

Abstract
Mass spectrometry‐based proteomics experiments typically start with the digestion of proteins using trypsin, chosen because of its high specificity, availability, and ease of use. It has become apparent that the sole use of trypsin may impose certain limits on our ability to grasp the full proteome, missing out particular sites of post‐translational modifications, protein segments, or even subsets of proteins. To tackle this problem, alternative proteases have been introduced and shown to lead to an increase in the detectable (phospho)proteome. Here, we argue that there may be further room for improvement and explore the protease EndoPro. For optimal peptide identification rates, we explored multiple peptide fragmentation techniques (HCD, ETD, and EThcD) and employed Byonic as search algorithm. We obtain peptide IDs for about 40% of the MS2 spectra (66% for trypsin). EndoPro cleaves with high specificity at the C‐terminal site of Pro and Ala residues and displays activity in a broad pH range, where we focused on its performance at pH = 2 and 5.5. The proteome coverage of EndoPro at these two pH values is rather distinct, and also complementary to the coverage obtained with trypsin. As about 40% of mammalian protein phosphorylations are proline‐directed, we also explored the performance of EndoPro in phosphoproteomics. EndoPro extends the coverable phosphoproteome substantially, whereby both the, at pH = 2 and 5.5, acquired phosphoproteomes are complementary to each other and to the phosphoproteome obtained using trypsin. Hence, EndoPro is a powerful tool to exploit in (phospho)proteomics applications.
Keywords: (phospho)proteomics, EndoPro, mass spectrometry, proline effect, protease
To improve (phospho)proteome coverage, we characterized EndoPro, a proline‐specific protease. The protease exhibits high specificity for cleavage C‐terminal to proline and alanine and is interestingly not hindered by phosphorylations near the cleavage site. EndoPro is complementary to trypsin and enabled us to detect over 2200 unique proteins not observed by trypsin and contributed 49% of the uniquely identified phosphosites.
Abbreviations
- ETD
electron transfer dissociation
- EThcD
electron transfer higher‐energy collision dissociation
- HCD
higher‐energy collision dissociation
- LC
liquid chromatography
- MS
mass spectrometry
- ON
overnight
- pI
isoelectric point
- PSM
peptide spectrum match
- PTM
post‐translational modification
Introduction
Proteins are involved in nearly all biological processes. Their functionality can be regulated extensively, through the formation of complexes, changes in expression levels, and widespread post‐translational modifications, such as acetylation and phosphorylation. Proteins must be tightly regulated as undesired changes at the protein level can cause disease and other unintended biological effects 1, 2. Owing to their crucial role, identifying and quantifying proteins present in certain biological states is of great importance and can improve our understanding of the mechanisms underlying health and disease. To this end, the field of proteomics aims to measure all proteins expressed by a certain organism or cell type 3, 4. Proteomics comes in a range of different workflows 3. In the more standard bottom‐up workflow, proteins are extracted from the material of interest and subjected to proteolysis, which results in a complex mixture of peptides that originated from the proteins present in the targeted cells. Through LC‐MS/MS analysis, these peptides are separated, fragmented, and analyzed. Then, the collected empirical spectra are correlated to peptide and thus protein sequences through the use of theoretical in silico fragmentation spectra 5, 6. However, due to the indirect nature of this assignment, how peptides are generated from the intact protein is of critical importance.
Most proteomic studies use trypsin for the protein digestion as it cleaves with very high specificity proteins C terminally to only arginine (Arg) and lysine (Lys) residues. As both amino acids are basic, the resulting peptides have basic C termini. This, combined with the free amine at the peptide N terminus, ensures that tryptic peptides carry a positive charge at either end of the peptide, making them very suitable for fragmentation‐based sequencing 6. In addition, trypsin's high specificity reduces the complexity of the subsequent database searches as they can be restricted to peptides ending with Arg or Lys, which reduces computational requirements of the search. However, the use of trypsin also has limitations and is not the optimal enzyme for all analyses.
Owing to the high specificity of trypsin, the spacing of Arg and Lys amino acids across the proteome dictates the length of peptides, and thus the number of unique peptides. For standard intracellular proteins, Arg and Lys occur at a high frequency (5.6% and 5.7%, respectively), which leads to the fact that roughly 50% of the peptides produced by trypsin are too short (<6 amino acids) to be nicely fragmented and uniquely assigned to a protein 7. Conversely, some proteins, notably membrane proteins, exhibit few tryptic cleavage sites and extreme hydrophobicity, resulting in poor coverage of this class of proteins in trypsin‐based proteomics 8. These combined effects all contribute to undetected, less visible areas of the proteome. To illustrate this, we have performed an in silico digestion of the human proteome using the specificity listed in Table 1 and asked what the upper limit of detection was for each protease using the search and mass spectrometry settings employed in this study (Table 1). For trypsin, a maximum of 87% of the proteome would in theory be detectable using this proteomics setup, assuming every peptide of suitable characteristics is actually fragmented and identified. To improve on this boundary, efforts have been made to utilize different proteases within bottom‐up workflows. Several groups have shown that by using proteases that cleave at different amino acid motifs, the number of unique peptides identified, and thus the proteome coverage, can be substantially improved 7, 9, 10, 11, 12.
Table 1.
Cleavage specificities reported for some of the most commonly used proteases in bottom‐up proteomics 9, 10, 13, 14, 15, 16. Xnp indicates any amino acid except proline.
| Protease | Cleavage site (↓) | Optimal pH | Max proteome coverage |
|---|---|---|---|
| AspN | ↓D | 8 | 78% |
| LysargiNase | ↓K/R | 7.5 | 87% |
| LysC | K↓Xnp | 8 | 79% |
| LysN | ↓KXnp | 8 | 78% |
| ArgC | R↓Xnp | 8 | 82% |
| GluC | E↓ | 8 | 86% |
| Chymotrypsin | F/W/Y↓ | 8 | 87% |
| Trypsin | K/R↓Xnp | 8–9 | 87% |
| Sap‐9 | K/R↓Xnp | 6–7 | 87% |
Numerous alternative proteases have been used for the digestion of proteins from a lysate, whereby each has its own cleavage specificity and optimal conditions (Table 1). By combining the proteases either in parallel or sequentially, one is able to improve the proteome coverage through combining the results of individual proteases together. For instance, work by Swaney et al. 7 nicely illustrated that expanding beyond a single protease can yield a roughly 20% increase in protein identifications and achieved double the proteome sequence coverage. Similarly, our group has shown that the use of multiple proteases in parallel for phosphoproteomics gives rise to highly complementary sets of phosphosites, where only 27% of all identified sites were found in more than one protease dataset 17.
While the combination of proteases has already been shown to aid in expanding the proteome sequence coverage, the presence of (multiple) proline residues presents a particular challenge for many proteases. Proline is a unique amino acid in peptides/proteins as it is the only cyclic amino acid, giving rise to a tertiary amide, limiting hydrogen donating properties and imposing rigid structural constraints on peptide bonds 18, 19. Because of its unique properties, proline often leads to missed proteolytic events during digestion 9, increasing the resultant peptide length and database search complexity. Moreover, proline also effects the fragmentation step during mass analysis, known as the ‘proline effect’ 20, where fragmentation shows enhanced production of y‐ions spanning from the proline to the peptide C terminus due to the enhanced basicity of the proline nitrogen, restricting the peptide sequence coverage 20, 21, 22.
To overcome these limitations, research efforts have been directed toward finding a proline‐directed protease as such a protease would decrease database search complexity by well defining the proline position, as well as substantially improve proteome sequence coverage due to its high complementarity to Arg‐ and Lys‐directed proteases. In 2009, Šebela et al. 23 evaluated an acidic prolyl endoprotease from Aspergillus niger, called An‐PEP, for its use in proteomics and found that the enzyme has potential for in‐solution digestion studies. Moreover, our laboratory showed that An‐PEP, also termed EndoPro, exhibited maximum activity at pH = 2 and is active at moderately high urea concentrations and low temperatures, making it very suitable for use in mass spectrometry‐based hydrogen–deuterium exchange experiments 24. In addition, work published on another prolyl endopeptidase originally from Nepenthes ventrata, termed neprosin, showed that almost half of the sequence coverage achieved by the proline‐directed protease on proteins detected in both tryptic and neprosin digests were not observed when digestion was performed with trypsin 25. Collectively, these works suggest huge potential of proline‐directed proteases to shed light on previously undetectable areas of the proteome. In phosphoproteomics, however, proline‐induced complications are even more prevalent as in eukaryotic systems around 40% of the phosphorylation events detected are proline‐directed, dominated by so‐called SP or TP motifs 26. Hence, in most eukaryotic phosphoproteomics experiments, prolines are highly enriched and even more prevalent than in a standard proteomics analysis.
Here, we extend substantially on previous work using proline‐directed proteases. We first benchmark EndoPro versus trypsin, thereby generating large proteomics datasets on HeLa lysates digested by EndoPro at pH = 2, EndoPro at pH = 5.5, and trypsin at pH = 8.5. We optimize the peptide ID rates using multiple peptide fragmentation techniques, and the search engine Byonic, allowing us to increase the ID rate substantially to about 40% and 66% of all PSMs for EndoPro and trypsin, respectively. When using EndoPro at these two different pH values, we find the specificities and activities to be similar. However, our datasets reveal a substantial difference between the peptides generated with EndoPro at pH = 2, EndoPro at pH = 5.5, and trypsin, indicating the cleavage of different proteins and/or sites at different pH values. Overall, EndoPro enabled us to detect over 2200 unique proteins not observed in our tryptic digests and contributed 49% of the total unique phosphosites detected, making it a protease almost equally powerful as, and complementary, to trypsin.
Results
To assess the benefit of EndoPro in shotgun proteomics 25, we decided to evaluate and optimize the performance of this proline‐directed protease on a complex HeLa cell lysate, first focusing on nonmodified peptides. To characterize the specificity of EndoPro in a full proteome, we performed a quadruplicate digestion of a HeLa lysate with EndoPro for 1 h at pH = 5.5. For comparison, we also performed a standard trypsin overnight digestion at pH = 8.5. These data were subjected to a nonspecific search in Byonic, and subsequently, the environment of all cleaved sites was analyzed using an in‐house R script. The distribution of amino acids following the residue cleaved by either EndoPro (purple) or trypsin (orange) is shown in Fig. 1A. EndoPro showed a strong specificity for cleavage C‐terminal to proline (49.1%) and alanine (35.5%), resulting in an overall cleavage specificity close to that observed for trypsin (84.6% Ala/Pro versus 89.6% Arg/Lys in our datasets). Inspection of the cleavage site environment of EndoPro (Fig. 1B) revealed a disfavor for cleaving when the cleavage site is preceding a proline. In these cases, only the last proline is cleaved. In addition, positively charged residues appear disfavored in the P + 2 position (Fig. 1B).
Figure 1.
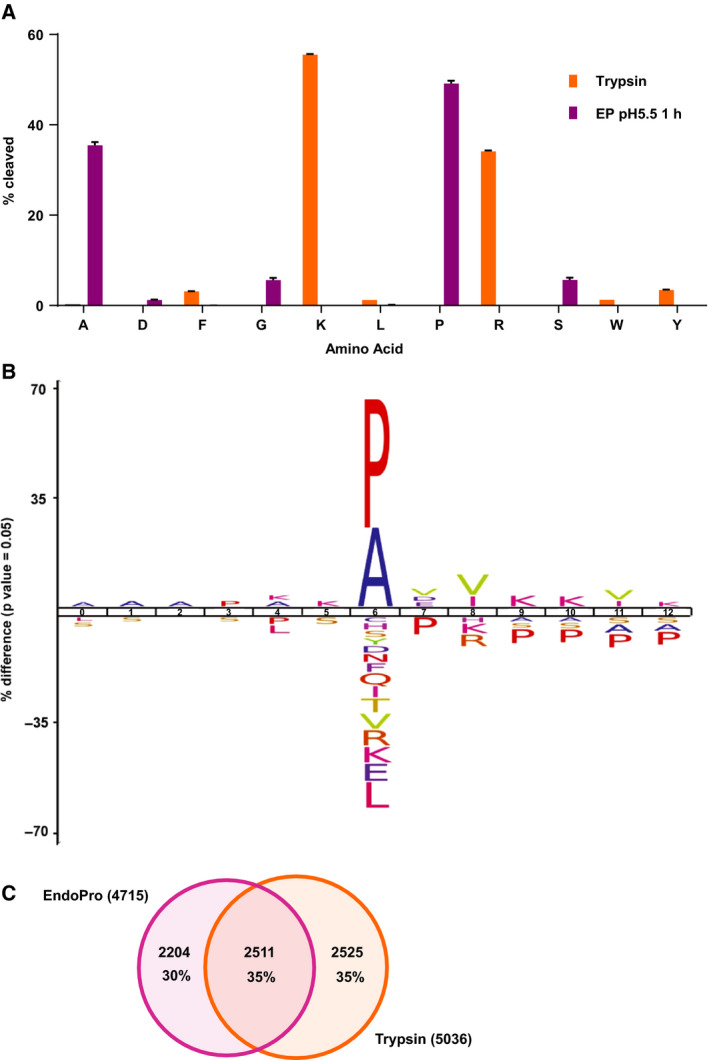
Characterization of EndoPro cleavage specificity. (A) Overview of amino acids after which was cleaved by EndoPro (n = 4, purple) and trypsin (n = 4, orange), based on a nonspecific search, revealing a high specificity of 84.6% A/P and 89.6% R/K for EndoPro and trypsin, respectively. Only amino acids with a cleavage frequency of 1% or higher were included. Data are represented as mean percentage of total cleavages per protease ± SEM. (B) An iceLogo showing the differences between the EndoPro cleavage site environment (17 032 unique environments from nonspecific search) and the human proteome, illustrating a disfavor for R/K on the +2 position and a reluctance to cleave between proline residues. (C) Overlap of unique proteins identified by EndoPro or trypsin using a semispecific search. Although the sizes of the identified proteomes are roughly equal, the overlap between the two is only 35%.
Since EndoPro reached almost 85% specificity, we subsequently used less computationally heavy semispecific database searches (allowing one side of the peptide to result from nonspecific cleavages), which saves data analysis time and is inherently less error‐prone. Doing these two searches on the same dataset, we observed that we still captured nearly all the peptides formed (97.6%).
Performance evaluation of EndoPro at pH = 2 and pH = 5.5
Next, we set out to compare the performance of EndoPro and trypsin. Thereby, we took into account that EndoPro exhibits several maxima in its activity profile, with maxima at pH = 2 and 5.5, as also reported earlier 24. Therefore, HeLa cell lysates were digested with either EndoPro at pH = 2 and pH = 5.5 and digested for 1 h or overnight (ON), and additionally, for benchmarking, the same HeLa cell lysate sample was digested with trypsin using conventional conditions (i.e., pH = 8.5, ON). The resulting peptides were analyzed by LC‐MS/MS on a Fusion hybrid mass spectrometer using in parallel ETD, EThcD, and HCD as peptide fragmentation methods. Spectra were searched with Byonic. A global overview of the search outcomes is shown in Table S1. The different fragmentation methods resulted in a highly similar number of protein identifications (Table S1). It was therefore decided to pool all the data acquired with different fragmentation techniques to assess the performance of EndoPro across the different digestion conditions, independent of the used fragmentation method. An overview of the pooled datasets is shown in Table 2. In terms of unique proteins detected, an overlap of 35% was observed between the two proteases (Fig. 1C). The four different EndoPro digestion conditions resulted in comparable identification rates, with slightly more PSMs and unique peptides in the experiments performed at pH = 5.5 when compared to pH = 2. Under all four tested conditions roughly, the same number of peptides (≈ 15 000) and proteins (≈ 2600) could be identified (Table 2). As expected, the peptide identification rate achieved with trypsin (67%) could not be reached with EndoPro (≈ 40%). Still, the EndoPro ID rates of ≈ 40% are better than what has been reported for many other alternative enzymes (e.g., LysN, AspN, chymotrypsin typically reach 20–30%) 10, 24, 25.
Table 2.
Characteristics of measured and analyzed EndoPro and trypsin datasets.
| Protease | pH | Digestion time | Fragmentation | Byonic semispecific search | ||||
|---|---|---|---|---|---|---|---|---|
| # MS2 scans | #PSMs 0.1 FDR | # unique peptides | # unique proteins | % identification | ||||
| Trypsin | 8.5 | ON | ETD/EThcD/HCD | 163 823 | 109 682 | 35 330 | 5036 | 67% |
| EndoPro | 2 | 1 h | ETD/EThcD/HCD | 152 064 | 54 251 | 13 631 | 2633 | 36% |
| EndoPro | 2 | ON | ETD/EThcD/HCD | 151 115 | 57 722 | 15 264 | 2439 | 38% |
| EndoPro | 5.5 | 1 h | ETD/EThcD/HCD | 155 229 | 68 902 | 18 268 | 2810 | 44% |
| EndoPro | 5.5 | ON | ETD/EThcD/HCD | 155 565 | 60 318 | 17 378 | 2621 | 39% |
| EndoPro cumulative | 613 973 | 241 193 | 38 004 | 4715 | 39% | |||
Characteristics of EndoPro peptides generated at pH = 2 and 5.5
For a more in‐depth exploration of the type of peptides produced by EndoPro, we compared general peptide characteristics such as peptide length, mass, amino acid content, and cleavage specificity as observed in the unique peptides identified from the EndoPro and, for comparison, tryptic digests, as depicted in Fig. 2. In terms of peptide length and charge, the four explored different EndoPro cleavage conditions produced similar peptides. We observed a substantially broader peptide length distribution for EndoPro peptides than for trypsin, revealing more peptides with a length of more than 20 amino acids and a tail toward peptides with a length of 50 or more amino acids (Fig. 2A). This already indicates that EndoPro generates peptides with more missed cleavages than trypsin. In terms of peptide charge, an average of about 33% of the unique EndoPro peptides carried four or more charges, compared to only 5% for the tryptic peptides (Fig. 2B). This difference in charge distribution could not be explained by the increase in peptide length, as the average number of amino acids to charge ratio of the different EndoPro conditions was lower than we found for trypsin (5.75 and 6.22 amino acids per positive charge, respectively).
Figure 2.
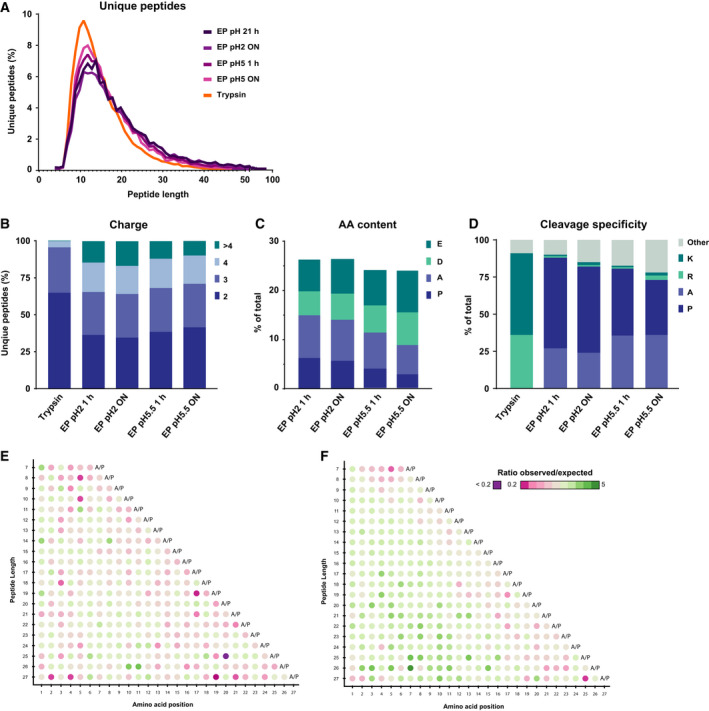
Comparison of peptide characteristics in EndoPro and tryptic digests. (A) Peptide length distribution of identified unique peptides following digestion with trypsin or EndoPro. All four EndoPro conditions probed here reveal a similar distribution, exhibiting a long tail toward peptides with more than 50 amino acids, which was not observed for tryptic peptides. (B) Charge distribution of all unique peptides identified following the different digestion conditions, where digestion with EndoPro results in more highly charged peptides (z ≥ 4). (C) Amino acid content of the peptides identified in the EndoPro digests under various digestion conditions. With increase in pH and digestion duration, negatively charged amino acids are more frequently observed and the A/P content of the peptides is reduced. (D) Cleavage specificity of the identified peptides. Digestion with EndoPro yields highly specific proline and alanine C‐terminal peptides, especially at pH = 2, with a Pro/Ala specificity close to that of trypsin for Arg/Lys. (E, F) Location of Asp on peptides digested ON with EndoPro at (E) pH = 2 and (F) pH = 5.5. At pH = 5.5, the negatively charged amino acid is disfavored at the C terminus of the generated peptides. This was not observed for peptides produced at pH = 2, indicating that two distinct sets of peptides are formed at these pH values.
The identified unique peptide length and number of charges found after EndoPro digestions did not vary much with digestion time. In contrast, the amino acid content of the peptides as well as the cleavage specificity of EndoPro appeared to be sensitive to the digestion conditions. In total, four amino acids (alanine, aspartic acid, glutamic acid, and proline) showed a substantial change in abundance when comparing EndoPro digests prepared at pH = 2 and 5.5 (Fig. 2C). With the increase in pH, the contribution of alanine and proline to the total amino acid content of the peptides decreased, whereas the contribution of the negatively charged aspartic acid and glutamic acid increased. Although clearly visible after 1 h of digestion, this effect is even more pronounced after ON digestion with EndoPro, where the proline content of the peptides at pH = 5.5 decreased to less than half of the value observed at pH = 2. The cleavage specificity of EndoPro also slightly decreased with increasing pH (Fig. 2D). Interestingly, the location of Asp on the peptides also changed with pH (Fig. 2E,F). This indicates that a different set of peptides is generated, depending on the digestion condition used.
The complete overview of amino acid content of the peptides generated by EndoPro under the four evaluated conditions and trypsin is shown in Fig. S1. For reference, the natural occurrence of each amino acid within the human proteome is indicated with a dashed line. Due to the Arg/Lys‐specific cleavage by trypsin, these tryptic peptides clearly underrepresent the abundance of Arg/Lys in the human proteome. Peptides generated by EndoPro do not impose limits on the number of Arg/Lys residues and hence are richer in these positively charged residues, which is in agreement with the on average higher charges we observe for EndoPro peptides. In addition, at low pH these basic amino acids carry a positive charge, which may help to prevent aggregation and therefore aid protein solubility. Similarly, the observed increase in Asp/Glu content with pH may also be related to their charge, as the presence of negatively charged amino acids has been correlated to an increase in solubility 32. At pH = 2, virtually none of the carbonic acid side chains will be negatively charged due to the excess in protons. At pH = 5.5, however, these amino acids would be predominantly negatively charged and essentially all would be charged at pH = 8.5. Therefore, this Asp/Glu rich subset of the proteome may have a better solubility over other proteins at increasing pH, which could explain why they are more abundantly represented on the peptide level.
As indicated above, we found the peptide dataset generated by EndoPro to be sensitive to both the cleavage time and pH (Fig. 2D). This could be partly attributed to more subtle changes in specificity. After 1‐h digestion at pH = 2, 26% of the peptides were cleaved after alanine and 68% of the cleavages were proline‐specific. This decreased slightly to 25% alanine and 62% proline‐specific cleavages following ON digestion. At pH = 5.5, however, the percentage of cleavages C‐terminal to alanine increased to 36% (1 h) and 37% (ON), with only 49% (1 h) and 39% (ON) proline‐specific cleavages. Furthermore, the percentage of nonspecific cleavages (i.e., not C‐terminal of Ala/Pro) observed after EndoPro digestion increased with an increase in pH from pH = 2–5.5. Therefore, the specificity of EndoPro can be to some extent controlled via the pH in the digestion step. Notably, EndoPro reaches up to 87% specificity for Pro/Ala at pH = 2 and 1‐h digestion, thereby achieving a very high specificity, on par with trypsin that reaches 91% specificity for Arg/Lys in our data. The ability of EndoPro to perform proteome‐wide digestion with such high specificity could be advantageous in downstream data analysis, as specific searches are far less computationally demanding. Hence, we conclude that EndoPro may be used as a high‐performance protease for proteomics, as in many aspects its performance is comparable to that of trypsin.
Performance of EndoPro versus trypsin
Comparing the search input and output characteristics for all EndoPro and tryptic digests, we found that all digestion conditions generated a similar number of MS2 scans (Table S1), indicating that a similar number of peptides with suitable charge states were produced by EndoPro and trypsin. However, we observed a lower conversion of MS/MS events to peptide identifications for EndoPro (around 40%) than for trypsin (67%). Still, the 40% ID rate, which we obtained using Byonic, is well above what has been typically reported for other proteases (i.e., ~ 15–30%) than trypsin 9, 10, 25. To objectively compare the characteristics and performance of EndoPro and trypsin, the peptide and protein identification datasets should ideally be of similar size. Therefore, we decided to accumulate all nonredundant peptide and protein IDs obtained by EndoPro under the four tested digestion conditions, which resulted in a dataset in numbers comparable with that acquired following tryptic digestion (see Table 2).
Using these equally large datasets (around 5000 proteins and 35 000 peptides each, see Table 2), we compared the overlap of unique proteins identified following digestion by either EndoPro or trypsin (Fig. 3A). Of the 7240 unique proteins identified in total, only 35% were identified by both proteases, whereas 30% and 35% were uniquely identified in tryptic and EndoPro digests, respectively (Fig. 3B,C). Typically, in our laboratory (and in line with many other laboratories), the overlap between proteome analyses on digests acquired under exactly identical digestion conditions is around 65% (Fig. 3B,C), largely due to the undersampling problem which cannot be avoided in shotgun proteomics 33. Hence, we consider this to be the maximum achievable protein overlap. The overlap in protein ID between the datasets obtained following digestion at pH = 2, comparing 1‐h and ON digestions, was 61%, slightly superior to the overlap between the datasets obtained following digestion at pH = 5.5, for either 1 h or ON (49%). The overlap between the datasets acquired either at pH = 2 or at pH = 5.5 was found to be only 44%. Even more strikingly, the overlap between peptides generated with EndoPro and trypsin was even much lower, namely only 35% (Fig. 3A). We conclude that this low overlap is not simply due to the stochastic nature of shotgun mass spectrometry, as the increase in protein identifications when adding a replicate of the same protease is significantly smaller than when using another protease and 30% of the proteins reproducibly identified in EndoPro were not identified using trypsin (Fig. 3D).
Figure 3.
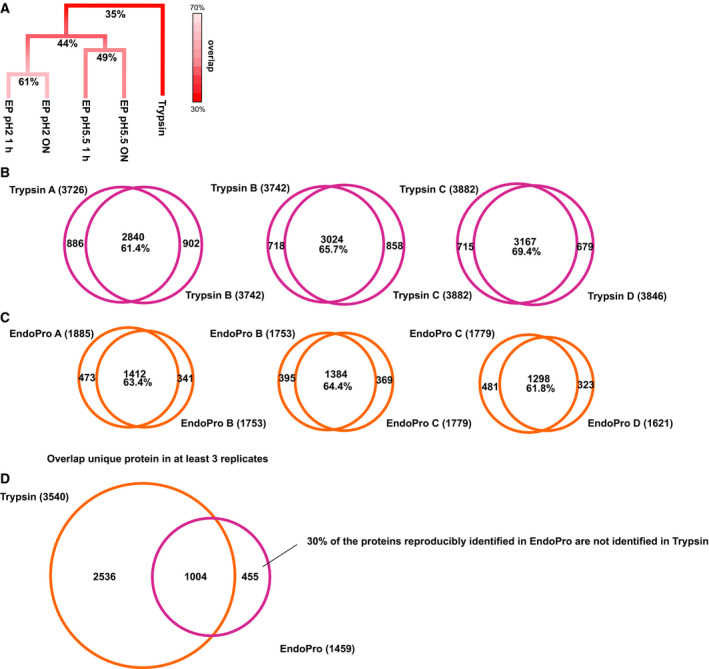
Highly complementary protein identifications observed by using EndoPro or trypsin. (A) Overview of the overlap in proteins identified by using the different proteases and varying digestion conditions as listed in Table 2, illustrating how complementarity increases when cleaving with EndoPro at different conditions. The smallest overlap, 35%, is observed between EndoPro and trypsin. (B, C) Reproducibility of (B) Trypsin and (C) EndoPro technical replicate analyses, revealing a robust overlap of around 65%. (D) When comparing all unique protein groups identified in at least three out of four technical replicates, 30% of the proteins that are reproducibly identified using EndoPro are not identified in tryptic lysates.
Next, we set out to assess what kind of characteristics form the basis for the complementarity in proteome coverage we observed between EndoPro and trypsin. To this end, we compared proteins for which one protease clearly outperformed the other. As a metric, we focused on proteins whose obtained sequence coverage with EndoPro was at least 50% higher than with trypsin, or vice versa (Fig. 4A). These data proved to be very consistent in all four biological replicates, as demonstrated in Fig. S2. Although we identified many proteins with a sufficient sequence coverage in both EndoPro and tryptic digests, our data also revealed large clusters of proteins that remain seemingly undetectable by using trypsin. These data nicely illustrate the increase in proteome depth that can be achieved when digesting with a protease other than trypsin.
Figure 4.
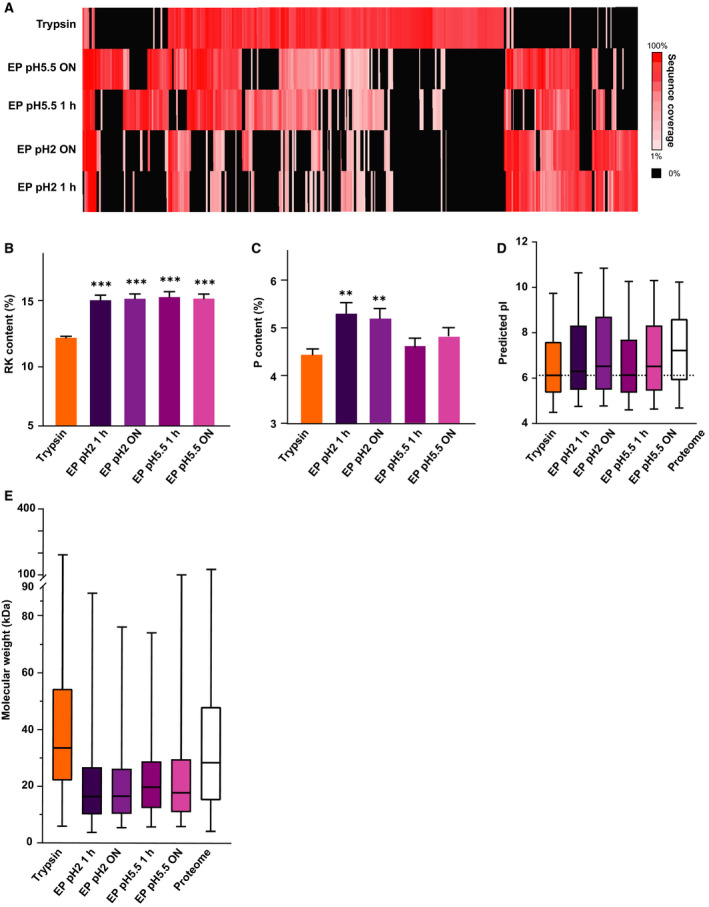
Proteome Characteristics. (A) Comparison of the sequence coverage achieved by using trypsin and EndoPro (the latter under 4 different digestion conditions) for in total 380 selected proteins. Only these 380 proteins showing at least 50% more sequence coverage in one of the datasets were considered in B–E. For clarity, proteins for which the two proteases performed comparably were not included. Black indicates no coverage of a protein in a certain condition. (B) Comparison of the arginine and/or lysine content, which is significantly higher in EndoPro peptides. (C–E) Comparison of the proline content (C), isoelectric point (D), and molecular weight (E) of proteins identified using EndoPro (at 4 different conditions) or trypsin. Notably, as shown in (E) EndoPro favors smaller proteins; trypsin shows a bias for larger proteins. Significance was determined using one‐way ANOVA, with α = 0.05. **P < 0.01, and ***P < 0.001; error bars represent SEM.
Since the digestions with EndoPro and trypsin are performed at distinct pH values, the source of the low overlap could be due to differences in protein solubility and thus accessibility to the protease (i.e., different proteins precipitate at pH = 2, 5.5, and 8.5, removing them from the possible substrate pool), or on the proteases' substrate preferences. We considered various protein characteristics that might cause the complementarity between the two proteases (Fig. 4B–E). Following expectations, EndoPro resulted in better sequence coverage for proteins that have a high arginine and/or lysine content (see Fig. 4B), as these proteins likely give rise to very small and potentially ambiguous peptides when digested with trypsin. With regard to the proline content, however, this trend is not observed (Fig. 4C). No significant difference in proline content was found between trypsin and EndoPro at pH = 5.5 and at pH = 2; EndoPro even outperformed trypsin on proteins with a high proline content. This distinction might be caused by frequent occurrence of proline‐rich regions. These Pro‐Pro bonds are not cleaved by EndoPro; hence, the protease likely produced less short, ambiguous peptides. In most cases, we only observed cleavage C‐terminal to the last proline in a proline repeat. Following GO term analysis, no clear differences in protein function or localization were found between the proteins identified with EndoPro or trypsin.
Subsequently, we evaluated whether the observed complementarity stems from the use of different proteases or is influenced significantly by the different digestion conditions, such as pH. Although the solubility of a protein is influenced by many factors, a key feature is its isoelectric point (pI), the pH where the protein carries no net charge. A comparison of the pI values of the identified proteins is shown in Fig. 4D. For reference, we also included the distribution of pIs found in the total human proteome 34. Despite the large pH difference between the five different conditions (i.e., four distinct EndoPro digestions and a trypsin digestion), the pI distributions all have a median well below the median for the complete human proteome. Although some differences may be observed between the five conditions, it seems they differ more from the complete proteome than from each other. Hence, we conclude that solubility is not likely the cause of the increase in proteome depth that can be achieved by utilizing EndoPro.
Finally, we evaluated whether there was a size bias within the subset of proteins for which one of the proteases outperformed the other (Fig. 4E). When compared to the whole human proteome, trypsin preferred slightly larger proteins, whereas EndoPro favored smaller substrates. Evaluation of protein function or localization yielded no clear preferences for either of the two proteases. Taken together, these data reveal that at the protein level, EndoPro and trypsin perform comparable and give highly complementary results. The source of complementarity could be solubility based due to the large pH range spanned in these experiments, but this hypothesis is not supported by the distribution of pIs. Therefore, it is likely that enzyme specificity drives the observed complementarity. Interestingly, EndoPro digests also show clear differences based on the cleavage conditions used to generate them, making EndoPro a remarkably flexible proline‐specific protease with great potential in bottom‐up proteomic studies.
Phosphoproteomics with EndoPro
In addition to changes in its abundance, a proteins' function and/or activity can also be regulated by post‐translational modifications (PTMs), such as phosphorylation. These phosphorylation events can be challenging to study due to their low stoichiometry compared to their nonmodified counterparts and instability of the modification itself. The field of phosphoproteomics specializes in the analysis of this modification, usually employing enrichment of phosphorylated peptides prior to their analysis by LC‐MS/MS. A common problem, however, is that many conventional proteases (e.g., trypsin) have difficulties cleaving near a phosphorylated amino acid, leading to increased missed cleavages around phosphosites 17, 35, 36. Using first several synthetic (phospho)peptides, however, we observed that EndoPro does not exhibit a significant decrease in cleavage rate when cleaving phosphorylated peptides when compared to their nonphosphorylated counterparts (data not shown). We hypothesized that this feature, combined with the high proline content present near phosphorylation sites, could make EndoPro an enzyme very well suitable for phosphoproteomics.
To assess how EndoPro performs in phosphoproteomics, we enriched peptides generated by digestion with EndoPro at pH = 2 and 5 for 1 h or ON using Fe(III)‐NTA cartridges in an automated fashion using the AssayMAP Bravo Platform 27. To benchmark the performance of EndoPro, phosphorylated tryptic peptides were enriched in parallel. For comparison, a general overview of the resulting datasets is shown in Table 3 and an extended overview of the contribution of each fragmentation technique is available in Table S2.
Table 3.
Search input and outcome characteristics for EndoPro and tryptic phospho‐enriched digests.
| Protease | pH | Digestion time | Fragmentation | # MS2 scans | Byonic semispecific search | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| #PSMs FDR < 0.1 | #PSMs dmod > 20 | Phospho PSMs | % identification | % phos | Total phos sites | Unique phos sites | |||||
| Trypsin | 8.5 | ON | ETD/EThcD/HCD | 96 641 | 51 502 | 44 933 | 35 319 | 46% | 79% | 39 905 | 8898 |
| EndoPro | 2 | 1 h | ETD/EThcD/HCD | 87 736 | 25 285 | 20 532 | 14 918 | 23% | 73% | 15 422 | 3275 |
| EndoPro | 2 | ON | ETD/EThcD/HCD | 87 415 | 25 254 | 19 895 | 16 423 | 23% | 83% | 17 489 | 3794 |
| EndoPro | 5.5 | 1 h | ETD/EThcD/HCD | 90 021 | 27 805 | 23 213 | 19 406 | 26% | 84% | 20 471 | 4326 |
| EndoPro | 5.5 | ON | ETD/EThcD/HCD | 93 374 | 26 370 | 22 658 | 17 667 | 24% | 78% | 19 070 | 4316 |
| EndoPro cumulative | 104 714 | 86 298 | 68 414 | 24% | 79% | 72 452 | 8486 | ||||
Since the main goal of looking beyond trypsin as a protease in (phospho)proteomics is to increase our coverage of the phosphorylation sites present in the human proteome, we first set out to assess whether EndoPro is complementary to trypsin in terms of phosphoprotein and unique phosphosite coverage. Using EndoPro, we identified 2937 unique phosphoproteins, which is comparable to the 3124 unique phosphoproteins identified using trypsin, see Fig. 5A. Interestingly, just 37% of the 4409 unique proteins identified in total were identified by both proteases. If we delve deeper into these shared phosphoproteins, it becomes evident that the two proteases mostly reveal different phosphosites on these shared proteins, see Fig. 5B. On the 1652 proteins identified by both EndoPro and trypsin, 9279 phosphosites were identified of which only 30% were found by both proteases. The remaining 6500 sites were identified by only one of the two enzymes; 3095 sites were uniquely identified by EndoPro and 3405 sites by trypsin; therefore, the proteases appear extremely orthogonal and employing EndoPro in this setting yields a large increase in attainable information. To evaluate the coverage of phosphosites more thoroughly, we plotted the number of spectral counts we observed for each phosphosite, see Fig. 5C (or Fig. S3 for more extended heatmaps). This figure revealed that many phosphosites consistently identified with EndoPro (in at least 2 out of 3 biological replicates) were not found at all when digesting with trypsin and vice versa, highlighting further the complementarity of the enzymes and the importance of extending phosphoproteomics analysis beyond the use of just a single protease9.
Figure 5.
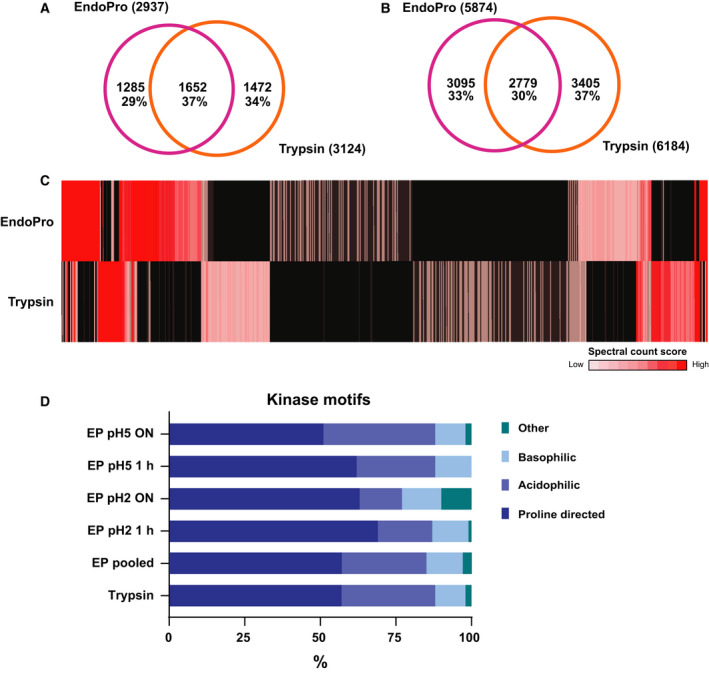
EndoPro is highly complementary to trypsin in the identification of site‐specific phosphorylation events. (A) Comparison of identified unique phosphoproteins between EndoPro and trypsin, revealing a 37% overlap. (B) Overlap in identified unique phosphosites on 1652 phosphoproteins identified by both proteases, indicating that on these shared phosphoproteins, only 30% of the phosphosites could be identified by both proteases. (C) Heatmap displaying phosphosite spectral count scores of 13 762 phosphosites from low (1) to high (> 10), revealing that EndoPro is highly complementary to trypsin in identification of phosphosites. Black indicated not identified. (D) Global kinase classification analysis of all identified phosphopeptides, dividing them into 4 categories: proline‐directed, acidophilic, basophilic, or other. Although in all analyses the SP/TP motif encompasses over 50% of the detected sites, short digestion with EndoPro results in a further increase of this proline‐directed motif to about 70%.
Localization of phosphorylation and motif analysis
Since we expected EndoPro to cleave after prolines and these are extremely frequently occurring in mammalian phosphorylation motifs, we evaluated both the phosphorylated motifs present in our datasets and the location of the phosphorylation sites on the identified phosphopeptides. To assess the different types of kinase motifs present in the dataset, we isolated the environment of each phosphosite identified (seven amino acids up‐ and downstream of the phosphorylated amino acid) and assessed the relative contribution of known motifs to the EndoPro and tryptic datasets. For clarity, the motifs were classified in only four categories: proline‐directed, acidophilic, basophilic, or other (Fig. 5D). Markedly, the contribution of proline‐directed motifs is even larger for EndoPro digestions than we observe for trypsin, most notably under the short digestion conditions (1 h). This observation is in line with the decrease in relative proline content observed at longer digestion times as depicted in Fig. 2C. As expected, we see an increase in motifs containing arginine and lysine after EndoPro digestion. Overall, our findings are in agreement with previous work from this laboratory, in which a thorough examination of multiple proteases for phosphoproteomics revealed that each protease exhibits a bias toward different classes of phosphorylation sites 17.
As EndoPro precisely cleaves after prolines, which are found in the most frequently occurring Ser‐Pro/Thr‐Pro phosphorylation sites, and since it is well known that a phosphorylation close to an Arg/Lys hampers the cleavage activity of trypsin, we queried whether the phosphorylation on these motifs would prevent cleavage of the following proline residue. To assess this, we evaluated the location of the phosphorylation on unique phosphopeptides. We computed the frequency of phosphorylations for each position of the phosphopeptide, with the exception of the last amino acid, as we expect this to be Ala/Pro and Arg/Lys for EndoPro and trypsin, respectively. The frequency of the phosphorylation site was compared to the frequency expected if phosphorylations would have been randomly distributed across the amino acids of the phosphopeptide (Fig. 6). The under‐ or overrepresentation of a phosphorylation location on the peptides is shown by a color gradient, and extreme underrepresentation (at least fivefold lower than expected) was indicated in purple. These ‘dot‐plots’ display several very interesting features.
Figure 6.
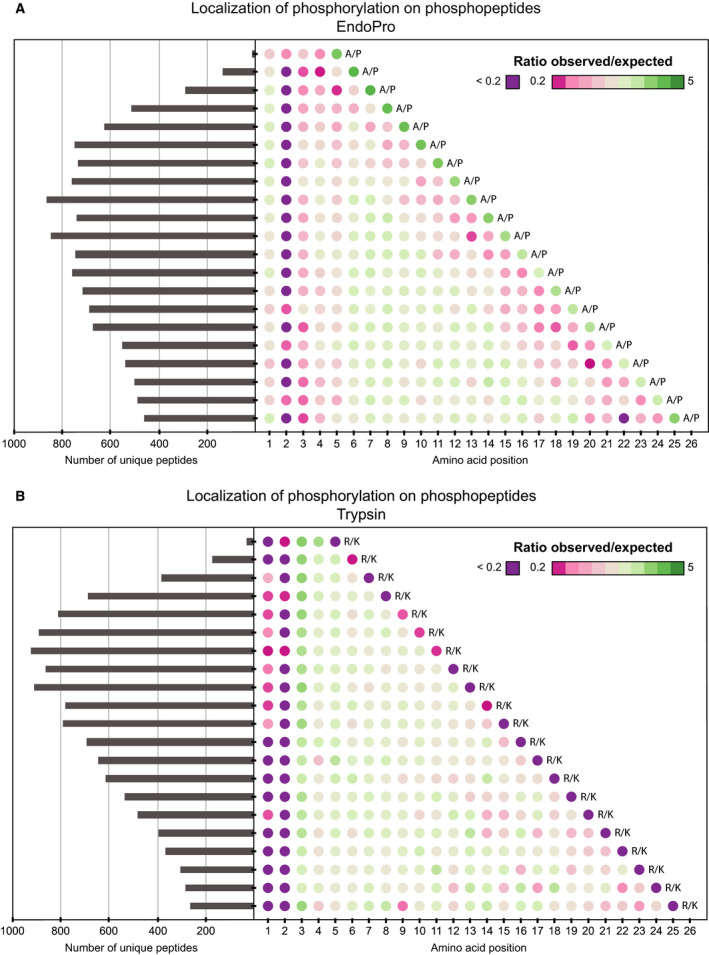
Amino acid length and localization of phosphorylation sites on the identified phosphopeptides. (A) Localization of the phosphorylation on unique phosphopeptides from EndoPro, showing the highly preferred phosphorylation on the second to last amino acid on the peptide (i.e., Ser‐Pro or Thr‐Pro), and the disfavor for phosphorylation on the penultimate N‐terminal amino acid on the EndoPro peptides. (B) Localization of phosphorylation on unique phosphopeptides following trypsin digestion at pH = 8.5, revealing a strong disfavor for phosphorylation on the ultimate and penultimate N‐terminal amino acids on the peptides, and preferential phosphorylation on the third amino acid of the identified phosphopeptides.
For the EndoPro phosphopeptides, the highly preferred phosphorylation on the penultimate C‐terminal amino acid is very evident, as is the disproportion for phosphorylation on the penultimate amino acid at the N terminus (Fig. 6A). Interestingly, EndoPro also seems to disfavor positively charged amino acids on this position (Fig. 1B), which implies no charge is tolerated at this position in the substrate‐binding pocket. Repulsion of both charges would suggest steric hindrance to be the source of this lack of activity. For trypsin‐like proteases, the N + 2 position is reported to be situated in a hydrophobic pocket prior to cleavage 37. Based on our data, this might also be the case for EndoPro. For the tryptic phosphopeptides, the dot‐plot reveals a strong disfavor for phosphorylation on the ultimate and penultimate N‐terminal amino acids and the penultimate C‐terminal amino acid (Fig. 6B), confirming that phosphorylation near the Arg/Lys hinders cleavage by trypsin. Trypsin displays a preferential phosphorylation on the third N‐terminal amino acid (likely representing the well‐known RXXS/T basophilic kinase motif). These findings largely explain the increase in missed cleavages on phosphopeptides observed 36.
In contrast to what is observed with trypsin, the activity of EndoPro seemed unaffected by a phosphorylation directly preceding the cleavage site, resulting in an overrepresentation of phosphorylation events on the second to last amino acid of the phosphopeptides, see Fig. 5A. In total, of all detected singly phosphorylated EndoPro phosphopeptides, 19% had their phosphorylation on the C‐terminal SP/TP. See Fig. S4 for the phosphosite localization of all specific EndoPro digestion conditions employed.
Discussion
Although still not frequently used, the use of proline‐directed proteases in a mass spectrometry‐based proteomics setting has been explored previously 23, 24, 25. Schräder et al. 25 recognized the potential of proline‐directed proteases in proteomics characterizing neprosin, a protease originally from Nepenthes ventrata. In their work, the ON digestion of a HeLa cell lysate at pH = 2.5 yielded 61% proline‐specific cleavages for neprosin, which is comparable to our findings for EndoPro (62% after ON digestion at pH = 2). Additionally, they nicely illustrated the potential of proline‐specific proteases for the mapping of PTMs on a histone sample. Due to the high activity of EndoPro at low pH, the protease has also found applications in the food industry, where EndoPro was assessed for its ability to degrade gluten and the debittering of protein hydrolysates, as well as in structural studies based on hydrogen–deuterium exchange mass spectrometry, where a low pH is essential to reduce the rate of deuterium back exchange 24, 38, 39, 40. Thus, proline‐directed proteases are versatile proteases that can be used orthogonally to the more conventional proteases in various mass spectrometry‐based studies.
Here, we have evaluated EndoPro for its use in bottom‐up (phospho)proteomics, with the aim to boost its performance by optimizing different digestion conditions, peptide fragmentation methods, and scoring algorithms. We showed that the protease has a capacity to generate peptides from proteins comparable to trypsin, evidenced by similar numbers of MS/MS events. When the proper digestion conditions are chosen, the cleavage specificity for alanine and proline is very high. Interestingly, EndoPro cleavage patterns appear influenced by the pH during the digestion, with a lower overall specificity observed using EndoPro at pH = 5.5 than at pH = 2. The mechanism underlying this pH‐dependency was not studied thoroughly here; however, we did find that the overall Pro content of the proteins identified at pH = 5.5 was significantly lower than at pH = 2. Hence, it might be possible that fewer proline residues were available for cleavage, possibly due to the occurrence of a different pool of soluble proteins at pH = 5.5 when compared to pH = 2. This hypothesis is supported by the limited overlap (44%) in identified proteins between the two EndoPro conditions. We did not observe an effect on the length of the peptides identified, which also implies proline residues were not missed during digestion but likely not present as frequently.
Through this work, we show that the performance of EndoPro as protease for proteomics applications is already very good, but its full potential is still not reached. As observed also with other proteases, EndoPro also suffers from the tryptic bias that has been created in the conventional proteomics pipelines, both in the peptide separation, fragmentation, and scoring segments of the proteomics experiment. Despite that, our data show that EndoPro is a protease very suitable for producing peptides for proteomics analysis. One should keep in mind, however, that the obtainable proteome coverage is rather distinct when the digestion is performed at pH = 2 or at pH = 5.5. In addition, the proteome coverage generated with EndoPro is highly complementary to the coverage that can be reached using trypsin. Finally, EndoPro provides one of the most complementary proteases for phosphoproteomics, delivering a large subset of phosphosites not easily covered by trypsin. Furthermore, in contrast to trypsin, cleavage by EndoPro is not hampered by the presence of a neighboring phosphorylation.
Identifying nontryptic MS/MS spectra
One of the main concerns when using less conventional proteases in proteomics‐type experiments is that the resulting datasets almost always give a lower peptide identification rate than the tryptic datasets. For ArgC, AspN, chymotrypsin, GluC, LysC, and LysN, average identification rates of 22%, 11%, 17%, 13%, 23%, and 11% have been reported, compared to a 37% identification rate for trypsin 10. Similarly, Schräder et al. 25 previously reported identification rates of 20%, 46%, and 52% for neprosin, LysC, and trypsin, respectively. These findings are in agreement with our finding, where EndoPro identification rates are also about half of the trypsin identification rate. The lower rates associated with nontryptic digestions are probably not caused by a lack of good peptides, as the number of MS2 scans for these runs is similar. Hence, the number of peptides with suitable mass‐to‐charge ratio is expected to be similar for all digests.
This leaves several other sources likely responsible for the reduced identification rates. Firstly, the peptides produced by each of these proteases may have characteristics that make them less suitable for current fragmentation‐based sequencing methods by mass spectrometry. They may, for instance, carry less positive charges, reducing the likelihood of observing charged fragment ions that can be used for database matching. For our dataset, however, this is not the case as the peptides generated by EndoPro have even more positive charges than the tryptic peptides. Secondly, the peptides' chemical composition may lead to fragmentations patterns or cleavages at positions that are unexpected for trypsin. For instance, EndoPro peptides do not carry an arginine or lysine residue at their C terminus, which likely leads to a less extended sequence informative y‐ion series. Thirdly, database search and peptide scoring algorithms have mainly been optimized for tryptic peptides. Any fragmentation behavior not observed in tryptic peptides, therefore, is likely penalized by the conventional scoring algorithms, resulting in lower scores. Notably, using standard conditions with other search engines such as Mascot, Andromeda, and Sequest gave us even lower identification rates than those reported here (by a factor 2, data not shown), evidently depending also on the fragmentation method employed.
Due to EndoPro's high preference to cleave C‐terminal to proline, many of the peptides generated with this protease are expected to have a Pro residue at their C terminus, making them very dissimilar to the typical tryptic peptides that carry a Lys or Arg at their C terminus. Indeed, we observed a clear C‐terminal proline effect in their fragmentation spectra. During HCD fragmentation, more than 95% of the EndoPro MS2 spectra contained a very prominent y1 ion at 116.07 m/z, corresponding to the preferential gas‐phase cleavage of the bond preceding the proline. Assuming that the presence of a 116.07 m/z ion is diagnostic for a peptide ending in C‐terminal proline, we noticed in our LC‐MS runs many more MS2 spectra likely originating from EndoPro peptides in which sequence could not be assigned. This could possibly be improved by optimizing the MS parameters such as the collision energy, to maintain the diagnostic y1 ion while also allowing sufficient fragmentation in other parts of the peptide. In EThcD spectra, we observed significantly less proline y1 ion formation, only about 50% of the recorded MS2 spectra, which allows for the detection and assignment of other fragment ions and hence a better scoring of the PSMs. This is also reflected in the higher ID rate observed with the EThcD/HCD DT method.
Taken together, many factors contribute to a lower score for the EndoPro, illustrating a deeply rooted tryptic bias in proteomic workflows, resulting in lower PSMs for nontryptic peptides. This argues especially for a better optimization of MS methods and search algorithms toward nontryptic peptides.
Fragmenting with a C‐terminal phosphorylation and phosphosite localization
Given the large proportion of phosphopeptides that carry their phosphorylation on the penultimate amino acid of the EndoPro peptide, we hypothesize that these phosphopeptides may have a negatively charged C terminus. Again, this is in sharp contrast to tryptic phosphopeptides, which have a positively charged C terminus and for the most part carry their phosphorylation somewhere in the middle of the peptide. Phosphorylation of the amino acid before the C‐terminal proline seemed to reduce the proline effect observed for the EndoPro peptides, resulting in a better fragmentation ion coverage than observed for nonphosphorylated peptides. In addition, since the EndoPro phosphopeptides predominantly carry their phosphorylation at the C terminus, this affects the probability of having multiple potential phosphorylation sites directly adjacent to each other. When a phosphorylation site is directly preceding the Ala/Pro on the C terminus, there cannot be a second potential phosphorylation site on that end of the peptide; hence, the odds of having many potential sites side by side on a phosphopeptide is lower than when phosphorylations are located more toward the middle of a peptide. This could potentially boost phosphosite localization certainty, especially in peptides that harbor multiple putative phosphate acceptors, such as Ser, Thr, and Tyr. Since one of the major remaining issues in phosphoproteomics is the confident assignment of the exact site of phosphorylation, much computational effort has been invested in improving fragmentation methods and algorithms to boost confident site assignments. Knowledge about the natural occurrence of phosphorylation sites for each used protease, as depicted graphically in Fig. 6, can be used to further improve scoring algorithms and boost the confidence in site localization.
Conclusion
Here, we evaluated EndoPro and show it is a versatile protease with a very high proline‐ and alanine‐directed specificity. Its activity can be influenced by adjusting the pH of the digestion buffer, whereby it largely retains its specificity but seemingly samples a different part of the proteome. By benchmarking its performance against trypsin, we observed that over 30% of all unique HeLa proteins were solely identified by EndoPro, as well as 5705 phosphosites that were not observed in the tryptic digests, illustrating EndoPro's high complementarity to trypsin. This complementarity allows EndoPro to expand our coverage of the various proteomes and sheds light on previously dark, invisible stretches of (phospho)proteins. Since EndoPro clearly outperforms trypsin on arginine‐ and lysine‐rich proteins, we see potential for EndoPro in studying proteins involved in nucleotide and chromatin binding, which are often enriched in these positively charged amino acids 41. In addition, the longer peptides generated by EndoPro and its ability to cleave close to modifications make the enzyme an interesting candidate for middle‐down approaches allowing for more combinatorial PTM information 42, 43. Compared to other alternative proteases, such as LysC, chymotrypsin, ArgC, EndoPro performs better and is in our view one of the most complementary alternatives to trypsin, due to its completely different activity profile and specificity. It is rather unique in effectively targeting proline residues in (phospho)proteomics that are often causing complications for the other proteases.
Materials and methods
In silico proteome coverage
Human proteins deposited in the Swissprot database (20 417 reviewed proteins, downloaded July 25, 2019) were digested in silico using the specificity requirements listed in Table 1. Zero, one, or two missed cleavages were allowed for each peptide, resulting in a database with all possible peptides formed by each of the nine listed proteases. Subsequently, these peptides were filtered on precursor m/z (375 ≤ m/z ≤ 1500); mass (m ≤ 10 000 Da) and only fully specific peptides were taken into account. All peptides passing these filters were mapped to the proteome to find the theoretical upper limit of proteome coverage possible.
Cell culture
HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and 10 mm glutamine (Lonza, Basel, Switzerland) at 37°C/5%CO2. One hour prior to harvesting, the medium was refreshed to stimulate phosphorylation. Cells were washed with ice‐cold PBS, and cell pellets were collected by mild centrifugation (150 xg) for 3 min, and stored at −80°C until lysis.
Sample preparation
Cell pellets were lysed in a boiling lysis buffer containing 6 m guanidinium HCl (GuHCl), 5 mm Tris (2‐carboxyethyl)phosphine (TCEP), 10 mm chloroacetamide, 100 mm Tris/HCl pH 8.5, supplemented with protease inhibitor (cOmplete mini EDTA‐free, Roche, Woerden, The Netherlands). Pellets were boiled for 10 min at 99 °C, sonicated for 30 rounds of 5 s (Bioruptor Plus, Diagenode, Seraing, Belgium), and spun down at 20 000 g for 15 min. Protein concentration was determined using Pierce™ BCA protein assay kit. Equal amounts of protein per condition were diluted to a final concentration of 2 m GuHCl, and pH was adjusted to pH = 2 and 5.5 for EndoPro, or pH = 8.5 for trypsin, using formic acid (FA) (Merck, Zwijndrecht, The Netherlands). Finally, proteins were digested with EndoPro (1:100, DSM, Delft, The Netherlands) or trypsin (1:100, Sigma‐Aldrich, Zwijndrecht, The Netherlands) for 1 h or overnight at 37 °C. The resulting peptides were acidified to a final concentration of 1% FA, cleaned up using Sep‐Pak cartridges (Waters, Etten‐Leur, The Netherlands), and dried in vacuo.
Phosphopeptide enrichment
Phosphorylated peptides were enriched using Fe(III)‐NTA cartridges (Agilent Technologies) in an automated fashion using the AssayMAP Bravo Platform (Agilent Technologies) 27. The cartridges were primed with 0.1% TFA in ACN and equilibrated with loading buffer (80% ACN/0.1% TFA). Samples were suspended in loading buffer and loaded onto the cartridge. The peptides bound to the cartridges were washed with loading buffer, and the phosphorylated peptides were eluted with 1% ammonia directly into 10% formic acid. Samples were dried in vacuo and stored at −80 °C until LC‐MS/MS analysis.
LC‐MS/MS analysis
Peptide samples were resuspended in 20 mm citric acid with 2% FA and analyzed with an UHPLC 1290 system (Agilent Technologies) coupled to an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Peptides were trapped (Dr Maisch Reprosil C18, 3 µm, 2 cm × 100 µm) and then separated on an analytical column (Agilent Poroshell EC‐C18, 2.7 µm, 50 cm × 75 µm). All columns were made in‐house. Trapping was performed for 5 min in solvent A (0.1% FA), followed by a gradient of the following: 0–8% solvent B (0.1% FA in 80% ACN) in 10 s, 8–32% in 100 min, 32–100% in 5 min, hold for 5 min, 100–0% in 1 min, and hold for 4 min. Flow was passively split to 300 nL·min−1.
The mass spectrometer was operated in data‐dependent mode. Full scan MS spectra from m/z 375 to 1500 were acquired at a resolution of 60 000 after accumulation to a target value or 4e5 or a maximum injection time of 50 ms. The most intense precursor ions were selected for fragmentation for a duration of 3 s with a 24‐s dynamic exclusion duration. Target peaks were isolated in a 1.6 Da isolation window and subjected to either higher‐energy collision‐induced dissociation (HCD), electron transfer dissociation (ETD), or electron transfer higher‐energy collision‐induced dissociation (EThcD) 28. MS/MS spectra were acquired with a resolution of 30 000 using an AGC target of 1e5 ions with a maximum injection time of 125 ms. Charge state screening was enabled, and precursors with an unknown charge state or a charge state of 1+ were excluded. For the decision tree strategy, HCD and EThcD fragmentation were performed with normalized collision energies of 35% and 40%, respectively. Fragmentation was done based on charge state. HCD was selected for peptide ions with charge states of 2+ and 3+; and for EThcD, charge states 4+ to 20+ were selected.
Data analysis
The resulting mass spectra were searched using Byonic (Protein Metrics Inc., Cupertino, CA, USA, v.3.3.11) in a fully nonspecific or semispecific search (C‐terminal cleavage on Arg/Lys or Ala/Pro for trypsin or EndoPro, respectively). The number of missed cleavages was not restricted. Mass tolerance was set at 10 and 20 ppm for precursor and fragment ions, respectively. Carbamidomethylation was set as a fixed cysteine modification, oxidation of methionine, deamidation of asparagine, and sodium adducts of aspartate, glutamate, serine, and threonine were set as common modifications. The formation of pyro‐glutamine from N‐terminal glutamine or glutamate, loss of ammonia, and acetylation was set as rare modifications. Overall, one common and one rare modification were allowed in the standard bottom‐up workflow. For the phospho‐enriched peptides, phosphorylation on serine or threonine was included as a common modification, and in total, 3 common and 1 rare modification were allowed.
Using Byonic Viewer (Protein Metrics Inc., v.3.3‐421), the PSMs were filtered by a PEP 2D < 0.001 resulting in a 0.1% PSM level FDR. In the phospho‐enriched dataset, we also asked that the delta mod. score (dmod) was larger than 20, to only include the more confident phosphosite localizations for each PSM. The resulting PSMs from different fragmentation methods were combined prior to further data analysis.
Peptide and/or protein characteristics such as peptide length, charge, amino acid content, and location of phosphosites on the peptide were determined using in‐house R scripts (available upon request), Venn diagrams were made using both venny (BioinfoGP v.2.1.0) 29 and biovenn 30, and the bar graphs, boxplots, and heatmaps were visualized using graphpad prism 8.0.1. IceLogo was generated as described in Colaert et al. 31.
In Fig. 5D, the phosphorylation motifs were assigned to one of four categories: proline‐directed phosphosites p(S/T)P, acidophillic (D/E after the phosphosite), basophillic (R/K before phosphosite), or other. Assignment was hierarchical, meaning that a phosphosite exhibiting both proline‐directed and basophilic characteristic was only included in the proline‐directed group.
Conflict of interest
The authors declare the following competing financial interest(s): M.A. and M.O. are DSM employees. DSM sells An‐PEP for food applications. MB is the founder and employee of ProteinMetrics. ProteinMetrics develops and commercializes the Byonic software.
Author contributions
SAML, CAGHG, and AJRH designed experiments and wrote the manuscript, SAML and CAGHG performed the (phospho)proteomics experiments. SAML wrote the scripts for data analysis; SAML and CAGHG analyzed and interpreted the data supported by MB; and MA and MO provided EndoPro as well as critical comments to the manuscript. All authors read and approved the manuscript.
Supporting information
Fig. S1. Amino acid content of PSMs following different digestion conditions.
Fig. S2. Extended heatmap of proteome dataset.
Fig. S3. Extended heatmap of the phosphoproteomics data and assessment of reproducibility.
Fig. S4. Phosphosite localization as extracted from the unique phosphopeptides for the four different EndoPro digestion conditions.
Table S1. Search input and outcome characteristics for EndoPro and tryptic digests.
Table S2. Search input and outcome characteristics for EndoPro and tryptic phosphopeptides.
Acknowledgements
We acknowledge support from the Netherlands Organization for Scientific Research (NWO) funding the large‐scale proteomics facility Proteins@Work (project 184.032.201) and X‐omics (project 184.034.019) embedded in the Netherlands Proteomics Centre. A.J.R.H. acknowledges further support by the NWO TOP‐Punt Grant 718.015.003 and the EU Horizon 2020 program INFRAIA project Epic‐XS (Project 823839). We thank Simone Lemeer and Liana Tsiatsiani for the critical evaluation of the manuscript.
Saar A. M. van der Laarse and Charlotte A. G. H. van Gelder contributed equally to this article.
References
- 1. Cohen P (2001) The role of protein phosphorylation in human health and disease. Eur J Biochem 268, 5001–5010. [DOI] [PubMed] [Google Scholar]
- 2. Hanash S (2003) Disease proteomics. Nature 422, 226–232. [DOI] [PubMed] [Google Scholar]
- 3. Zhang Y, Fonslow BR, Shan B, Baek M‐C & Yates JR (2013) Protein analysis by shotgun/bottom‐up proteomics. Chem Rev 113, 2343–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mann M, Kulak NA, Nagaraj N & Cox J (2013) The coming age of complete, accurate, and ubiquitous proteomes. Mol Cell 49, 583–590. [DOI] [PubMed] [Google Scholar]
- 5. Steen H & Mann M (2004) The abc's (and xyz's) of peptide sequencing. Nat Rev Mol Cell Biol 5, 699–711. [DOI] [PubMed] [Google Scholar]
- 6. Vandermarliere E, Mueller M & Martens L (2013) Getting intimate with trypsin, the leading protease in proteomics. Mass Spectrom Rev 32, 453–465. [DOI] [PubMed] [Google Scholar]
- 7. Swaney DL, Wenger CD & Coon JJ (2010) Value of using multiple proteases for large‐scale mass spectrometry‐based proteomics. J Proteome Res 9, 1323–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eichacker LA, Granvogl B, Mirus O, Müller BC, Miess C & Schleiff E (2004) Hiding behind hydrophobicity. J Biol Chem 279, 50915–50922. [DOI] [PubMed] [Google Scholar]
- 9. Tsiatsiani L & Heck AJR (2015) Proteomics beyond trypsin. FEBS J 282, 2612–2626. [DOI] [PubMed] [Google Scholar]
- 10. Giansanti P, Tsiatsiani L, Low TY & Heck AJR (2016) Six alternative proteases for mass spectrometry–based proteomics beyond trypsin. Nat Protoc 11, 993–1006. [DOI] [PubMed] [Google Scholar]
- 11. Biringer RG (2006) Enhanced sequence coverage of proteins in human cerebrospinal fluid using multiple enzymatic digestion and linear ion trap LC‐MS/MS. Brief Funct Genomic Proteomic 5, 144–153. [DOI] [PubMed] [Google Scholar]
- 12. Gauci S, Helbig AO, Slijper M, Krijgsveld J, Heck AJR & Mohammed S (2009) Lys‐N and trypsin cover complementary parts of the phosphoproteome in a refined SCX‐based approach. Anal Chem 81, 4493–4501. [DOI] [PubMed] [Google Scholar]
- 13. Huesgen PF, Lange PF, Rogers LD, Solis N, Eckhard U, Kleifeld O, Goulas T, Gomis‐Rüth FX & Overall CM (2015) LysargiNase mirrors trypsin for protein C‐terminal and methylation‐site identification. Nat Methods 12, 55–58. [DOI] [PubMed] [Google Scholar]
- 14. Wu Z, Huang J, Huang J, Li Q & Zhang X (2018) Lys‐C/Arg‐C, a more specific and efficient digestion approach for proteomics studies. Anal Chem 90, 9700–9707. [DOI] [PubMed] [Google Scholar]
- 15. Raijmakers R, Neerincx P, Mohammed S & Heck AJR (2010) Cleavage specificities of the brother and sister proteases Lys‐C and Lys‐N. Chem Commun 46, 8827. [DOI] [PubMed] [Google Scholar]
- 16. Burkhart JM, Schumbrutzki C, Wortelkamp S, Sickmann A & Zahedi RP (2012) Systematic and quantitative comparison of digest efficiency and specificity reveals the impact of trypsin quality on MS‐based proteomics. J Proteomics 75, 1454–1462. [DOI] [PubMed] [Google Scholar]
- 17. Giansanti P, Aye TT, van den Toorn H, Peng M, van Breukelen B & Heck AJR (2015) An augmented multiple‐protease‐based human phosphopeptide Atlas. Cell Rep 11, 1834–1843. [DOI] [PubMed] [Google Scholar]
- 18. Schimmel PR & Flory PJ (1968) Conformational energies and configurational statistics of copolypeptides containing L‐proline. J Mol Biol 34, 105–120. [DOI] [PubMed] [Google Scholar]
- 19. MacArthur MW & Thornton JM (1991) Influence of proline residues on protein conformation. J Mol Biol 218, 397–412. [DOI] [PubMed] [Google Scholar]
- 20. Vaisar T & Urban J (1996) Probing the proline effect in CID of protonated peptides. J Mass Spectrom 31, 1185–1187. [DOI] [PubMed] [Google Scholar]
- 21. Raulfs MDM, Breci L, Bernier M, Hamdy OM, Janiga A, Wysocki V & Poutsma JC (2014) Investigations of the mechanism of the “proline effect” in tandem mass spectrometry experiments: the “pipecolic acid effect”. J Am Soc Mass Spectrom 25, 1705–1715. [DOI] [PubMed] [Google Scholar]
- 22. Huo D, Qin T & Zu L (2019) Energetic switch of the proline effect in collision‐induced dissociation of singly and doubly protonated peptide Ala‐Ala‐Arg‐Pro‐Ala‐Ala. J Mass Spectrom 54, 55–65. [DOI] [PubMed] [Google Scholar]
- 23. Šebela M, Řehulka P, Kábrt J, Řehulková H, Oždian T, Raus M, Franc V & Chmelík J (2009) Identification of N‐glycosylation in prolyl endoprotease from Aspergillus niger and evaluation of the enzyme for its possible application in proteomics. J Mass Spectrom 44, 1587–1595. [DOI] [PubMed] [Google Scholar]
- 24. Tsiatsiani L, Akeroyd M, Olsthoorn M & Heck AJR (2017) Aspergillus niger prolyl endoprotease for hydrogen‐deuterium exchange mass spectrometry and protein structural studies. Anal Chem 89, 7966–7973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schräder CU, Lee L, Rey M, Sarpe V, Man P, Sharma S, Zabrouskov V, Larsen B & Schriemer DC (2017) Neprosin, a selective prolyl endoprotease for bottom‐up proteomics and histone mapping. Mol Cell Proteomics 16, 1162–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen X, Wu D, Zhao Y, Wong BHC & Guo L (2011) Increasing phosphoproteome coverage and identification of phosphorylation motifs through combination of different HPLC fractionation methods. J Chromatogr B 879, 25–34. [DOI] [PubMed] [Google Scholar]
- 27. Post H, Penning R, Fitzpatrick MA, Garrigues LB, Wu W, MacGillavry HD, Hoogenraad CC, Heck AJR & Altelaar AFM (2017) Robust, sensitive, and automated phosphopeptide enrichment optimized for low sample amounts applied to primary hippocampal neurons. J Proteome Res 16, 728–737. [DOI] [PubMed] [Google Scholar]
- 28. Frese CK, Zhou H, Taus T, Altelaar AFM, Mechtler K, Heck AJR & Mohammed S (2013) Unambiguous phosphosite localization using electron‐transfer/higher‐energy collision dissociation (EThcD). J Proteome Res 12, 1520–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oliveros JC (2007) VENNY. An interactive tool for comparing lists with Venn Diagrams. https://bioinfogp.cnb.csic.es/tools/venny/index.html.
- 30. Hulsen T, de Vlieg J & Alkema W (2008) BioVenn – a web application for the comparison and visualization of biological lists using area‐proportional Venn diagrams. BMC Genom 9, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Colaert N, Helsens K, Martens L, Vandekerckhove J & Gevaert K (2009) Improved visualization of protein consensus sequences by iceLogo. Nat Methods 6, 786–787. [DOI] [PubMed] [Google Scholar]
- 32. Trevino SR, Scholtz JM & Pace CN (2007) Amino acid contribution to protein solubility: Asp, Glu, and Ser contribute more favorably than the other hydrophilic amino acids in RNase Sa. J Mol Biol 366, 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tabb DL, Vega‐Montoto L, Rudnick PA, Variyath AM, Ham A‐JL, Bunk DM, Kilpatrick LE, Billheimer DD, Blackman RK, Cardasis HL et al (2010) Repeatability and reproducibility in proteomic identifications by liquid chromatography‐tandem mass spectrometry. J Proteome Res 9, 761–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kozlowski LP (2017) Proteome‐ pI: proteome isoelectric point database. Nucleic Acids Res 45, D1112–D1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schlosser A, Pipkorn R, Bossemeyer D & Lehmann WD (2001) Analysis of protein phosphorylation by a combination of elastase digestion and neutral loss tandem mass spectrometry. Anal Chem 73, 170–176. [DOI] [PubMed] [Google Scholar]
- 36. Molina H, Horn DM, Tang N, Mathivanan S & Pandey A (2007) Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci USA 104, 2199–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huber R & Bode W (1978) Structural basis of the activation and action of trypsin. Acc Chem Res 11, 114–122. [Google Scholar]
- 38. Mitea C, Havenaar R, Drijfhout JW, Edens L, Dekking L & Koning F (2008) Efficient degradation of gluten by a prolyl endoprotease in a gastrointestinal model: implications for coeliac disease. Gut 57, 25–32. [DOI] [PubMed] [Google Scholar]
- 39. König J, Holster S, Bruins MJ & Brummer RJ (2017) Randomized clinical trial: effective gluten degradation by Aspergillus niger‐derived enzyme in a complex meal setting. Sci Rep 7, 13100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Edens L, Dekker P, van der Hoeven R, Deen F, de Roos A & Floris R (2005) Extracellular prolyl endoprotease from Aspergillus niger and its use in the debittering of protein hydrolysates. J Agric Food Chem 53, 7950–7957. [DOI] [PubMed] [Google Scholar]
- 41. Chandana T & Venkatesh YP (2016) Occurrence, functions and biological significance of arginine‐rich proteins. Curr Protein Pept Sci 17, 507–516. [DOI] [PubMed] [Google Scholar]
- 42. Pandeswari PB & Sabareesh V (2019) Middle‐down approach: a choice to sequence and characterize proteins/proteomes by mass spectrometry. RSC Adv 9, 313–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cristobal A, Marino F, Post H, van den Toorn HWP, Mohammed S & Heck AJR (2017) Toward an optimized workflow for middle‐down proteomics. Anal Chem 89, 3318–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Amino acid content of PSMs following different digestion conditions.
Fig. S2. Extended heatmap of proteome dataset.
Fig. S3. Extended heatmap of the phosphoproteomics data and assessment of reproducibility.
Fig. S4. Phosphosite localization as extracted from the unique phosphopeptides for the four different EndoPro digestion conditions.
Table S1. Search input and outcome characteristics for EndoPro and tryptic digests.
Table S2. Search input and outcome characteristics for EndoPro and tryptic phosphopeptides.