

Abstract
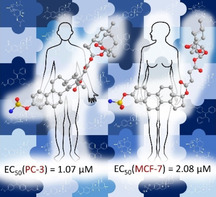
In the search for new and effective treatments of breast and prostate cancer, a series of hybrid compounds based on tamoxifen, estrogens, and artemisinin were successfully synthesized and analyzed for their in vitro activities against human prostate (PC‐3) and breast cancer (MCF‐7) cell lines. Most of the hybrid compounds exhibit a strong anticancer activity against both cancer cell lines – for example, EC50 (PC‐3) down to 1.07 μM, and EC50 (MCF‐7) down to 2.08 μM – thus showing higher activities than their parent compounds 4‐hydroxytamoxifen (afimoxifene, 7; EC50=75.1 (PC‐3) and 19.3 μM (MCF‐7)), dihydroartemisinin (2; EC50=263.6 (PC‐3) and 49.3 μM (MCF‐7)), and artesunic acid (3; EC50=195.1 (PC‐3) and 32.0 μM (MCF‐7)). The most potent compounds were the estrogen‐artemisinin hybrids 27 and 28 (EC50=1.18 and 1.07 μM, respectively) against prostate cancer, and hybrid 23 (EC50=2.08 μM) against breast cancer. These findings demonstrate the high potential of hybridization of artemisinin and estrogens to further improve their anticancer activities and to produce synergistic effects between linked pharmacophores.
Keywords: antitumor agents, artemisinin, estrogen, hybrids, tamoxifen, breast cancer, prostate cancer
Stronger together: The synthesis and biological investigation of four tamoxifen‐artemisinin and 16 estrogen‐artemisinin hybrid molecules are presented. In most cases, the anticancer activities of these new compounds exceed those of their parent compounds and reference drugs. The developed hybrids exhibit excellent in vitro activity against human prostate (PC‐3) and breast cancer (MCF‐7) cell lines.
Introduction
Cancer is a leading cause of death worldwide, with breast cancer being the most common cancer type in women and prostate cancer being the second most frequent malignancy (after lung cancer) in men.1 The fact that anticancer therapeutics have the lowest clinical trial success rate of all major diseases,2 shows that the cure of these cancer types remains a challenging health problem.
Natural product artemisinin (1, Figure 1) and its semisynthetic derivatives dihydroartemisinin (DHA, 2), artesunic acid (3; Figure 1) initially known to possess a high activity against malaria,3 were found to also have an anticancer potency,4 including activities against breast and prostate cancer.5
Figure 1.
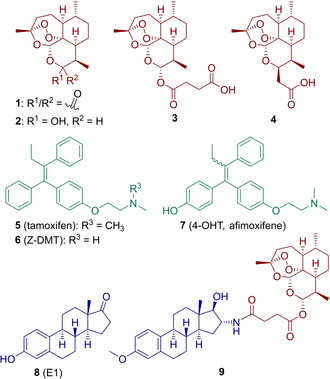
Structures of artemisinin (1) and its semisynthetic derivatives (dihydroartemisinin (DHA, 2), artesunic acid (3) and artemisinin‐derived acid 4), SERMs (tamoxifen (5), Z‐DMT (6), 4‐OHT (7)), estrone (8) and the first estrogen–artemisinin hybrid 9.15
Estrogens have remarkable and diverse pharmacological properties, including anticancer activities.6, 7 For instance, the estrogen fulvestrant is an efficient estrogen receptor antagonist, which was approved for the treatment of hormone‐related breast cancer,8 or abiraterone, which is a known steroidal therapeutic against prostate cancer.9 Another class of anticancer drugs are selective estrogen receptor modulators (SERMs), including tamoxifen (5, Figure 1), which has been used for over 30 years in treatment of hormone‐dependent breast cancer.10 Nonetheless, the development of new effective medications for the treatment of breast and prostate cancer is urgently needed, as most of the currently available drugs are no longer effective due to enhanced drug resistance in tumors (e. g., resistance to tamoxifen11 and to abiraterone12) and many unwanted side effects.1a, 13
One of the most promising, time‐ and cost‐effective drug discovery approaches to obtain new anticancer compounds with improved pharmacological properties is the hybridization of existing drugs. The concept of hybridization means covalent binding via a linker of two or more bioactive compounds with different mechanisms of action, which can result in the discovery of new and potent drug candidates, able to exceed parent compounds in the corresponding activities (by possible synergistic effects of subunits in a multifunctional hybrid drug), even able to overcome drug resistance and being an alternative to the standard drugs.14
Notably, several successful attempts have been made already to apply the method of hybridization in the development of new medications against hormone‐dependent breast16 and prostate17 cancer.18 Recently, our group reported the synthesis of the first five estrogen–artemisinin hybrids (e. g., hybrid 9, Figure 1), which were investigated for their in vitro anticancer potential, and indeed, most of them were highly potent against a panel of human malignant cells of gynecological origin containing breast (MCF‐7, MDA‐MB‐231, MDA‐MB‐361, T47D) and cervical tumor cell lines (HeLa, SiHa, C33A).15 These hybrid molecules can be regarded as potential lead compounds for development of new improved anticancer agents. Following this investigation and in order to obtain additional compounds for structure‐activity relationship (SAR) studies, we herein present the synthesis of the first four tamoxifen–artemisinin hybrids and 16 estrogen–artemisinin hybrids and their investigation against breast (MCF‐7) and prostate (PC‐3) cancer.
Results and Discussion
Chemistry
Although tamoxifen (5, Figure 1) has been used since its approval to treat millions of women diagnosed with hormone‐receptor‐positive breast cancer,10 tamoxifen resistance was recently reported.11 We therefore hypothesized that linking artemisinins with tamoxifen derivatives might result in novel therapeutic agents with a decreased chance of developing resistance.
(Z)‐N‐desmethyl‐tamoxifen (Z‐DMT, 6) and 4‐hydroxytamoxifen (4‐OHT, 7), which were used for the synthesis of novel hybrids 10–13 (Figure 2), are active metabolites of tamoxifen (5). Estrone (8) and ten of its synthetic analogues 35–42, 48 and 53 (Scheme 2) were chosen to present estrogenic moieties of the designed estrogen‐artemisinin hybrid molecules 14–29 (Figure 3). The estrogen analogues were chosen due to their different substituents in their skeleton to elucidate the structure‐activity relationship.
Figure 2.
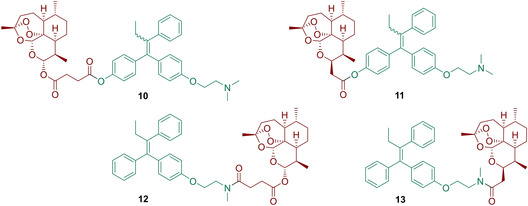
Structures of the synthesized tamoxifen‐artemisinin hybrids 10–13.
Scheme 2.
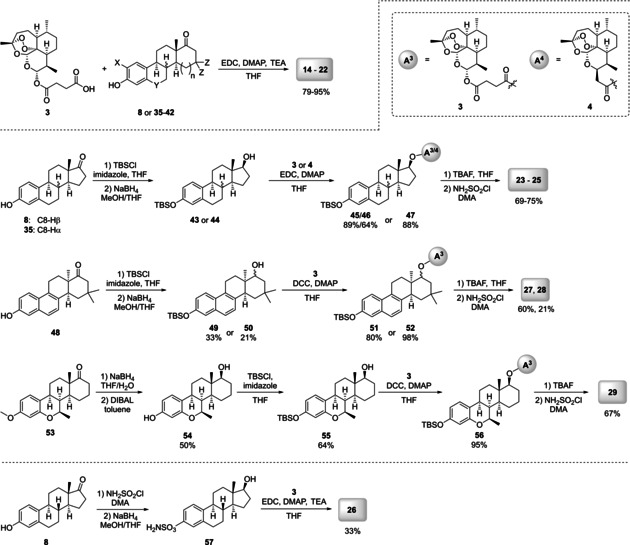
Synthesis of estrogen‐artemisinin hybrids 14–29 by using enantiopure estrone (8) and racemic estrogens 35–42, 48, 53.
Figure 3.
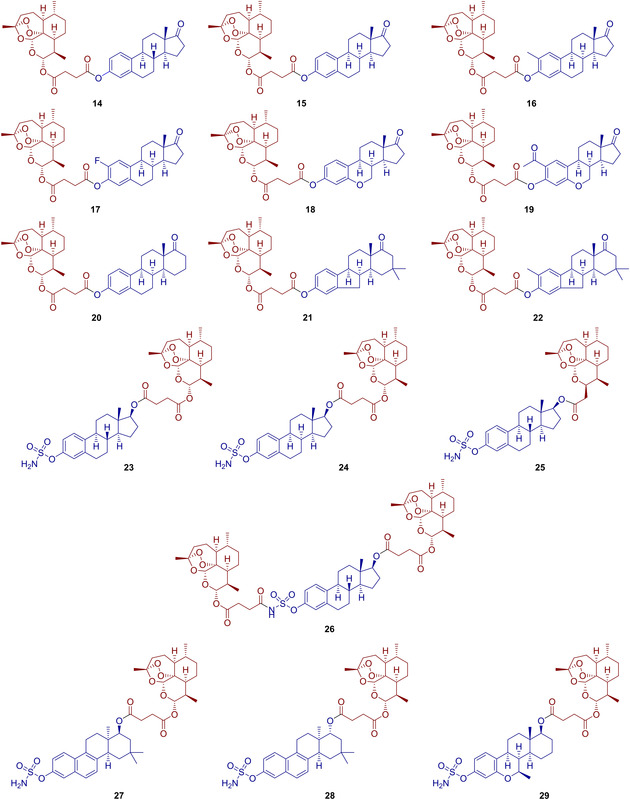
Structures of the synthesized estrogen‐artemisinin hybrids 14–29.
We recently reported a new fluorescent artemisinin‐derived antiviral hybrid, containing an ester linker, which was studied for its cellular uptake using confocal laser‐scanning microscopy.19 No degradation of ester‐linked hybrid under assay conditions was observed. Another new artemisinin based hybrid with ester linker was recently evaluated by us in vitro against P. falciparum strains and in vivo in P. berghei‐infected mice.20 Notably, this ester‐linked hybrid was not only more potent in comparison to its derivatives with amide and/or triazole linker, it has also demonstrated high thermal, hydrolytic, and enzymatic stability.20 Other groups also reported in vivo studies of artemisinin‐based hybrids containing an ester linker where no problems of stability or cleavage were observed.21 Based on these previous results with artemisinin‐derived hybrids with ester linkers and ease of ester synthesis, we chose an ester linkage for the preparation of most new hybrids in this work.
Z‐DMT (6) and 4‐OHT (7) were first synthesized using a McMurry reductive coupling22 as a key step (Scheme 1). Thus, propiophenone (31) was coupled with either benzophenone derivative 30 or 33, yielding the triphenylethylenes 32 and 34. Compound 32 was then alkylated in order to obtain a 1 : 1 E/Z mixture of 4‐OHT (7).22 As pure Z‐4‐OHT is known to equilibrate rapidly into E/Z equimolar mixture under physiological conditions,16a the separation of E/Z isomers was considered unnecessary, and, therefore the mixture was used for further coupling reactions. On the contrary, compound 34 was isolated by crystallization from iPrOH as pure Z‐isomer (such derivatives are much more stable towards isomerization under physiological conditions) and reacted with methylamine, yielding Z‐DMT (6).23 The compounds 6 and 7 were suitable for coupling with the artemisinin derivatives 3 and 4 using carbodiimide activation. Thus, starting from 7, hybrids 10 and 11 were obtained by Steglich esterification with DCC.24 However, later EDC was shown to have some superiority concerning yields and simplicity of final purification, and it was used in all subsequent couplings. The amidation reactions involving 3 and 4, activated by this reagent gave the corresponding amide‐bond linked hybrids 12 and 13.25
Scheme 1.
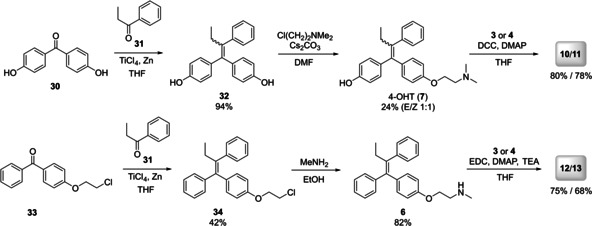
Synthesis of tamoxifen‐artemisinin hybrids 10–13.
Hence, the diverse set of tamoxifen‐based hybrids was synthesized (Figure 2). All of them are supposed to have different chemical and enzymatic stability depending on the type of linker. The next class of hybrids was based on estrogen‐derivatives. As such, new hybrids 14–22 (Figure 3) were synthesized by reacting artesunic acid (3) in an esterification reaction, using EDC as carbodiimide activator, with the corresponding estrogen derivatives 8 or 35–42 (Scheme 2).24 Hybrids 14–22 were obtained in good to excellent yields of 79 to 95 %. Furthermore, estrogen‐derivatives bearing a sulfamate moiety were used as building blocks for hybridization with artemisinin derivatives providing the new hybrid molecules 23–29 (Figure 3). Sulfamate based estrogen analogues are a well‐known new class of potential medications against hormone‐dependent diseases,26 and for that reason we decided to evaluate the potency of their hybrids with artemisinin against breast and prostate cancer. Starting with a protection reaction by transforming the phenolic hydroxy group in 8, 35 and 48 to a tert‐butyldimethylsilyl ether,27 the subsequent reduction of the carbonyl group with NaBH4 gives the hydroxyl moiety which is necessary for the following coupling reaction with either artemisinin derivative 3 or 4. Finally, the phenolic hydroxyl is deprotected using TBAF and converted to the desired sulfamate group, yielding the five different hybrids 23–25 and 27–28.
This route had to be slightly modified in order to obtain hybrid 29 (Scheme 2), because in this case the starting material 53 bears a methoxy moiety instead of the phenolic hydroxy group as the other starting materials 8, 35 and 48. Therefore, the first step involves the reduction of the carbonyl group and demethylation to give a phenolic hydroxy group which is then protected by silylation in the next step. The newly formed aliphatic hydroxy group is then used in an esterification with artesunic acid (3) and again, the final sulfamate containing artemisinin‐estrogen hybrid 29 is obtained by deprotection and conversion of the phenolic hydroxy group to a sulfamate moiety. This procedure was further improved for the synthesis of hybrid 26, where no protection of the phenolic hydroxy group was performed. Thus, hybrid 26 was obtained after only three steps, instead of five or six as previously described, involving firstly, a conversion of the alcohol to the sulfamate group and secondly, reduction of the carbonyl group,28 followed by an esterification reaction24 between 57 and artesunic acid (3). The stability of the hybrid compounds 10–29 was assessed by exposition to heat (65 °C/60 °C/40 °C) for 24 h to 48 h (see the Supporting Information for more details). After applying these conditions, 1H NMR showed less than 5 % decomposition indicating a sufficient stability of all hybrids.
Biological evaluation
Following the synthesis of hybrid molecules, we intended to evaluate their chemotherapeutic potential. To evaluate the activities of hybrid molecules in the context of breast cancer, we aimed to assess the cellular fitness of MCF‐7 cells in response to compound exposure. MCF‐7 is a common and estrogen receptor‐positive breast cancer in vitro model that has been used for more than 40 years in breast cancer research.29 To analyze the cell line specificity of the hybrid molecules, we aimed to assess their cytotoxic activity also in PC‐3 cells. Notably, PC‐3 cells do not respond to androgens, glucocorticoids, or epidermal or fibroblast growth factors and are thus, suitable to identify estrogen receptor‐dependent cytotoxic activity of the synthesized molecules.30 To assess the chemotherapeutic potential, i. e. the cytotoxic activity of the hybrid molecules, we employed a common assay type, which is based on quantification of the intracellular ATP concentration [ATP]i. Noteworthy, [ATP]i serves as an energy carrier that basically drives all cell functions. Persistent depletion of [ATP]i causes a cell to die and, in turn, cell death is indicated by low ATP levels. Due to its simple accessibility, for example, by an ATP‐dependent luciferase‐luciferin reaction, intracellular ATP has been a long‐serving indicator of cellular fitness.31 Cell fitness screening in MCF‐7 and PC‐3 cells was conducted in 384‐well plates as described in the Methods section. Each chemical was tested in increasing concentrations (0.001, 0.01, 0.1, 1, 10, 100 and 300 μM) and was incubated for 24–36 h with at least three replicates per concentration. The cytotoxic activity was assessed based on measurement of intracellular ATP content. Average (mean±SD) agonist dose–responses were constructed by pooling results from wells exposed to different solutions and are shown in the Supporting Information in Figures S2 and S3. The determined half‐maximal inhibition concentration (EC50) for afimoxifene (4‐OHT, 7), DHA (2), artesunic acid (3), estrogen precursor 57 and the hybrids 10–29 are shown in Table 1. These data demonstrate that most hybrids are more active against PC‐3 and MCF‐7 than their parent compounds DHA (2), artesunic acid (3) and selected estrogen 57 (precursor of hybrid 26). Additionally, a combination of estrogen 57 and artesunic acid (3) was analyzed, which showed an activity similar to that of artesunic acid (3) alone.
Table 1.
EC50 values for DHA (2), artesunic acid (3), afimoxifene (4‐OHT, 7), precursor 57 and hybrids 10–29 against human prostate (PC‐3) and breast cancer (MCF‐7) cell lines.
|
Compound |
MW [g/mol] |
PC‐3 |
MCF‐7 |
||
|---|---|---|---|---|---|
|
EC50 [μM] |
SD |
EC50 [μM] |
SD |
||
|
afimoxifene (7) |
387.52 |
75.1 |
81.5 |
19.3 |
17.5 |
|
DHA (2) |
284.35 |
263 |
82.5 |
49.3 |
6.33 |
|
artesunic acid (3) |
384.43 |
195 |
72.4 |
32.0 |
5.67 |
|
10 |
753.93 |
45.6 |
4.29 |
20.0 |
1.65 |
|
11 |
695.90 |
16.0 |
4.58 |
15.7 |
4.95 |
|
12 |
723.90 |
6.38 |
8.90 |
22.1 |
57.1 |
|
13 |
665.87 |
2.74 |
2.17 |
3.91 |
4.15 |
|
14 |
636.78 |
14.3 |
4.01 |
27.7 |
7.54 |
|
15 |
636.78 |
20.7 |
19.3 |
>1000 |
– |
|
16 |
650.81 |
41.8 |
119 |
14.8 |
3.75 |
|
17 |
654.77 |
179 |
176 |
17.4 |
5.22 |
|
18 |
638.75 |
10.5 |
14.4 |
51.8 |
44.6 |
|
19 |
680.79 |
114 |
31.6 |
23.8 |
5.04 |
|
20 |
650.81 |
111 |
70.2 |
56.7 |
61.2 |
|
21 |
664.84 |
14.1 |
10.0 |
7.03 |
4.19 |
|
22 |
678.86 |
5.62 |
0.82 |
5.23 |
3.76 |
|
23 |
717.87 |
3.23 |
1.72 |
2.08 |
0.45 |
|
24 |
717.87 |
2.04 |
0.61 |
3.09 |
0.85 |
|
25 |
659.84 |
3.76 |
0.85 |
2.84 |
1.52 |
|
26 |
1084.28 |
33.7 |
1.72 |
22.3 |
3.48 |
|
57 |
351.46 |
>1000 |
– |
165 |
50.6 |
|
57+3 (1 : 1) |
351.46+384.43 |
156 |
45.4 |
33.7 |
6.03 |
|
27 |
755.92 |
1.18 |
1.44 |
52.7 |
154 |
|
28 |
755.92 |
1.07 |
0.18 |
>1000 |
– |
|
29 |
747.90 |
3.94 |
1.44 |
3.86 |
1.85 |
In PC‐3 prostate cancer cells, most hybrids are more active than afimoxifene (7; EC50=75.1 μM), except for hybrids 17, 19 and 20, which are based on steroidal phenols. Comparing the tamoxifen‐based hybrids, it is interesting that the hybrids 12 (EC50=6.38 μM) and 13 (2.74 μM), containing Z‐DMT (6), are more active than the hybrids 10 (EC50=45.6 μM) and 11 (EC50=16.0 μM) containing 4‐OHT (7) as building block. Furthermore, the hybrids 23–25 and 29, with EC50 values ranging from 3.94 to 2.04 μM, are very active and 27 (EC50=1.18 μM) and 28 (EC50=1.07 μM), containing a naphthalene moiety, are most active.
Besides PC‐3, all hybrids and parent compounds were also tested towards their biological activity against MCF‐7 breast cancer cells. Within the experiments, the two hybrids 15 and 28 showed no activity at all against MCF‐7, whereas hybrid 28 exhibited a very high activity against PC‐3. Nevertheless, many compounds showed higher activity than their parent compounds artesunic acid (3) and DHA (2), and ten of them are even more active than afimoxifene (7; EC50=19.3 μM) with EC50 values between 17.4 and 2.08 μM. The highest activity was seen with the hybrids 13, 23–25 and 29 (EC50=3.91–2.08 μM). Although we observed an overall high reproducibility in our in vitro study with MCF‐7 and PC‐3 cells, some of the conducted experiments, depicted in the supporting information, showed relatively high SD values. Due to the fact that the averaged data with relatively higher SD values confirm the dose‐dependent effects of the tested molecules, those compounds were not subjected to re‐testing. The reason for the observed higher SD values was not evaluated in detail but may be due to errors during liquid handling using multi‐channel pipetting devices.
Conclusion
In conclusion, we synthesized the first tamoxifen–artemisinin hybrids 10–13 and were able to expand upon our previously published results on estradiol‐artemisinin based hybrids by synthesizing sixteen new estrogen–artemisinin hybrids 14–29. The biological activity of all hybrids was analyzed in human prostate (PC‐3) and human breast (MCF‐7) cancer cell lines. Most of them exhibit a strong anticancer activity against PC‐3 cell lines with EC50 values of 1.07–45.6 μM (hybrids 10–16, 18 and 21–29) and were therefore more active than reference compound afimoxifene (7; EC50=75.1 μM). Furthermore, the hybrids 10–13, 16, 17, 19, 21–26, and 29 were also highly active against MCF‐7 cancer cell lines, showing EC50 values of 2.08–23.8 μM, where nine of them outperformed the activity of afimoxifene (4‐OHT, 7). In terms of anti‐prostate cancer activity, hybrids 28 (EC50=1.07 μM) and 27 (EC50=1.18 μM) were the most active compounds and hybrid 23 (EC50=2.08 μM) showed the highest anti‐breast cancer activity. These promising results further underline the high potential of the hybridization concept for further investigations and hybrid‐based drug design.
Experimental Section
Purity data, stability experiments, experimental section, additional figures illustrating inhibition data, superpositions, and alignment of IspC from different organisms, details of multiple reaction monitoring, PAINS, recorded NMR spectra and Molecular Formula Strings are available in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG) through grant TS 87/16‐3. We also thank the Interdisciplinary Center for Molecular Materials (ICMM), the Graduate School Molecular Science (GSMS) for research support, as well as Emerging Fields Initiative (EFI) “Chemistry in Live Cells” supported by Friedrich‐Alexander‐Universität Erlangen‐Nürnberg for funding.
T. Fröhlich, C. Mai, R. P. Bogautdinov, S. N. Morozkina, A. G. Shavva, O. Friedrich, D. F. Gilbert, S. B. Tsogoeva, ChemMedChem 2020, 15, 1473.
Contributor Information
Dr. Daniel F. Gilbert, Email: daniel.gilbert@fau.de.
Prof. Dr. Svetlana B. Tsogoeva, Email: svetlana.tsogoeva@fau.de.
References
- 1.
- 1a. Joshi D., Int. J. Innovative Sci. Technol. 2019, 34; [Google Scholar]
- 1b. Ferlay J., Colombet M., Soerjomataram I., Mathers C., Parkin D. M., Piñeros M., Znaor A., Bray F., Int. J. Cancer 2019, 144, 1941. [DOI] [PubMed] [Google Scholar]
- 2. Cagan R., Meyer P., Dis. Model. Mech. 2017, 10, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Robert A., Benoit-Vical F., Meunier B., Coord. Chem. Rev. 2005, 249, 1927; [Google Scholar]
- 3b. Meunier B., Robert A., Acc. Chem. Res. 2010, 43, 1444. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Efferth T., Dunstan H., Sauerbrey A., Miyachi H., Chitambar C. R., Int. J. Oncol. 2001, 18, 767; [DOI] [PubMed] [Google Scholar]
- 4b. Efferth T., Curr. Drug Targets 2006, 7, 407; [DOI] [PubMed] [Google Scholar]
- 4c. Wondrak G. T., Antioxid. Redox Signaling 2009, 11, 3013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Chaturvedi D., Goswami A., Pratim Saikia P., Barua N. C., Rao P. G., Chem. Soc. Rev. 2010, 39, 435; [DOI] [PubMed] [Google Scholar]
- 4e. Liu W. M., Gravett A. M., Dalgleish A. G., Int. J. Cancer 2011, 128, 1471; [DOI] [PubMed] [Google Scholar]
- 4f. Li Z., Li Q., Wu J., Wang M., Yu J., Molecules 2016, 21, 1331; [Google Scholar]
- 4g. Chen G.-Q., Benthani F. A., Wu J., Liang D., Bian Z.-X., Jiang X., Cell Death Differ. 2020, 27, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Michaelsen F. W., Saeed M. E., Schwarzkopf J., Efferth T., Phytomedicine 2015, 22, 1223; [DOI] [PubMed] [Google Scholar]
- 5b. Slezakova S., Ruda-Kucerova J., Anticancer Res. 2017, 37, 5995; [DOI] [PubMed] [Google Scholar]
- 5c. Paccez J. D., Duncan K., Sekar D., Correa R. G., Wang Y., Gu X., Bashin M., Chibale K., Libermann T. A., Zerbini L. F., Oncogenesis 2019, 8, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arenas-Gonzalez A., Mendez-Delgado L. A., Merino-Montiel P., Padron J. M., Montiel-Smith S., Vega-Baez J. L., Meza-Reyes S., Steroids 2016, 116, 13. [DOI] [PubMed] [Google Scholar]
- 7. Alsayari A., Kopel L., Ahmed M. S., Pay A., Carlson T., Halaweish F. T., Steroids 2017, 118, 32. [DOI] [PubMed] [Google Scholar]
- 8. Liu J., Li J., Wang H., Wang Y., He Q., Xia X., Hu Z.-Y., Ouyang Q., J. Transl. Med. 2019, 17, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Malikova J., Brixius-Anderko S., Udhane S. S., Parween S., Dick B., Bernhardt R., Pandey A. V., J. Steroid Biochem. Mol. Biol. 2017, 174, 192. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Lu W. J., Desta Z., Flockhart D. A., Breast Cancer Res. Treat. 2012, 131, 473; [DOI] [PubMed] [Google Scholar]
- 10b. Ekholm M., Bendahl P. O., Fernö M., Nordenskjöld B., Stål O., Rydén L., Eur. J. Cancer 2019, 110, 53. [DOI] [PubMed] [Google Scholar]
- 11. Xue X., Yang Y. A., Zhang A., Fong K. W., Kim J., Song B., Li S., Zhao J. C., Yu J., Oncogene 2016, 35, 2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pal S. K., Patel J., He M., Foulk B., Kraft K., Smirnov D. A., Twardowski P., Kortylewski M., Bhargava V., Jones J. O., Cancer 2018, 124, 1216. [DOI] [PubMed] [Google Scholar]
- 13. Shagufta, Ahmad I., Eur. J. Med. Chem. 2018, 143, 515. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Tietze L. F., Bell H. P., Chandrasekhar S., Angew. Chem. Int. Ed. 2003, 42, 3996; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 4128; [Google Scholar]
- 14b. Gademann K., Chimia 2006, 60, 841; [DOI] [PubMed] [Google Scholar]
- 14c. Meunier B., Acc. Chem. Res. 2008, 41, 69; [DOI] [PubMed] [Google Scholar]
- 14d. Muregi F. W., Ishih A., Drug Dev. Res. 2010, 71, 20; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14e. Tsogoeva S. B., Mini-Rev. Med. Chem. 2010, 10, 773; [DOI] [PubMed] [Google Scholar]
- 14f. Decker M., Curr. Med. Chem. 2011, 18, 1464; [DOI] [PubMed] [Google Scholar]
- 14g.“Towards Antimalarial Hybrid Drugs”, B. Meunier in Polypharmacology in Drug Discovery (Ed.: J. U. Peters), John Wiley & Sons, 2012;
- 14h. Fröhlich T., Çapci Karagöz A., Reiter C., Tsogoeva S. B., J. Med. Chem. 2016, 59, 7360. [DOI] [PubMed] [Google Scholar]
- 15. Fröhlich T., Kiss A., Wölfling J., Mernyák E., Kulmány Á. E., Minorics R., Zupkó I., Leidenberger M., Friedrich O., Kappes B., Hahn F., Marschall M., Schneider G., Tsogoeva S. B., ACS Med. Chem. Lett. 2018, 9, 1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Patri A. K., Kukowska-Latallo J. F., J. R. Baker, Jr. , Adv. Drug Delivery Rev. 2005, 57, 2203; [DOI] [PubMed] [Google Scholar]
- 16b. Hawco C. L., Marchal E., Uddin M. I., Baker A. E., Corkery D. P., Dellaire G., Thompson A., Bioorg. Med. Chem. 2013, 21, 5995; [DOI] [PubMed] [Google Scholar]
- 16c. Gryder B. E., Rood M. K., Johnson K. A., Patil V., Raftery E. D., Yao L. P., Rice M., Azizi B., Doyle D. F., Oyelere A. K., J. Med. Chem. 2013, 56, 5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Madasu C., Karri S., Sangaraju R., Sistla R., Uppuluri M. V., Eur. J. Med. Chem. 2020, 188, 111974. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Hassan A. H. E., Choi E., Yoon Y. M., Lee K. W., Yoo S. Y., Cho M. C., Yang J. S., Kim H. I., Hong J. Y., Shin J.-S., Chung K.-S., Lee J.-H., Lee K.-T., Lee Y. S., Eur. J. Med. Chem. 2019, 161, 559; [DOI] [PubMed] [Google Scholar]
- 18b. Gao F., Sun Z., Kong F., Xiao J., Eur. J. Med. Chem. 2020, 188, 112044. [DOI] [PubMed] [Google Scholar]
- 19. Held F. E., Guryev A. A., Fröhlich T., Hampel F., Kahnt A., Hutterer C., Steingruber M., Bahsi H., von Bojničić-Kninski C., Mattes D. S., Foertsch T. C., Nesterov-Mueller A., Marschall M., Tsogoeva S. B., Nat. Commun. 2017, 8, 15071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Çapcı A., Lorion M. M., Wang H., Simon N., Leidenberger M., Borges Silva M. C., Moreira D. R. M., Zhu Y., Meng Y., Chen J. Y., Lee Y. M., Friedrich O., Kappes B., Wang J., Ackermann L., Tsogoeva S. B., Angew. Chem. Int. Ed. 2019, 58, 13066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Jung M., Tak J., Chung W. Y., Park K. K., Bioorg. Med. Chem. Lett. 2006, 16, 1227; [DOI] [PubMed] [Google Scholar]
- 21b. Galal A. M., Gul W., Slade D., Ross S. A., Feng S., Hollingshead M. G., Alley M. C., Kaur G., ElSohly M. A., Bioorg. Med. Chem. 2009, 17, 741. [DOI] [PubMed] [Google Scholar]
- 22. Gauthier S., Mailhot J., Labrie F., J. Org. Chem. 1996, 61, 3890. [DOI] [PubMed] [Google Scholar]
- 23. Jarman M., Mccague R., J. Chem. Res. 1985, 116. [Google Scholar]
- 24. Reiter C., Capci Karagöz A., Fröhlich T., Klein V., Zeino M., Viertel K., Held J., Mordmüller B., Emirdağ Öztürk S., Anil H., Efferth T., Tsogoeva S. B., Eur. J. Med. Chem. 2014, 75, 403. [DOI] [PubMed] [Google Scholar]
- 25. Reiter C., Herrmann A., Capci A., Efferth T., Tsogoeva S. B., Bioorg. Med. Chem. 2012, 20, 5637. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Thomas M. P., Potter B. V., J. Med. Chem. 2015, 58, 7634; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Potter B. V. L., J. Mol. Endocrinol. 2018, 61, T233. [DOI] [PubMed] [Google Scholar]
- 27. Hosoda H., Yamashita K., Ikegawa S., Nambara T., Chem. Pharm. Bull. 1977, 25, 2545. [Google Scholar]
- 28. Schwarz S., Thieme I., Richter M., Undeutsch B., Henkel H., Elger W., Steroids 1996, 61, 710. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Sugarman B. J., Aggarwal B. B., Hass P. E., Figari I. S., M. A. Palladino, Jr. , Shepard H. M., Science 1985, 230, 943; [DOI] [PubMed] [Google Scholar]
- 29b. Comsa S., Cimpean A. M., Raica M., Anticancer Res. 2015, 35, 3147. [PubMed] [Google Scholar]
- 30. Kaighn M. E., Narayan K. S., Ohnuki Y., Lechner J. F., Jones L. W., Invest. Urol. 1979, 17, 16. [PubMed] [Google Scholar]
- 31. Gilbert D. F., Erdmann G., Zhang X., Fritzsche A., Demir K., Jaedicke A., Muehlenberg K., Wanker E. E., Boutros M., PLoS One 2011, 6, e28338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary