

Abstract
NIR exposure at 790 nm activated photopolymerization of monomers comprising UV‐absorbing moieties by using [CuII/(TPMA)]Br2 (TPMA=tris(2‐pyridylmethyl)amine) in the ppm range and an alkyl bromide as initiator. Some of them comprised structural elements selected either from those showing proton transfer or photocycloaddition upon UV excitation. Polymers obtained comprise living end groups serving as macroinitiator for controlled synthesis of block copolymers with relatively narrow molecular weight distributions. Chromatographic results indicated formation of block copolymers produced by this synthetic approach. Free‐radical polymerization of monomers pursued for comparison exhibited the expected broader dispersity of molecular weight compared to photo‐ATRP. Polymerization of these monomers by UV photo‐ATRP failed on the contrary to NIR photo‐ATRP demonstrating the UV‐filter function of the monomers. This work conclusively provides a new approach for the polymerization of monomers comprising UV‐absorbing moieties through photo‐ATRP in the NIR region. This occurred in a simple and efficient pathway. However, studies also showed that not all monomers chosen successfully proceeded in the NIR photo‐ATRP protocol.
Keywords: block copolymers, copper catalysis, cyanine, near-infrared radiation, photo-ATRP
Does it work or not? NIR‐sensitized photo‐ATRP facilitates controlled radical polymerization of monomers comprising UV‐absorbing moieties. New block copolymers were synthesized with special functionalities. UV ATRP failed with UV‐absorbing monomers.

Introduction
Block copolymers have been one of the most interesting classes of materials due to the novel properties from their special chemical structures. Possible applications for block copolymers cover a wide range of areas involving the fields of chemistry, physics, materials science, medicine, or biological sciences.1, 2, 3, 4, 5 Specifically, they have been used in drug delivery, solar cells, holography, patterning, or stimuli responsive materials6, 7, 8, 9, 10—just to name a few of them. Traditionally, block copolymers have been synthesized by cationic or anionic living polymerization procedures.11 Various coupling strategies related to click chemistry, such as the copper‐catalyzed cycloaddition (CuAAC),12 Diels–Alder reaction,13 and thiol‐ene chemistry14 give also access to block copolymers. Nevertheless, the development of modern radical polymerization methods with focus to tailor‐made materials has been moved to this area.15 Controlled/living polymerization of structurally suitable monomers facilitates preparation of block copolymers with well‐defined structures and chain lengths. Particularly atom transfer radical polymerization (ATRP) has received big interest.15 This reaction protocol bases on CuI catalysts requesting a relatively large amount of heavy metal. This point and the fact that these catalysts sensitively respond to air brings some issues regarding the universal use of the traditional ATRP.
Therefore, more of interest could be the use of catalytic amounts of less sensitive CuII catalysts or metal‐free systems.16, 17, 18, 19 The latter depict interesting systems in combination with light as reagent and tool facilitating the access to green technologies. This gave birth of a new methodology in the ATRP frameworks; that is the photo‐ATRP.15 Whereas metal‐free photoredox systems disclosed their operational functionality in systems requesting either UV20 or visible‐light excitation,21 NIR excitation requested catalytic amounts of CuII in the ppm range.18 Metal‐free approaches failed upon excitation at 790 nm by using heptamethine‐based cyanines as sensitizer in combination with an amine.18 This occurred similarly as in previous metal‐free studies by using either UV15, 20 or visible light15, 21 for excitation. The polymers obtained by NIR exposure followed rather a free‐radical polymerization (FRP) protocol. Replacement of the amine by an iodonium salt as co‐initiator resulted in similar high molecular weights with broad dispersities (MWD).18 On the other hand, the new system introduced based on CuII catalyst with amounts in the ppm range worked well for tailor‐made synthesis of block copolymers.18 A general mechanism was developed explaining the catalytic activity, which was recently also copied to another approach where carbon dots served as sensitizer in an ATRP framework applying blue light LEDs for excitation.22 In addition, oxygen tolerance in controlled/living radical polymerization was recently reported for some examples.15d
NIR radiation may facilitate the use of monomers comprising structural patterns absorbing either in the UV/visible part in contrast to previous published UV‐ or visible‐light‐based ATRP protocols. Such materials can be seen of interest as additives for coatings23 or UV‐absorbing filter materials.24 Interesting are also monomers comprising UV‐reactive groups facilitating cycloaddition reactions upon UV exposure.25, 26 This definitively explains the need to extend studies providing more opportunities for controlled polymer synthesis to give access to more materials comprising either UV‐absorbing or UV‐active moieties. The existing redox equilibria between CuII/CuI and the participating reagents address the question whether certain monomers may interfere the photo‐ATRP excited at 800 nm considering the respective redox data of the participating monomers. This question was put in the focus of this contribution to find answers and also perhaps limitations of the photo‐ATRP operating in the NIR at around 800 nm.
Consequently, this study explores feasibilities to polymerize distinct monomers comprising UV‐absorbing components functioning either as UV stabilizer where intramolecular proton transfer prevents further undesired photoreactions, UV‐reactive groups following a photocycloaddition protocol, and some more to compliment the investigations given below. The results obtained provide a deeper understanding of NIR‐sensitized photo‐ATRP at around 800 nm.18 NIR excitation also brings the benefit that light can travel even through thick samples.27 Photo‐reversible addition‐fragmentation chain transfer (RAFT) polymerization also facilitated polymerization of monomers showing photocleavage upon UV excitation resulting in small dispersities.28 Obviously, the photoreactive UV moiety of the monomer did not significantly interfere the RAFT polymerization mechanism.
Results and Discussion
Compounds M1–M8 (Scheme 1) served as monomers comprising a UV absorbing moiety up‐taking the function as a UV filter (M1 and M2), carbazole as electron‐donating moiety (M3), photoactive moieties showing either [4+4] photocycloaddition25 (M4) or [2+2] photocycloaddition26 (M5), and more (meth)acrylate monomers used to synthesize block copolymers. Compound M7 provides the opportunity to synthesize a block in which the tert‐butyl group can easily hydrolyze resulting in formation of an acidic polymer whose increased acid number facilitates easier dissolution in lithographic applications.29 Alternatively, M8 operated just as monomer to form a hydrophobic block. Phenylacrylate (M6) complements the investigations because its polymerization activity in traditional free‐radical polymerization can be seen as more or less modest.30
Scheme 1.

Structures of the monomers investigated in this study.
Figure 1 depicts the absorption studies of the monomers M1–M8 comprising an aromatic substituent. M1, M2, and M4 predominantly cover the absorption of the complete UV range indicating that their use in photo‐ATRP protocols based on UV excitation would definitively fail. One of the examples can be seen vide infra. It has been clear that none of the aforementioned monomers with UV absorbing moiety should show any polymerization activity following the reactions shown in Scheme 2.
Figure 1.

UV/Vis spectrum of monomers studied in this work. All spectra were taken in DMF (arrow indicates the wavelength where the intrinsic absorption of the monomer becomes negligible at high concentration).
Scheme 2.

UV‐initiated photo‐ATRP by using DPMA (Irgacure 651) to generate nucleophilic radicals reducing CuII resulting in CuI. This simple sketch shows the reactions from a simplest point of view.
On the other hand, M8 resulted in a polymer exhibiting relatively narrow dispersity following the protocol of Scheme 2. It resulted in a dispersity of 1.2 and an acceptable average M n value of 19 000 g mol−1 when using M8 as monomer. In this mechanism, DPMA cleaved upon UV exposure as shown in Scheme 2 resulting in formation of benzoyl radicals and nucleophilic dialkoxy radicals, which easily oxidize by electron‐accepting moiety by using an onium ion as acceptor.31 (CuIIL)Br2 can also function as electron acceptor because its reduction potential of −0.24 V32a favors the oxidation of the nucleophilic radical.31 This serves as the source delivering continuously CuI catalyst into the ATRP equilibrium in Scheme 2. This reaction neither worked with any of the monomers selected from M1–M6. Only M8 showed acceptable polymerization results in the ATRP protocol according to Scheme 2. Surprisingly, the benzoyl radicals formed do not interfere the controlled polymerization. One could expect a considerable contribution of free‐radical polymerization, which was not observed as long as CuII catalyst was available in the reaction mixture. No polymerization occurred with a monomer covering the emission of the UV‐LED used.
To overcome the disadvantages of intrinsic monomer absorption in the UV range, further polymerization studies were pursued in the NIR at 790 nm according to a previous introduced system. Figure 2 summarizes the mechanism leading to polymerization of M8. It worked well with the NIR sensitizer (Sens) shown in this figure comprising a barbiturate group at the meso position. This moiety can interact with CuII as earlier reported for analytical detection of this metal ion.33 Indeed, a change of absorption also occurred upon addition of CuII into a solution comprising Sens (see Figure SI1 in the Supporting Information). The absorption of Sens strongly decreases whereas a new absorption builds between 400–500 nm confirming the CuII–barbiturate interaction.32 This reaction explains why particularly such cyanines worked and others with no barbital group in the molecular skeleton did not following this reaction route.18 Thus, the barbital group brings CuII to the place where it should react. After excitation, the excited state of Sens (Sens*) transfers an electron to the copper(II) complex (Figure 2, step b) resulting in formation of its cation radical (Sens +.) and the respective CuI complex, which operates in the equilibrium (Figure 2, step c) shown in Scheme 2. This leads to polymer formation (Figure 2, step d). This mechanism also forms back Sens (Figure 2, step e) formally by reaction between Sens +. and Br−. The bromine atom formed in step f fast recombines with the polymer radical. Nevertheless, Br− can be found in equilibria such as in step g. The concentration of bromide also affects the dispersity.32a
Figure 2.

Redox mechanism of the used NIR‐induced photo‐ATRP.
According to this protocol depicted in Figure 2 and to overcome the aforementioned disadvantages of UV‐based controlled photo‐ATRP, NIR‐sensitized polymerization was pursued with monomers M8 and M1–M6. Figure 3 depicts the kinetics of M1 following a first order confirming the expected kinetics. The polymerization proceeded similarly as with M8 where after 6, 12, 18, and 24 h exposure similar conversion was reported18 as concluded by comparison the slopes being 0.025 and 0.023 h−1 for M8 and M1, respectively.
Figure 3.
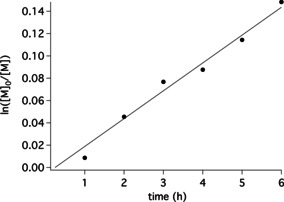
NIR‐sensitized photo‐ATRP by using the cyanine Sens as sensitizer, TPMA as ligand, and EBPA as initiator (for structures see Figure 2). Polymerization proceeded in DMF.
However, other monomers did not proceed so successful as M1 and M8 in the NIR‐sensitized photo‐ATRP. Table 1 summarizes the results obtained for the remaining monomers. Compound M2 exhibits in the same time frame a significant smaller conversion (x), which may be caused by hydrogen bonding of the OH group of the benzophenone comprising monomer. Such OH‐containing benzophenones were described in photochemistry as proton‐transfer agents resulting often in an improvement of UV‐light stability.34 Therefore, interactions between the OH‐comprising benzophenone‐based monomer M2 and initiator components, such as the Sens* and/or the CuII/L component may be possible. This might have an impact on the initiation of the ATRP and, therefore, on the residual molecular weight of the polymer. The thermally initiated free‐radical polymerization may rule out a discussion that the aromatic OH group might interfere the radical polymerization mechanism. Most of the monomers showed acceptable conversion using AIBN for thermal initiation. Also, M2 exhibited the highest conversion in the time frame considered whereas the reference M8 as well as M1, M4, and M6 showed lower conversions. Moreover, low conversion of the cinnamoyl comprising monomer M5 was low for both the NIR‐sensitized photo‐ATRP and the thermally initiated free‐radical polymerization.
Table 1.
Photo‐initiated and free‐radical polymerization of different (meth)acrylate‐based monomers. Photo‐ATRP used sensitizers absorbing in the UV (DMPA) and NIR (S2265) under different experimental conditions (NIR light‐induced ATRP: [M]/[EBPA]/[CuIIBr2]/[TPMA]/[S2265]=300:1:0.03:0.135:0.3, t=24 h; UV light‐induced ATRP: [M]/[EBP]/[CuIIBr2]/[PMDETA]/[DMPA]=4000:5:1:3:1, t=5 h; thermal‐initiated free‐radical polymerization: T=70 °C, [AIBN]=0.3 mol %, t=6 h). Average molecular weight M n (in kg mol−1) and dispersity Ð (MWD) determined by gel permeation chromatography by using PMMA standards. Conversion (x in %) was gravimetrically determined.
|
Monomer |
NIR photo‐ATRP |
UV photo‐ATRP |
FRP |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
M n (theory)[a] [g mol−1] |
M n [g mol−1] |
Ð |
x |
M n [g mol−1] |
Ð |
x |
M n [g mol−1] |
Ð |
x |
|
M8 |
13.8 |
9.7 |
1.2 |
46 |
19.0 |
1.2 |
46 |
19.8 |
2.1 |
53 |
|
M1 |
50.4 |
9.4 |
1.5 |
52 |
|
|
0 |
22.0 |
3.0 |
50 |
|
M2 |
5.6 |
3.3 |
1.4 |
6 |
|
|
0 |
9.3 |
2.5 |
97 |
|
M3 |
|
|
|
≪1 |
|
|
0 |
6.3 |
3.0 |
82 |
|
M4 |
|
|
|
≪1 |
|
|
0 |
5.5 |
3.5 |
69 |
|
M5 |
1.8 |
2.8 |
1.4 |
3 |
|
|
0 |
7.2 |
2.5 |
5 |
|
M6 |
3.6 |
2.2 |
1.2 |
8 |
|
|
0 |
19.6 |
2.6 |
77 |
[a] Calculated as follows: M n=[M]0/[EPB]0 DP n, DP n=average number degree of polymerization.
The vinylcarbazole‐comprising monomer M3 and the anthracene‐containing monomer M4 showed very low conversion in the photo‐ATRP. The polymer isolated exhibited a molecular weight located below the specified value of the column material used. Both monomers carry an adjacent aromatic moiety (Ar) with respect to the vinyl group. Thus, addition of a radical R. to Ar−CH=CH2 results in the radical Ar−C.H−CH2−R possessing electron‐donating properties because the π electron of the C‐centered radical facilitates easy oxidation by electron acceptors similarly as in the case of the ether radical shown in Scheme 2. Furthermore, both monomers showed acceptable conversion in thermal initiated polymerizations ruling out a discussion that chain transfer to Ar might mainly cause the low molecular weight. Though NIR‐sensitized photo‐ATRP showed promising results with monomer M1, the remaining monomers exhibited more or less modest results caused either by hydrogen bonding interactions (M2) or side reactions (M2–M5). These results should be considered for future project planning tailor made synthesis based on light sensitized photo‐ATRP. Furthermore, a photo‐RAFT reaction protocol27a should be applied to check whether these monomers, namely M2–M5, would interfere the photo‐RAFT initiation mechanism. Previous investigations showed successful working with monomers comprising reactive groups. This included reaction with singlet oxygen28b and monomers comprising a UV‐sensitive moiety showing bond cleavage upon UV excitation.28a The results question whether photo‐RAFT or photo‐ATRP would be the better approach to pursue living radical polymerization with functional monomers comprising monomers with UV‐active moieties. Thus, a transfer of M2–M5 to a photo‐RAFT reaction protocol27 will bring an answer.
Cyclic voltammetry studies (Table 2) may also exclude any discussion that the redox potentials of the monomer might interfere the initiator system. Considering that Sens exhibits an excitation energy of 1.57 eV18 and an oxidation potential of 0.48 V,18 the respective free enthalpy of the electron transfer might reside slightly below 0 eV in the case of M3 and M4 whereas all other monomers possess significant lower reduction potentials resulting in a more pronounced shift towards endothermal conditions regarding the photoinduced electron transfer. Thus, this interaction does not seem to be likely. Cu(TPMA)2+ (TPMA=tris(2‐pyridylmethyl)amine) exhibited a reduction potential of −0.24 V.32a
Table 2.
Cyclic voltammetry (CV) measurements for redox potentials of the used UV‐active monomers (taken against Ag/AgCl as reference electrode).
|
Monomer |
E ox [V] |
E red [V] |
|---|---|---|
|
M1 |
1.70 |
−1.08 |
|
M2 |
1.13 |
−1.25 |
|
M3 |
1.31 |
−0.73 |
|
M4 |
1.26 |
−0.85 |
|
M5 |
1.12 |
−1.00 |
|
M6 |
1.21 |
−1.02 |
Further polymerization studies focused on the influence of the macroinitiator. Thus, a block copolymer was synthesized by using either a macroinitiator based on M1 or M8 to add a second block, which was M8 and M1, respectively, resulting in the block copolymers Ini‐(M1)n‐(M8)m‐Br and Ini‐(M8)n‐(M1)m‐Br, respectively (Ini=C2H5O‐CO‐CH(Ph)‐). Figure 4 depicts the results obtained. Dispersities remain in a range typical for a controlled radical polymerization. Polymerization of the first block M1 resulted in a molecular weight of 9400 g mol−1 whereas the block extension with M8 resulted in and final M n value of 11 700 g mol−1. This corresponds to 28 and 23 polymerized monomer units in the polymer blocks (M1)n and (M8)n, respectively. However, this pattern changed if photo‐ATRP started with M8 whereas the second polymer segment was pursued with M1 resulting in M n values of 9700 g mol−1 for the first segment and 67 300 g mol−1 for the second segment in the block copolymer. This corresponds to 97 and 203 polymerized monomer units in the polymer blocks (M8)n and (M1)n, respectively. This was unexpected and might be caused by a distinct reactivity of the macroinitiators. Macroinitiator M1 dissolved less in DMF compared to macroinitiator M8 resulting in distinct availability of initiating bromine groups of the respective macroinitiator. The macroinitiator based on M8, however, showed no issues considering the solubility in the solvent chosen.
Figure 4.

GPC traces of precursor (M8)n‐Br and block copolymer (M8)n‐b‐(M1)n‐Br in comparison with the precursor (M8)n‐Br and the block copolymer (M8)n‐b‐(M1)n‐Br, both reactions proceeded in 50 vol % DMF at room temperature applying 790 nm exposure.
In addition, block copolymerization studies with the monomers M1 and M7 resulted in the block copolymer Ini‐(M1)n‐(M7)m‐Br showing a M n of 17 600 g mol−1 and a dispersity of 1.7. This corresponds to 28 and 62 polymerized monomer units in the polymer blocks (M1)n and (M7)n, respectively, showing again that it matters from which side architectural growth of the block copolymer begins. The same also holds for discussion of reactivity given above. Furthermore, the tert‐butyl group of M7 can easily hydrolyze under mild alkaline conditions where other acrylic based esters typically do not show significant hydrolysis. It results in this case in a block copolymer comprising a hydrophobic block exhibiting UV filter properties and an additional block with functional groups promoting the adhesion on substrates comprising oxides at the surface. This gives new impetus in coating sciences for the manufacture of special additives useful for surface protection.
Conclusions
NIR‐sensitized photo‐ATRP successfully worked with some (meth)acrylates comprising an UV‐absorbing moiety. Particular a methacrylic ester with benzotriazole (M1) showed similar reactivity compared to methylmethacrylate in the NIR‐sensitized photo‐ATRP. Replacement of benzotriazole in M1 by hydroxy(benzophenone), which relates to M2, yields a system with less reactivity. However, comparison with redox potentials of the methacrylates used with the available data of the respective CuII complex showed no point to discuss that these quantities might interfere the ATRP process except monomer M3. Its reduction potential of −0.73 V is comparable to that of iodonium cations showing acceptable reactivity with the excited state of the sensitizer used in this study considering the formation of initiating radicals for radical photopolymerization in the NIR. Discussion of different OH interaction of M2 with the Cu catalyst as well as the formation of reactive intermediates by M4, which forms an easy oxidizable anthryl‐methyl radical by addition of a radical to vinylanthracene during polymerization, may be main points diminishing the efficiency of the NIR‐sensitized photo‐ATRP. This concludes by comparison of the data obtained by thermal initiated polymerization resulting in a similar high degree of polymerization as other monomers except M5. The double bond of the cinnamoyl group might copolymerize with methacrylic groups explaining the low reactivity.
Nevertheless, the general drawback of this study gives positive directions for future research in this field. More studies with distinct vinyl monomers will give deeper impetus in this field. It will also show limitations regarding the use of monomers to make tailor‐made block copolymers. Nevertheless, the use of monomers forming intermediates interfering the redox mechanism or macroinitiators diminished solubility should be avoided. Additional characterizations regarding the properties of the polymer will clarify how side reactions of the polymers may affect their properties.
Finally, such photocatalytic systems possesses big potential for the development of future technologies based on information recording, such as lithography or holography, 3D and 4D printing, tailor‐made polymer synthesis—just to name a few of them.
Experimental Section
Materials
Tris(2‐pyridylmethyl)amine (TPMA, 98 %), copper(II)bromide (CuBr2, 99 %, Aldrich), ethyl α‐bromophenylacetate (EBPA, 97 %), ethyl 2‐bromopropionate (EBP, 99 %), N,N,N′,N′′,N′‐pentamethyldiethylentriamine (PMDETA, 99 %), and α,α‐dimethoxy‐α‐phenylacetophenone (DMPA, 99 %) were purchased from Sigma–Aldrich and used as received. Methyl methacrylate (M8, 99 %, Sigma–Aldrich), phenylacrylate (M6, 98 %, TCI) and cinnamyl methacrylate (M5, Polysciences) are passed through a plug of basic alumina before use to remove the inhibitor. 2‐[3‐(2H‐Benzotriazol‐2‐yl)‐4‐hydroxyphenyl]ethyl methacrylate (M1, 99 %), 2‐(4‐benzoyl‐3‐hydroxyphenoxy)ethyl acrylate (M2, 98 %), 9‐vinylcarbazole (M3, 99 %) and 9‐vinylanthracene (M4, 97 %) were purchased from Sigma–Aldrich and used as received. The NIR sensitizer Sens (5‐(6‐(2‐(3‐ethyl‐1,1‐dimethyl‐1H‐benzo[e]indol‐2(3H)‐ylidene)ethylidene)‐2‐(2‐(3‐ethyl‐1,1‐dimethyl‐1H benzo[e]‐indol‐3‐ium‐2‐yl)vinyl)cyclohex‐1‐en‐1‐yl)‐1,3‐dimethyl‐2,6‐dioxo‐1,2,3,6 tetrahydropyrimidin‐4‐olate) was received from FEW Chemicals GmbH as S2265. N,N‐Dimethylformamide (DMF, anhydrous, 99.8 %) was purchased from Acros Organics and stored over activated molecular sieves. Methanol (99.9 %, Bernd Kraft), tetrahydrofurane (THF, 99.9 %, Carl Roth) and 2‐butanone (99.0 %, Sigma Aldrich) were used as received.
Instrumentation
Photoreactor (NIR): The sample was exposed with NIR radiation in a Schlenk tube (diameter 18.0 mm) with four NIR‐LEDs (LED 790‐66‐60 from Roithner Lasertechnik) arranged in an angle of 90° between each LED and a distance of 11 mm around the tube. The light intensity of each LED was 100 mW cm−2 within the exposed area in the middle height on the surface of the tube. This photoreactor was sealed in a lightproof can and the sample was cooled with an airflow around the tube to have ambient temperature during exposure. The mixture was stirred with a magnetic mixer during irradiation.
Photoreactor (UV): The sample was exposed with UV radiation in a Schlenk tube (diameter 18.0 mm) with one UV‐LED at 395 nm arranged in a distance of 11 mm to the tube. The light intensity of the LED was 91 mW cm−2 within the exposed area in the middle height on the surface of the tube. This photoreactor was sealed with a light proof foil and the sample was cooled with an air flow around the tube to have ambient temperature during exposure. The mixture was stirred with a magnetic mixer during irradiation.
Gel permeation chromatography (GPC): GPC was used to determine number average molecular weight (M n) and dispersity Đ, (Đ=M w/M n). GPC measurements were conducted with a GPC Viscotek 270 max using TGuard Col 10×4.6 mm and two T6000M General Mixed 3000×7.8 mm columns, a column temperature of 30 °C, an RI detector, and tetrahydrofuran (THF) as an eluent at a flow rate of 1 mL min−1. The column system was calibrated with seven linear poly(methyl methacrylate) standards received from Shodex (1850 g mol−1; 6380 g×mol−1; 20 100 g×mol−1; 73 200 g×mol−1; 218 000 g×mol−1; 608 000 g×mol−1; and 1050 000 g×mol−1). GPC data were analyzed using Omni SEC 4.6.2:GPC. H‐NMR of polymers isolated and purified indicated no residual monomer.
NMR spectroscopy: A Fourier 300 from Bruker was used for all 1H‐NMR measurements. 15 mg sample were dissolved in 0.7 mL solvent.
Cyclic voltammetry (CV): Oxidation and reduction potentials of the UV‐active monomers were recorded by cyclic voltammetry (VERSASTAT4‐400 from AMETEK served as potentiostat) in acetonitrile (c=10−3 m) with tetrabutylammonium hexafluorophosphate from Sigma–Aldrich (0.1 mol/l) as a supporting electrolyte against ferrocene as an external standard. The data were taken with a scanning rate of 0.015 V s−1 using a platinum disc as a working electrode and Ag/AgCl as reference electrode.35
Polymerization
General Procedure for NIR light‐induced ATRP of monomers: A vial equipped with a magnetic stir bar and fitted with a Teflon screw cap septum was charged with Sens (32.9 mg, 0.045 mmol), 25 μL of a 180 mm CuBr2 stock solution in DMF (4.5 μmol) and 75 μL of a 270 mm TPMA stock solution in DMF (20.3 μmol). A mixture of monomer (45 mmol) and DMF (50 vol%), and ethyl α‐bromophenylacetate (36.5 mg, 0.150 mmol) were added and homogenized with stirring. The solution was transferred to a Schlenk Flask with a magnetic stirrer and degassed by four freeze–pump–thaw cycles. The reaction mixture was placed in a photoreactor irradiating at the desired wavelength (NIR‐LED exposure at 790 nm, 24 h). At the end of the irradiation, the resulting polymers were precipitated in cold methanol and then dried under reduced pressure. Conversion was determined gravimetrically.
Block copolymerization experiment (M8) n ‐b‐(M1) n ‐Br: A vial equipped with a magnetic stir bar and fitted with a Teflon screw cap septum was charged with M8 based macroinitiator (585 mg, 0.045 mmol), 7.5 μL of a 180 mm CuBr2 stock solution in DMF (1.35 μmol) and 45 μL of a 270 mm TPMA stock solution in DMF (6.09 μmol). M1 (1.6 g, 13.5 mmol) and DMF (5.1 mL) were added to this mixture, and the resulting solution was homogenized by vigorous stirring. The solution was transferred to a Schlenk Flask with a magnetic stirrer and degassed by four Freeze‐Pump‐Thaw cycles. The reaction mixture was placed in a photoreactor irradiating at the desired wavelength (NIR‐LED exposure at 790 nm, 24 h). At the end of the irradiation, the polymers obtained were precipitated in cold methanol and then dried under reduced pressure. Conversion was determined gravimetrically.
This procedure was transferred for synthesis of the remaining block copolymers using the disclosed molar concentrations of the reaction components.
General procedure for UV light‐induced ATRP of UV‐absorbing monomers: A vial equipped with a magnetic stir bar and fitted with a Teflon screw cap septum was charged with PMDETA (58.18 μL, 0.028 mmol), CuBr2 (20.75 mg, 0.093 mmol), 2 (1.2 mg, 0.0047 mmol), EBP (3 μL, 0.023 mmol) and methanol (1 mL, 24 mmol). To this mixture 18.6 mmol monomer was added and the solution was homogenized by stirring. The solution was transferred to a Schlenk Flask with a magnetic stirrer and degassed by four Freeze‐Pump‐Thaw cycles. The reaction mixture was placed in a photoreactor irradiating at the desired wavelength (UV‐LED exposure at 365 nm, 5 h). At the end of the irradiation was put in cold n‐hexane. Precipitated polymer was collected and then dried under reduced pressure. Conversion was determined gravimetrically.
Free radical polymerization of UV‐absorbing monomers: An amount of 5 g of monomer was dissolved as a 15 % solution in butanone. The solution was charged in a 100 mL reaction vessel under nitrogen atmosphere. The solution was heated to 70 °C and stirred with a magnetic mixer during reaction. AIBN (0.3 mol %) was added to the mixture and reaction starts for 6 hours at 70 °C. At the end of reaction, the resulted solution was filtrated and concentrated to a 60 % solution in rotary vaporizer. The resulted polymers were precipitated in cold methanol and then dried under reduced pressure. Conversion was determined gravimetrically.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
B.S. thanks the county of North Rhine–Westphalia for funding of the project REFUBELAS (grant 005‐1703‐0006, managed by P.T.J.) and the project D‐NL‐HIT carried out in the framework of INTERREG–Program Deutschland–Nederland, which is co‐financed by the European Union, the MWIDE NRW, the Ministerie van Economische Zaken en Klimaat and the provinces of Limburg, Gelderland, Noord‐Brabant, und Overijssel.
C. Kütahya, N. Meckbach, V. Strehmel, J. S. Gutmann, B. Strehmel, Chem. Eur. J. 2020, 26, 10444.
ATRP=atom transfer radical polymerization.
References
- 1. Kim H.-C., Park S.-M., Hinsberg W. D., Chem. Rev. 2010, 110, 146–177. [DOI] [PubMed] [Google Scholar]
- 2. Feng H., Lu X., Wang W., Kang N.-G., Mays W. J., Polymers 2017, 9, 494 (31 pages). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. T. H. Epps III , O'Reilly R. K., Chem. Sci. 2016, 7, 1674–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jennings J., He G., Howdle S. M., Zetterlund P. B., Chem. Soc. Rev. 2016, 45, 5055–5084. [DOI] [PubMed] [Google Scholar]
- 5. Blanazs A., Armes S. P., Ryan A. J., Macromol. Rapid Commun. 2009, 30, 267–277. [DOI] [PubMed] [Google Scholar]
- 6. Adams M. L., Lavasanifar A., Kwon G. S., J. Pharm. Sci. 2003, 92, 1343–1355. [DOI] [PubMed] [Google Scholar]
- 7. Sun S.-S., So. Energy Mater. Sol. Cells 2003, 79, 257–264. [Google Scholar]
- 8. Häckel M., Kador L., Kropp D., Schmidt H. W., Adv. Mater. 2007, 19, 227–231. [Google Scholar]
- 9. Bates C. M., Maher M. J., Janes D. W., Ellison C. J., Willson C. G., Macromolecules 2014, 47, 2–12. [Google Scholar]
- 10. Yin J., Chen Y., Zhang Z.-H., Han X., Polymers 2016, 8, 268–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang Y., Liu X., Zhang F., Dong J., Luo Y., Huang C., Polymer J. 2013, 45, 125–128. [Google Scholar]
- 12.
- 12a. Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B., Angew. Chem. Int. Ed. 2002, 41, 2596–2599; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 2708–2711; [Google Scholar]
- 12b. Kolb H. C., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed. 2001, 40, 2004–2021; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 2056–2075. [Google Scholar]
- 13. Tasdelen M. A., Polym. Chem. 2011, 2, 2133–2145. [Google Scholar]
- 14.
- 14a. Lowe A. B., Hoyle C. E., Bowman C. N., J. Mater. Chem. 2010, 20, 4745–4750; [Google Scholar]
- 14b. Hoyle C. E., Bowman C. N., Tasdelen M. A., Yagci Y., Angew. Chem. Int. Ed. 2010, 49, 1540–1573; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 1584–1617; [Google Scholar]
- 14c. Wu P., Feldman A. K., Nugent A. K., Hawker C. J., Scheel A., Voit B., Pyun J., Frechet J. M. J., Sharpless K. B., Fokin V. V., Angew. Chem. Int. Ed. 2004, 43, 3928–3932; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 4018–4022. [Google Scholar]
- 15.
- 15a. Dadashi-Silab S., Doran S., Yagci Y., Chem. Rev. 2016, 116, 10212–10275; [DOI] [PubMed] [Google Scholar]
- 15b. Pan X., Tasdelen M. A., Laun J., Junkers T., Yagci Y., Matyjaszewski K., Prog. Polym. Sci. 2016, 62, 73–125; [Google Scholar]
- 15c. Discekici E. H., Anastasaki A., Read de Alaniz J., Hawker C. J., Macromolecules 2018, 51, 7421–7434; [Google Scholar]
- 15d. Yeow J., Chapman R., Gormley A. J., Boyer C., Chem. Soc. Rev. 2018, 47, 4357–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ribelli T. G., Lorandi F., Fantin M., Matyjaszewski K., Macromol. Rapid Commun. 2019, 40, 1800616. [DOI] [PubMed] [Google Scholar]
- 17. Treat N. J., Sprafke H., Kramer J. W., Clark P. G., Barton B. E., Read de Alaniz J., Fors B. P., Hawker C. J., J. Am. Chem. Soc. 2014, 136, 16096–16101. [DOI] [PubMed] [Google Scholar]
- 18. Kütahya C., Schmitz C., Strehmel V., Yagci Y., Strehmel B., Angew. Chem. Int. Ed. 2018, 57, 7898–7902; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 8025–8030. [Google Scholar]
- 19. Ribelli T. G., Konkolewicz D., Bernhard S., Matyjaszewski K., J. Am. Chem. Soc. 2014, 136, 13303–13312. [DOI] [PubMed] [Google Scholar]
- 20. Kutahya C., Allushi A., Isci R., Kreutzer J., Ozturk T., Yilmaz G., Yagci Y., Macromolecules 2017, 50, 6903–6910. [Google Scholar]
- 21.
- 21a. Ciftci M., Tasdelen M. A., Yagci Y., Polym. Chem. 2014, 5, 600; [Google Scholar]
- 21b. Konkolewicz D., Schröder K., Buback J., Bernhard S., Matyjaszewski K., ACS Macro Lett. 2012, 1, 1219–1223. [DOI] [PubMed] [Google Scholar]
- 22. Kütahya C., Wang P., Li S., Liu S., Li J., Chen Z., Strehmel B., Angew. Chem. Int. Ed. 2020, 59, 3166–3171; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 3192–3197. [Google Scholar]
- 23.
- 23a. Berndt E., Behnke S., Dannehl A., Gajda A., Wingender J., Ulbricht M., Polymer 2010, 51, 5910–5920; [Google Scholar]
- 23b. Wang Z., Li N., Wang M., Wang X., Li D., Havas D., Zhang H., Wu G., RSC Adv. 2015, 5, 83931–83935. [Google Scholar]
- 24. Farkas R., Lhiaubet-Vallet V., Corbera J., Törincsi M., Gorchs O., Trullas C., Jiménez O., Miranda M. A., Novak L., Molecules 2010, 15, 6205–6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manhart J., Ayalur-Karunakaran S., Radl S., Oesterreicher A., Moser A., Ganser C., Teichert C., Pinter G., Kern W., Griesser T., Schlögl S., Polymer 2016, 102, 10–20. [Google Scholar]
- 26. Yamazaki S., Sugiura H., Ohashi S., Ishizuka K., Saimu R., Mikata Y., Ogawa A., J. Org. Chem. 2016, 81, 10863–10886. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Wu Z., Jung K., Boyer C., Angew. Chem. Int. Ed. 2020, 59, 2013–2017; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 2029–2033; [Google Scholar]
- 27b. Corrigan N., Yeow J., Judzewitsch P., Xu J., Boyer C., Angew. Chem. Int. Ed. 2019, 58, 5170–5189; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5224–5243; [Google Scholar]
- 27c. Shanmugam S., Xu J., Boyer C., Angew. Chem. Int. Ed. 2016, 55, 1036–1040; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1048–1052. [Google Scholar]
- 28.
- 28a. Bagheri A., Yeow J., Arandiyan H., Xu J., Boyer C., Lim M., Macromol. Rapid Commun. 2016, 37, 905–910; [DOI] [PubMed] [Google Scholar]
- 28b. Xu S., Yeow J., Boyer C., ACS Macro Lett. 2018, 7, 1376–1382. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Baumann H., Hoffmann-Walbeck T., Wenning W., Lehmann H.-J., Simpson C. D., Mustroph H., Stebani U., Telser T., Weichmann A., Studenroth R., in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2015, pp. 1–51; [Google Scholar]
- 29b. Baumann H., Chem. Unserer Zeit 2015, 49, 14–29; [Google Scholar]
- 29c. Strehmel B., Ernst S., Reiner K., Keil D., Lindauer H., Baumann H., Z. Phys. Chem. (Muenchen Ger.) 2014, 228, 129–153. [Google Scholar]
- 30. Demirelli K., Kaya E., Coşkun M., J. Appl. Polym. Sci. 2006, 99, 3344–3354. [Google Scholar]
- 31. Baumann H., Müller U., Pfeifer D., Timpe H. J., J. Prakt. Chem. 1982, 324, 217–226. [Google Scholar]
- 32.
- 32a. Ribelli T. G., Fantin M., Daran J.-C., Augustine K. F., Poli R., Matyjaszewski K., J. Am. Chem. Soc. 2018, 140, 1525–1534; [DOI] [PubMed] [Google Scholar]
- 32b. Qiu J., Matyjaszewski K., Thouin L., Amatore C., Macromol. Chem. Phys. 2000, 201, 1625–1631. [Google Scholar]
- 33. Zwikker J. J. L., Pharm. Weekbl. 1933, 70, 551–559. [Google Scholar]
- 34.
- 34a. Karaca Balta D., Karahan Ö., Avci D., Arsu N., Prog. Org. Coat. 2015, 78, 200–207; [Google Scholar]
- 34b. Klesse W., Knebel J., Saal D., Roehm GmbH Evonik, 2016 (Evonik Roehm GmbH), US 2016/0297738 A1.
- 35. Schmitz C., Halbhuber A., Keil D., Strehmel B., Prog. Org. Coat. 2016, 100, 32–46. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary