

Abstract
Peficitinib (ASP015K) is a novel Janus kinase inhibitor developed for the treatment of rheumatoid arthritis (RA). The impact of hepatic impairment on the peficitinib pharmacokinetic (PK) and safety profile was investigated in non‐RA subjects (n = 24) in an open‐label, parallel‐group, multicenter comparative study in Japan. Subjects received a single, clinically relevant, oral dose of a peficitinib 150 mg tablet under fasting conditions. Plasma PK parameters were measured for peficitinib and its metabolites H1 (sulfate and methylated metabolite), H2 (sulfate metabolite), and H4 (methylated metabolite) in subjects with normal hepatic function, mild hepatic impairment, or moderate hepatic impairment. The peficitinib area under the plasma‐concentration–time curve from time 0 to infinity (AUCinf) and maximum observed concentration (Cmax) were not markedly different in subjects with mild hepatic impairment versus normal hepatic function. In subjects with moderate hepatic impairment versus normal hepatic function, the geometric mean ratios for peficitinib AUCinf and Cmax, were 1.92 (90% CI: 1.39, 2.66) and 1.82 (90% CI: 1.24, 2.69), respectively. Five treatment‐emergent adverse events (TEAEs) were experienced by 3 subjects, 1 in each group. There were no deaths, no serious TEAEs, and no TEAEs leading to withdrawal. In summary, the PK profile was unaltered in subjects with mild hepatic impairment after a single clinically relevant dose of peficitinib, but exposure almost doubled in subjects with moderate hepatic impairment. Peficitinib dose reduction may be considered in RA patients with moderate hepatic impairment.
Keywords: ASP015K, hepatic impairment, Janus kinase inhibitor, peficitinib, pharmacokinetics
Peficitinib is an orally bioavailable once‐daily Janus kinase (JAK) inhibitor that targets various cytokine signaling pathways to suppress the activation and proliferation of inflammatory cells in rheumatoid arthritis (RA).1, 2, 3, 4, 5 The efficacy and safety of peficitinib, as monotherapy or in combination with disease‐modifying antirheumatic drugs, in patients with moderate‐to‐severe RA have been demonstrated in randomized, double‐blind, placebo‐controlled studies.1, 6, 7, 8, 9 These included studies conducted in Japan,1, 8, 9 where the Ministry of Health, Labour and Welfare reports there are an estimated 0.7 million people with RA. In 2019 peficitinib was approved in Japan as a new treatment option for patients with RA who had an inadequate response to conventional therapy.10
The pharmacokinetic (PK) and pharmacodynamic profiles of single‐ and multiple‐dose peficitinib have been assessed in healthy subjects.11 Peficitinib was absorbed rapidly, and plasma levels reached steady state by day 3 of multiple dosing. Dose proportionality was shown for a single peficitinib dose from 3 to 300 mg and for multiple peficitinib doses from 30 mg twice daily to 100 mg twice daily.11 Urinary excretion of peficitinib accounted for 9% to 15% of the oral dose, and 3 conjugated metabolites (H1 [M1], H2 [M2], and H4 [M4]) were produced that showed very weak in vitro pharmacological action.11, 12 The H2 and H4 metabolites are produced by sulfuric acid conjugation and methylation of peficitinib, respectively, and may undergo further metabolic transformation to H1 (a sulfated and methylated metabolite).12
In clinical studies of RA patients, peficitinib was effective and well tolerated at daily doses of 100 mg and 150 mg,1, 6, 7, 8, 9 which formed the basis for the recent approval of peficitinib (50 mg and 100 mg tablets) in Japan.13 The usual clinical dosage for adult patients with RA is 150 mg per day, which can be reduced to 100 mg per day depending on the patient's condition.13
The peficitinib PK profile has been characterized in healthy subjects,11 and a recent drug interaction study with rosuvastatin (a probe substrate for the organic anion transporting polypeptide 1B1) did not demonstrate any clinically significant changes in the PK profile of either agent during coadministration.14 However, a recent study of peficitinib in combination with verapamil (P‐glycoprotein inhibitor) showed increased exposure of peficitinib and its metabolites, suggesting that P‐glycoprotein may be involved in peficitinib transport.15
While the peficitinib PK profile has been described in healthy subjects,11 it is important to determine the impact of potential comorbidities, such as liver disease, on peficitinib exposure. The burden of liver disease related to hepatitis infection remains high,16 and there is a rise in nonalcoholic fatty liver disease and its histological phenotype, nonalcoholic steatohepatitis (NASH), which are related to increased obesity and can progress to chronic liver disease.17 Recent estimates suggest the global prevalence of nonalcoholic fatty liver disease/NASH is 24%, with the highest rates reported from South America and the Middle East, followed by Asia, the United States, and Europe.17 In Japan it is estimated that around 1 million people have NASH,18 and, owing to diet and lifestyle alterations and the global trend for obesity, NASH is likely to become an increasingly important cause of hepatic disease in Japan and across the world.18, 19, 20
To determine whether peficitinib exposure is affected by the level of hepatic functioning, this study assessed the PK and safety profile of a single oral dose of peficitinib 150 mg in subjects with normal and impaired hepatic function.
Methods
Study Design and Participants
This study was conducted in compliance with the Declaration of Helsinki, Good Clinical Practice, International Committee on Harmonisation guidelines, and all applicable laws and regulations. The final protocol, amendments, and informed consent documentation were reviewed and approved by the institutional review board at each study center. The names and location of the individual institutional review boards are shown in Supplementary Table S1. All participants provided written, informed consent.
This was an open‐label, single oral dose, parallel‐group comparison study conducted between December 2015 and September 2016 at 6 sites in Japan (Supplementary Table S1). The objective was to evaluate plasma PK parameters and safety for peficitinib in subjects with and without hepatic impairment.
The planned sample size was 8 subjects in each group: normal hepatic function, mild hepatic impairment, and moderate hepatic impairment. This sample size was based on an accrual potential of subjects with impaired hepatic function according to PK studies in subjects with impaired hepatic function and normal hepatic function. The sample size determination for this study was not based on statistical power.
Eligible subjects were aged 20–75 years, with body mass index ≥17.0 and <30.0 kg/m2. Hepatic impairment was defined at screening using Child‐Pugh classification: Class A, mild (5–6 points); Class B, moderate (7–9 points). Subjects with severe hepatic impairment (Child‐Pugh classification Class C, 10–15 points) were excluded. The PK analysis was based on all subjects who received the study drug and who had at least 1 PK assessment. The safety analysis was based on all subjects who received the study drug.
Administration and Monitoring
Subjects were required to fast overnight before dosing. On the morning of day 1, subjects received 1 peficitinib 150 mg tablet (Astellas Pharma Inc, Tokyo, Japan) with 150 mL of water and were requested to swallow without chewing. Other drinking water was prohibited 1 hour before and after drug administration. Subjects were discharged on day 4 and had a follow‐up examination at day 7 (Figure 1).
Figure 1.
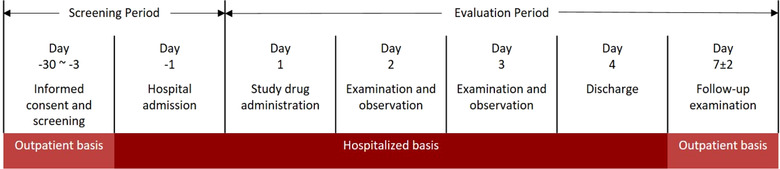
Overview of study design. Follow‐up investigation was to be performed as needed after the follow‐up examination.
Sample Analysis Method for Peficitinib and Metabolite H2
The concentrations of plasma peficitinib and metabolite H2 (sulfate metabolite) were measured using validated liquid chromatography‐tandem mass spectrometry (LC‐MS/MS) methods. The lower limit of quantification was 0.25 ng/mL when 25 µL of plasma was used. The analysis method was previously published using rat plasma.21 For clinical sample analysis, the method was developed using human plasma. Calibration curves for peficitinib and H2 in human plasma were linear over the concentration range 0.25–500 ng/mL, with correlation coefficients ≥0.9988. The intra‐day accuracies were within ±15% of the nominal concentration, and the intra‐day precision did not exceed 15%.
Sample Analysis Method for Metabolites H1 and H4
The concentrations of metabolites H1 (sulfate and methylated metabolite) and H4 (methylated metabolite) were measured separately using 2 validated LC‐MS/MS methods. The lower limit of quantification was 0.25 ng/mL, when 25 µL of plasma was used.
Extraction of H1 and H4 was carried out separately by solid‐phase extraction. Deuterated H1 (d3‐H1) was used for the internal standard (IS) of H1, and a stereoisomer of peficitinib was used for the IS of H4. Plasma samples (25 µL) were mixed with 100 mmol/L phosphate solution (pH 7) (1 mL), acetonitrile‐water (1:1, v/v) (25 µL), and IS working solution (25 µL) and loaded onto a preconditioned Oasis® HLB cartridge (30 mg/1 cc; Waters Co, Milford, Massachusetts). For H1 extraction, the cartridge was washed with 1 mL of methanol‐water (5:95, v/v) 3 times, and analytes were eluted with 0.5 mL of methanol 2 times. For H4 extraction, the cartridge was washed 3 times with 1 mL of methanol‐water (25:75, v/v), and analytes were eluted twice with 0.5 mL of methanol‐formic acid (100:0.1, v/v). Eluted solvent was evaporated at 40°C under a stream of nitrogen gas. Residues were reconstituted with a 0.5‐mL aliquot of reconstitution solution (10 mmol/L ammonium acetate‐methanol [65:35, v/v] or [60:40, v/v]) for H1 or H4, respectively. An aliquot of each sample was injected into the LC system for H1 or H4 analysis (10 µL and 20 µL, respectively).
LC‐MS/MS Condition for H1 Analysis
LC separation was performed on an Inertsil Ph‐3 column (4.6 mm I.D. × 100 mm [GL Sciences, Tokyo, Japan]) for 5 µm particle size with a mobile phase consisting of 10 mmol/L ammonium acetate and methanol. H1 was eluted using mobile phase A (10 mmol/L ammonium acetate solution) and mobile phase B (methanol) in the following linear gradient conditions: 0–3.50 minutes, 55% (B); 3.51–4.00 minutes, 90% (B); 4.01–6.00 minutes, 90% (B); 6.01–7.00 minutes, 55% (B); and 7.01–7.50 minutes, 55% (B). Flow rate was set at 0.8 mL/min except for 4.01–7.00 minutes (1.5 mL/min), and column temperature was set at 40°C.
MS/MS detection was performed using an API4000 (AB SCIEX, Framingham, Massachusetts). The ion‐spray voltage was maintained at 3000 V, and temperature maintained at 700°C. Ion source gas 1 (air) was set at 60 psi, and ion source gas 2 (air) was set at 60 psi. Curtain gas (nitrogen) was set at 10 psi. Selected reaction monitoring was conducted in positive ion electrospray mode. The first quadrupole selected for the cationic moiety of each compound was as follows: H1, m/z 421; IS d3‐H1, m/z 424. Product ions were generated by collision‐induced dissociation within the second quadrupole (collision gas nitrogen) and detected at the electron multiplier as follows: H1, m/z 341; IS d3‐H1, m/z 344. Data were processed using Analyst Software (version 1.5, AB SCIEX).
Calibration curves for H1 in human plasma were linear over the concentration range 0.25–100 ng/mL, with correlation coefficients ≥0.9991. The intra‐ and interday accuracies were within ±15% of the nominal concentration, and the intra‐ and inter‐day precision did not exceed 15%.
LC‐MS/MS Condition for H4 Analysis
LC separation was performed on an Inertsil Ph‐3 column (2.1 mm I.D. × 33 mm [GL Sciences, Tokyo, Japan]) for 5 µm particle size with mobile phase A (10 mmol/L ammonium acetate‐methanol [60:40, v/v]) and mobile phase B (acetonitrile‐water [70:30, v/v]) in the following isocratic conditions: 0–6.00 minutes, 0% (B); 6.01–9.00 minutes, 100% (B); 9.01–10.5 minutes, 0% (B). Flow rate was set at 0.8 mL/min, and column temperature was set at 40°C.
MS/MS detection was performed using an API4000. The ion‐spray voltage was maintained at 5000 V, and temperature maintained at 650°C. Ion source gas 1 (air) was set at 60 psi, and ion source gas 2 (air) was set at 70 psi. Curtain gas (nitrogen) was set at 10 psi. Selected reaction monitoring was conducted in positive‐ion electrospray mode. The first quadrupole selected for the cationic moiety of each compound was as follows: H4, m/z 341; IS, a stereoisomer of peficitinib, m/z 327. Product ions were generated by collision‐induced dissociation within the second quadrupole (collision gas nitrogen) and detected at the electron multiplier as follows: H4, m/z 174; IS, a stereoisomer of peficitinib, m/z 91. Data were processed using Analyst Software.
Calibration curves for H4 in human plasma were linear over the concentration range 0.25–100 ng/mL, with correlation coefficients ≥0.9976. The intra‐ and interday accuracies were within ±15% of the nominal concentration, and the intra‐ and inter‐day precision did not exceed 15%.
Pharmacokinetics
Blood samples for the PK analysis were collected before study drug administration, at 15 minutes after dosing, every 30 minutes until 2 hours after dosing, and then at postdosing hour 3, 4, 5, 6, 8, 10, 12 (day 1), 24, 36 (day 2), 48, 60 (day 3), 72 (day 4/discharge).
The PK parameters for plasma peficitinib were area under the concentration‐time curve (AUC) from the time of dosing extrapolated to time infinity (AUCinf), AUC from the time of dosing to the last measurable concentration (AUClast), maximum concentration (Cmax), apparent total systemic clearance after oral dosing (CL/F), time to attain Cmax (tmax), and terminal elimination half‐life (t½). The PK parameters for plasma metabolites H1, H2, and H4 were AUCinf, AUClast, Cmax, tmax, t½, and metabolite‐to‐parent ratio of AUClast (MPR). The relationship between peficitinib AUCinf and measures of hepatic impairment (serum albumin and prothrombin time) were analyzed across hepatic function groups. Prothrombin time was expressed as a percentage of standard human plasma clotting times, where a lower percentage corresponds to prolonged clotting times.
Safety
Treatment‐emergent adverse events (TEAEs) were defined as any adverse events that started or worsened in severity after administration of the study drug. Safety was assessed throughout the study. All events were classified using the Japanese Medical Dictionary for Regulatory Activities (J16.0).
Further assessments included vital signs, ECG, hematology, biochemistry, and urinalysis. Liver function tests included alkaline phosphatase, alanine aminotransferase, γ‐glutamyltranspeptidase, total bilirubin, and aspartate aminotransferase.
Statistical Analyses
Plasma PK parameters for peficitinib and metabolites H1, H2, and H4 were described using summary statistics, and AUCinf was also described using scatter plots. The GMR and 90% CI for the impaired groups versus the normal group were calculated using SAS® (Cary, North Carolina) mixed procedure with hepatic function group as a fixed effect.
Results
Subjects
A total of 24 subjects were enrolled and received study drug; all subjects completed the study. There were 16 subjects with impaired hepatic function (n = 8 with mild impairment and n = 8 with moderate impairment) and 8 subjects with normal hepatic function.
The mean age of the subjects was 62.3 years, and the mean weight was 65.5 kg. In the mild and moderate impairment groups, respectively, the mean prothrombin time was 95.9% and 69.7%, and the mean albumin concentration was 40.6 g/L and 32.1 g/L. All other demographic and baseline characteristics were similar across the normal, mild, and moderate impairment groups (Table 1). The cohorts were balanced for mean age (normal, 60.0 years; mild, 67.4 years; moderate, 59.4 years) and mean weight (normal, 66.3 kg; mild, 65.2 kg; moderate, 65.0 kg), with a median ±10% across the cohorts. Most subjects (70.8%) were male (normal, 75.0%; mild, 62.5%; moderate, 75.0%).
Table 1.
Baseline Demographics and Clinical Characteristics by Hepatic Function Group in All Subjects
| Normal Function (n = 8) | Mild Impairment (n = 8) | Moderate Impairment (n = 8) | Total(N = 24) | |
|---|---|---|---|---|
| Sex, n (%) | ||||
| Male | 6 (75.0) | 5 (62.5) | 6 (75.0) | 17 (70.8) |
| Female | 2 (25.0) | 3 (37.5) | 2 (25.0) | 7 (29.2) |
| Age, years | ||||
| Mean (SD) | 60.0 (4.7) | 67.4 (8.2) | 59.4 (6.7) | 62.3 (7.4) |
| Weight, kg | ||||
| Mean (SD) | 66.3 (9.8) | 65.2 (11.5) | 65.0 (13.4) | 65.5 (11.2) |
| BMI, kg/m2 | ||||
| Mean (SD) | 24.0 (2.6) | 26.0 (3.5) | 23.3 (3.7) | 24.4 (3.4) |
| Prothrombin time, % | ||||
| N | 0 | 8 | 8 | 16 |
| Mean (SD) | – | 95.9 (14.1) | 69.7 (16.5) | 82.8 (20.1) |
| Albumin, g/L | ||||
| N | 8 | 8 | 8 | NA |
| Mean (SD) | 40.4 (1.8) | 40.6 (2.3) | 32.1 (3.9) | NA |
BMI indicates body mass index.
Hepatic impairment defined according to Child‐Pugh classifications: Class A, mild (5–6 points); Class B, moderate (7–9 points).
Pharmacokinetics
Peficitinib
The mean plasma peficitinib concentration from dosing to 72 hours by degree of hepatic impairment is shown in Figure 2. An overview of the plasma PK parameters by hepatic impairment group is shown in Table 2.
Figure 2.
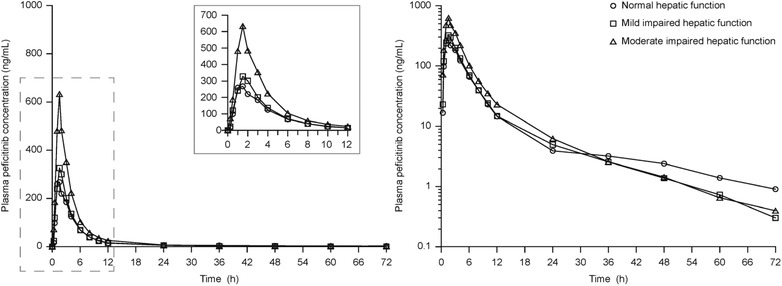
Peficitinib plasma concentration–time profiles by degree of hepatic impairment group. Values are means shown on linear (left) and semi‐logarithmic (right) scales. Subjects who received the study drug and provided 1 or more estimable pharmacokinetic parameters (PKAS). Plasma concentrations below the quantification limit were treated as 0. Hepatic impairment defined according to Child‐Pugh classifications: Class A, mild (5–6 points); Class B, moderate (7–9 points).
Table 2.
Plasma Pharmacokinetic Parameters of Peficitinib and Its Metabolites by Hepatic Function Group in All Subjects
| Hepatic Function | ||||
|---|---|---|---|---|
| Parameter | Normal (n = 8) | Mild Impairment (n = 8) | Moderate Impairment (n = 8) | |
| AUCinf, ng·h/mL | Peficitinib | 1149 (231.1) | 1435 (525.1) | 2332 (895.6) |
| H1 | 372.1 (111.8) | 887.9 (453.0) | 1138 (1259) | |
| H2 | 2707 (557.4) | 4393 (2075) | 2489 (2358) | |
| H4 | 357.2 (143.1) | 571.8 (281.1) | 1346 (1011) | |
| AUClast, ng·h/mL | Peficitinib | 1298 (528.8) | 1413 (525.1) | 2316 (899.4) |
| H1 | 412.1 (182.6) | 876.1 (454.5) | 1131 (1260) | |
| H2 | 2920 (910.1) | 4369 (2089) | 2480 (2361) | |
| H4 | 402.0 (211.5) | 563.4 (283.3) | 1331 (1012) | |
| Cmax, ng/mL | Peficitinib | 350.4 (129.1) | 371.6 (146.5) | 673.8 (331.6) |
| H1 | 44.79 (23.87) | 90.64 (50.46) | 110.3 (107.2) | |
| H2 | 641.3 (174.0) | 808.9 (247.1) | 485.0 (426.3) | |
| H4 | 35.57 (26.23) | 53.46 (34.03) | 110.5 (80.01) | |
| tmax, h | Peficitinib | 1.25 (1.00–3.00) | 1.47 (0.50–2.00) | 1.50 (1.00–2.83) |
| Median (min, max) | H1 | 3.00 (3.00–4.00) | 3.00 (1.93–4.00) | 3.00 (2.00–4.00) |
| H2 | 1.50 (1.00–3.00) | 1.75 (1.00–3.00) | 1.50 (1.50–2.83) | |
| H4 | 3.50 (3.00–4.00) | 4.00 (1.00–6.00) | 4.00 (3.00–4.02) | |
| t½, h | Peficitinib | 10.43 (6.22) | 13.70 (9.93) | 11.16 (8.88) |
| H1 | 12.50 (7.98) | 9.94 (5.50) | 14.11 (7.02) | |
| H2 | 10.77 (6.10) | 7.74 (5.09) | 12.24 (9.03) | |
| H4 | 12.24 (6.96) | 10.71 (4.77) | 15.13 (7.50) | |
| CL/F, L/h | Peficitinib | 134.9 (25.77) | 121.3 (54.89) | 74.51 (32.28) |
| H1 | … | … | … | |
| H2 | … | … | … | |
| H4 | … | … | … | |
| MPR | Peficitinib | … | … | … |
| H1 | 0.25 (0.06) | 0.52 (0.28) | 0.32 (0.31) | |
| H2 | 1.88 (0.42) | 2.56 (0.76) | 0.75 (0.53) | |
| H4 | 0.29 (0.06) | 0.37 (0.09) | 0.51 (0.24) | |
AUCinf indicates area under the curve from the time of dosing extrapolated to time infinity; AUClast, area under the curve from the time of dosing to the last measurable concentration; CL/F, apparent total systemic clearance after oral dosing; Cmax, maximum concentration; MPR, metabolite‐to‐parent ratio of AUClast; tmax, time to attain Cmax; t½, terminal elimination half‐life.
Data are mean (SD) unless stated otherwise.
Hepatic impairment defined according to Child‐Pugh classifications: Class A, mild (5–6 points); Class B, moderate (7–9 points).
In subjects with mildly impaired hepatic function versus normal hepatic function, the GMRs for peficitinib AUCinf and Cmax were 1.19 (90% CI: 0.86, 1.64) and 1.04 (90% CI: 0.71, 1.53), respectively (Table 3). In moderately impaired subjects versus subjects with normal hepatic function, the GMRs for peficitinib AUCinf and Cmax were 1.92 (90% CI: 1.39, 2.66) and 1.82 (90% CI: 1.24, 2.69), respectively (Table 3). Scatter plots of AUCinf versus measures of hepatic function (serum albumin and prothrombin time) are shown in Figure 3.
Table 3.
Statistical Assessment of the Effect of Hepatic Impairment on Peficitinib Plasma Pharmacokinetics
| GMR,a % | |||
|---|---|---|---|
| Parameter | Hepatic Function Group | n | Impaired/Normal (90% CI) |
| AUCinf (ng·h/mL) | Normal | 7 | |
| Mild | 8 | 1.19 (0.86, 1.64) | |
| Moderate | 8 | 1.92 (1.39, 2.66) | |
| AUClast (ng·h/mL) | Normal | 8 | |
| Mild | 8 | 1.07 (0.76, 1.51) | |
| Moderate | 8 | 1.76 (1.25, 2.47) | |
| Cmax (ng/mL) | Normal | 8 | |
| Mild | 8 | 1.04 (0.71, 1.53) | |
| Moderate | 8 | 1.82 (1.24, 2.69) |
AUCinf indicates area under the curve from the time of dosing extrapolated to time infinity; AUClast, area under the curve from the time of dosing to the last measurable concentration; Cmax, maximum concentration; GMR, geometric mean ratio.
Hepatic impairment defined according to Child‐Pugh classifications: Class A, mild (5–6 points); Class B, moderate (7–9 points).
GMRs and CIs were transformed back to the raw scale.
Figure 3.
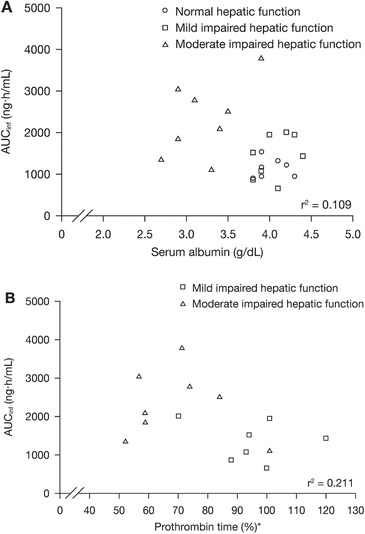
The relationship between peficitinib AUCinf and measures of hepatic function, (A) serum albumin and (B) prothrombin time. AUCinf, AUC from the time of dosing extrapolated to time infinity. Hepatic impairment defined according to Child‐Pugh classifications: Class A, mild (5–6 points); Class B, moderate (7–9 points). *Subjects with impaired hepatic function only.
Metabolites
The 3 conjugated metabolites of peficitinib, H2 (major metabolite), H1, and H4 (minor metabolites) were analyzed.12 Compared with measurements in subjects with normal hepatic function, there was a trend for greater exposure to H1 and H4 metabolites in subjects with mild or moderate hepatic impairment; there was no clear trend for H2 exposure (Table 2). MPRs were 0.25 (H1), 1.88 (H2), and 0.29 (H4) in subjects with normal hepatic function, 0.52 (H1), 2.56 (H2), and 0.37 (H4) in subjects with mildly impaired hepatic function, and 0.32 (H1), 0.75 (H2), and 0.51 (H4) in subjects with moderately impaired hepatic function (Table 2).
Safety
Three subjects, including 1 in each group, reported 5 TEAEs (Table 4). The TEAEs were diarrhea, blood urine present, back pain, hypotension, and nausea. All TEAEs were considered drug‐related except for back pain. All TEAEs were mild, except for nausea, which was considered moderate in severity. There were no serious TEAEs, no deaths, and no subjects withdrew from the study due to a TEAE.
Table 4.
Treatment‐Emergent Adverse Events in All Subjects by Hepatic Function Group
| n (%) | Normal Function (n = 8) | Mild Impairment (n = 8) | Moderate Impairment (n = 8) | Total (N = 24) |
|---|---|---|---|---|
| Overall | 1 (12.5)a | 1 (12.5)b | 1 (12.5) | 3 (12.5) |
| Diarrhea | 0 | 1 (12.5) | 0 | 1 (4.2) |
| Nausea | 1 (12.5) | 0 | 0 | 1 (4.2) |
| Blood urine present | 0 | 1 (12.5) | 0 | 1 (4.2) |
| Back pain | 1 (12.5) | 0 | 0 | 1 (4.2) |
| Hypotension | 0 | 0 | 1 (12.5) | 1 (4.2) |
Hepatic impairment defined according to Child‐Pugh classifications: Class A, mild (5–6 points); Class B, moderate (7–9 points).
Adverse events of nausea and back pain were observed in a single subject with normal hepatic function.
Adverse events of diarrhea and presence of blood in urine were observed in a single subject with mild hepatic impairment.
There were no clinically significant changes from baseline in vital signs, ECG, or clinical laboratory parameters. One subject in the mild impairment group had blood urine present on day 8, which was considered by the investigator to be grade 1 in severity and possibly related to the study drug.
Discussion
Peficitinib is a novel oral JAK inhibitor that is efficacious for the treatment of moderate‐to‐severe RA.1, 8, 9 In this PK study we evaluated and compared the 72‐hour PK profile of a single oral dose of peficitinib 150 mg in subjects with normal hepatic function and with mildly or moderately impaired hepatic function. A single 150‐mg oral dose was selected as the “usual” clinical dose based on the daily dose of peficitinib in phase 3 studies in patients with RA (100 mg and 150 mg).8, 9, 10 In previous dose‐ranging studies in healthy adult volunteers, single doses of peficitinib up to 450 mg did not cause any toxicity or significant safety findings that required dose limitation. Thus, a single 150‐mg dose was considered acceptable for subjects with hepatic impairment. Peficitinib (100‐mg and 50‐mg tablets) has recently been approved in Japan for adult RA patients at a usual dosage of 150 mg once daily, which can be reduced to 100 mg once daily depending on the patient's condition.13
The results demonstrate that the PK profile of single‐dose peficitinib in subjects with normal hepatic function was consistent with that observed in the previous PK study in healthy subjects.11 In the previous study the median tmax was 1 and 1.8 hours in subjects who received peficitinib 3 mg and 300 mg, respectively, and the mean t½ ranged from 2.8 to 13 hours.11 In subjects with normal hepatic function in our study, peficitinib was absorbed rapidly with a median tmax of 1.25 hours, and the mean t½ ranged from 3.3 to 18.4 hours. The median tmax and mean t½ were comparable between subjects with normal hepatic function and those with mild and moderate hepatic impairment.
There were no marked differences in the AUCinf and Cmax of plasma peficitinib between subjects with normal hepatic function and those with mild hepatic impairment. However, compared with subjects with normal hepatic function, subjects with moderately impaired hepatic function had higher exposure to plasma peficitinib. These findings are consistent with those observed in a PK study of the pan‐JAK inhibitor, tofacitinib, conducted in a non‐Asian sample of subjects in the USA.22 Tofacitinib AUCinf and Cmax were not altered in subjects with mild hepatic impairment, but, in those with moderate hepatic impairment, the geometric mean AUCinf and Cmax were increased by approximately 65% and 49%, respectively.22 In our study slightly greater increases in peficitinib geometric mean AUCinf and Cmax, of 92% and 82%, respectively, were observed in subjects with moderate hepatic impairment versus normal hepatic function. However, consistent with the tofacitinib safety findings, peficitinib was generally well tolerated by all subjects, regardless of hepatic function.
The 3 main peficitinib metabolites (H2, H4, and H1) are produced via 2 proposed pathways (Figure 4).12 There were no marked differences in the AUCinf and Cmax of H1, H2, or H4 between subjects with normal hepatic function and those with mild hepatic impairment. However, subjects with moderately impaired hepatic function had higher exposure to H1 and H4 metabolites and a somewhat lower exposure to the H2 metabolite. The median MPR for H2 in subjects with moderately impaired hepatic function was 0.56, compared with 1.83 and 2.70 for subjects with normal and mildly impaired hepatic function, respectively; this suggests that the metabolic clearance of H2 was decreased with moderately impaired hepatic function, which may have caused the increase in exposure to plasma peficitinib.
Figure 4.

Proposed metabolic pathways of peficitinib. H1, H2, and H4 are peficitinib metabolites; NNMT, nicotinamide N‐methyltransferase; SULT2A1, sulfotransferase 2A1.
The sulfotransferase (SULT) isozyme responsible for the formation of H2 is SULT2A1 (Figure 4).12 Although the information on SULT2A1 activity in patients with hepatic impairment is limited, Pacifici et al examined in vitro sulfation activity using liver samples from patients with liver damage. The study showed that the sulfation activity of ethinyl‐estradiol, a substrate of SULT2A1, in the cytosol prepared from the liver of patients with liver damage was lower than that of healthy donors.23 Thus, reduced conjugating capacity in subjects with moderately impaired hepatic function may be the cause of the observed reduction in metabolic clearance of H2.
In the moderate hepatic impairment group not only peficitinib but also H1 and H4 showed high exposure compared with their exposure in both normal and mild groups. The methyltransferase isozyme responsible for the formation of both compounds is suggested to be nicotinamide N‐methyltransferase (NNMT).12 Although the reasons for the observed elevation of methyl‐conjugated metabolites in the moderate group remain unclear, it may have been associated with enhanced NNMT activity in these subjects. Increased NNMT gene expression has been detected in a mouse model of severe hepatic impairment,24 and enhanced NNMT activity has been observed in patients with cirrhosis.25, 26 Indeed, the increased AUCinf and MPR of H4 in the moderate hepatic impairment group compared with the normal group suggest that the metabolism of peficitinib by NNMT has been increased.
There was no clear relationship between measures of hepatic function (albumin and prothrombin time) and the PK parameters of peficitinib.
There were 5 TEAEs reported in 3 subjects, and all events were deemed to be mild or moderate in intensity. Four of the TEAEs (nausea, diarrhea, blood urine present, and hypotension) were considered to be related to peficitinib. Additionally, there were no clinically meaningful changes from baseline in any of the clinical laboratory parameters measured during the study.
The limitation of the study was the small sample of subjects (8 subjects per group), which may affect the generalizability of the findings. In addition, the study did not include subjects with severe hepatic impairment; information about the peficitinib PK profile and safety in this group is therefore limited. It should also be noted that this was a single‐dose study in non‐RA subjects and does not provide information about peficitinib tolerance with extended use in patients.
In conclusion, this study showed that, following a single dose of peficitinib 150 mg, subjects with mild hepatic impairment (Child‐Pugh Class A) had no marked difference in peficitinib exposure compared with subjects with normal hepatic function. In contrast, in subjects with moderately impaired hepatic function (Child‐Pugh Class B) exposure to peficitinib approximately doubled compared with subjects with normal hepatic function. Peficitinib was well tolerated in (non‐RA) subjects with mild or moderate hepatic impairment as well as (non‐RA) subjects with normal hepatic function after a single oral dose. However, the starting dose of peficitinib in RA patients with moderate hepatic impairment will need to be reduced compared with the dose used in an RA patient without hepatic impairment, in accordance with the package insert.
Author Contributions
All authors met the following criteria for authorship: substantial contributions to the acquisition, analysis, and interpretation of data for the work; contribution to drafting the work and revising it critically; final approval of the version submitted; and acceptance of accountability for all aspects of the work.
Conflicts of Interest
M.S. reports grants from AbbVie Inc and MSD KK, a subsidiary of Merck & Co, Inc; lecturing fees from Gilead Sciences, Inc, Eisai Co, Sumitomo Dainippon Pharma Co, TUMURA & Co, ASKA Pharmaceutical Co, Japan Blood Products Organization, and Kyowa Hakko Kirin Co. K.I. reports grants for acute liver failure and lecturing fees for artificial liver support. D.M., T.S., J.T., Y.K., T.N., and M.K. are employees of Astellas Pharma Inc. K.O. is an employee of Astellas Research Institute of America LLC. A.U., T.I., N.U., and K.F. have nothing to declare.
Previous Publication
These data were presented in part as a poster at the European League Against Rheumatism, June 12–15, 2019, Madrid, Spain. An abstract, including data from this study, has been accepted as an oral presentation to the 2019 Japanese Society for Clinical Rheumatology and Related Research annual meeting.
Data‐Sharing Statement
Access to anonymized individual participant level data will not be provided for this trial as it meets 1 or more of the exceptions described on clinicalstudydatarequest.com under “Sponsor Specific Details for Astellas.”
Funding and Editorial Support
This study was funded by Astellas Pharma Inc. Medical writing support was provided by Annick Moon, PhD, and Lisa O'Rourke, PhD, for Cello Health MedErgy (Europe) and funded by Astellas Pharma Inc.
Supporting information
Supplementary Table S1. Study Site and Institutional Review Board Information
Acknowledgments
This study was performed at Souseikai Hakata Clinic in addition to the 5 other clinical sites under the supervision of Dr Megumi Inoue. We would like to thank the investigators, staff, study team, and the volunteers who were involved in the study.
Clinical trial registration identifier: NCT02586194
References
- 1. Takeuchi T, Tanaka Y, Iwasaki M, Ishikura H, Saeki S, Kaneko Y. Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: a 12‐week, randomised, double‐blind, placebo‐controlled phase IIb study. Ann Rheum Dis. 2016;75(6):1057‐1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hamaguchi H, Amano Y, Moritomo A, et al. Discovery and structural characterization of peficitinib (ASP015K) as a novel and potent JAK inhibitor. Bioorg Med Chem. 2018;26(18):4971‐4983. [DOI] [PubMed] [Google Scholar]
- 3. Qiu Q, Feng Q, Tan X, Guo M. JAK3‐selective inhibitor peficitinib for the treatment of rheumatoid arthritis. Expert Rev Clin Pharmacol. 2019;12(6):547‐554. [DOI] [PubMed] [Google Scholar]
- 4. Roskoski R Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol Res. 2016;111:784‐803. [DOI] [PubMed] [Google Scholar]
- 5. Iwata S, Tanaka Y. Progress in understanding the safety and efficacy of Janus kinase inhibitors for treatment of rheumatoid arthritis. Expert Rev Clin Immunol. 2016;12(10):1047‐1057. [DOI] [PubMed] [Google Scholar]
- 6. Genovese MC, Greenwald M, Codding C, et al. Peficitinib, a JAK inhibitor, in combination with limited conventional synthetic disease‐modifying antirheumatic drugs in the treatment of moderate‐to‐severe rheumatoid arthritis. Arthritis Rheum. 2017;69(5):932‐942. [DOI] [PubMed] [Google Scholar]
- 7. Kivitz AJ, Gutierrez‐Ureña SR, Poiley J, et al. Peficitinib, a JAK inhibitor, in the treatment of moderate‐to‐severe rheumatoid arthritis in patients with an inadequate response to methotrexate. Arthritis Rheum. 2017;69(4):709‐719. [DOI] [PubMed] [Google Scholar]
- 8. Tanaka Y, Takeuchi T, Tanaka S, et al. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to conventional DMARDs: a randomised, double‐blind, placebo‐controlled phase III trial (RAJ3). Ann Rheum Dis. 2019;78(10):1320‐1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takeuchi T, Tanaka Y, Tanaka S, et al. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III randomised, double‐blind, placebo‐controlled trial (RAJ4) in Japan. Ann Rheum Dis. 2019;78(10):1305‐1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Markham A, Keam SJ. Peficitinib: first global approval. Drugs. 2019;79(8):887‐891. [DOI] [PubMed] [Google Scholar]
- 11. Cao YJ, Sawamoto T, Valluri U, et al. Pharmacokinetics, pharmacodynamics, and safety of ASP015K (peficitinib), a new Janus kinase inhibitor, in healthy subjects. Clin Pharmacol Drug Dev. 2016;5(6):435‐449. [DOI] [PubMed] [Google Scholar]
- 12. Oda K, Cao YJ, Sawamoto T, et al. Human mass balance, metabolite profile and identification of metabolic enzymes of [14C]ASP015K, a novel oral Janus kinase inhibitor. Xenobiotica. 2015;45(10):887‐902. [DOI] [PubMed] [Google Scholar]
- 13. Astellas Pharma Inc. Oral JAK inhibitor Smyraf® tablets approved in Japan for the treatment of rheumatoid arthritis (including prevention of structural joint damage) in patients who have an inadequate response to conventional therapies [Press release]. https://www.astellas.com/en/news/14651. Published 2019. Accessed June 12, 2019.
- 14. Zhu T, Parker B, Wojtkowski T, et al. Drug interactions between peficitinib, an orally administered, once‐daily Janus kinase inhibitor, and rosuvastatin in healthy subjects. Clin Pharmacokinet. 2017;56(7):747‐757. [DOI] [PubMed] [Google Scholar]
- 15. Zhu T, Howieson C, Wojtkowski T, et al. The effect of verapamil, a P‐glycoprotein inhibitor, on the pharmacokinetics of peficitinib, an orally administered, once‐daily JAK inhibitor. Clin Pharmacol Drug Dev. 2017;6(6):548‐555. [DOI] [PubMed] [Google Scholar]
- 16. Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol. 2019;70(1):151‐171. [DOI] [PubMed] [Google Scholar]
- 17. Younossi Z, Anstee QM, Marietti M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2017;15(1):11‐20. [DOI] [PubMed] [Google Scholar]
- 18. Kim SR, Kim KI. An overview of NAFLD/NASH in Japan. Yakugaku Zasshi. 2016;136(4):565‐572. [DOI] [PubMed] [Google Scholar]
- 19. Michitaka K, Nishiguchi S, Aoyagi Y, et al. Etiology of liver cirrhosis in Japan: a nationwide survey. J Gastroenterol. 2010;45(1):86‐94. [DOI] [PubMed] [Google Scholar]
- 20. Kang J‐H, Matsui T. Changing etiology in liver cirrhosis in Sapporo, Japan. Ozkan H, ed. Euroasian J Hepato‐Gastroenterol. 2018;8(1):77‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oda K, Mera K, Nagasaka Y, Tokoro K. Simultaneous determination of a novel oral Janus kinase inhibitor ASP015K and its sulfated metabolite in rat plasma using LC‐MS/MS. Biomed Chromatogr. 2015;29(7):967‐969. [DOI] [PubMed] [Google Scholar]
- 22. Lawendy N, Lamba M, Chan G, Wang R, Alvey CW, Krishnaswami S. The effect of mild and moderate hepatic impairment on the pharmacokinetics of tofacitinib, an orally active Janus kinase inhibitor. Clin Pharmacol Drug Dev. 2014;3(6):421‐427. [DOI] [PubMed] [Google Scholar]
- 23. Pacifici GM, Viani A, Franchi M, et al. Conjugation pathways in liver disease. Br J Clin Pharmacol. 1990;30(3):427‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dong H, Toyoda N, Yoneyama H, et al. Gene expression profile analysis of the mouse liver during bacteria‐induced fulminant hepatitis by a cDNA microarray system. Biochem Biophys Res Commun. 2002;298(5):675‐686. [DOI] [PubMed] [Google Scholar]
- 25. Pumpo R, Sarnelli G, Spinella A, Budillon G, Cuomo R. The metabolism of nicotinamide in human liver cirrhosis: a study on N‐methylnicotinamide and 2‐pyridone‐5‐carboxamide production. Am J Gastroenterol. 2001;96(4):1183‐1187. [DOI] [PubMed] [Google Scholar]
- 26. Cuomo R, Dattilo M, Pumpo R, Capuano G, Boselli L, Budillon G. Nicotinamide methylation in patients with cirrhosis. J Hepatol. 1994;20(1):138‐142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Study Site and Institutional Review Board Information