmed I., Fruk L., Mol. Biosyst. 2013, 9, 565–570; [DOI] [PubMed] [Google Scholar]
- 1b. Ankenbruck N., Courtney T., Naro Y., Deiters A., Angew. Chem. Int. Ed. 2018, 57, 2768–2798; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2816–2848; [Google Scholar]
- 1c. Brieke C., Rohrbach F., Gottschalk A., Mayer G., Heckel A., Angew. Chem. Int. Ed. 2012, 51, 8446–8476; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8572–8604; [Google Scholar]
- 1d. Lerch M. M., Hansen M. J., van Dam G. M., Szymanski W., Feringa B. L., Angew. Chem. Int. Ed. 2016, 55, 10978–10999; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11140–11163; [Google Scholar]
- 1e. Lubbe A. S., Szymanski W., Feringa B. L., Chem. Soc. Rev. 2017, 46, 1052–1079; [DOI] [PubMed] [Google Scholar]
- 1f. Kim B., Lin M. Z., Biochem. Soc. Trans. 2013, 41, 1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Ackroyd R., Kelty C., Brown N., Reed M., Photochem. Photobiol. 2001, 74, 656–669; [DOI] [PubMed] [Google Scholar]
- 2b. Dolmans D. E. J. G. J., Fukumura D., Jain R. K., Nat. Rev. Cancer 2003, 3, 380–387. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Huang Z., Technol. Cancer Res. Treat. 2005, 4, 283–293; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Sharman W. M., Allen C. M., van Lier J. E., Drug Discovery Today 1999, 4, 507–517; [DOI] [PubMed] [Google Scholar]
- 3c. Yano S., Hirohara S., Obata M., Hagiya Y., Ogura S., Ikeda A., Kataoka H., Tanaka M., Joh T., J. Photochem. Photobiol. C 2011, 12, 46–67. [Google Scholar]
- 4.
- 4a. Agostinis P., Berg K., Cengel K. A., Foster T. H., Girotti A. W., Gollnick S. O., Hahn S. M., Hamblin M. R., Juzeniene A., Kessel D., Korbelik M., Moan J., Mroz P., Nowis D., Piette J., Wilson B. C., Golab J., Ca-Cancer J. Clin. 2011, 61, 250–281; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Li X., Kwon N., Guo T., Liu Z., Yoon J., Angew. Chem. Int. Ed. 2018, 57, 11522–11531; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11694–11704; [Google Scholar]
- 4c. Triesscheijn M., Baas P., Schellens J. H., Stewart F. A., Oncologist 2006, 11, 1034–1044. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Wang J., Higgins S. L., Winkel B. S., Brewer K. J., Chem. Commun. 2011, 47, 9786–9788; [DOI] [PubMed] [Google Scholar]
- 5b. Fan J.-X., Liu M.-D., Li C.-X., Hong S., Zheng D.-W., Liu X.-H., Chen S., Cheng H., Zhang X.-Z., Nanoscale Horiz. 2017, 2, 349–355. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Pibiri I., Buscemi S., Palumbo Piccionello A., Pace A., ChemPhotoChem 2018, 2, 535–547; [Google Scholar]
- 6b. Ogilby P. R., Photochem. Photobiol. Sci. 2010, 9, 1543–1560. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Clo E., Snyder J. W., Ogilby P. R., Gothelf K. V., ChemBioChem 2007, 8, 475–481; [DOI] [PubMed] [Google Scholar]
- 7b. Durantini A. M., Greene L. E., Lincoln R., Martinez S. R., Cosa G., J. Am. Chem. Soc. 2016, 138, 1215–1225; [DOI] [PubMed] [Google Scholar]
- 7c. Lovell J. F., Liu T. W., Chen J., Zheng G., Chem. Rev. 2010, 110, 2839–2857; [DOI] [PubMed] [Google Scholar]
- 7d. Majumdar P., Nomula R., Zhao J. Z., J. Mater. Chem. C 2014, 2, 5982–5997; [Google Scholar]
- 7e. Turksoy A., Yildiz D., Akkaya E. U., Coord. Chem. Rev. 2019, 379, 47–64; [Google Scholar]
- 7f. Wu W., Shao X., Zhao J., Wu M., Adv. Sci. 2017, 4, 1700113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Lau J. T., Lo P. C., Jiang X. J., Wang Q., Ng D. K., J. Med. Chem. 2014, 57, 4088–4097; [DOI] [PubMed] [Google Scholar]
- 8b. Tian J., Ding L., Xu H. J., Shen Z., Ju H., Jia L., Bao L., Yu J. S., J. Am. Chem. Soc. 2013, 135, 18850–18858; [DOI] [PubMed] [Google Scholar]
- 8c. Xiong H., Zhou K., Yan Y., Miller J. B., Siegwart D. J., ACS Appl. Mater. Interfaces 2018, 10, 16335–16343; [DOI] [PubMed] [Google Scholar]
- 8d. Zhu X. C., Lu W. T., Zhang Y. Z., Reed A., Newton B., Fan Z., Yu H. T., Ray P. C., Gao R. M., Chem. Commun. 2011, 47, 10311–10313. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. He H., Lo P. C., Ng D. K., Chem. Eur. J. 2014, 20, 6241–6245; [DOI] [PubMed] [Google Scholar]
- 9b. Kolemen S., Isik M., Kim G. M., Kim D., Geng H., Buyuktemiz M., Karatas T., Zhang X. F., Dede Y., Yoon J., Akkaya E. U., Angew. Chem. Int. Ed. 2015, 54, 5340–5344; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5430–5434; [Google Scholar]
- 9c. Turan I. S., Cakmak F. P., Yildirim D. C., Cetin-Atalay R., Akkaya E. U., Chem. Eur. J. 2014, 20, 16088–16092. [DOI] [PubMed] [Google Scholar]
- 10. Chen J., Jarvi M., Lo P. C., Stefflova K., Wilson B. C., Zheng G., Photochem. Photobiol. Sci. 2007, 6, 1311–1317. [DOI] [PubMed] [Google Scholar]
- 11. Ozlem S., Akkaya E. U., J. Am. Chem. Soc. 2009, 131, 48–49. [DOI] [PubMed] [Google Scholar]
- 12. Chan M. H., Chen C. W., Lee I. J., Chan Y. C., Tu D., Hsiao M., Chen C. H., Chen X., Liu R. S., Inorg. Chem. 2016, 55, 10267–10277. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Cló E., Snyder J. W., Voigt N. V., Ogilby P. R., Gothelf K. V., J. Am. Chem. Soc. 2006, 128, 4200–4201; [DOI] [PubMed] [Google Scholar]
- 13b. Zheng G., Chen J., Stefflova K., Jarvi M., Li H., Wilson B. C., Proc. Natl. Acad. Sci. USA 2007, 104, 8989–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu Z., Tang Z. W., Phillips J. A., Yang R. H., Wang H., Tan W. H., J. Am. Chem. Soc. 2008, 130, 10856–10857. [DOI] [PubMed] [Google Scholar]
- 15. Hou L. L., Zhang X. Y., Pijper T. C., Browne W. R., Feringa B. L., J. Am. Chem. Soc. 2014, 136, 910–913. [DOI] [PubMed] [Google Scholar]
- 16. Linden G., Zhang L., Pieck F., Linne U., Kosenkov D., Tonner R., Vazquez O., Angew. Chem. Int. Ed. 2019, 58, 12868–12873; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13000–13005. [Google Scholar]
- 17. Verwilst P., David C. C., Leen V., Hofkens J., de Witte P. A., De Borggraeve W. M., Bioorg. Med. Chem. Lett. 2013, 23, 3204–3207. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Wang W., Lorion M. M., Martinazzoli O., Ackermann L., Angew. Chem. Int. Ed. 2018, 57, 10554–10558; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10714–10718; [Google Scholar]
- 18b. Banappagari S., McCall A., Fontenot K., Vicente M. G., Gujar A., Satyanarayanajois S., Eur. J. Med. Chem. 2013, 65, 60–69; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18c. Zhao N., Williams T. M., Zhou Z., Fronczek F. R., Sibrian-Vazquez M., Jois S. D., Vicente M. G. H., Bioconjugate Chem. 2017, 28, 1566–1579; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18d. Mendive-Tapia L., Zhao C., Akram A. R., Preciado S., Albericio F., Lee M., Serrels A., Kielland N., Read N. D., Lavilla R., Vendrell M., Nat. Commun. 2016, 7, 10940; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18e. Milroy L. G., Rizzo S., Calderon A., Ellinger B., Erdmann S., Mondry J., Verveer P., Bastiaens P., Waldmann H., Dehmelt L., Arndt H. D., J. Am. Chem. Soc. 2012, 134, 8480–8486. [DOI] [PubMed] [Google Scholar]
- 19. Almeida-Marrero V., van de Winckel E., Anaya-Plaza E., Torres T., de la Escosura A., Chem. Soc. Rev. 2018, 47, 7369–7400. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Stewart K. M., Horton K. L., Kelley S. O., Org. Biomol. Chem. 2008, 6, 2242–2255; [DOI] [PubMed] [Google Scholar]
- 20b. Lostalé-Seijo I., Montenegro J., Nat. Rev. Chem. 2018, 2, 258–277; [Google Scholar]
- 20c. Méndez-Ardoy A., Lostale-Seijo I., Montenegro J., ChemBioChem 2019, 20, 488–498; [DOI] [PubMed] [Google Scholar]
- 20d. Cerrato C. P., Kunnapuu K., Langel U., Expert Opin. Drug Delivery 2017, 14, 245–255. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Oliveira C. S., Turchiello R., Kowaltowski A. J., Indig G. L., Baptista M. S., Free Radical Biol. Med. 2011, 51, 824–833; [DOI] [PubMed] [Google Scholar]
- 21b. Castano A. P., Demidova T. N., Hamblin M. R., Photodiagn. Photodyn. Ther. 2004, 1, 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. McKinlay C. J., Waymouth R. M., Wender P. A., J. Am. Chem. Soc. 2016, 138, 3510–3517; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Wender P. A., Mitchell D. J., Pattabiraman K., Pelkey E. T., Steinman L., Rothbard J. B., Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22c. Vázquez O., Blanco-Canosa J. B., Vazquez M. E., Martinez-Costas J., Castedo L., Mascarenas J. L., ChemBioChem 2008, 9, 2822–2829. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Horton K. L., Stewart K. M., Fonseca S. B., Guo Q., Kelley S. O., Chem. Biol. 2008, 15, 375–382; [DOI] [PubMed] [Google Scholar]
- 23b. Jean S. R., Ahmed M., Lei E. K., Wisnovsky S. P., Kelley S. O., Acc. Chem. Res. 2016, 49, 1893–1902; [DOI] [PubMed] [Google Scholar]
- 23c. Zhao K., Zhao G. M., Wu D., Soong Y., Birk A. V., Schiller P. W., Szeto H. H., J. Biol. Chem. 2004, 279, 34682–34690. [DOI] [PubMed] [Google Scholar]
- 24. Radford R. J., Chyan W., Lippard S. J., Chem. Sci. 2014, 5, 4512–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang C., Jin S., Yang K., Xue X., Li Z., Jiang Y., Chen W. Q., Dai L., Zou G., Liang X. J., ACS Appl. Mater. Interfaces 2014, 6, 8971–8975. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Koniev O., Wagner A., Chem. Soc. Rev. 2015, 44, 5495–5551; [DOI] [PubMed] [Google Scholar]
- 26b. Anderson G. W., Callahan F. M., Zimmerman J. E., J. Am. Chem. Soc. 1967, 89, 178. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Henker J., Wirmer-Bartoschek J., Bendel L. E., Xiang Y. G., Fu C., Harms K., Schwalbe H., Meggers E., Eur. J. Inorg. Chem. 2016, 5161–5170; [Google Scholar]
- 27b. Zhang X. L., Xiao Y., Qian X. H., Org. Lett. 2008, 10, 29–32. [DOI] [PubMed] [Google Scholar]
- 28. Yogo T., Urano Y., Ishitsuka Y., Maniwa F., Nagano T., J. Am. Chem. Soc. 2005, 127, 12162–12163. [DOI] [PubMed] [Google Scholar]
- 29. Wozniak M., Tanfani F., Bertoli E., Zolese G., Antosiewicz J., Biochim. Biophys. Acta 1991, 1082, 94–100. [DOI] [PubMed] [Google Scholar]
- 30. Redmond R. W., Gamlin J. N., Photochem. Photobiol. 1999, 70, 391–475. [PubMed] [Google Scholar]
- 31. Lovell J. F., Chen J., Jarvi M. T., Cao W. G., Allen A. D., Liu Y., Tidwell T. T., Wilson B. C., Zheng G., J. Phys. Chem. B 2009, 113, 3203–3211. [DOI] [PubMed] [Google Scholar]
- 32. Raben N., Shea L., Hill V., Plotz P., Methods Enzymol. 2009, 453, 417–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Farrelly E., Amaral M. C., Marshall L., Huang S. G., Anal. Biochem. 2001, 293, 269–276. [DOI] [PubMed] [Google Scholar]
- 34. Shiraishi I., Takamatsu T., Minamikawa T., Fujita S., Anat. Embryol. 1992, 185, 401–408. [DOI] [PubMed] [Google Scholar]
- 35. Bode S. A., Timmermans S., Eising S., van Gemert S. P. W., Bonger K. M., Lowik D., Chem. Sci. 2019, 10, 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.
- 36a. Futaki S., Suzuki T., Ohashi W., Yagami T., Tanaka S., Ueda K., Sugiura Y., J. Biol. Chem. 2001, 276, 5836–5840; [DOI] [PubMed] [Google Scholar]
- 36b. Stanzl E. G., Trantow B. M., Vargas J. R., Wender P. A., Acc. Chem. Res. 2013, 46, 2944–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schneider C. A., Rasband W. S., Eliceiri K. W., Nat. Methods 2012, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bolte S., Cordelieres F. P., J. Microsc. 2006, 224, 213–232. [DOI] [PubMed] [Google Scholar]
- 39.
- 39a. Moan J., J. Photochem. Photobiol. 1990, 6, 343–347; [Google Scholar]
- 39b. Skovsen E., Snyder J. W., Lambert J. D. C., Ogilby P. R., J. Phys. Chem. B 2005, 109, 8570–8573; [DOI] [PubMed] [Google Scholar]
- 39c. Snyder J. W., Skovsen E., Lambert J. D. C., Ogilby P. R., J. Am. Chem. Soc. 2005, 127, 14558–14559. [DOI] [PubMed] [Google Scholar]
- 40.
- 40a. MacDonald I. J., Morgan J., Bellnier D. A., Paszkiewicz G. M., Whitaker J. E., Litchfield D. J., Dougherty T. J., Photochem. Photobiol. 1999, 70, 789–797; [PubMed] [Google Scholar]
- 40b. Indig G. L., Anderson G. S., Nichols M. G., Bartlett J. A., Mellon W. S., Sieber F., J. Pharm. Sci. 2000, 89, 88–99. [DOI] [PubMed] [Google Scholar]
- 41. Wang C., Qian Y., Org. Biomol. Chem. 2019, 17, 8001–8007. [DOI] [PubMed] [Google Scholar]
- 42. Haupt S., Malik Z., Ehrenberg B., Photochem. Photobiol. Sci. 2014, 13, 38–47. [DOI] [PubMed] [Google Scholar]
- 43. Kochevar I. E., Bouvier J., Lynch M., Lin C. W., Biochim. Biophys. Acta Biomembr. 1994, 1196, 172–180. [DOI] [PubMed] [Google Scholar]
- 44. Bulina M. E., Chudakov D. M., Britanova O. V., Yanushevich Y. G., Staroverov D. B., Chepurnykh T. V., Merzlyak E. M., Shkrob M. A., Lukyanov S., Lukyanov K. A., Nat. Biotechnol. 2006, 24, 95–99. [DOI] [PubMed] [Google Scholar]
- 45. Williams D. B., Lawton M., J. Org. Chem. 2010, 75, 8351–8354. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary