

Abstract
Lanthanides (Ln) are critical raw materials, however, their mining and purification have a considerable negative environmental impact and sustainable recycling and separation strategies for these elements are needed. In this study, the precipitation and solubility behavior of Ln complexes with pyrroloquinoline quinone (PQQ), the cofactor of recently discovered lanthanide (Ln) dependent methanol dehydrogenase (MDH) enzymes, is presented. In this context, the molecular structure of a biorelevant europium PQQ complex was for the first time elucidated outside a protein environment. The complex crystallizes as an inversion symmetric dimer, Eu2PQQ2, with binding of Eu in the biologically relevant pocket of PQQ. LnPQQ and Ln1Ln2PQQ complexes were characterized by using inductively coupled plasma mass spectrometry (ICP‐MS), infrared (IR) spectroscopy, 151Eu‐Mössbauer spectroscopy, X‐ray total scattering, and extended X‐ray absorption fine structure (EXAFS). It is shown that a natural enzymatic cofactor is capable to achieve separation by precipitation of the notoriously similar, and thus difficult to separate, lanthanides to some extent.
Keywords: coordination chemistry, lanthanides, PQQ, rare earth elements, separations
No co‐ligands required: The precipitation and solubility behavior of lanthanide complexes with pyrroloquinoline quinone (PQQ), is presented. Complexes with early lanthanides are preferentially formed with PQQ and rapidly precipitate out of aqueous solutions. The first molecular structure of a biorelevant europium PQQ complex outside a protein environment is reported and shows a Eu2PQQ2 dimer.
Introduction
Rare earth elements (REE) include the elements 21Sc, 39Y and 57La, in addition to the 14 lanthanides (Ln) from 58Ce to 71Lu. Due to their extensive usage in modern technologies, they are also called “vitamins, or seeds of technology” and the global demand of rare earth oxides is growing steadily.1 Unlike their name suggests, REE are not particularly rare and the occurrence of the two least abundant ones, Tm and Lu, is even higher than the one of silver.2 Mining of those elements is, however, a challenge, due to their dispersion and low concentrations in REE containing ores. In addition, extraction methods include strong acids or bases and produce large scales of radioactive and heavy metal contaminated waste.3 Separations of the chemically similar REE are energy‐intensive and challenging.4 However, several exciting new directions for REE separation have been presented recently. The group of Schelter used the size‐sensitive ligand TriNOx3− (Figure 1 B), which is able to form a self‐associative equilibrium out of REE mixtures and can be used for REE separation by leaching.5 Sun, Bünzli and co‐workers used a supramolecular approach with a tris‐tridentate ligand, which forms 4‐nuclear cages preferentially with the smaller, late REE (Figure 1 A).6
Figure 1.
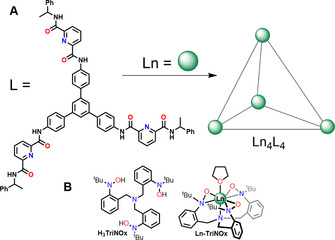
A) Tris‐tridentate ligand and supramolecular Ln complex reported by Sun, Bünzli et al.6 and B) H3TriNOx ligand and Ln TriNOx THF complex from Schelter et al.7
With a modification of the ligand, using long alkyl chains, the group was able to perform a liquid‐liquid extraction of late REE, while the early ones remained in the aqueous phase.8 Recently, also magnetic field driven REE separations have been reported.9 Among the REE, the early lanthanides (La–Eu) are now recognized as biorelevant for methylotrophic bacteria habituating a number of different ecosystems (plant phyllospheres, volcanic mudpots, soil and aquatic environments).10 Those bacteria use Ln containing enzymes (methanol dehydrogenases, MDH, active site shown in Figure 2 A) in their C1 metabolism. The active site of the Ln‐containing enzymes includes redox cofactor PQQ (Figure 2 B) that coordinates the central metal in a tridentate fashion.11
Figure 2.
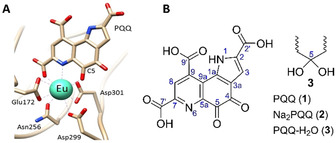
A) Active site of a Ln‐dependent MDH (PDB 6FKW). B) Structure of PQQ and related species. Water adduct 3 forms readily in aqueous solution. Numbering scheme according to Unkefer et al.12
Remarkably, early lanthanides are taken up more quickly and preferentially by bacteria than the later ones. It was shown that Methylorubrum extorquens AM1 can even grow with Nd‐containing hard‐drive magnets as the only source of Ln, making those bacteria interesting for bioleaching or biomining.13 With early Ln, bacteria grow faster and their respective MDH enzymes are more efficient in turning over methanol. Seemingly, natural systems have been tuned specifically by evolution to work best with the earlier, larger, and more abundant lanthanides. The reasons for the preference of natural systems for early lanthanides remain somewhat elusive. However, factors such as changing coordination numbers across the Ln‐series, lack of efficient activation and negative impact on redox cycling of PQQ by certain Ln in the active site have been proposed.14 PQQ is one of the few pincer ligands existing in nature and coordinates via a carboxylic acid moiety, a pyridyl nitrogen and a quinone oxygen atom.15 Similar binding motifs have been used in ligands employed in the separation of lanthanides and actinides. 2,6‐pyridine dicarboxylic acid (PDCA) and derivatives have been widely used for solvent extraction and ion chromatography.16 The tris‐tridentate ligand shown in Figure 1 A also features an ONO binding pocket. Here, we show that the MDH‐cofactor PQQ by itself preferentially forms complexes with early Ln and we evaluate whether this could be exploited for Ln‐separation. In addition, we report the first crystal structure of PQQ with a biorelevant metal ion (Eu) outside of a MDH protein environment and without synthetic co‐coordinating ligands or modified PQQ precursors.
Results and Discussion
Ln1PQQ complexes: characterization
In MDH, PQQ acts as a tridentate ligand for lanthanides, using 7’‐O; 6‐N and 5‐O (see numbering in Figure 2 B). Without the protein environment, all carboxyl groups, as well as 4‐O can participate in metal coordination, often complicating analysis.17 From aqueous solutions of the sodium salt of the cofactor (Na2PQQ, 2, isolated from vitamin capsules) complexes with lanthanides rapidly precipitate after mixing, showing 1:1 stoichiometry, even with lanthanides added in excess (6 equiv).17a Single crystals from the first Eu2PQQ2 complex outside the MDH protein environment were obtained for X‐ray structural analysis (Figure 3). To this end, very few crystal structures of PQQ complexes have been reported18 and none of them contained metals relevant for MDH activity (Ca, Ln). A structure of a 1:1 complex with a PQQ derivative (here the trimethylester PQQMe3 and copper(I) were used) was reported by Kaim.18a Kojima reported a structure of PQQMe3 with Ru2+ bearing a terpyridine coligand. A structure of PQQ (1) with Cu2+ and the same coligand was reported by Suzuki.18b, 18c Crystals of a Eu2PQQ2 complex were derived after several days from a mixture of aqueous Na2PQQ and EuCl3 solutions at 80 °C, which was allowed to slowly cool down to room temperature (see Supporting Information). The Eu‐structure is consistent with a 1:1 stoichiometry but surprisingly reveals a dimer with head to tail coordination of PQQ. While no other co‐coordinating ligands other than water were necessary to crystallize the complex, a carboxylic acid of a second PQQ molecule is needed to complete the coordination sphere. Modified PQQ derivatives, where the carboxylic acid moiety at the pyrrole ring is either blocked by alkylation to yield an ester or replaced entirely by for example, a methyl group, could favor the formation of mononuclear complexes that could find use as biomimetics for MDH. The ligand is fully deprotonated (PQQ3−), which is not surprising as the pK a values of the three carboxylic acids are all below the pH of the experiment. In addition to the coordination in the biologically relevant pocket (using 7’‐O; 6‐N and 5‐O with numbering scheme in Figure 2) Eu is coordinated with 2’‐O by a second PQQ molecule. The coordination sphere is completed by five water molecules (consistent with elemental analysis of the bulk material) yielding a final coordination number (CN) of 9. The PQQ‐water adduct (C5‐diol, 3), which is present to some extend in aqueous PQQ solutions, is not observed in the Eu2PQQ2 complex.19 To gain more insight into the coordination chemistry and exclude redox processes throughout complex formation, we recorded a 151Eu Mössbauer spectrum of the precipitated bulk material. The experimental Mössbauer spectrum of Eu2PQQ2 at 5 K is shown in Figure 4 together with the transmission integral fit. The spectrum clearly indicates the presence of only Eu3+ in this sample, supporting the oxidation state assigned in the crystal structure. Due to increased electron density at the core, Eu2+ complexes commonly display negative isomer shifts around −10 to −14 mm s−1.19 Here, the isomer shift of Eu2PQQ2 of δ=0.32(2) mm s−1 is well in the range for reported Eu3+‐complexes with N‐ and O donors such as [Eu(acac)3(H2O)2] (δ=0.36 mm s−1, at 80 K), [Eu(NO3)2(phen)2] (δ=0.41 mm s−1, at 80 K) or H[Eu(dcta)]⋅4 H2O (δ=0.30 mm s−1, at rt) and Ba[Eu(dtpa)]⋅7.5 H2O (δ=0.38 mm s−1, at rt).20 The fitting of the Eu2PQQ2 data further reveals an electric quadrupole splitting of ΔE Q=1.6(4) mm s−1 and an experimental line width of Γ=2.5(1) mm s−1. The small experimental line width indicates one single europium species rather than multiple Eu3+ ions in crystallographically distinct sites.19b, 20a Usually for Eu3+ complexes (and other Eu3+ solids), the range of observed isomer shifts is only within 0.8 mm s−1 and largely insensitive to the nature and number of coordinating ligands.20a However, the Mössbauer spectroscopic data supports overall the presence of only one Eu3+ species in the bulk material.
Figure 3.
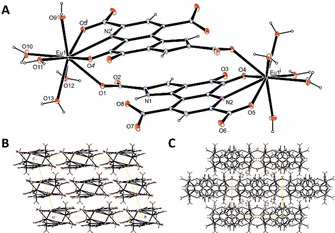
A) Structure of the inversion symmetric Eu2PQQ2 complex drawn at the 50 % ellipsoid probability level (symmetry code i=1−x, 1−y, 1−z, free water molecule has been omitted for clarity). The packing of the dimers is depicted in viewing direction [010] (B) and [001] (C). The latter illustrates the formation of AB‐stacked layers along [100] caused by C‐centering.
Figure 4.
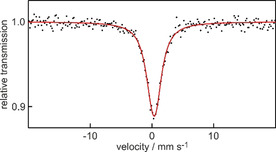
Experimental (black data points) and simulated (red line) 151Eu Mössbauer spectrum of the Eu2PQQ2 complex at 5 K.
To analyze whether the structure shown in Figure 3 is representative for the bulk material that precipitated and to compare coordination numbers (CN) and metal‐ligand distances of different LnPQQ complexes (Ln=La, Eu, Tb, Lu), we turned to EXAFS spectroscopy and pair distribution functions analysis (PDF). The results are shown in Table S2, Figure 5 and in the Supporting Information. Among four evaluated monometallic species, only EuPQQ showed good crystallinity with relatively sharp diffraction lines in the powder X‐ray diffraction (Figure 5 A). The diffractogram characteristic for bulk material can be completely indexed based on structural data obtained using single‐crystal data (Figure S7). LnPQQ species (Ln=La, Tb, Lu) do not show any crystallinity and can be considered as X‐ray amorphous (Figure 5 A). Pair distribution functions can be reconstructed up to experimental Q max <23 Å−1. Nevertheless, interatomic correlations for LuPQQ, TbPQQ and LaPQQ (Figure 5 B) can be detected only up to r=5–8 Å, which is typical for amorphous materials without long‐range order and without detectable M⋅⋅⋅M correlations at approximately 12–14 Å obtained for Eu2PQQ2. Qualitatively, the short‐range structure characteristic for TbPQQ and LuPQQ is similar. Average Ln−L distances (Table S2) are similar and the coordination number is comparable for all three species. The LaPQQ local structure seems to differ slightly from EuPQQ with visibly longer La−L distances.
Figure 5.
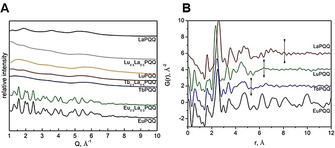
A Powder X‐ray diffraction data for LnPQQ species after background subtraction (P02.1 beamline at the PETRA III; λ=0.20714 Å; empty capillaries were used for background subtraction). B Pair distribution functions for EuPQQ, TbPQQ, LuPQQ and LaPQQ species (vertical marks show qualitative cut offs in interatomic correlations for amorphous species).
Analysis of the EXAFS data showed that from early to late Ln, both the CN as well as average Ln−L distances are reduced (Table S2 and Figure 6). The determination of the coordination number was achieved by using the well‐established data reduction procedures presented by Ravel et al.21 A first shell fit was performed between the measured LnPQQ EXAFS and the model based on Eu2PQQ2 single‐crystal data. The first shell fit entails the scattering paths from all neighboring atoms presented in Table S2, obtained from the used model. This comprises one Ln−O10 at 2.36 Å, two Ln−O9 at 2.423 Å, three Ln−O1 at 2.45 Å, one Ln−O12 at 2.48 Å, one Ln−O3 at 2.57 Å and one Ln−N1 at 2.65 Å. This results in a total coordination number of 9. In the case of LaPQQ and EuPQQ this fitted well, but in the case of TbPQQ and LuPQQ a total coordination number of seven yielded a better fit. The total coordination number results in the first peak observed experimentally (Figure 6), entailing all scattering paths, supported by the model. Furthermore, the position of this peak delivers information about the Ln−L distance. Measured EXAFS supported by the model show different average ligand distances as in Table S2. La−L is at 2.64 Å, Eu−L at 2.48 Å, Tb−L at 2.44 Å and Lu−L at 2.35 Å.
Figure 6.
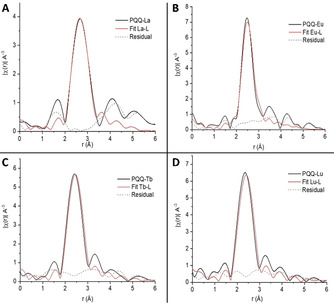
Fourier transformations from the extended part of the EXAFS signal of PQQ‐Ln species (black line), fitted with the solved structure (red line) and the difference between them (residual, dashed grey line). A LaPQQ, B EuPQQ, C TbPQQ, D LuPQQ.
The coordination sphere of Eu (CN=9) in the 1:1 EuPQQ and LaPQQ complex would therefore consist of five equiv of water and four times coordinated PQQ. For the later Ln, Tb and Lu, the CN is reduced concurrent to the number of coordinated water molecules. This agrees with the first shell fit by EXAFS, where a CN of 7 is obtained (for further details, see Methods in Supporting Information). The decrease of the preferred CN throughout the lanthanide series is a common feature in lanthanide coordination chemistry.22 Analytical data of a previously reported LaPQQ sample was in best agreement with five equiv of water.17a A correlation is further recognized between the determined CN with XAS and the calculated water equivalents based on elemental analysis on the one hand and the IR intensity of the large O−H stretching feature between 3600‐2500 cm−1 (Figure S1).
By comparing IR spectra of the 1:1 LnPQQ complexes (Ln=La, Eu, Tb, Lu) with Na2PQQ⋅H2O large similarities are revealed, which indicate participation of at least two of the three carboxyl groups in Ln coordination (Figure S1) as also found in the Eu2PQQ2 structure. In comparison with PQQ (1, >20 main peaks) the complexes exhibited IR spectra with only eight main features resolved: 3630–2500, 1735, 1580, 1507, 1351, 1241, 1195 and 1149 cm−1. Especially the seven quinone and carboxyl related vibrations of PQQ,23 merge to a broad, poorly resolved area between 1735–1580 cm−1.17a, 24 Further, the IR spectra of the LnPQQ complexes reveal more similarities to Na2PQQ than to free PQQ (Figure S1). More importantly, the IR spectra of bulk material and single crystals of the Eu2PQQ2 dimers show almost identical IR spectra (Figure S2), further confirming that the obtained crystal structure is representative for the bulk material.
Ln1Ln2PQQ complexes: structure and lanthanide preference
The property of aqueous solutions of Na2PQQ to instantly precipitate one equivalent of lanthanide prompted us to study whether there would be a preferred Ln when mixtures of Ln were offered. Considering the demand of new separation techniques for REE and the preference of bacteria for certain lanthanides, we were curious about the ability of PQQ to separate mixtures of Ln. Importantly, PQQ does not have to be made synthetically but can be obtained in kg amounts by fermentation without the use of any organic solvents and can further be fully recovered and separated from Ln by addition of concentrated HCl. This will destroy the complex and lead to precipitation of fully protonated PQQ (1) which is extremely stable against acids. In fact, it needed a mixture of boiling concentrated nitric and hydrochloric acid to fully dissolve (and possibly decompose) 1 in our studies. Stock solutions of Na2PQQ in water were mixed with aqueous solutions of the lanthanide chlorides, each consisting of three equiv of lanthanum chloride and three equiv of a second lanthanide chloride. All samples showed similar IR spectra compared to the monometallic LnPQQ complexes (Figure S3). And like the LnPQQ species, small differences appeared in the height of the large IR‐absorption band between 3630–2500 cm−1, stemming from different equiv of coordinated (or cocrystallized) water (Figure S3, Table S3). Analysis of the received precipitated material still revealed a 1:1 PQQ/metal stoichiometry by elemental analysis and ICP‐MS, but the metal content was now distributed between lanthanum and the additional Ln (see Supporting Information). The experiments were repeated three times (experimental replicates) and the distribution between the two added metals in the 1:1 LnPQQ complexes is given in Figure 7 in percent.
Figure 7.
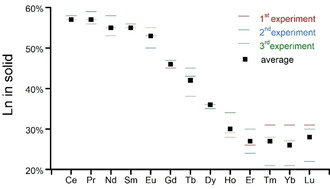
Amount of Ln vs. La in 1:1 PQQ–metal complexes, precipitated from aqueous Na2PQQ (51 μm) solutions at room temperature in percent. Values of three experiments are shown as bars, the averaged values of all three experiments are shown as squares.
Remarkably, when the additional metal is an early lanthanide (Ce–Eu) its uptake into the complex is preferred over lanthanum to up to 57 % for Ce, but slowly decreases over the series to 53 % for Eu. Preference for the second lanthanide then further decreases, falling below La, starting from Gd (46 %) to 26–28 % for the last three lanthanides (Tm‐Lu). In general, the slightly different properties of Ln caused by lanthanide contraction usually cause increasing complex stabilities from La to Lu,22b concurrent with higher affinities to ligands such as EDTA.4 Exceptions with rigid macrocycles such as functionalized aza‐crown ethers, where the larger Ln form more stable complexes are rare but have been reported.25 It is interesting that the “break” between favoring early Ln (Ce–Eu) over La versus favoring La over late Ln (Gd–Lu) occurs at Gd. Notably, in activity measurements of PQQ‐ and Ln‐dependent MDH isolated from M. fumariolicum SolV, it was also shown that activity was not stimulated anymore with the later lanthanides starting with Gd.14
Due to their different Lewis acidities, the lanthanide stock solutions exhibited varying pH values (pH 4.8–6.1, MilliQ water pH 5.9). Hence, to rule out an effect of varying pH, an additional experiment with fixed pH at 3.5 was performed with the Ln series at 25 °C. ICP‐MS of the precipitated solids revealed a similar pattern along the series as without controlled pH, although the percentage of lanthanum was overall slightly increased (Figure S4). Thus, variation of the pH of the Ln stock solutions did not influence the separation outcome significantly. In addition to monitoring enrichment in the solid phase we also investigated the separation factors of selected lanthanide pairs when an equimolar mixture of lanthanum and the following 13 lanthanides (excluding Pm) were mixed with PQQ at pH 2.5, 3.5 and 4.5 (Table S5). A lower pH was not investigated as it caused rapid precipitation of (partially) protonated PQQ. In the experiment with lanthanum and all lanthanides, separation factors (and enrichment in the solid phase) of directly adjacent pairs were hardly affected by a change in pH, while pairs (Ln2/Ln1) such as Nd/Dy and La/Lu showed a slight enhancement of the SFLn2/Ln1 at pH 2.5 (Table S5). Schelter and co‐workers recently described a hexadentate H3Tren‐1,2,3‐HOPO ligand that precipitated late lanthanides such as Dy at very low pH (2 m HCl) but not Nd and La under the same conditions.26 However, given that PQQ is mainly deprotonated at pH 3.5 (pK a=0.30 (N6), 1.60 (C7‐CO2H), 2.20 (C9‐CO2H), 3.30 (C2‐CO2H), 10.30 (N1), see Figure 2 B for numbering scheme)27 and that the late lanthanides were disfavored in our experiment, a different mechanism might be at work here. Another factor, which has to be considered, is the equilibrium between 1 and 3 that might shift upon lanthanide‐binding in solution.17a Furthermore, the impact of temperature on the lanthanide preference was tested with the two extremes of the lanthanide series (La and Lu). Aqueous mixtures of the two metal chlorides were prepared and were added to the Na2PQQ solution at four different temperatures (approximately 4, 25, 50 and 80 °C, Figure S5). At room temperature, La is preferred over Lu to 78 %, comparable with the solid enrichment data shown in Figure 7. However, when the metal mixtures are added to the PQQ solution at higher temperatures, the incorporation of Lu increased slightly in all cases. We also noted a small amount of Lu incorporation in the solid phase over time when a pure LaPQQ complex was heated with a solution of Lu at 80 °C for several hours. As demonstrated in Figure 7, a turn in selectivity occurred between La/Eu and La/Gd, hence Eu and Gd were tested against each other, as well as the Gd/Tb pair. The La/Ce couple was tested again in this series as control, as well as additional industrially attractive REE couples (Nd/Dy, Eu/Y) as they occur in End of Life (EoL) products such as permanent magnets or fluorescent materials. Here, Ce is again slightly preferred over La. For the other couples, the larger, earlier Ln is preferred in all cases (Figure S6). Both Eu in the Eu/Gd couple and Gd in the Gd/Tb couple show a ≈10 % higher occurrence over the following Ln in the series. With larger size differences between the two REE, the difference in incorporation in the solid phase increases. With Eu and Y, the two main elements of YOX, the precipitated EuYPQQ solid contains only 19 % of yttrium. With equimolar amounts of Nd and Dy, only 29 % of the smaller Dy gets coordinated by, and precipitates with PQQ. For the latter pair a separation factor SFDy/Nd of 2.4 was determined.
To investigate whether the separation is possibly based on structural preference, preorganization and size of the Ln/REE, the structures of the Ln1Ln2PQQ bulk materials were analyzed with the same techniques used for the monometallic LnPQQ complexes discussed above. The bimetallic EuLaPQQ complex shows high crystallinity with a powder X‐ray diffraction pattern similar to monometallic EuPQQ. The diffraction pattern of EuLaPQQ can also be completely indexed using the cell parameters and space group obtained for the complex shown in Figure 2. TbLaPQQ and LuLaPQQ are amorphous (Figure 5 A). Their pair distribution functions do not show correlations above r=6–8 Å (Figure 8).
Figure 8.
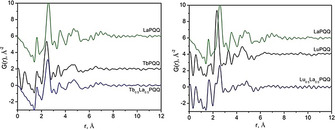
Pair distribution functions for TbLaPQQ and LuLaPQQ species in comparison with monometallic LaPQQ, LuPQQ, and TbPQQ.
The PDF of TbLaPQQ is similar to TbPQQ but does not fit with LaPQQ, while the PDF for LuLaPQQ seems to be a superposition of profiles characteristic for LaPQQ and LuPQQ, indicating that LuLaPQQ might be a mixture of two species or might have completely different structure in comparison with LaPQQ and LuPQQ with possible ordering of metals in structure or islands occupied by different metals.
Conclusions
In this study, we obtained the first crystal structure of PQQ with a lanthanide ion (Eu3+) and without the need of any co‐ligands. Different spectroscopic methods were used for extensive characterization of this and other LnPQQ complexes. Here, we also find that complexes with early lanthanides are preferentially formed with PQQ and rapidly precipitate out of aqueous solutions. It is very likely that the size of the Ln plays a role in the observed separation. While separation of Ln with PQQ is not competitive with existing processes at present, it is nevertheless noteworthy that a natural enzymatic cofactor is capable to achieve separation of the notoriously similar and thus difficult to separate Ln to some extent. Remarkably, Ln‐utilizing bacteria also show a preference for early lanthanides (uptake and growth), and isolated methanol dehydrogenases are more active with earlier Ln. It has been reasoned that the size and coordination of the Ln in the active site might be responsible for this, corroborated both by molecular dynamics simulations combined with fragment molecular orbital as well as with DFT calculations.14, 28 We hope that the thorough structural analysis described here will also help to characterize the exciting, recently discovered natural lanthanide enzymes to shed light into the many open questions in this emerging field of lanthanide‐dependent bacterial metabolism.
Experimental Section
Crystallographic data: Deposition number 1991651 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Lena J. Daumann obtained her Diploma in Chemistry in 2010 from the University of Heidelberg working with Peter Comba. After an internship at BASF she completed her PhD in 2013 with Lawrie Gahan at the University of Queensland in Australia working on pesticide‐degrading enzymes and biomimetic complexes. Her postdoctoral work as a Feodor‐Lynen fellow with Ken Raymond at UC Berkeley involved luminescent lanthanide complexes with siderophore‐inspired ligands. In 2016, she took up a position as W2 Professor for bioinorganic and coordination chemistry at the Ludwig‐Maximilians‐Universität Munich, where she is exploring the bioinorganic chemistry of lanthanides and the role of high valent iron species in epigenetic processes. She has won numerous Awards for her research and teaching. Among them the Prinzessin Therese von Bayern‐Preis and the Ars legendi‐Fakultätenpreis in Chemistry, both in 2019.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors wish to thank Christine Benning for ICP‐MS measurements. L.J.D., H.L. and A.M. would like to acknowledge a grant from the Deutsche Forschungsgemeinschaft (DFG)‐392552271 as well as support from the L.M.U. The authors thank the P02.1 beamline at the PETRA III synchrotron facility (DESY, Hamburg) and BL10 beamline at the DELTA synchrotron facility (Technische Universität Dortmund) for providing us measurement time and technical support. The authors also thank Dr. Alexander Schökel (PETRA III) and Dr. Ralph Wagner (Bergische Universität Wuppertal, Wuppertal) for their help with measurements at the beamlines.
H. Lumpe, A. Menke, C. Haisch, P. Mayer, A. Kabelitz, K. V. Yusenko, A. Guilherme Buzanich, T. Block, R. Pöttgen, F. Emmerling, L. J. Daumann, Chem. Eur. J. 2020, 26, 10133.
References
- 1.
- 1a. Howanietz R., China's Virtual Monopoly of Rare Earth Elements: Economic, Technological and Strategic Implications, 1st ed., Routledge, New York, 2018; [Google Scholar]
- 1b. Goodenough K. M., Wall F., Merriman D., Nat. Resour. Res. 2018, 27, 201–216. [Google Scholar]
- 2. Hans Wedepohl K., Geochim. Cosmochim. Acta 1995, 59, 1217–1232. [Google Scholar]
- 3.A. Walters, P. Lusty, A. Hill, British Geological Survey: Rare Earth Elements, Natural Environment Research Council, http://www.bgs.ac.uk/mineralsuk/, 2011.
- 4. Cheisson T., Schelter E. J., Science 2019, 363, 489–493. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Bogart J. A., Lippincott C. A., Carroll P. J., Schelter E. J., Angew. Chem. Int. Ed. 2015, 54, 8222–8225; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8340–8343; [Google Scholar]
- 5b. Cole B. E., Falcones I. B., Cheisson T., Manor B. C., Carroll P. J., Schelter E. J., Chem. Commun. 2018, 54, 10276–10279; [DOI] [PubMed] [Google Scholar]
- 5c. Cheisson T., Cole B. E., Manor B. C., Carroll P. J., Schelter E. J., ACS Sustainable Chem. Eng. 2019, 7, 4993–5001. [Google Scholar]
- 6. Li X.-Z., Zhou L.-P., Yan L.-L., Dong Y.-M., Bai Z.-L., Sun X.-Q., Diwu J., Wang S., Bünzli J.-C., Sun Q.-F., Nat. Commun. 2018, 9, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bogart J. A., Cole B. E., Boreen M. A., Lippincott C. A., Manor B. C., Carroll P. J., Schelter E. J., Proc. Natl. Acad. Sci. USA 2016, 113, 14887–14892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li X.-Z., Zhou L.-P., Yan L.-L., Dong Y.-M., Bai Z.-L., Sun X.-Q., Diwu J., Wang S., Bünzli J.-C., Sun Q.-F., Nat.Commun. 2018, 9, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Higgins R. F., Cheisson T., Cole B. E., Manor B. C., Carroll P. J., Schelter E. J., Angew. Chem. Int. Ed. 2020, 59, 1851–1856; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 1867–1872. [Google Scholar]
- 10. Daumann L. J., Angew. Chem. Int. Ed. 2019, 58, 12795–12802; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 12926–12933. [Google Scholar]
- 11. Pol A., Barends T. R. M., Dietl A., Khadem A. F., Eygensteyn J., Jetten M. S. M., Op den Camp H. J. M., Environ. Microbiol. 2014, 16, 255–264. [DOI] [PubMed] [Google Scholar]
- 12. Houck D. R., Hanners J. L., Unkefer C. J., van Kleef M. A. G., Duine J. A., Antonie Van Leeuwenhoek 1989, 56, 93–101. [DOI] [PubMed] [Google Scholar]
- 13. Martinez-Gomez N. C., Vu H. N., Skovran E., Inorg. Chem. 2016, 55, 10083–10089. [DOI] [PubMed] [Google Scholar]
- 14. Lumpe H., Pol A., Op den Camp H. J. M., Daumann L. J., Dalton Trans. 2018, 47, 10463–10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nevarez J., Turmo A., Hu J., Hausinger R. P., ChemCatChem 2020, 10.1002/cctc.202000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Roach B. D., Fenske E. K., Ilgner R. H., Hexel C. R., Haverlock T. J., Giaquinto J. M., J. Chromatogr. A 2019, 1587, 155–165; [DOI] [PubMed] [Google Scholar]
- 16b. Ganjali M. R., Gupta V. K., Faridbod F., Norouzi P., Lanthanides Series Determination by Various Analytical Methods, Elsevier, Amsterdam, 2016; [Google Scholar]
- 16c. Borrini J., Favre-Reguillon A., Lemaire M., Gracia S., Arrachart G., Bernier G., Hérès X., Hill C., Pellet-Rostaing S., Solvent Extr. Ion Exch. 2015, 33, 224–235. [Google Scholar]
- 17.
- 17a. Lumpe H., Daumann L. J., Inorg. Chem. 2019, 58, 8432–8441; [DOI] [PubMed] [Google Scholar]
- 17b. Nakamura N., Kohzuma T., Kuma H., Suzuki S., Inorg. Chem. 1994, 33, 1594–1599; [Google Scholar]
- 17c. Schwederski B., Kasack V., Kaim W., Roth E., Jordanov J., Angew. Chem. Int. Ed. Engl. 1990, 29, 78–79; [Google Scholar]; Angew. Chem. 1990, 102, 74–76. [Google Scholar]
- 18.
- 18a. Wanner M., Sixt T., Klinkhammer K.-W., Kaim W., Inorg. Chem. 1999, 38, 2753–2755; [Google Scholar]
- 18b. Mitome H., Ishizuka T., Shiota Y., Yoshizawa K., Kojima T., Inorg. Chem. 2013, 52, 2274–2276; [DOI] [PubMed] [Google Scholar]
- 18c. Nakamura N., Kohzuma T., Kuma H., Suzuki S., Inorg. Chem. 1994, 33, 1594–1599. [Google Scholar]
- 19.
- 19a. Concas G., Congiu F., Spano G., Bettinelli M., Speghini A., Flint C. D., Z. Naturforsch. A 2001, 56a, 789–793; [Google Scholar]
- 19b. Dietel A. M., Döring C., Glatz G., Butovskii M. V., Tok O., Schappacher F. M., Pöttgen R., Kempe R., Eur. J. Inorg. Chem. 2009, 1051–1059. [Google Scholar]
- 20.
- 20a. Glentworth P., Nichols A. L., Newton D. A., Large N. R., Bullock R. J., J. Chem. Soc. Dalton Trans. 1973, 546–550; [Google Scholar]
- 20b. Kaneko M., Miyashita S., Nakashima S., Dalton Trans. 2015, 44, 8080–8088. [DOI] [PubMed] [Google Scholar]
- 21. Ravel B., Newville M., J. Synchrotron Radiat. 2005, 12, 537–541. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Cotton S. A., C. R. Chim. 2005, 8, 129–145; [Google Scholar]
- 22b. Cotton S. A., Raithby P. R., Coord. Chem. Rev. 2017, 340, 220–231. [Google Scholar]
- 23.
- 23a. Seitz M., Pluth M. D., Raymond K. N., Inorg. Chem. 2007, 46, 351–353; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Nakamoto K., Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, 6th ed., Wiley, Hoboken, 2009. [Google Scholar]
- 24.
- 24a. Dimitrijevic N. M., Poluektov O. G., Saponjic Z. V., Rajh T., J. Phys. Chem. B 2006, 110, 25392–25398; [DOI] [PubMed] [Google Scholar]
- 24b. Tommasi L., Shechter-Barloy L., Varech D., Battioni J. P., Donnadieu B., Verelst M., Bousseksou A., Mansuy D., Tuchagues J. P., Inorg. Chem. 1995, 34, 1514–1523. [Google Scholar]
- 25.
- 25a. Peters J. A., Djanashvili K., Geraldes C. F. G. C., Platas-Iglesias C., Coord. Chem. Rev. 2020, 406, 213146; [Google Scholar]
- 25b. Roca-Sabio A., Mato-Iglesias M., Esteban-Gómez D., Tóth É., d. Blas A., Platas-Iglesias C., Rodríguez-Blas T., J. Am. Chem. Soc. 2009, 131, 3331–3341. [DOI] [PubMed] [Google Scholar]
- 26. Nelson J. J. M., Cheisson T., Rugh H. J., Gau M. R., Carroll P. J., Schelter E. J., Commun. Chem. 2020, 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a. Kano K., Mori K., Uno B., Kubota T., Ikeda T., Senda M., J. Electroanal. Chem. Interfacial Electrochem. 1990, 299, 193–201; [Google Scholar]
- 27b. Kano K., Mori K., Uno B., Kubota T., Ikeda T., Senda M., J. Electroanal. Chem. Interfacial Electrochem 1990, 298, 227–238; [Google Scholar]
- 27c. Zhang Z. P., Tillekeratne L. M. V., Kirchhoff J. R., Hudson R. A., Biochem. Biophys. Res. Commun. 1995, 212, 41–47. [DOI] [PubMed] [Google Scholar]
- 28. Tsushima S., Phys. Chem. Chem. Phys. 2019, 21, 21979–21983. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary