

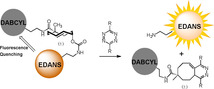
Abstract
The inverse electron demand Diels–Alder pyridazine elimination reaction between tetrazines and allylic substituted trans‐cyclooctenes (TCOs) is a key player in bioorthogonal bond cleavage reactions. Determining the rate of elimination of alkylamine substrates has so far proven difficult. Here, we report a fluorogenic tool consisting of a TCO‐linked EDANS fluorophore and a DABCYL quencher for accurate determination of both the click and release rate constants for any tetrazine at physiologically relevant concentrations.
Keywords: bioorthogonal chemistry, Diels–Alder reactions, fluorescent probes, kinetics, nitrogen heterocycles
Click‐to‐release! A new fluorogenic trans‐cyclooctene probe was has been designed and synthesized to determine the elimination kinetics of the click‐to‐release reaction. The reagent enables accurate detection of the decaging potential for any tetrazine at physiologically relevant concentrations.
Introduction
Bioorthogonal chemistry is broadening its scope to include bond cleavage reactions alongside known ligation methods,1, 2, 3, 4 with many new reactions steadily emerging in this area.5, 6, 7, 8, 9, 10, 11, 12 Among these is the inverse electron demand Diels–Alder (IEDDA) pyridazine elimination reaction,13 the first of the bioorthogonal processes known as “click‐to‐release” (Figure 1 A). In the IEDDA decaging sequence, the reaction of a tetrazine and a trans‐cyclooctene (TCO) bearing a leaving group at the allylic position14, 15 results in a 4,5‐dihydropyridazine intermediate. This adduct tautomerizes to yield a 1,4‐dihydropyridazine species, which induces elimination of the allylic payload. The biocompatibility of the tetrazine and TCO components, combined with their selectivity and overall deprotection rate, has culminated in various applications, such as the activation of cytotoxic (pro)drugs,16, 17, 18 proteins,19, 20 and immune cells.21 The technique is also compatible with reagents that enable spatiotemporal control within biological systems.16, 17, 18
Figure 1.
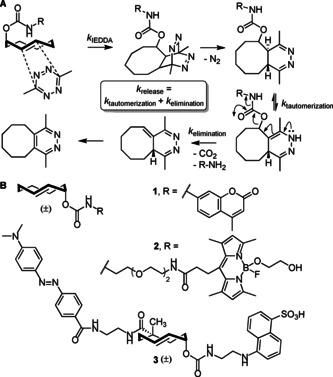
(A) Overview of the IEDDA pyridazine elimination reaction, which is also known as the ‘click‐to‐release’ reaction. (B) Fluorogenic TCOs to determine click‐to‐release kinetics: TCO reporters 1 and 2 (Fan et al.22 and Carlston et al.,23 respectively) and bifunctional TCO‐reporter‐quencher pair 3 designed in this study.
In order to successfully apply click‐to‐release in biology, the identification of optimized tetrazines is crucial. Altering tetrazine substituents can have drastic effects on both the rate and extent of the release obtained.13, 22, 23, 24 Fluorogenic reporters, such as the caged coumarin 1 22 (Figure 1 B), are important tools to characterize tetrazine release behavior. These are supported by NMR13, 25 and LC‐MS22, 23, 24 analyses. Among the difficulties associated with these studies are the profound effect of pH and buffer concentration on elimination.23, 25 Furthermore, Chen and co‐workers22 noted that 1 displays a lower decaging rate than a caged compound in which the coumarin (aniline release) is substituted by Fmoc‐lysine ϵ‐amine (alkylamine release). Carlson et al.23 described caged reporters that enable the determination of primary amine release kinetics by LC‐MS (e.g., 2, Figure 1 B). Compound 2 was also evaluated in a quenched fluorescence assay23 that employed a tetrazine bearing a black hole quencher. The click reaction of 2 with 1 mm black‐hole‐quencher‐tetrazine led to the rapid formation of a 4,5‐dihydropyridazine adduct in which the caged MayaFluor dye is quenched. The detection of fluorescence thereby provided a direct measurement of k release (constituting the sum of k tautomerization and k elimination) without the influence of cycloaddition (k IEDDA). However, this method does not allow the characterization of (unmodified) tetrazines at lower concentrations.
A method based on fluorescence quenching to rapidly determine the rate constants of both the cycloaddition (k IEDDA) and alkylamine release (k release) caused by tetrazines without stringent concentration requirements would be of great importance. With this in mind, we hereby report on the design, synthesis, and evaluation of quenched reporter 3 (Figure 1 B) based on the bifunctional TCO introduced by Robillard and co‐workers.16 By linking the 4‐[4‐(dimethylamino)phenylazo]benzoyl (DABCYL) quencher to an 5‐(2‐aminoethylamino)naphthalene‐1‐sulfonic acid (EDANS) fluorophore through the bifunctional TCO scaffold, the rate constants for cycloaddition and alkylamine release can be quantified in a 96‐well plate reader format. Pseudo‐first‐order rate constants were determined for a series of tetrazines, and DFT calculations were employed to probe the differences in the initial cycloaddition step between mono‐ and bifunctional TCOs.
Results and Discussion
The synthesis of bifunctional TCO reagent 4 was based on the published route (Scheme 1).16 However, to both simplify and accelerate the procedure, the initial functionalization of cyclooctadiene 5 to yield the α‐methylated carboxylic acid 6 (bromination, cyanide substitution, oxidation, basic hydrolysis, and methylation) was carried out using the crude reaction mixtures obtained after aqueous workup (see Scheme S1 in the Supporting Information). The crystallization of iodolactone 7 from EtOH resulted in a yield of 23 % over five steps from 5 on the 500 mmol scale, without the need for the previously reported distillations.16
Scheme 1.
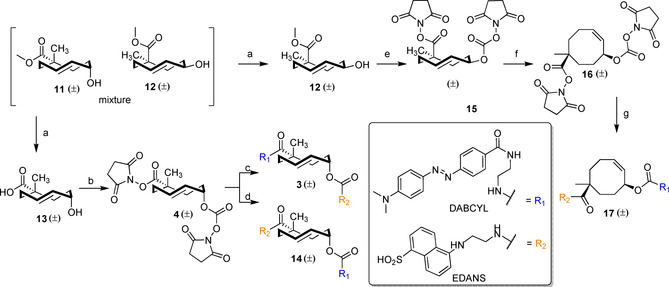
Synthesis of bifunctional TCO‐reporter‐quencher pairs 3 and 4 and CCO‐reporter‐quencher pair 17 from cyclooctadiene 5. Reagents and conditions for the synthesis of 11 and 12 from cyclooctadiene 5 can be found in Scheme S1 in the Supporting Information. Reagents and conditions: (a) KOH, MeOH, H2O, 4 °C, 43 % (12), 42 % (13); (b) N,N′‐disuccinimidyl carbonate, N,N‐diisopropylethylamine (DIPEA), DMAP, MeCN, RT, 72 %; (c) i. EDANS‐NH2, DIPEA, DMF, RT, 79 %; ii. DABCYL‐NH2, DIPEA, DMF, RT, 60 %; (d) i. DABCYL‐NH2, DIPEA, DMF, RT, 96 %; ii. EDANS‐NH2, DIPEA, DMF, RT, 88 %; (e) i. KOH, MeOH, H2O, 50 °C, 79 %; ii. N,N′‐disuccinimidyl carbonate, DIPEA, MeCN, RT, 66 %; (f) hν (CFL), CDCl3, RT, 74 %; (g) i. DABCYL‐NH2, DIPEA, DMF, RT, 60 %; ii. EDANS‐NH2, DIPEA, DMF, RT, 50 %.
The transesterification procedure starting from bicyclic lactone 8 (64 h, 48 % yield)16 was replaced by a one‐pot, two‐step procedure to improve the overall conversion and reaction time. Saponification afforded monocyclic carboxylic acid 9, which was directly methylated to obtain methyl ester 10 in a yield of 77 % over two steps. Photoisomerization26 resulted in a 1:1.4 mixture of axial isomer 11 (axial hydroxy, equatorial methyl ester) and equatorial isomer 12 (equatorial hydroxy, axial methyl ester), respectively (see Scheme S1 in the Supporting Information). The isomeric mixture was treated with potassium hydroxide at 4 °C to selectively hydrolyze the axial isomer 11, followed by acid–base extraction to obtain carboxylic acid 13 and ester 12 separately. The disuccinimidyl functionalization of 13 to give 4 (3 days, 46 % yield)16 was accelerated through nucleophilic catalysis (4‐dimethylaminopyridine (DMAP)) and hydrolysis of the obtained product during chromatographic purification was prevented by employing neutralized silica gel, resulting in a yield of 72 %. Compound 4 reacted with EDANS (fluorophore) or DABCYL (quencher) moiety with complete regioselectivity towards the carbonate due to the steric hindrance induced by the methyl group. This was followed by functionalization with the complementary moiety to obtain axial TCO‐reporter‐quencher pairs 3 and 14.
We also synthesized a bifunctional cis‐cyclooctene (CCO)‐reporter‐quencher pair. However, attempts to functionalize carboxylic acid 9 resulted in the formation of cyclic lactone 8 (see Scheme S1 in the Supporting Information). Instead, the equatorial isomer 12 was hydrolyzed and functionalized to obtain the equatorial TCO reagent 15, followed by trans‐to‐cis isomerization in the presence of visible light (26 W CFL bulb, see Figures S1–S3 in the Supporting Information) to form bifunctional CCO reagent 16. Functionalization with EDANS and DABCYL occurred with the same degree of regioselectivity to yield CCO‐reporter‐quencher pair 17.
We used 3 to assess a series of tetrazines for their ability to induce the IEDDA pyridazine elimination. The axial TCO‐reporter‐quencher pair 3 is fluorogenic by virtue of intramolecular fluorescence quenching until the EDANS fluorophore is released from the post‐ligation construct (Figure 2), and could thus serve to determine the overall properties of the deprotection reaction. This was confirmed by treating 3 and 17 with 3,6‐dimethyltetrazine (18, see Figure S4 in the Supporting Information). The reaction between 3 and 18 was characterized by quantifying the fluorescence emitted from the liberated EDANS fluorophore (see Figure S5). This experiment showed an EDANS release yield of 80 % after 1 h, which corresponds with other reports.13, 23, 25 A panel of tetrazines was then selected from the literature (Figure 3 A, 18–28), including key substrates reported by Chen,22 Weissleder,23 and ourselves.24 The release of EDANS from TCO‐reporter‐quencher pair 3 (10 μm) was measured over time using an excess of tetrazine (100 and 400 μm) in a phosphate‐buffered solution (0.2 m PO4 2−, 10 % DMSO in H2O, see Figure S6). The functional groups of (asymmetric) tetrazines direct the regiochemistry of the initial Diels–Alder ligation reaction to form the 4,5‐dihydropyridazine adduct. Tautomerization to the 1,4‐dihydropyridazine enables fast release whereas tautomerization to the 2,5‐dihydropyridazine results in a slow releasing tautomer (which proceeds by tautomerization back to the 4,5‐dihydropyidazine). Part of the 2,5‐dihydropyridazine population can also convert into the oxidation product without releasing the allylic EDANS.23, 25 The release rate constant k obs is therefore the sum of the rate constants of these processes, which we fitted with a biphasic decay line. The pseudo‐first‐order rate constants for the “fast‐releasing” adduct (k obs, Figure 3 B) and elimination after 4 h (Figure 3 C, normalized for [18]=400 μm) were determined. For several tetrazines (18, 20, 24, and 26) with comparable k obs values at 100 and 400 μm, the measurements were repeated with 100 nm 3 and 10–100 μm tetrazine (see Figure S7). We also repeated the initial measurements for the TCO‐reporter‐quencher pair 14 to determine the release of DABCYL over time (Figure 3 D, see Figures S8 and S9). In this case, the formed pyridazine adduct appeared to act as a fluorescence quencher after IEDDA, limiting the fluorescence emitted by EDANS compared with the results obtained for 3. These results show 14 to be unfit for elimination efficiency analysis.
Figure 2.
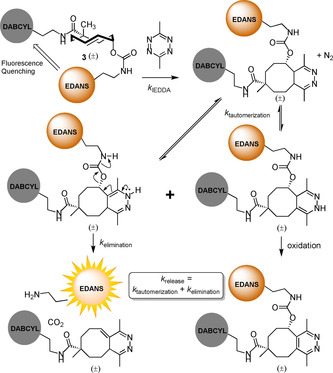
Schematic representation of the TCO‐quenched fluorescence assay. The bifunctional TCO‐reporter‐quencher pair 3 does not display fluorescence in its native state due to fluorescence quenching between the EDANS fluorophore and DABCYL quencher. Upon cycloaddition of a tetrazine, the 4,5‐dihydropyridazine adduct may tautomerize to the 1,4‐dihydropydazine intermediate. This species can eliminate CO2 and the EDANS fluorophore, thereby disabling the fluorescence quenching and enabling a fluorescent readout for the elimination step. Alternatively, the 2,5‐dihydropyridazine intermediate is formed, which may tautomerize back to the 4,5‐dihydropyridazine adduct or undergo oxidation.
Figure 3.
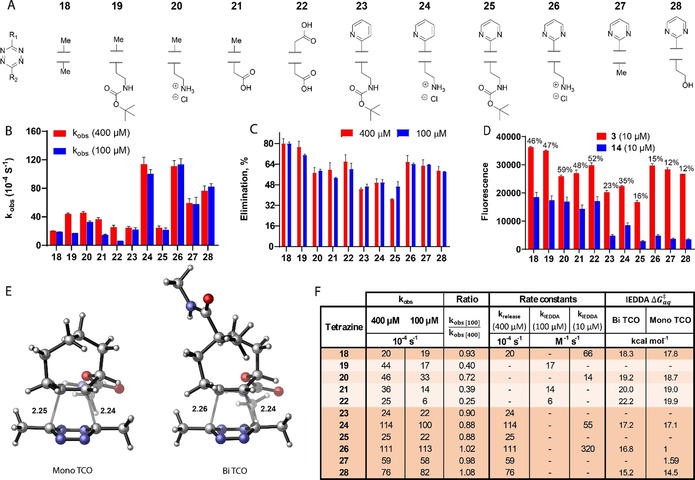
(A) Tetrazines 18–28 selected from the literature and investigated in this study. (B) Pseudo‐first‐order rate constants (k obs) determined by treating TCO‐reporter‐quencher pair 3 (10 μm) with tetrazines 18–28 (100 or 400 μm, N=2) and measuring the fluorescence intensity of released EDANS (λ ex=340 nm, λ em=495 nm). (C) Elimination efficiency observed for TCO‐reporter‐quencher pair 3 (10 μm) using tetrazines 18–28 (100 or 400 μm, N=2, t=4 h), normalized to tetrazine 18 (100 μm, t=1 h, 80 %, see Figure S5 in the Supporting Information). (D) Fluorescence observed for TCO‐reporter‐quencher pairs 3 (10 μm) and 14 (10 μm) using tetrazines 18–28 (100 μm, N=2, t=4 h). The fluorescence detected for 14 relative to the corresponding result for 3 is given as percentages. (E) Representative examples of the PCM(H2O)‐M06‐2X/6‐31+G(d)‐optimized transition‐state structures for the reactions of tetrazines (18 shown in the figure) and model axial mono‐ and bifunctional TCO. Bond lengths are in Å. (F) Summary of the kinetic investigations with TCO‐reporter‐quencher pair 3, including pseudo‐first‐order rate constants (k obs, for 10 μm 3 and 400 or 100 μm tetrazine, Figure 3 B), the ratio of k obs values (k obs at 100 μm/k obs at 400 μm≈1, orange; k obs at 100 μm/k obs at 400 μm<1, white), estimated rate constants k release and k IEDDA based on the ratio of the k obs values or on a separate experiment at 100 nm 3 with 10–100 μm tetrazine, Figure S6) and calculated energies of the transition states formed between tetrazines 18–28 and mono‐ and bifunctional TCO model compounds.
Within the panel of tetrazines that we experimentally examined with 10 μm 3, tetrazines 19, 21, and 22 displayed reduced k obs values when switching from 400 to 100 μm, which indicates that for these tetrazines the initial cycloaddition step plays a significant role in the overall reaction rate (k obs≈k IEDDA[tetrazine], Figure 3 F). For tetrazines 18 and 23–28, the k obs values remained similar for both concentrations examined, thereby demonstrating that the overall reaction rate is controlled by the release rate (k obs≈k release, Figure 3 F). Additionally, the experiments performed at 100 nm 3 with tetrazines 18, 20, 24, and 26 (10–100 μm) allowed us to determine k IEDDA for these tetrazines as well, because in this concentration range the k obs values decreased when reducing the tetrazine concentration (k obs≈k IEDDA[tetrazine], Figure 3 F). The total elimination yield for a given tetrazine is difficult to predict on the basis of the functional groups present, and no clear correlation between release rate and elimination yield can be determined, highlighting the importance of the kinetic quantification described here. Furthermore, it should be noted that the 4 h timepoint does not always show the absolute endpoint of a given reaction.
To rationalize the rate constants for IEDDA cycloaddition determined with 3, DFT calculations were performed. The transition state of the IEDDA cycloaddition step was studied for two model TCOs (mono TCO and bi TCO) with tetrazines 18, 20–22, 24, 26, and 28 (Figure 3 E) by using PCM(H2O)‐M06‐2X/6‐31+G(d) (see the Supporting Information). The results revealed a slight destabilization of the transition state for the cycloaddition of the bifunctional TCO compared with the monofunctional TCO for all tetrazines, which results in a minor increase in the reaction barrier (Figure 3 F). The transition‐state structures for both model TCOs are very similar in terms of geometry (see the Supporting Information), and the trends in reactivity between tetrazines were maintained. Therefore, these results suggest that the steric bulk introduced into the bifunctional TCO does not significantly hamper the initial cycloaddition step.
Conclusions
We have developed a new method based on fluorescence quenching to determine the rate constants for both the cycloaddition and alkylamine release steps of the IEDDA pyridazine elimination in a 96‐well plate reader format. The TCO‐reporter‐quencher pair 3 was synthesized by functionalization of bisuccinimidyl‐TCO reagent 4 with an EDANS fluorophore and a DABCYL quencher. The new method reported herein was used to determine the click‐to‐release kinetics and yields for 3 with tetrazines 18–28. For tetrazines 19–22, the results indicated a greater concentration dependence compared with the other tetrazines studied. We were able to determine both rate constants (k IEDDA and k release) for tetrazines 18, 24, and 26. Furthermore, the DFT calculations described here indicated similar IEDDA reactivity for both mono‐ and bifunctional TCOs, which suggests that the observations made with probe 3 can be translated to the deprotection of mono‐functionalized TCOs. However, predicting the tetrazine behavior in IEDDA pyridazine elimination remains challenging. We therefore recommend this new fluorescence assay to rapidly screen tetrazines for click‐to‐release potential.
Conflict of interest
Wolter ten Hoeve: employment at Syncom; Marc Robillard: cofounder of Tagworks Pharmaceuticals.
Biographical Information
Sander van Kasteren earned his degree in Chemistry and Medicinal Chemistry from the University of Edinburgh. In 2007, he completed his PhD under the supervision of Prof. B. G. Davis at the University of Oxford, working on the synthesis and application of complex carbohydrate imaging probes. Using his Henry Wellcome Fellowship, he moved to the University of Dundee to study antigen processing with Prof. C. Watts. He next worked with Profs. H. Ovaa and J. Neefjes at the Netherlands Cancer Institute on a NWO‐Veni‐Fellowship. In 2012, he won the Early Career Award of the British Biochemical Society and took up an independent position at Leiden University. His group works on synthetic methodology to study information transfer in the adaptive immune response.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
S.I.V.K. was the recipient of an ERC Starting Grant from the European Research Council (ERC‐2014‐StG‐639005). A.J.C.S., D.V.F., H.S.O., and S.I.V.K. were funded by the Institute of Chemical Immunology (NWO Zwaartekracht). The authors thank SURFsara for use of the Lisa supercomputer and kindly acknowledge Mark Somers for technical support.
M. A. R. de Geus, E. Maurits, A. J. C. Sarris, T. Hansen, M. S. Kloet, K. Kamphorst, W. ten Hoeve, M. S. Robillard, A. Pannwitz, S. A. Bonnet, J. D. C. Codée, D. V. Filippov, H. S. Overkleeft, S. I. van Kasteren, Chem. Eur. J. 2020, 26, 9900.
References
- 1. Li J., Chen P. R., Nat. Chem. Biol. 2016, 12, 129–137. [DOI] [PubMed] [Google Scholar]
- 2. Neumann K., Gambardella A., Bradley M., ChemBioChem 2019, 20, 872–876. [DOI] [PubMed] [Google Scholar]
- 3. Tu J., Xu M., Franzini R. M., ChemBioChem 2019, 20, 1615–1627. [DOI] [PubMed] [Google Scholar]
- 4. Devaraj N. K., ACS Cent. Sci. 2018, 4, 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li J., Yu J., Zhao J., Wang J., Zheng S., Lin S., Chen L., Yang M., Jia S., Zhang X., Chen P. R., Nat. Chem. 2014, 6, 352–361. [DOI] [PubMed] [Google Scholar]
- 6. Jiménez-Moreno E., Guo Z., Oliveira B. L., Albuquerque I. S., Kitowski A., Guerreiro A., Boutureira O., Rodrigues T., Jiménez-Osés G., Bernardes G. J. L., Angew. Chem. Int. Ed. 2016, 55, 243–247; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 249–253. [Google Scholar]
- 7. Pérez-López A. M., Rubio-Ruiz B., Sebastián V., Hamilton L., Adam C., Bray T. L., Irusta S., Brennan P. M., Lloyd-Jones G., Sieger D., Santamaría J., Unciti-Broceta A., Angew. Chem. Int. Ed. 2017, 56, 12548–12552; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12722–12726. [Google Scholar]
- 8. Bernard S., Audisio D., Riomet M., Bregant S., Sallustrau A., Plougastel L., Decuypere E., Gabillet S., Kumar R. A., Elyian J., Trinh M. N., Koniev O., Wagner A., Kolodych S., Taran F., Angew. Chem. Int. Ed. 2017, 56, 15612–15616; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15818–15822. [Google Scholar]
- 9. Tu J., Xu M., Parvez S., Peterson R. T., Franzini R. M., J. Am. Chem. Soc. 2018, 140, 8410–8414. [DOI] [PubMed] [Google Scholar]
- 10. Matikonda S. S., Orsi D. L., Staudacher V., Jenkins I. A., Fiedler F., Chen J., Gamble A. B., Chem. Sci. 2015, 6, 1212–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lelieveldt L. P. W. M., Eising S., Wijen A., Bonger K. M., Org. Biomol. Chem. 2019, 17, 8816–8821. [DOI] [PubMed] [Google Scholar]
- 12. Wu H., Alexander S. C., Jin S., Devaraj N. K., J. Am. Chem. Soc. 2016, 138, 11429–11432. [DOI] [PubMed] [Google Scholar]
- 13. Versteegen R. M., Rossin R., ten Hoeve W., Janssen H. M., Robillard M. S., Angew. Chem. Int. Ed. 2013, 52, 14112–14116. [DOI] [PubMed] [Google Scholar]
- 14. Sauer J., Wiest H., Angew. Chem. 1962, 74, 353. [Google Scholar]
- 15. Blackman M. L., Royzen M., Fox J. M., J. Am. Chem. Soc. 2008, 130, 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rossin R., van Duijnhoven S. M. J., ten Hoeve W., Janssen H. M., Hoeben F. J. M., Versteegen R. M., Robillard M. S., Bioconjugate Chem. 2016, 27, 1697–1706. [DOI] [PubMed] [Google Scholar]
- 17. Rossin R., Versteegen R. M., Wu J., Khasanov A., Wessels H. J., Steenbergen E. J., ten Hoeve W., Janssen H. M., van Onzen A. H. A. M., Hudson P. J., Robillard M. S., Nat. Commun. 2018, 9, 1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mejia Oneto J. M., Khan I., Seebald L., Royzen M., ACS Cent. Sci. 2016, 2, 476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li J., Jia S., Chen P. R., Nat. Chem. Biol. 2014, 10, 1003–1005. [DOI] [PubMed] [Google Scholar]
- 20. Zhang G., Li J., Xie R., Fan X., Liu Y., Zheng S., Ge Y., Chen P. R., ACS Cent. Sci. 2016, 2, 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van der Gracht A. M. F., de Geus M. A. R., Camps M. G. M., Ruckwardt T. J., Sarris A. J. C., Bremmers J., Maurits E., Pawlak J. B., Posthoorn M. M., Bonger K. M., Filippov D. V., Overkleeft H. S., Robillard M. S., Ossendorp F., van Kasteren S. I., ACS Chem. Biol. 2018, 13, 1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fan X., Ge Y., Lin F., Yang Y., Zhang G., Ngai W. S. C., Lin Z., Zheng S., Wang J., Zhao J., Li J., Chen P. R., Angew. Chem. Int. Ed. 2016, 55, 14046–14050; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14252–14256. [Google Scholar]
- 23. Carlson J. C. T., Mikula H., Weissleder R., J. Am. Chem. Soc. 2018, 140, 3603–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sarris A. J. C., Hansen T., de Geus M. A. R., Maurits E., Doelman W., Overkleeft H. S., Codée J. D. C., Filippov D. V., van Kasteren S. I., Chem. Eur. J. 2018, 24, 18075–18081. [DOI] [PubMed] [Google Scholar]
- 25. Versteegen R. M., ten Hoeve W., Rossin R., de Geus M. A. R., Janssen H. M., Robillard M. S., Angew. Chem. Int. Ed. 2018, 57, 10494–10499; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10654–10659. [Google Scholar]
- 26. Royzen M., Yap G. P. A., Fox J. M., J. Am. Chem. Soc. 2008, 130, 3760–3761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary