

Abstract
We investigated metabolic changes during brain death (BD) using hyperpolarized magnetic resonance (MR) spectroscopy and ex vivo graft glucose metabolism during normothermic isolated perfused kidney (IPK) machine perfusion. BD was induced in mechanically ventilated rats by inflation of an epidurally placed catheter; sham‐operated rats served as controls. Hyperpolarized [1‐13C]pyruvate MR spectroscopy was performed to quantify pyruvate metabolism in the liver and kidneys at 3 time points during BD, preceded by injecting hyperpolarized[1‐13C]pyruvate. Following BD, glucose oxidation was measured using tritium‐labeled glucose (d‐6‐3H‐glucose) during IPK reperfusion. Quantitative polymerase chain reaction and biochemistry were performed on tissue/plasma. Immediately following BD induction, lactate increased in both organs (liver: eµd0.21, 95% confidence interval [CI] [−0.27, −0.15]; kidney: eµd0.26, 95% CI [−0.40, −0.12]. After 4 hours of BD, alanine production decreased in the kidney (eµd0.14, 95% CI [0.03, 0.25], P < .05). Hepatic lactate and alanine profiles were significantly different throughout the experiment between groups (P < .01). During IPK perfusion, renal glucose oxidation was reduced following BD vs sham animals (eµd0.012, 95% CI [0.004, 0.03], P < .001). No differences in enzyme activities were found. Renal gene expression of lactate‐transporter MCT4 increased following BD (P < .01). In conclusion, metabolic processes during BD can be visualized in vivo using hyperpolarized magnetic resonance imaging and with glucose oxidation during ex vivo renal machine perfusion. These techniques can detect differences in the metabolic profiles of the liver and kidney following BD.
Keywords: animal models, basic (laboratory) research/science, donors and donation: donation after brain death (DBD), graft survival, kidney (allograft) function/dysfunction, kidney transplantation/nephrology, liver allograft function/dysfunction, liver transplantation/hepatology, organ procurement and allocation, translational research/science
Short abstract
Differences in glucose metabolism in the liver and kidney can be visualized in vivo during brain death using hyperpolarized magnetic resonance spectroscopy and ex vivo following brain death during normothermic kidney machine perfusion using glucose oxidation.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate transaminase
- ATP
adenosine triphosphate
- BD
brain death
- eµd
estimated mean difference
- ICP
intracranial pressure
- IPK
isolated perfused kidney
- IRR
intrarenal vascular resistance
- LDH
lactate dehydrogenase
- MAP
mean arterial pressure
- MDA
malondialdehyde
- MRI
magnetic resonance imaging
- PDH
pyruvate dehydrogenase
- Sham
sham‐operated
- TBARS
thiobarbituric acid reactive substances
1. INTRODUCTION
The global shortage of donor organs results in almost 115 000 people in the United States and 15 000 people in Europe on the organ waiting list. 1 , 2 For most of these patients, organ transplantation would mean a significant improvement of their survival and quality of life. 3 Most organs worldwide are obtained from brain‐dead organ donors. 3 However, brain death (BD) causes major changes in donor physiology, 4 , 5 , 6 , 7 leading to impaired graft function, higher rejection rates, and inferior long‐term outcomes after transplantation compared to living‐donor transplantations. 7 , 8 Therefore, it is important to improve our understanding of BD‐induced injuries. This would allow us to treat or precondition organs, in the donor or during organ preservation.
Donor BD affects systemic as well as organ‐specific metabolism. In plasma, glucose levels decrease yet lactate and fatty acid levels increase. 9 , 10 , 11 In both the liver and kidneys, tissue injury increases while adenosine triphosphate (ATP) levels decline. 9 , 12 , 13 , 14 , 15 However, a prior study by our group suggests that the underlying mechanisms vary. 9 In the kidneys, anaerobic metabolism increased, resulting in more pyruvate breakdown and decreased glucose production. 9 As BD causes decreased renal perfusion and increased oxidative stress, 9 , 12 this is likely the result of renal hypoxia. Meanwhile in the liver, glycogen stores declined while glycolysis and oxygen consumption increased 9 , 10 (Figure 1). Also, several mitochondrial peptides involved in fatty acid oxidation and substrate transport increased. In contrast to the kidneys, the liver seems to respond to increasing energy demands by altering its metabolic profile. 9 These metabolic changes are potentially significant, as the pretransplant metabolic status of the liver and kidney has been correlated to posttransplant graft viability. 16 , 17 , 18 , 19 , 20 This might be explained by the fact that (changes in) metabolism can influence cell survival, cell growth, and cell death pathways including autophagy and apoptosis. 21 , 22 , 23
FIGURE 1.
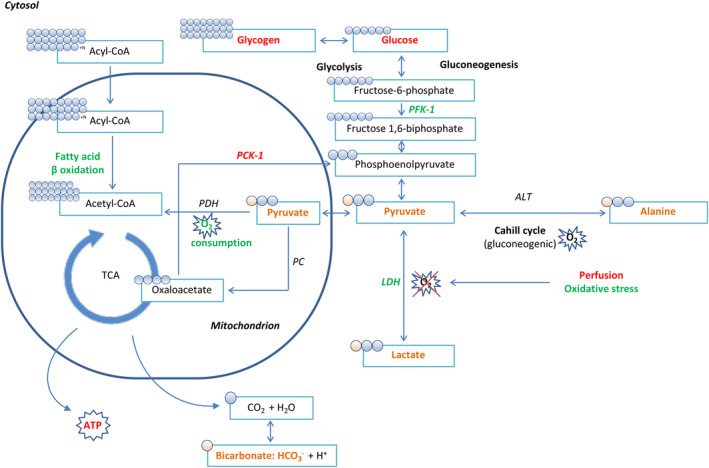
Overview of metabolic changes in the liver and kidney following brain death. All changes depicted in green represent an increase in activity or level, whereas all changes marked in red denote decreased activity or levels. 9 All compounds marked in orange are involved in the metabolization of pyruvate into its metabolites: the pyruvate‐alanine (or Cahill) cycle involving enzyme ALT, the pyruvate‐lactate cycle involving LDH as the last step of glycolysis in the absence of oxygen, pyruvate decarboxylation involving the transformation of pyruvate to acetyl‐CoA involving PDH, and finally the formation of bicarbonate (HCO3) from carbon dioxide (CO2) as an end‐product of the mitochondrial TCA cycle. ALT, alanine aminotransferase; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase; TCA, tricarboxylic acid
Cellular metabolism of 13C‐labeled (enriched) molecules can be monitored noninvasively using a novel technique called hyperpolarized magnetic resonance imaging (MRI). Hyperpolarization increases the sensitivity of the MRI system more than 10 000‐fold, using techniques such as dynamic nuclear polarization. 24 , 25 Labeling of 13C‐pyruvate allows for real‐time metabolic mapping of pyruvate as well as its metabolites, including lactate, alanine, and bicarbonate (Figure 1). 26 Visualization of these metabolites using hyperpolarized 13C‐pyruvate MRI has already shown promise in diagnosing and monitoring early renal functional changes 27 , 28 and has recently been translated to humans. 29
There are currently no good quantitative biomarkers to be used before transplantation that correlate with organ quality. Furthermore, most clinically tested interventions in the deceased donor have not improved organ quality or survival after transplantation. 30 , 31 As mentioned, previous reports have demonstrated that organ‐specific, metabolic changes already occur within the brain‐dead donor. 9 , 10 , 11 , 12 , 13 , 14 , 15 Here we investigated metabolic pathophysiological changes in the brain‐dead donor using hyperpolarized MR spectroscopy in the liver and kidney and ex vivo graft glucose metabolism during normothermic kidney machine perfusion. We hypothesized that metabolism would differ between BD animals and controls.
2. MATERIALS AND METHODS
2.1. BD model
Twenty‐two male, Fisher 344 rats were randomized into 3 groups: a sham‐operated (sham) (n = 8), a BD (n = 8), and a BD control group (represented as BD [T = 0], n = 6). The BD and sham groups were exposed to an experimental duration of 4 hours of BD, whereas the BD control group was terminated immediately after BD induction (Figure 2). The BD control group was added to correlate differences in MRI measurements to changes such as enzyme of gene expression in tissue. The BD model previously described by Kolkert et al was used 32 with several adaptations to ensure MRI compatibility, 9 described in more detail in the Supplementary Material. Briefly, animals were anesthetized with sevoflurane, intubated via a tracheostomy and ventilated (MR‐compatible Small Animal Ventilator; SA Instrument, Inc, New York, NY). Through an arterial and venous cannula, volume was replaced and the mean arterial pressure (MAP) was measured. BD was induced by inflation of a Fogarty catheter in the epidural space (Edwards Lifesciences Co, Irvine, CA). Intracranial pressure (ICP) was monitored through a contralaterally placed 24G cannula. BD was confirmed when the ICP superseded the MAP and animals were solely ventilated with a FIO2 of 50%. Sham animals underwent an identical procedure, except for insertion of the Fogarty catheter while anesthesia lasted the entire 4 hours of the experiment. After 4 hours, the experiment was terminated and the organs, plasma, and urine were retrieved.
FIGURE 2.
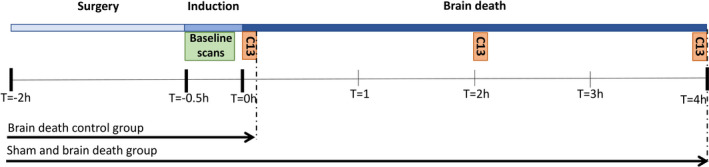
Experimental overview, where T = −2 marks the beginning of surgery, T = −0.5 the beginning of BD induction, T = 0 the beginning of the BD period, and the subsequent hours of the total BD duration. At T = 0, T = 2, and T = 4, hyperpolarized [1‐13C]pyruvate MR spectroscopy was performed following an injection of [1‐13C]pyruvate at each time point. The BD control group was terminated following the first round of MR spectroscopy (shortly after T = 0). BD, brain death; MR, magnetic resonance
2.2. MRI assessment of 13C‐pyruvate metabolism
We used a 9.4 T preclinical MR system (Agilent, Santa Clara, CA) with VnmrJ 4.0A (Agilent) with a dual tuned 13C/1H volume rat coil (Doty Scientific, Columbia, MO). 33 , 34 After a 1H anatomical MR scan to determine the location of the liver and kidneys, hyperpolarized [1‐13C]pyruvate MR spectroscopy was performed to quantify pyruvate metabolism in the liver and kidneys at 3 different time points: at the start of BD (T = 0), after 2 hours (T = 2), and after 4 hours (T = 4, Figure 2), preceded each time by an injection of 1.5 mL of a 85 mmol hyperpolarized [1‐13C]pyruvate, pH neutral and isotonic solution (details can be found in the Supplementary Material). [1‐13C]pyruvate was polarized in a SpinLab (GE Healthcare). 33 , 35 Each time, 1.5 mL [1‐13C]pyruvate solution was injected into the femoral vein in 10 seconds, after which scanning was initiated. The area under the curve of the spectral peaks of [1‐13C]pyruvate and each of the metabolites was calculated for the total signal using MATLAB. More details can be found in the Supplementary Material.
2.3. Reperfusion model and glucose oxidation in the IPK
Following 4 hours of BD, the left kidney was normothermically perfused for 90 minutes in an isolated perfused kidney (IPK) device (Figure S1), 36 described in more detail in the Supplementary Material. Briefly, the IPK system is a pressure‐ (100 mm Hg) and temperature‐ (37°C) controlled perfusion system that allows for pulsatile perfusion of the kidney via arterial and venous cannulas with an oxygenated (95% O2, 95% CO2) perfusion medium. During the experiment, flow and (pre‐ and postrenal) oxygenation were recorded at 10‐minute intervals. Sampling of perfusate and urine was done after 0, 15, 30, 60, and 90 minutes of perfusion. After 90 minutes, the kidney was disconnected and stored at −80°C for further analyses.
Rates of glucose oxidation were measured during reperfusion using a tritium‐labeled glucose isotope (d‐6‐3H‐glucose) in accordance with a protocol modified from isolated heart perfusions 37 , 38 and described in more detail in the Supplementary Material. A buffer volume of 100 mL (5 μL d‐6‐3H‐glucose/100 mL KH buffer) was recirculated. The oxidation of exogenous glucose was assessed measuring the amount of 3H2O, liberated from the oxidation of d‐[6‐3H]‐glucose in the citric acid cycle. 3H2O was separated using anion exchange chromatography on AG 1‐X8 resin columns (Bio‐Rad, Hercules, CA ) and quantified using beta‐scintillation on a TriCarb 2900TR liquid scintillation analyzer (Packard, Shelton, CT ) in disintegrations per minute (dpm). The final glucose concentration was presented as the baseline activity of the perfusate divided by the dpm, corrected for the wet weight (mass) of each kidney and the flow.
2.4. Plasma and urine injury markers
Plasma levels of aspartate transaminase (AST), alanine aminotransferase (ALT), creatinine, urea, lactate dehydrogenase (LDH), glucose, and lactate, and urine creatinine were determined at the clinical chemistry laboratory of the University Medical Centre Groningen according to standard procedures.
2.5. Enzyme activity assays and gene expression
The enzyme activities of LDH, pyruvate dehydrogenase (PDH), and ALT from liver and kidney tissue were determined using enzyme activity assay kits according to the manufacturer's instructions 39 , 40 , 41 , 42 (Sigma‐Aldrich, Zwijndrecht, The Netherlands).
For gene expression, RNA was isolated from whole liver and kidney sections using TRIzol (Life Technologies, Carlsbad, CA) using a method described in more detail in the Supplementary Material. 9 Primer sets used to amplify LDHA, MCT1, and MCT4 are outlined in Table S1. Gene expression was normalized to the mean mRNA β‐actin content and pooled cDNA from brain‐dead rats used as an internal reference. Real‐time polymerase chain reaction was done according to methods described previously 9 and in more detail in the Supplementary Material. Results are expressed as 2−△△CT (CT: threshold cycle).
2.6. Tissue ATP levels
ATP content was determined as a measure of the organ energy status in 50 mg of frozen hepatic and renal tissue slices, using standard methods described in detail in the Supplementary Material. 9
2.7. Oxidative stress markers
Oxidative damage was determined through estimation of the lipid peroxidation product malondialdehyde (MDA) in plasma as well as tissue homogenates (each 20 µL). Levels of MDA were measured fluorescently after binding of MDA to thiobarbituric acid reactive substances (TBARS), which are formed as a byproduct of lipid peroxidation when reactive oxygen species react with lipids in plasma membranes. TBARS in plasma and tissue were determined using methods described in more detail in the Supplementary Material. 9
2.8. Statistical analysis
For the MRI data, a mixed‐effects regression model with a restricted maximum likelihood method with repeated measures over time was used to analyze the impact of treatment (BD or sham) on different metabolites or IPK parameters. Data were checked for a normal distribution of variances. If this assumption was not met, data transformations were applied or extreme values removed from the analysis (see below). The model included fixed effects of time, treatment, the interaction between time and treatment, and a random effect that took between‐rat variations into account (Stata/IC 12.1 for Mac, Stata‐Corp LP, College Station, TX). For the IPK data, a mixed‐effects analysis of variance was performed to check for significant interaction effects. If the interaction was not significant, we checked for main effects of group (BD or sham) or time. If significant differences were found, Bonferroni correction for 5 multiple comparisons was done to test for differences between individual groups (IBM SPSS Statistics 23, Armonk, NY). Results are presented as the estimated mean difference between groups (model coefficient, eµd), with the 95% confidence interval (CI) and the P value.
For the enzyme activity assay, ATP, MDA, and gene expression analyses, data were checked for a normal distribution. When variances of the dependent variable were equal across groups and there were no outliers, a pooled Student t test was done. If data were not normally distributed, Mann‐Whitney tests were done to compare between 2 groups individually (IBM SPSS Statistics 23). When multiple groups were compared, Bonferroni corrections for 3 multiple comparisons was done. Each separate animal is presented as a dot or square in each of the figures (with the exception of Figure S1). Extreme outliers were identified according to Tukey's hinges as xi ≥ Q3+2 × step or xi ≤ H1‐2 × step and removed from the analyses. Statistical tests were 2‐tailed and P < .05 was regarded as statistically significant. Results are presented as mean ± standard deviation.
3. RESULTS
3.1. BD parameters
Throughout the experiment, the MAP was measured continuously. BD was confirmed based on a rise in MAP following the drop in blood pressure that is characteristic for the slow‐induction BD model. The induction of BD showed a uniform MAP pattern in all animals that was in agreement with prior BD studies, 9 , 13 , 32 , 43 with a mean time to declare BD of 25 ± 3.1 minutes (Figure S2). To maintain a MAP higher than 80 mm Hg, the use of colloid HAES was required in 3 brain‐dead and 3 sham animals (0.38 ± 0.55 vs 0.44 ± 0.63 mL, P = .85), and the use of noradrenaline was required in 2 brain‐dead animals (0.50 ± 0.87 mL, P = .17).
3.2. Plasma injury markers
Several hepatic (AST, ALT, and LDH) and renal injury markers (urea and creatinine) were assessed to validate our model and to evaluate the effect of BD on tissue injury. In plasma of brain‐dead animals, AST after 4 hours (T = 4) and creatinine immediately after BD induction (T = 0) were significantly higher compared to sham animals (AST: 186 ± 87 vs 104 ± 76 U/L, P = .017 and creatinine: 43.3 ± 2.7 vs 28.4 ± 5.3, P < .001, Figure 3). Urine creatinine was significantly lower in brain‐dead animals at T = 4 compared to sham animals (4.0 ± 0.7 vs 8.8 ± 1.2 mmol/L, P < .001, Figure 3). Plasma urea was significantly lower in BD animals at T = 0 compared to sham animals (7.7 ± 0.6 vs 10.6 ± 0.9 mmol/L, P < .001, Figure 3). Finally, AST, ALT, LDH, and urea were significantly higher in brain‐dead animals at T = 4 (vs brain‐dead animals at T = 0, AST: 186 ± 86.9 vs 73.8 ± 76.3 U/L, P = .01; ALT: 144 ± 101 vs 43.8 ± 64.2 U/L, P < .01, LDH: 580 ± 514 vs 265 ± 216 U/L, P = .01; urea: 11.1 ± 1.8 vs 7.7 ± 0.9 mmol/L, P < .001, Figure 3), whereas urine creatinine was significantly lower than in brain‐dead animals at T = 0 (4.0 ± 0.7 vs 8.2 ± 1.2 mmol/L, P < .01, Figure 3).
FIGURE 3.
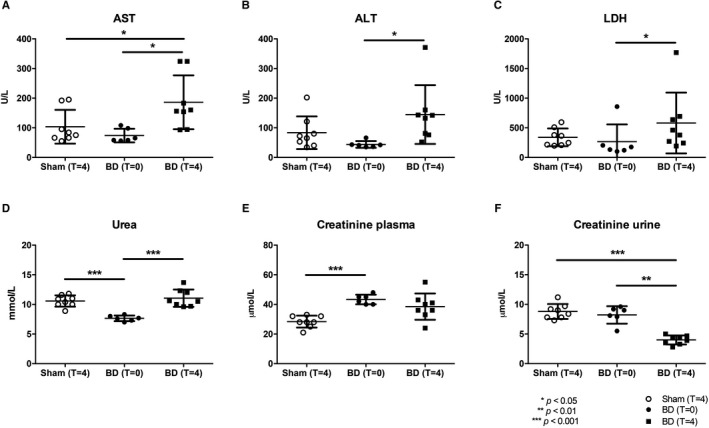
BD reduced hepatic and renal function. Plasma levels of (A) AST, (B) ALT, (C) LDH, (D) urea, and (E) plasma and (F) urine creatinine, determined after 4 h (at T = 4) of experimental time or immediately after BD induction (at T = 0). Results are presented as mean ± SD, n = 8 in the sham and BD (T = 4) group, and n = 6 in the BD (T = 0) group. ALT, alanine transaminase; AST, aspartate transaminase; BD, brain death; LDH, lactate dehydrogenase.
3.3. MRI assessment of 13C‐pyruvate metabolism
Enriched 13C‐pyruvate was injected repetitively to evaluate the metabolic profile at T = 0, T = 2, and T = 4 hours by assessing the relative conversion of pyruvate into lactate, alanine, and bicarbonate. The metabolic profiles of the liver and kidneys are portrayed in Figure 4. In order to determine the relative metabolic shift, each metabolite was normalized to the signal intensity of the sum of the products (ie, lactate, alanine, and bicarbonate). At T = 0, brain‐dead animals showed significantly higher lactate levels, and significantly lower alanine levels in both the liver and kidney compared to sham animals (liver lactate T = 0: eµd −0.21, 95% CI [−0.27, −0.15], P < .001; liver alanine T = 0: eµd0.14, 95% CI [0.07, 0.23], P < .001; kidney lactate T = 0: eµd−0.26, 95% CI [−0.40; −0.12], P < .001; kidney alanine T = 0: eµd0.24, 95% CI [0.06; 0.42], P < .001, Figure 4). In the kidney, alanine levels were significantly lower in brain‐dead compared to sham animals at T = 4 (eµd0.14 [0.03; 0.25], P = .02), and bicarbonate levels were significantly lower at T = 0 and T = 4 hours (T = 0: eµd2.36, 95% CI [1.62; 3.10], P < .001, T = 4: eµd0.62, 95% CI [0.05; 1.19], P = .04, Figure 4). Furthermore, the interaction term showed a significantly different effect of time on lactate and alanine levels in the liver (P < .001 and P < .01, respectively, Figure 4). In the kidney, the interaction term was significantly different for bicarbonate, indicating this metabolite differs between groups (BD vs sham) over the course of time.
FIGURE 4.
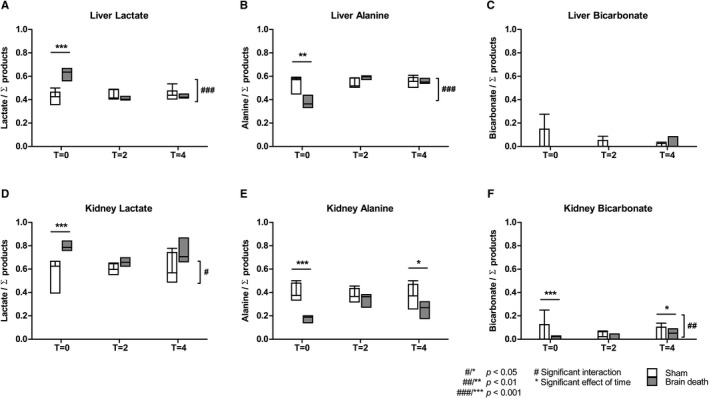
Pyruvate metabolites in the liver and kidneys of sham and BD animals. The signal of each metabolite—A,D, lactate; B,E, alanine; or C,F, bicarbonate—is represented as the fraction of the sum of the metabolites (lactate + alanine +bicarbonate). Results are presented as the minimum, first quartile, median, third quartile, for each group at each of the different time points, where T = 0 represents the start of the BD period, and T = 2 and T = 4 2 and 4 hours later, n = 8 per group. BD, brain‐dead
3.4. Kidney function and glucose oxidation in isolated perfused kidney model
Renal function was assessed in an isolated perfused kidney model following BD by assessing the flow, ultrafiltrate production, intrarenal vascular resistance (IRR), 44 and GFR (creatinine clearance). We found a significant interaction of flow and IRR, indicating a significantly different effect of time on flow and IRR in brain‐dead versus sham animals (flow: eµd 0.93, 95% CI [−1.14, 3.00], P < .01; IRR: eµd2.02, 95% CI [−0.48, 4.53], P < .001, Figure 5). In brain‐dead animals, the IRR was significantly lower at T = 0 compared to sham animals (IRR: 14.7 ± 7.0 vs 35.0 ± 21.0 mm Hg/(mL/min), P < .001, Figure 5). Finally, glucose oxidation was significantly different between the 2 groups, as well as over the course of time, between brain‐dead and sham animals (eµd 0.012, 95% CI [0.004, 0.03], main effect group P < .01, main effect time: P < .001, Figure 5), highlighting the significantly lower glucose oxidation levels during the reperfusion period in brain‐dead animals.
FIGURE 5.
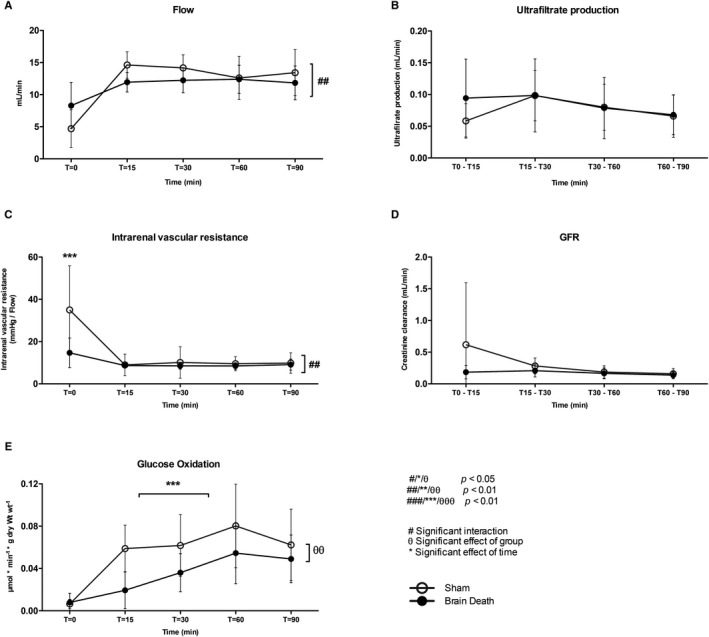
Reduced glucose oxidation following brain death in the isolated perfused kidney. A, Perfusate flow. B, Ultrafiltrate production. C, Intrarenal vascular resistance. D, GFR (creatinine clearance). E, Glucose oxidation rates in the kidney of brain‐dead and sham animals. Results are presented as mean ± SD, n = 6 per group
3.5. Enzyme activities and gene expression
We evaluated the different enzymes involved in pyruvate metabolism to assess whether the observed changes could be explained by changes in enzyme activities of LDH, PDH, or ALT (Figure 6) or gene expression of LDHA and lactate transporters monocarboxylate transport (MCT)1, responsible for transferring lactate into cells and MCT4, which transfers lactate out of glycolytic cells (Figure 6). At T = 4 hours, a significant reduction in LDH enzyme activity was observed in the kidney of brain‐dead animals (sham vs BD [T = 0], 0.50 ± 0.15 vs 0.61 ± 0.27 units/mg, P = .04, Figure 6). The activities of the remaining enzymes were not significantly different between the groups. The renal gene expression of MCT4, which transfers lactate out of glycolytic cells, was increased in brain‐dead animals (sham vs BD: 0.73 ± 0.16 vs 1.2 ± 0.36, P < .01). No differences in gene expression of LDHA, MCT1, or MCT4 in the liver were found between groups (Figure 7).
FIGURE 6.
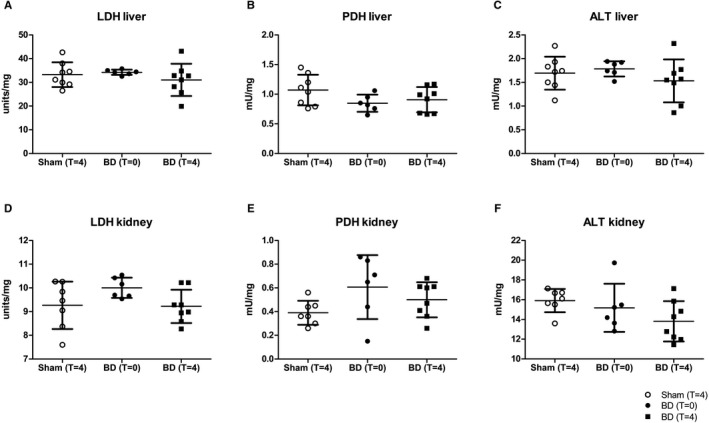
No difference in LDH, PDH, and ALT enzyme activities following brain death. Enzyme activities of A,B, LDH; C,D, PDH; and E,F, alanine transaminase (ALT) in the liver and kidney of sham and BD animals, determined after 4 h (in the case of sham and BD animals at T = 4) of experimental time or immediately after BD induction (BD animals at T = 0). Results are presented as mean ± SD, n = 8 in the sham and BD (T = 4) group, and n = 6 in the BD (T = 0) group. ALT, alanine transaminase; BD, brain‐dead; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase
FIGURE 7.
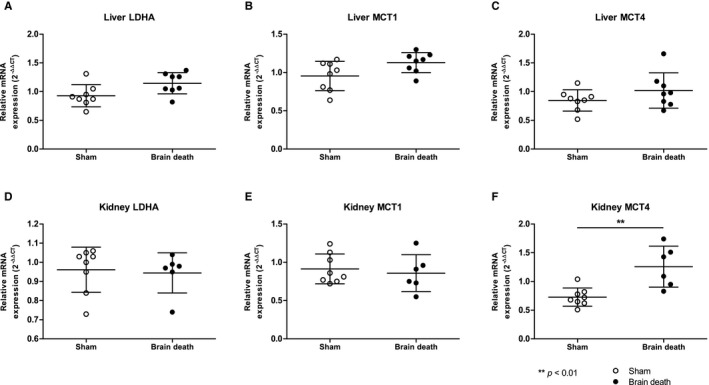
No difference in LDHA, MCT1, and hepatic MCT4 gene expression following BD, yet increased MCT4 expression in the kidney. Gene expression of A,D LDHA; B,E MCT1; and C,F MCT4 in the liver and kidney of sham and brain‐dead animals, determined after 4 h (in the case of sham and brain‐dead animals at T = 4) of experimental time or immediately after BD induction (brain‐dead animals at T = 0). Results are presented as mean ± SD, n = 8. BD, brain death; LDHA, lactate dehydrogenase‐A; MCT1, monocarboxylate transport 1
3.6. Decreased ATP levels in the liver following BD
To estimate cellular energy status in the liver and kidney, cellular ATP content was measured. In the liver of brain‐dead animals, ATP levels were reduced at T = 0 and T = 4 compared to sham animals (T = 0:29.0 ± 11.5 vs 47.9 ± 7.5 µmol/g protein, P = .01; T = 4:30.8 ± 15.4 µmol/g protein, P = .04, Figure 8). ATP levels in the kidney were not different between groups (sham: 26.3 ± 10.8 µmol/g protein, T = 0:26.9 ± 11.3 µmol/g protein, T = 4:21.1 ± 8.9 µmol/g protein, Figure 8). These results suggest a decreased bio‐energetic efficiency in the liver following BD.
FIGURE 8.
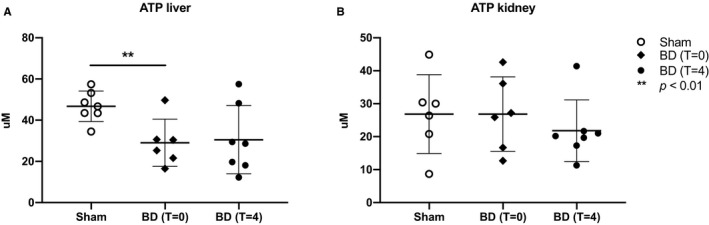
Decreased ATP levels in the liver but not the kidneys following BD. A, ATP content in liver and B, kidney tissue determined after 4 h (in the case of sham and brain‐dead animals at T = 4) of experimental time or immediately after BD induction (brain‐dead animals at T = 0). Results are presented as mean ± SD, n = 7 in the sham and BD (T = 4) group, and n = 6 in the BD (T = 0) group. ATP, adenosine triphosphate; BD, brain death
3.7. Oxidative stress markers in tissue and plasma
Oxidative stress marker MDA was significantly increased in the plasma of brain‐dead animals after 4 hours, when compared to sham animals and brain‐dead animals at T = 0 (sham: 2.8 ± 0.41 mmol/L, BD T = 0:2.5 ± 0.33 mmol/L, BD T = 4:3.3 ± 0.38 mmol/L, P = .04 and P < .01, respectively, Figure 9). MDA levels and gene expression of injury marker KIM‐1, an early biomarker for BD‐induced injury in the kidney, of brain‐dead animals were not significantly different from sham animals (MDA ‐ sham: 0.14 ± 0.03 µmol/g protein, T = 0:0.20 ± 0.10 µmol/g protein, T = 4:0.16 ± 0.04 µmol/g protein; for KIM‐1 in the sham group: 0.14 ± 0.03 µmol/g protein, T = 0:0.20 ± 0.10 µmol/g protein, T = 4:0.16 ± 0.04 µmol/g protein, Figure 9).
FIGURE 9.
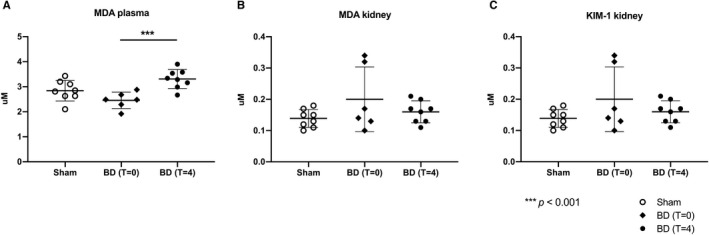
Increased oxidative stress in plasma but not the kidney following BD. (MDA) in (A) plasma, (B) the kidney determined after 4 h (in the case of sham and brain‐dead animals at T = 4) of experimental time or immediately after BD induction (brain‐dead animals at T = 0), and (C) refers to the gene expression of KIM‐1. Results are presented as mean ± SD, n = 8 in the sham and BD (T = 4) group, and n = 6 in the BD (T = 0) group. BD, brain death; MDA, malondialdehyde
4. DISCUSSION
The primary finding in this study was that the liver and kidney have a distinctly different metabolic profile during BD, with a preference for the production of alanine in the liver, yet lactate in the kidney. Secondly, lactate production increased in both organs immediately following BD induction, likely reflecting the hypoxic changes as a result of the hemodynamic instability before BD. Finally, metabolic assessment during machine perfusion of the kidneys showed a distinct reduction in glucose oxidation of brain‐dead compared to sham animals. Together, this suggests that hyperpolarization MRI and glucose oxidation measurements are promising methods to visualize metabolic profiles during BD and subsequent renal reperfusion and to highlight differences between brain‐dead and sham animals.
After BD induction, lactate increased and alanine decreased in the liver and kidneys. As BD induction is characterized by reduced perfusion of the abdominal organs, 45 this likely reflects this hemodynamic instability and hypoxic changes. Similarly, lactate production increased during liver surgery 42 and cardiac and renal ischemia‐reperfusion injury. 25 , 35 , 46 In each of these models, this likely results from a shift from aerobic to anaerobic glycolysis. 35 , 47 , 48 Increased lactate signals are potentially clinically relevant, as they have been correlated with a higher risk of early allograft dysfunction following liver 49 and delayed graft function following kidney transplantation. 30 Despite these studies, there is currently insufficient evidence that lactate or any other specific biomarker has a strong prognostic value in predicting transplantation outcomes. 30 , 50 With this in mind, it was interesting to observe that during machine perfusion, the level of glucose oxidation, and not renal creatinine clearance, was significantly altered in kidneys from brain‐dead animals. The majority of exogenous glucose, traced by d‐[6‐3H]glucose under aerobic conditions, undergoes oxidation, so quantification of 3H2O release is a reasonable marker of this final step in glucose metabolism in the tricarboxylic acid cycle. 51 , 52 However, it should be noted that 3H2O from d‐[6‐3H]glucose may be released during glycolysis because a proportion (6%‐14%) of the 3H may be lost from the C6‐position by pyruvate kinase activity. 53 Hence, a decreased 3H2O release may also reflect a diminished activity of pyruvate kinase in kidneys from BD donors. Nevertheless, liberation of 3H2O remains a valuable tool to detect differences in metabolic profiles between BD and sham kidneys.
Using 13C pyruvate in the MRI scan, we were able to visualize a distinctly different metabolic profile in the liver compared to the kidney. In sham animals, we observed a higher pyruvate‐to‐alanine conversion in the liver, yet increased pyruvate‐to‐lactate conversion rates in the kidneys. Interestingly, BD only enhanced this difference in both organs, with a trend towards increased lactate production in the kidneys (Figure 4). This was accompanied by an increase in gene expression of MCT4, which transfers lactate out of glycolytic cells. These changes could reflect a suppression of gluconeogenesis, increased lactate pools, and altered redox state as a result of BD‐induced hypoxia. 54 , 55 , 56 This is supported by prior studies on the effects of BD in the kidney, showing that gluconeogenesis‐related gene expression was decreased, while anaerobic, glycolytic‐gene expression was increased. 9 Furthermore, as BD progressed, oxidative stress levels increased, while renal perfusion declined. 9 , 12 , 57 Therefore, the altered metabolic profile in the kidney during BD likely reflects increased anaerobic production of lactate and lower gluconeogenesis. Conversely, our results indicate that the liver maintained a steady pyruvate‐to‐alanine conversion as well as unaltered enzyme activities. As increased alanine and ALT activity has been associated with adequate cellular oxygen availability, 58 these changes suggest that the liver maintains aerobic glycolysis. This idea is further supported by a prior study that showed increased expression of glycolysis‐related gene Pfk‐1 as well as higher oxygen consumption in the liver during BD. 9
Markers of hepatic and renal injury, oxidative stress, and tissue energy status were used to validate the BD model. Our results confirm that BD resulted in increased hepatic and renal injury, in line with prior animal and human BD studies. 7 , 9 , 12 , 13 , 39 Furthermore, levels of oxidative stress increased as evidenced by higher plasma MDA levels. Finally, the cellular energy status was decreased in the liver, immediately after BD as well as 4 hours later. Even though ATP and MDA levels were not significantly changed in the kidney of brain‐dead animals, our data show a trend of decreased energy levels and increased oxidative stress, similar to a prior study from our group. 9 Altogether, this confirms that the adjustments made to our model to allow for the administration of labeled pyruvate during BD did not impact the efficacy of our model.
This study has several limitations. As the main purpose of this study was to evaluate the potential of 2 metabolic assessment techniques, this study was not powered to statistically support some of the trends we observed in our data. Furthermore, we were unable to evaluate whether these metabolic changes can be correlated to organ quality or can be predictive of transplantation outcome. Finally, we observed no significant alterations in LDH, ALT, or PDH, indicating that changes in enzyme activities were probably not responsible for the changes in the metabolic profiles. Therefore, additional analyses on organ perfusion, oxygenation, and several transporter proteins will need to be conducted in a future study to assess whether the observed trends were affected by organ perfusion or limited by the uptake of any of the compounds.
In conclusion, this study shows that metabolic processes during BD in the liver and kidney can be visualized noninvasively using hyperpolarized MRI and also afterwards during ex vivo renal machine perfusion with assessment of glucose oxidation. Furthermore, these techniques can showcase the differences between metabolism in brain‐dead compared to sham animals. We therefore believe this study lays the basis for future studies aimed at investigating and treating BD‐induced metabolic changes and may be part of the key to improving transplantation outcomes.
5. DISCLOSURE
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
6. DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supporting information
Supplementary Material
van Erp AC, Qi H, Jespersen NR, et al. Organ‐specific metabolic profiles of the liver and kidney during brain death and afterwards during normothermic machine perfusion of the kidney. Am J Transplant. 2020;20:2425–2436. 10.1111/ajt.15885
[Correction added on 8 July 2020, after first online publication: The author's affiliation has been updated.]
REFERENCES
- 1. Organ Procurement and Transplantation Network . https://optn.transplant.hrsa.gov/. Accessed July 6, 2017.
- 2. Eurotransplant . https://www.eurotransplant.org/cms/. Accessed February 20, 2018.
- 3. Rudge C, Matesanz R, Delmonico FL, Chapman J. International practices of organ donation. Br J Anaesth. 2012;108(Suppl 1):i48‐i55. [DOI] [PubMed] [Google Scholar]
- 4. Mascia L, Bosma K, Pasero D, et al. Ventilatory and hemodynamic management of potential organ donors: an observational survey. Crit Care Med. 2006;34(2):321‐327. [DOI] [PubMed] [Google Scholar]
- 5. Fitzgerald RD, Hieber C, Schweitzer E, et al. Intraoperative catecholamine release in brain‐dead organ donors is not suppressed by administration of fentanyl. Eur J Anaesthesiol. 2003;20:952‐956. [DOI] [PubMed] [Google Scholar]
- 6. Adrie C, Monchi M, Fulgencio J‐P, et al. Immune status and apoptosis activation during brain death. Shock. 2010;33:353‐362. [DOI] [PubMed] [Google Scholar]
- 7. Weiss S, Kotsch K, Francuski M, et al. Brain death activates donor organs and is associated with a worse I/R injury after liver transplantation. Am J Transplant. 2007;7:1584‐1593. [DOI] [PubMed] [Google Scholar]
- 8. Floerchinger B, Oberhuber R, Tullius SG. Effects of brain death on organ quality and transplant outcome. Transplant Rev (Orlando). 2012;26:54‐59. [DOI] [PubMed] [Google Scholar]
- 9. Van Erp AC, Rebolledo RA, Hoeksma D, et al. Organ‐specific responses during brain death: increased aerobic metabolism in the liver and anaerobic metabolism with decreased perfusion in the kidneys. Sci Rep. 2018;8:4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roelsgaard K, Bøtker HE, Stødkilde‐Jørgensen H, et al. Effects of brain death and glucose infusion on hepatic glycogen and blood hormones in the pig. Hepatology. 1996;24:871‐875. [DOI] [PubMed] [Google Scholar]
- 11. Novitzky D, Cooper DKC, Rosendale JD, Kauffman HM. Hormonal therapy of the brain‐dead organ donor: experimental and clinical studies. Transplantation. 2006;82:1396‐1401. [DOI] [PubMed] [Google Scholar]
- 12. Hoeksma D, Rebolledo RA, Hottenrott M, et al. Inadequate anti‐oxidative responses in kidneys of brain‐dead rats. Transplantation. 2017;101(4):746‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rebolledo RA, Van Erp AC, Ottens PJ, et al. Anti‐apoptotic effects of 3,3',5‐triiodo‐L‐thyronine in the liver of brain‐dead rats. PLoS ONE. 2015;10:e0138749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nijboer WN, Schuurs TA, van der Hoeven J, et al. Effects of brain death on stress and inflammatory response in the human donor kidney. Transpl Proc. 2005;37:367‐369. [DOI] [PubMed] [Google Scholar]
- 15. Westendorp WH, Leuvenink HG, Ploeg RJ. Brain death induced renal injury. Curr Opin Organ Transplant. 2011;16:151‐156. [DOI] [PubMed] [Google Scholar]
- 16. Bretan PN, Baldwin NI, Novick AC, et al. Pretransplant assessment of renal viability by phosphorus‐31 magnetic resonance spectroscopy. Clinical experience in 40 recipient patients. Transplantation. 1989;48:48‐53. [DOI] [PubMed] [Google Scholar]
- 17. Hené RJ, van der Grond J, Boer WH, Mali WP, Koomans HA. Pre‐transplantation assessment of renal viability with 31P magnetic resonance spectroscopy. Kidney Int. 1994;46:1694‐1699. [DOI] [PubMed] [Google Scholar]
- 18. Wijermars LGM, Schaapherder AF, de Vries DK, et al. Defective postreperfusion metabolic recovery directly associates with incident delayed graft function. Kidney Int. 2016;90:181‐191. [DOI] [PubMed] [Google Scholar]
- 19. Nakatani T, Spolter L, Kobayashi K. Arterial ketone body ratio as a parameter of hepatic mitochondrial redox state during and after hemorrhagic shock. World J Surg. 1995;19:592‐596. [DOI] [PubMed] [Google Scholar]
- 20. Yamaoka Y, Taki Y, Gubernatis G, et al. Evaluation of the liver graft before procurement. Significance of arterial ketone body ratio in brain‐dead patients. Transpl Int. 1990;3:78‐81. [PubMed] [Google Scholar]
- 21. Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717‐1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231‐241. [DOI] [PubMed] [Google Scholar]
- 23. Van Erp AC, Hoeksma D, Rebolledo RA, et al. The crosstalk between ROS and autophagy in the field of transplantation medicine. Oxid Med Cell Longev. 2017;2017:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Golman K, in 't Zandt R, Thaning M. Real‐time metabolic imaging. Proc Natl Acad Sci USA. 2006;103(30):11270‐11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schroeder MA, Clarke K, Neubauer S, Tyler DJ. Hyperpolarized magnetic resonance: a novel technique for the in vivo assessment of cardiovascular disease. Circulation. 2011;124:1580‐1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. von Morze C, Chang GY, Larson PE, et al. Detection of localized changes in the metabolism of hyperpolarized gluconeogenic precursors 13C‐lactate and 13C‐pyruvate in kidney and liver. Magn Reson Med. 2016;77:1429‐1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hansen ESS, Stewart NJ, Wild JM, Jørgensen HS, Laustsen C. Hyperpolarized 13C,15N2‐urea MRI for assessment of the urea gradient in the porcine kidney. Magn Reson Med. 2016;76:1895‐1899. [DOI] [PubMed] [Google Scholar]
- 28. Mariager CØ, Nielsen PM, Qi H, Ringgaard S, Laustsen C. Hyperpolarized 13C, 15N2‐urea T2 relaxation changes in acute kidney injury. Magn Reson Med. 2017;5:198ra108. [DOI] [PubMed] [Google Scholar]
- 29. Kurhanewicz J, Vigneron DB, Ardenkjaer‐Larsen JH, et al. Hyperpolarized 13C MRI: path to clinical translation in oncology. Neoplasia. 2019;21:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bhangoo RS, Hall IE, Reese PP, Parikh CR. Deceased‐donor kidney perfusate and urine biomarkers for kidney allograft outcomes: a systematic review. Nephrol Dial Transplant. 2012;27:3305‐3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moers C, Varnav OC, van Heurn E, et al. The value of machine perfusion perfusate biomarkers for predicting kidney transplant outcome. Transplantation. 2010;90:966‐973. [DOI] [PubMed] [Google Scholar]
- 32. Kolkert JLP, t Hart NA, van Dijik A, et al. The gradual onset brain death model: a relevant model to study organ donation and its consequences on the outcome after transplantation. Lab Anim. 2007;41:363‐371. [DOI] [PubMed] [Google Scholar]
- 33. Qi H, Nørlinger TS, Nielsen PM, et al. Early diabetic kidney maintains the corticomedullary urea and sodium gradient. Physiol Rep. 2016;4:e12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bertelsen LB, Nielsen PM, Qi H, et al. Diabetes induced renal urea transport alterations assessed with 3D hyperpolarized 13C, 15N‐urea. Magn Reson Med. 2017;77:1650‐1655. [DOI] [PubMed] [Google Scholar]
- 35. Nielsen PM, Laustsen C, Bertelsen LB, et al. In situ lactate dehydrogenase activity: a novel renal cortical imaging biomarker of tubular injury? Am J Physiol Renal Physiol. 2017;312:F465‐F473. [DOI] [PubMed] [Google Scholar]
- 36. Mahboub P, Ottens P, Seelen M, et al. Gradual rewarming with gradual increase in pressure during machine perfusion after cold static preservation reduces kidney ischemia reperfusion injury. PLoS ONE. 2015;10:e0143859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hjortbak MV, Hjort J, Povlsen JA, et al. Influence of diabetes mellitus duration on the efficacy of ischemic preconditioning in a Zucker diabetic fatty rat model. PLoS ONE. 2018;13:e0192981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Støttrup NB, Løfgren BO, Birkler RD, et al. Inhibition of the malate–aspartate shuttle by pre‐ischaemic aminooxyacetate loading of the heart induces cardioprotection. Cardiovasc Research. 2010;88:257‐266. [DOI] [PubMed] [Google Scholar]
- 39. Lactate Dehydrogenase Activity Assay Kit . Technical bulletin. https://www.sigmaaldrich.com/content/dam/sigma‐aldrich/docs/Sigma/Bulletin/1/mak066bul.pdf. Accessed October 22, 2019.
- 40. Pyruvate Dehydrogenase Activity Assay Kit . Technical bulletin. https://www.sigmaaldrich.com/content/dam/sigma‐aldrich/docs/Sigma/Bulletin/1/mak183bul.pdf. Accessed October 22, 2019.
- 41. Alanine Aminotransferase Activity Assay Kit . Technical bulletin. https://www.sigmaaldrich.com/content/dam/sigma‐aldrich/docs/Sigma/Bulletin/1/mak052bul.pdf. Accessed October 22, 2019.
- 42. Clark MG, Bloxham DP, Holland PC, et al. Estimation of the fructose 1,6‐diphosphatase‐phosphofructokinase substrate cycle and its relationship to gluconeogenesis in rat liver in vivo. J Biol Chem. 1974;249(1):279‐290. [PubMed] [Google Scholar]
- 43. Rebolledo RA, Hoeksma D, Hottenrott CMV, et al. Slow induction of brain death leads to decreased renal function and increased hepatic apoptosis in rats. J Transl Med. 2016;14:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chudek J, Kolonko A, Król R, et al. The intrarenal vascular resistance parameters measured by duplex Doppler ultrasound shortly after kidney transplantation in patients with immediate, slow, and delayed graft function. Transpl Proc. 2006;38:42‐45. [DOI] [PubMed] [Google Scholar]
- 45. Herijgers P, Leunens V, Tjandra‐Maga TB, Mubagwa K, Flameng W. Changes in organ perfusion after brain death in the rat and its relation to circulating catecholamines. Transplantation. 1996;62:330‐335. [DOI] [PubMed] [Google Scholar]
- 46. Ball DR, Cruickshank R, Carr CA, et al. Metabolic imaging of acute and chronic infarction in the perfused rat heart using hyperpolarised [1‐13C]pyruvate. NMR Biomed. 2013;26:1441‐1450. [DOI] [PubMed] [Google Scholar]
- 47. Theodoraki K, Arkadopoulos N, Fragulidis G, et al. Transhepatic lactate gradient in relation to liver ischemia/reperfusion injury during major hepatectomies. Liver Transpl. 2006;12:1825‐1831. [DOI] [PubMed] [Google Scholar]
- 48. Hems DA, Brosnan JT. Effects of ischaemia on content of metabolites in rat liver and kidney in vivo. Biochem. J. 1970;120:105‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim DG, Lee JY, Jung YB, et al. Clinical significance of lactate clearance for the development of early allograft dysfunction and short‐term prognosis in deceased donor liver transplantation. Clin Transplant. 2017;31:e13136. [DOI] [PubMed] [Google Scholar]
- 50. Moers C, Smits JM, Maathuis MH, et al. Machine perfusion or cold storage in deceased‐donor kidney transplantation. N Engl J Med. 2009;360:7‐19. [DOI] [PubMed] [Google Scholar]
- 51. Butler P, Bell P, Rizza R. Choice and use of tracers. Horm Metab Res Suppl. 1990;24:20‐25. [PubMed] [Google Scholar]
- 52. Bolukoglu H, Goodwin GW, Guthrie PH, et al. Metabolic fate of glucose in reversible low‐flow ischemia of the isolated working rat heart. Am J Physiol. 1996;270(3 Pt 2):H817‐H826. [DOI] [PubMed] [Google Scholar]
- 53. Ben‐Yoseph O, Kingsley PB, Ross BD, et al. Metabolic loss of deuterium from isotopically labeled glucose. Magn Reson Med. 1994;32(3):405‐409. [DOI] [PubMed] [Google Scholar]
- 54. Qi H, Nielsen PM, Schroeder M, et al. Acute renal metabolic effect of metformin assessed with hyperpolarised MRI in rats. Diabetologia. 2018;61:445‐454. [DOI] [PubMed] [Google Scholar]
- 55. Lewis AJ, Miller JJ, McCallum C, et al. Assessment of metformin‐induced changes in cardiac and hepatic redox state using hyperpolarized [1‐13C]pyruvate. Diabetes. 2016;65:3544‐3551. [DOI] [PubMed] [Google Scholar]
- 56. Gerich JE, Meyer C, Woerle HJ, Stumvoll M. Renal gluconeogenesis: its importance in human glucose homeostasis. Diabetes Care. 2001;24:382‐391. [DOI] [PubMed] [Google Scholar]
- 57. Akhtar MZ, Huang H, Kaisar M, et al. Using an integrated ‐omics approach to identify key cellular processes that are disturbed in the kidney after brain death. Am J Transplant. 2016;16:1421‐1440. [DOI] [PubMed] [Google Scholar]
- 58. Laustsen C, Lipsø K, Østergaard JA, et al. Insufficient insulin administration to diabetic rats increases substrate utilization and maintains lactate production in the kidney. Physiol Rep. 2014;2:e12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.