

Abstract
The essential amino acid lysine is of great importance in the nutrition and pharmaceutical industries and is mainly produced in biorefineries by the fermentation of glucose. In biorefineries, downstream processing is often the most energy‐consuming step. Adsorption on hydrophobic adsorbents represents an energy, resource, and cost‐saving alternative. The results reported herein provide insights into the selective separation of l‐lysine from aqueous solution by liquid‐phase adsorption using tailored activated carbons. A variety of commercial activated carbons with different textural properties and surface functionalities is investigated. Comprehensive adsorbent characterization establishes structure–adsorption relationships that define the major roles of the specific surface area and oxygen functionalities. A 13‐fold increase of the separation of lysine and glucose is achieved through systematic modification of a selected activated carbon by oxidation, and lysine adsorption is enhanced by 30 %.
Keywords: adsorption, amino acids, carbon, oxidation, surface analysis
Made‐to‐measure: A variety of tailored activated carbons with different textural properties and surface functionalities is investigated to provide insights into the selective separation of l‐lysine and glucose from aqueous solution by liquid‐phase adsorption. A 13‐fold increase of the separation of lysine and glucose is achieved through systematic modification of a selected activated carbon and lysine adsorption is enhanced by 30 %.
Introduction
Biorefineries will be a fundamental part of the future sustainable bioeconomy because of the depletion of fossil resources and global climate change. Therefore, it is crucial to explore alternative, renewable sources of materials and develop innovative and efficient processes. A wide range of chemical building blocks is already produced from renewable feedstocks, and many recent studies established pathways to broaden the portfolio of application for bio‐based materials such as amino acids, aromatics, organic acids, and other monomers.1 Amino acids, l‐lysine in particular, play an essential role in the nutrition and pharmaceutical industries.2 Its demand has been growing at 5–10 % per year with a production of 2.4 million tons in 2015 and an amino acid market share of over 25 %.3 On an industrial scale, l‐lysine is mainly produced by microbial fermentation from sugar sources such as glucose, molasses, and sucrose using Corynebacterium glutamicum followed by suitable downstream processing that involves classical separation methods.1a, 2, 4 The selective separation and purification of l‐lysine from aqueous fermentation broths are key steps to reduce the investment and production costs, which generally account for 20–50 % of the total costs of biorefineries.5 Traditionally, chromatographic methods based on ion exchange resins are used to extract l‐lysine from fermentation solutions and thereafter concentrated by evaporation and spray drying.6
A major disadvantage of these methods is the resulting saline eluents, which leads to an additional cost‐intensive processing step, high amounts of wastewater, and high energy costs.7 In light of the climate crisis, the development of environmentally friendly, sustainable, and efficient separation processes are necessary. Adsorption on hydrophobic adsorbents is an energy‐, resource‐, and cost‐saving alternative for the recovery of components from aqueous media.8 Several studies have used zeolites for the selective liquid‐phase adsorption of amino acids.9 Faisal et al. used zeolite‐Y as an adsorbent for the recovery of l‐arginine from model solutions and real fermentation broths.9a Moreover, MOFs were used by Jonckheere et al. to separate amino acids such as l‐phenylalanine, l‐tryptophan, and l‐tyrosine by liquid‐phase adsorption.10 Most research on the adsorption of l‐lysine from aqueous solutions has been performed using adsorbents such as zeolites, minerals, silica, or lignin beads that result in low adsorption capacities of approximately 60 mg g−1.11 There have, however, only been a few studies in which activated carbons were used as adsorbents for amino acids, particularly for lysine.12 Activated carbons are widely used adsorbents because of their large porous volumes that result in high specific surface areas and their wide availability based on renewable carbonaceous organic materials.13 In addition, the physical and chemical properties of activated carbons can be tailored to enhance the affinity towards desired compounds and can be utilized for specific applications.14 The selective separation of lysine from glucose by activated carbons and the structure–adsorption relationship has, to date and to the best of our knowledge, not yet been discussed in literature. However, a fundamental understanding of adsorbent properties that influence adsorption capacity and selectivity is a prerequisite for targeted material design.
In this study, we investigate the selective separation of lysine and glucose by using tailored activated carbons as adsorbents. The influence of crucial parameters such as the pH value is investigated. A variety of commercial activated carbons are characterized by using N2 physisorption, water vapor sorption, Boehm titration, and temperature‐programmed desorption (TPD) MS to understand the influence of the textural properties and surface chemistry on the adsorption properties of lysine. The selective separation of lysine and glucose is evaluated as a crucial parameter for potential application. We evaluate a systematic modification of the activated carbon for a potential enhancement of the adsorption capacity and separation factor and confirm the stability of the modified form. Hereafter, lysine refers to the biologically active enantiomer l‐lysine.
Results and Discussion
Characterization of activated carbons
The N2 adsorption–desorption isotherms (see the Supporting Information; Figure S1 a) display, for most of the commercial activated carbon materials, a type I isotherm behavior with a fast uptake at a low relative pressure followed by a horizontal adsorption typical of microporous materials.15 For some samples, that is, PA4N and KB‐G, type IV and II isotherms are obtained because of their meso‐ and macroporosity.15 A narrow pore size distribution in the range of micropores (<20 Å) could be observed for the carbons (Figure S1 b). The results of N2 physisorption are summarized in Table 1.
Table 1.
Overview of textural properties of commercial activated carbons obtained by using N2 physisorption and the amounts of released CO and CO2 measured by using TPD‐MS.
|
Activated carbon |
S total [a] [m2 g−1] |
V total [cm3 g−1] |
V micro [b] [cm3 g−1] |
COdesorbed [c] [μmol g−1] |
CO2,desorbed [c] [μmol g−1] |
|---|---|---|---|---|---|
|
CW20 |
2023 |
1.83 |
0.49 |
2052 |
63 |
|
KB‐G |
1355 |
1.14 |
0.31 |
2157 |
144 |
|
PA4N |
1491 |
1.47 |
0.38 |
2134 |
73 |
|
CP6/400 |
1254 |
0.64 |
0.51 |
964 |
106 |
|
A Supra EUR |
1923 |
0.97 |
0.62 |
528 |
32 |
|
Blü 100562 |
2075 |
1.17 |
0.66 |
225 |
13 |
[a] BET method. [b] t‐plot method. [c] Amounts of CO and CO2 released in TPD‐MS experiments.
Information about the surface chemistry of the active carbon samples was obtained by using TPD‐MS. In this process, the relative thermal stability of surface functional groups is used to determine the concentration of these functionalities on the carbon surface. The O‐containing functional groups can be divided into two groups that decompose into CO2 and CO at different temperatures during the TPD experiment. The decomposition of carboxyl, anhydride, and lactone groups results mainly in CO2 formation, whereas the decomposition of phenol, ether, and carbonyl groups produces CO.16 The main oxygen surface groups on the activated carbons and their expected temperature ranges for CO and CO2 release in the TPD measurements are shown in Figure 1 a. A comprehensive assignment of CO and CO2 desorption profiles to some oxygen functionalities for an example carbon (Silcarbon CW20) is given in Figure 1 b. In the CO profile of the carbon, a large peak appears at elevated temperatures of 600–700 °C, mainly associated with phenol and ether groups. The measurements also show a tail that extends to 1000 °C related to the decomposition of carbonyl and quinone groups. CO desorbed at lower temperatures (400 °C) can be assigned to anhydride groups. The CO2 desorption profile emphasizes maximum CO2 release at 250 and 650 °C, which corresponds to carboxylic and lactone groups.
Figure 1.
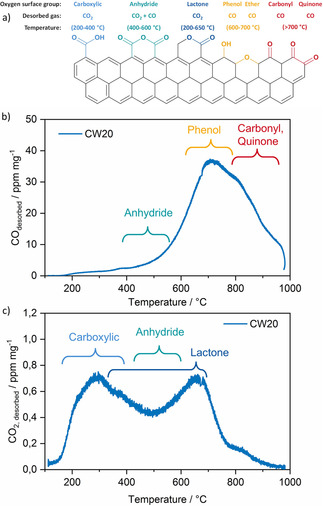
a) Oxygen surface groups on activated carbon and their temperature range of decomposition during TPD‐MS analysis. b, c) Rates of CO (b) and CO2 (c) desorption during TPD‐MS analysis of the activated carbon Silcarbon CW20 and the proposed corresponding functional groups.
CO2 evolution at temperatures of 400–600 °C is suggested to relate to anhydride groups. CO and CO2 desorption profiles of the different activated carbons measured by using TPD‐MS are provided in Figure S2. The cumulative amounts of CO and CO2 released for each activated carbon during the TPD experiments are shown in Table 1. The activated carbons 100562, A Supra EUR, and CP6/400 release the lowest amounts of CO and CO2, which indicates that they have a very low amount of O‐containing functional groups. Their main CO signal at approximately 700–900 °C is typical for quinone and carbonyl functional groups. Activated carbons PA4N, KB‐G, and CW20 possess significantly higher amounts of oxygen surface group functionalities. CO is mainly released at 500 °C, typical of anhydride groups, and 600–800 °C, which can be assigned to high amounts of phenol and ether functionalities. With regard to the CO2‐evolution profiles of these activated carbons, significant contributions occur at 180–300 °C assigned to carboxylic groups, 350–600 °C caused mainly by lactone groups, and 500–700 °C that originate from peroxide and carboxylic anhydride groups.16b, 16c Although the different functional groups can be assigned by the decomposition temperatures, interpretation of TPD spectra from the literature is somehow ambiguous because of the overlap of desorbed products.17
Therefore, and to complement these TPD‐MS results, Boehm titration was performed to determine the functional groups present on the surface of the commercial activated carbons.18
The results of Boehm titration are presented in Figure 2 and Table S1. Indeed, the results of Boehm titration agree with those obtained by TPD‐MS. Activated carbons that possess higher amounts of O‐containing surface groups also hold the highest content of acidic groups such as carboxylic, lactonic, and phenolic groups. The data indicate that all carbons have a large contribution of surface groups and differ significantly in their chemical nature. Notably, Boehm titration only considers the amount of acidic and not basic groups, whereas in TPD‐MS analysis both types of functional groups are measured. TPD‐MS results for CW20 and KB‐G showed almost the same amount of CO desorbed during the measurement but significantly differ in the results obtained by using Boehm titration. An explanation could relate to the much higher amount of basic functional groups such as ketone, pyrene, or chromene for CW20.
Figure 2.
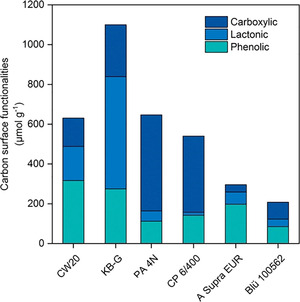
Activated carbon surface functionalities obtained by Boehm titration.
Water vapor sorption enables insights with regard to the surface polarity that result from the specific surface functionality and morphology of materials. The water vapor adsorption isotherms of the activated carbons are depicted in Figure S3. In the case of water vapor adsorption, type III and V isotherms are obtained for the studied activated carbons. This kind of adsorption is characteristic for weak interactions between a polar adsorbate and hydrophobic surfaces, especially at a low relative pressure (p/p 0<0.2), in which a very low uptake of water occurs.19 Furthermore, each adsorption isotherm shows an elevated water uptake at p/p 0=0.4–0.8 associated with the steady growth of water clusters. Differences between the respective isotherms are related to the different amounts of surface oxygen functional groups. Activated carbons with the highest amounts of CO and CO2 released during TPD‐MS, that is, CW20, KB‐G, and PA4N, also possess the highest water vapor uptake at a low relative pressure. This indicates more hydrophilic surface properties compared to the very hydrophobic materials 100562 and A Supra EUR. These results are in accordance with the results obtained by using TPD‐MS and Boehm titration.
Influence of pH on lysine adsorption
Lysine is a basic amino acid that can be found in four different dissociation states that depends on the pH value of the solution (Figure 3 a). Dissolved lysine in water has a pH value of 10, which makes it predominantly zwitterionic. pH values higher than 10 result in a change of the dissociation state towards the anionic form. For pH values of 1–8, lysine is mainly present in the cationic and dicationic form.20 As a result of the dissociation behavior of lysine, it shows various adsorption behaviors as the polarity of the adsorptive has a great impact on the adsorption performance.11c Furthermore, the pH value needs to be taken into consideration as fermentative processes for lysine production may differ in pH, and typical lysine production with Corynebacterium glutamicum is performed at pH 6–8.2 The influence of the pH value of the solution on the adsorption of lysine on activated carbon is shown in Figure 3 b. The adsorption capacity of lysine on activated carbon reached a maximum at pH 10, which enabled an adsorption of 249.4 mg g−1. At this pH value, lysine is primarily in the zwitterionic state, which is the most stable form in water. In this state, lysine also has its lowest solubility in water, which makes it less polar compared to the other dissociation states.20, 21 As AC is inherently hydrophobic, the dissociation state with the lowest polarity is the most favored as, in general, the adsorption of a nonpolar solute will be higher on adsorbents that are also nonpolar.22 The cationic and dicationic forms (pH 1–8) showed significantly lower adsorption capacities of 18–42 mg g−1. For higher pH values above 10, the anionic state becomes the major form and the adsorption capacity decreases significantly. If salts are added into the adsorption mixture, the polarity of the lysine solution changes, which affects the adsorption behavior. The protonated/deprotonated dissociation states are more polar compared to the zwitterionic state, which widens the gap in polarity between the adsorbent and adsorbate. Thus, the adsorption performance correlates directly with the properties of the molecule; the polarity increases with protonation/deprotonation to result in lower adsorption capacities. In addition, the surface state of the activated carbon is pH dependent. Functional groups of the activated carbon such as carboxyl, ketone, or phenol can be deprotonated in solutions with basic pH values, which results in stronger electrostatic interactions with the positively charged amino groups of lysine compared to an uncharged surface state at a lower pH value.23 As the best result was achieved at a solution pH of 10, the following experiments were conducted under these conditions.
Figure 3.

a) Changes as a function of pH in the distribution of different dissociation states of dissolved lysine, determined from reported dissociation constants.11c, 20 b) Influence of the pH value of the solution on the adsorption of lysine on activated carbon (Silcarbon CW20). T=30 °C, c Lys=30 g L−1, t ads=24 h. The error bars correspond to the standard deviation determined from triplicate experiments.
Lysine adsorption on different activated carbons
A set of different commercial activated carbons was tested by using a single‐solute aqueous solution with a lysine concentration of 30 g L−1. An adsorption temperature of 30 °C was chosen because temperatures between 30 and 34 °C are typically used for the fermentation process.6 The textural properties, surface functionalities, and polarity of the activated carbons are presented in Table 1, Figure 2, and Figures S1–S3 and discussed in the previous section. The lysine adsorption capacities of the different activated carbons are presented in Figure 4 a. The adsorption capacities are 206–256 mg g−1, which is, to date and to the best of our knowledge, the highest ever reported for lysine adsorption on carbon material.12c The activated carbon A Supra EUR has the highest adsorption capacity followed by Silcarbon CW20 and CP6/400. A correlation of the specific surface area to the adsorption performance indicates that a high surface area indeed favors a high adsorption capacity. However, this trend is not linear, and the experimental data scatter, which leads to the conclusion that there are more factors to consider to try to understand the adsorption performance. A Supra EUR, the activated carbon with the lowest surface functionality, has the highest affinity towards the least polar form of lysine (zwitterionic state). This adsorption behavior can be explained by the nonspecific enthalpy‐driven hydrophobic effect and van der Waals (vdW) forces. Nevertheless, CW20, a carbon with high amount of oxygen functional groups, shows a very high adsorption towards lysine, too. A possible explanation for this might be that, besides vdW forces and hydrophobic interactions, an increased amount of electrostatic interactions and hydrogen bonding are present and/or lysine is adsorbed according to an ion‐exchange mechanism.12c In general, for the single‐solute lysine adsorption, no correlation can be made between adsorption capacity and functionalities.
Figure 4.

a) Adsorbed amounts of lysine using commercial activated carbons with the corresponding surface areas. T=30 °C, c Lys=30 g L−1, t ads=24 h, pH 10. The error bars correspond to the standard deviation determined from triplicate experiments. b) Adsorption isotherm of lysine from aqueous solution fitted to a Langmuir adsorption isotherm model.
To gain more information on the interaction between the adsorbent material and lysine, a single‐component adsorption isotherm of lysine was measured for A Supra EUR (Figure 4 b). The lysine adsorption data were fitted to the Langmuir adsorption isotherm model. This model assumes monolayer adsorption, in which adsorption only occurs at a fixed number of definite localized sites, which are identical and equivalent, with no lateral interaction and steric hindrance between the adsorbed molecules.24 This model fits the data very well with a correlation coefficient (R 2) of 0.986. A characteristic plateau indicates an equilibrium saturation point in which once a molecule occupies a site, no further adsorption takes place.
Competitive adsorption of lysine and glucose
It is important to gain knowledge on the selectivity of the adsorption process because lysine is produced on industrial scale by the fermentation of glucose and a mixture of different components is present.2 As a model solution, a mixture of lysine and d‐glucose with equivalent masses was used (10 g L−1). The molarity was kept lower than that for the single‐model solution to inhibit the undesired Maillard reaction.25 The competitive adsorption performance for commercial activated carbons is shown in Figure 5. Single‐component adsorption showed high adsorption capacities towards lysine relatively independent of the respective characteristics of the activated carbons. In multicomponent adsorption, the influence of the activated carbon surface properties becomes more pronounced to consider the selectivity of adsorption. The selectivity was determined by the separation factor α calculated as the ratio of lysine to glucose adsorbed divided by the ratio of lysine and glucose that remains in solution. The separation factor α is 0.9–5.6 for the different commercial activated carbons. The highest selectivity towards lysine was for the activated carbons CW20 and KB‐G with α=4.4 and 5.9, respectively. This indicates that the selectivity correlates with the amount of surface oxygen groups as higher amounts of oxygen groups translate to a higher selectivity for lysine (Figure 6).
Figure 5.
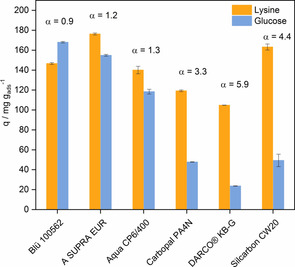
Adsorbed amounts of lysine and glucose in competitive adsorption experiments using commercial activated carbons. T=30 °C, c Lys/Glu=10 g L−1, t ads=4 h, pH 10. α=(q Lys/q Glu)/(c e,Lys/c e,Glu). Error bars correspond to the standard deviation determined from triplicate experiments.
Figure 6.
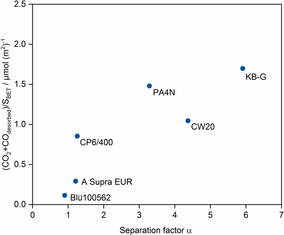
Separation factor as a function of cumulated amounts of CO2 and CO (normalized by specific surface area) released during TPD‐MS experiments for different activated carbons.
Although CW20 possesses slightly lower amounts of oxygen functional groups compared to, for example, KB‐G, the higher specific surface area coupled with high amount of oxygen results in a high separation factor. This shows that the interplay of both porosity and functionality presents the essential characteristics. The lowest selectivity towards lysine adsorption was found for the activated carbons Blü 100562, A Supra EUR, and Aqua CP6/400, which can be explained by their very low amounts of oxygen functional groups.
In single‐component adsorption, the dissociation state of lysine with the lowest polarity (at pH 10) was most favored for the adsorption on hydrophobic activated carbon. Surprisingly, competitive adsorption showed that a more hydrophilic carbon (caused by a higher content of oxygen surface groups) results in better selectivity towards lysine adsorption. We suggest that glucose (uncharged at pH 10) adsorption relies mainly on physical adsorption based on weaker vdW forces and hydrophobic interactions. As a result of the similar polarity of glucose and lysine, no separation based on physical adsorption can be made for low‐functionalized carbons. In contrast, an increasing content of oxygen functional groups enables enhanced electrostatic interactions and hydrogen bonding between the amine groups of lysine and oxygen surface groups of the activated carbon to result in a higher selectivity for lysine compared to glucose. To gain further insights into the origin of the high selectivity towards lysine in the lysine–glucose adsorption and to enhance the adsorption performance, an activated carbon was modified systematically to tailor the surface chemistry.
Modified activated carbon in lysine adsorption
The chemical structure of the activated carbon surface has a significant impact on the adsorption performance and especially on the selectivity as previous results revealed. Accordingly, the surface functionalities of the carbon were modified by using reduction and oxidation methods. For modification experiments, Silcarbon CW20 was chosen as a model carbon as it showed the most promising results in terms of selectivity and adsorption capacity. The textural properties of the modified carbons were characterized by using N2 physisorption (Figure S5); the polarity and surface functionalities were characterized by using water vapor sorption and TPD‐MS, respectively (Table S2). Comparison of the specific surface areas indicates that modification decreases the porosity. These results are in accordance with those of previous studies as harsh conditions can lead to a collapse of the pore walls and a decline in the specific surface area and total pore volume.26 The desorption of CO and CO2 from Silcarbon CW20 if it was heated from 120 to 1000 °C during TPD‐MS analysis is shown in Figure S5. Oxidative treatment with nitric acid led to a higher amount of CO and CO2 evolution from Silcarbon CW20. After oxidation, the increase of evolved CO2 was higher than that of CO, which indicates the generation of a higher proportion of acidic groups. Also, relevant peaks are observed between 175–400 °C in the CO2 evolution profile, which can be attributed to carboxylic acids and carboxylic anhydrides. The amount of evolved CO2 is also significantly higher above 600 °C compared to that of the untreated carbon. In addition, the amount of evolved CO at approximately 700 °C increased significantly. However, reduction under H2 results in exactly the opposite trend. The CO and CO2 desorption profiles decrease with an increase of the reduction temperature, and CW20 reduced at 500 °C showed the lowest amount of CO and CO2 evolved. This was expected as reduction removes most of the O‐containing functional groups from the surface.
Besides TPD‐MS, water vapor sorption was used to visualize the effect of the modification on the polarity of the activated carbon. The water vapor adsorption isotherms of untreated and modified Silcarbon CW20 are depicted in Figure S7. The shape of the isotherm describes the surface polarity of the activated carbon qualitatively. Indeed, the shape of the isotherms changed significantly upon the incorporation (through oxidation) and removal (through reduction) of surface oxygen functional groups. The O‐enriched activated carbon possessed a significantly higher water uptake at a low relative pressure (p/p 0=0.1–0.6). This observation is in good agreement with the TPD‐MS data. The reduction of the carbon, however, results in a lower water uptake at low relative pressures (p/p 0<0.7). A type III/V isotherm could be observed with a low water loading at a low relative pressure that increased at a higher relative pressure because of capillary condensation.19b These findings are in line with previous results, as fewer surface O‐containing functionalities relate to a more hydrophobic carbon compared to the as‐received Silcarbon CW20. It is expected that a higher amount of surface functional groups favors lysine over glucose adsorption. The adsorption and separation performance of the modified Silcarbon CW20 are shown in Figure 7. Through oxidation of the activated carbon, an outstanding 13‐fold increase in separation could be achieved, in which the lysine adsorption was enhanced by 30 %. Although the specific surface area declines by 40 %, surface functionalities seem to be key for the efficient separation of lysine from glucose (Figure S8). These results further support the idea of enhanced selectivity by electrostatic interactions and an increase of the amount of hydrogen bonding between the amine groups of lysine and oxygen surface groups of the activated carbon. Future studies will aim to gain a fundamental understanding of the governing surface interactions. Through oxidation, the CO content desorbed from the carbon could be doubled and the CO2 amount desorbed was eight times higher than that of the untreated carbon. Reduction of the activated carbon, in turn, led to lower separation factors and overall lower adsorption capacities, which confirms the crucial role of O‐containing surface groups.
Figure 7.
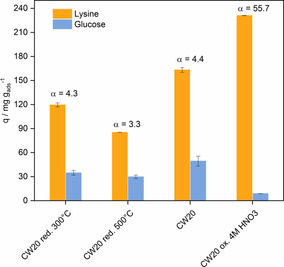
Competitive adsorption of lysine and glucose using modified Silcarbon CW20. T=30 °C, c Lys/Glu=10 g L−1, t ads=4 h, pH 10. Error bars correspond to the standard deviation determined from triplicate experiments.
To test the stability of the modified activated carbon and its recyclability, the oxidized carbon CW20 was tested in three consecutive adsorption runs. The recycling procedure is explained in detail in the Supporting Information. The results of the recycling test of the oxidized CW20 are shown in Figure 8. It became evident that after the first recycling, the adsorption capacity decreases from 211 to 191 mg g−1. This decline in the adsorbed amount of lysine could be related to a certain loss of surface functional groups. Overall, the recycling study proves the high stability and recyclability of the optimized activated carbon CW20.
Figure 8.
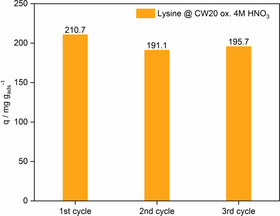
Adsorbed amounts of lysine by CW20 ox. 4 m HNO3 in three consecutive adsorption cycles. T=30 °C, c Lys=3 g L−1 pH 10.
Conclusions
A detailed insight into lysine adsorption and its separation from glucose on tailored activated carbon was performed. By testing a broad variety of different activated carbons, the necessity of a high specific surface area in combination with a large amount of oxygen surface functionalities was proven to achieve high lysine adsorption capacities of up to 256 mg g−1. The selectivity for lysine adsorption from an aqueous phase that contains both lysine and glucose depends strongly on the surface functionalities of the activated carbon rather than on the specific surface area. To gain further insights, an activated carbon was tailored by oxidation with nitric acid to result in a significantly enhanced lysine adsorption capacity by 30 % and a 13‐fold higher separation factor. Adsorption conditions such as pH had a significant impact on the lysine uptake. Recycling experiments were performed by testing the oxidized activated carbon CW20 in repetitive adsorption runs and showed only a minor decline in lysine adsorption capacity, which emphasizes the stability of the surface functionalization.
Experimental Section
Chemicals and materials
l‐Lysine (≥98 %) was supplied by abcr GmbH and d‐glucose (>99 %) by Merck. Activated carbon 100562 was provided by Blücher GmbH. Silcarbon CW20 was provided by Silcarbon Aktivkohle GmbH, and Aqua Air Adsorbens GmbH & Co.KG supplied the Aqua CP6/400. Activated carbons A Supra EUR and KB‐G were supplied by Cabot Corporation. Degassed Milli‐Q water from a Werner Reinstwasser system (>18.2 MΩ cm) was used in all the experiments.
Adsorption experiments
Before the adsorption experiments, the activated carbons were dried in a vacuum oven at 60 °C for 24 h. If not stated otherwise, 40 mg of adsorbent was added to 2 mL solution. The adsorption experiment was performed in a water shaking bath with a temperature control unit. Typically, the sample was kept in the shaking bath for 24 h at 30 °C. Afterwards, the loaded adsorbent was filtered, and the remaining solution was analyzed by using HPLC to determine the concentration. For each of the experiments, triplicate adsorption experiments were conducted to ensure accuracy. For the single adsorption, a solution of lysine (30 g L−1) in Milli‐Q water was used. For the competitive adsorption, a solution with equivalent masses of l‐lysine and d‐glucose (10 g L−1) was used. The amount adsorbed from the solutions on a specific mass of adsorbent in the equilibrium is represented by the adsorption capacity q and was calculated by using Equation (1).
| (1) |
In which c 0 is the initial liquid‐phase concentration of the adsorption solution, c eq is the concentration that remains at equilibrium, m sol. is the total mass of the solution, and m AC is the total mass of the activated carbon. For the pH‐dependent measurements, pH values from 1 to 12 were adjusted using 4 m NaOH and 6 m HCl.
Surface modification of activated carbon
Reduction of the activated carbon was performed in a tube oven with H2 at 300 and 500 °C, respectively. The sample was pretreated by heating to 100 °C for 1 h under N2 to remove impurities. Afterwards, the temperature was elevated at a heating rate of 10 °C min−1 under H2. After the temperature was held for 3 h, the sample was cooled overnight under N2 atmosphere. The activated carbon was oxidized by treating Silcarbon CW20 (1 g) with 4 m nitric acid (10 mL; Caution: corrosive acid and strong oxidizing agent). The mixture was heated to reflux at 150 °C for 20 min with stirring at 250 rpm. Nitrous gases produced by heating are neutralized by using a washing bottle that contained NaOH solution at the exit of the setup. The activated carbon was collected by filtration and washed with distilled water until pH neutral. Afterwards, the carbon was dried overnight at 80 °C.
Characterization methods
HPLC analysis
The aqueous phase of the adsorption experiments was analyzed by using a Shimadzu Prominence LC‐20 HPLC system. For the measurements of lysine and lysine/glucose mixtures, the system was equipped with a Primesep S 4,6×150 mm mixed zone column provided by SIELC Technologies and a refractive index detector. As the eluent, 80 mm ammonium formate in a water/acetonitrile mixture (60:40) was used. The temperature was kept at 50 °C, and the flow rate was 0.8 mL min−1.
N2 physisorption
A Quadrasorb SI was used to measure the N2 adsorption and desorption isotherms. Before the measurements, the samples were degassed at 150 °C under vacuum for at least 5 h by using a FloVac degasser. The temperature during the measurement was kept at −196 °C. The data were evaluated by using the software QuadraWin. The specific surface areas were determined by using the BET model in the range 0.05≤p/p 0≤0.2. For the analysis of the total pore volume, the highest relative pressure points p/p 0=0.95–0.98 were used. The micropore volumes were obtained by using the t‐plot model.
H2O vapor sorption
Before the measurement, the samples were treated in an analogous manner as the samples for the N2 physisorption measurements. The water vapor adsorption isotherms were measured by using an Autosorb® iQ, which works in a static and volumetric mode. Water vapor isotherms can be measured in the range of p/p 0=0.1–0.9. The isotherms were measured at 30 °C. The data were evaluated by using the software ASiQWin.
TPD‐MS
TPD‐MS experiments were performed by using a mass spectrometer (CIRRUS 2 device by MKS Instruments) equipped with a combined Faraday cage and electron multiplier detector. As an internal standard, 10 mL min−1 He was used. The experiments were conducted under an Ar stream of 90 mL min−1. The flow was regulated by using Labview, and the data were obtained from Process Eye™. Samples were pretreated at 120 °C for 3 h to ensure that all moisture and O2 was out of the system. The spectra were recorded by progressively heating the samples to 1000 °C at a heating rate of 20 °C min−1.
Boehm titration
The general titration procedure was performed by using the standard Boehm titration method.18, 27
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Noah Avraham and Jens Heller for HPLC analysis. This work was funded by the Bundesministerium für Bildung und Forschung (BMBF, Federal Ministry of Education and Research) within the Project “BioSorp” (FKZ 031B0678A). We gratefully thank Blücher GmbH, Silcarbon Aktivkohle GmbH, and Aqua Air Adsorbens GmbH & Co.KG for providing us the activated carbons. We also thank Jérôme Meyers, Moritz Litzel, Joel B. Mensah, F. Joschka Holzhäuser, Yannik Louven, Tobias Janke, and Jennifer Hanf for sparing their valuable time for discussion.
J. Deischter, N. Wolter, R. Palkovits, ChemSusChem 2020, 13, 3614.
References
- 1.
- 1a. Leuchtenberger W., Huthmacher K., Drauz K., Appl. Microbiol. Biotechnol. 2005, 69, 1–8; [DOI] [PubMed] [Google Scholar]
- 1b. Chen Z., Liu G., Zhang J., Bao J., Bioresour. Technol. 2019, 271, 196–201; [DOI] [PubMed] [Google Scholar]
- 1c. Meyers J., Mensah J. B., Holzhäuser F. J., Omari A., Blesken C. C., Tiso T., Palkovits S., Blank L. M., Pischinger S., Palkovits R., Energy Environ. Sci. 2019, 12, 2406–2411; [Google Scholar]
- 1d. Deischter J., Schute K., Neves D. S., Ebert B. E., Blank L. M., Palkovits R., Green Chem. 2019, 21, 1710–1717; [Google Scholar]
- 1e. Louven Y., Schute K., Palkovits R., ChemCatChem 2019, 11, 439–442. [Google Scholar]
- 2. Anastassiadis S., Recent Pat. Biotechnol. 2007, 1, 11–24. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Lee J.-H., Wendisch V. F., Bioresour. Technol. 2017, 245, 1575–1587; [DOI] [PubMed] [Google Scholar]
- 3b. Demain A. L., Ind. Biotechnol. 2007, 3, 269–283. [Google Scholar]
- 4. Tonouchi N., Ito H. in Amino Acid Fermentation (Eds.: A. Yokota, M. Ikeda), Springer Japan, Tokyo, 2017, pp. 3–14. [Google Scholar]
- 5.A. Aden, M. Ruth, K. Ibsen, J. Jechura, K. Neeves, J. Sheehan, B. Wallace, L. Montague, A. Slayton, J. Lukas, Lignocellulosic Biomass to Ethanol Process Design and Economics Utilizing Co-Current Dilute Acid Prehydrolysis and Enzymatic Hydrolysis for Corn Stover, NREL/TP-510-32438, National Renewable Energy Laboratory, Golden, CO, 2002, https://www.nrel.gov/docs/fy02osti/32438.pdf.
- 6. Hermann T., J. Biotechnol. 2003, 104, 155–172. [DOI] [PubMed] [Google Scholar]
- 7. Lee I., Lee K., Namgoong K., Lee Y.-S., Enzyme Microb. Technol. 2002, 30, 798–803. [Google Scholar]
- 8.
- 8a. Schute K., Detoni C., Kann A., Jung O., Palkovits R., Rose M., ACS Sustainable Chem. Eng. 2016, 4, 5921–5928; [Google Scholar]
- 8b. Detoni C., Gierlich C. H., Rose M., Palkovits R., ACS Sustainable Chem. Eng. 2014, 2, 2407–2415; [Google Scholar]
- 8c. Schute K., Louven Y., Detoni C., Rose M., Chem. Ing. Tech. 2016, 88, 355–362; [Google Scholar]
- 8d. Schroer G., Deischter J., Zensen T., Kraus J., Pöppler A.-C., Qi L., Scott S., Delidovich I., Green Chem. 2020, 22, 550–562; [Google Scholar]
- 8e. Rübenach L., Lins J., Koh E., Rose M., ChemSusChem 2019, 12, 3627–3634. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Faisal A., Holmlund M., Ginesy M., Holmgren A., Enman J., Hedlund J., Grahn M., ACS Sustainable Chem. Eng. 2019, 7, 8900–8907; [Google Scholar]
- 9b. Krohn J. E., Tsapatsis M., Langmuir 2005, 21, 8743–8750; [DOI] [PubMed] [Google Scholar]
- 9c. Munsch S., Hartmann M., Ernst S., Chem. Commun. 2001, 1978–1979. [DOI] [PubMed] [Google Scholar]
- 10. Jonckheere D., Steele J. A., Claes B., Bueken B., Claes L., Lagrain B., Roeffaers M. B. J., De Vos D. E., ACS Appl. Mater. Interfaces 2017, 9, 30064–30073. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Chen G., Liu M., BioResearch 2011, 7, 298–314; [Google Scholar]
- 11b. Yang Y., Wang S., Liu J., Xu Y., Zhou X., Langmuir 2016, 32, 4746–4754; [DOI] [PubMed] [Google Scholar]
- 11c. Kitadai N., Yokoyama T., Nakashima S., J. Colloid Interface Sci. 2009, 329, 31–37; [DOI] [PubMed] [Google Scholar]
- 11d. Kitadai N., Yokoyama T., Nakashima S., J. Colloid Interface Sci. 2009, 338, 395–401; [DOI] [PubMed] [Google Scholar]
- 11e. O'Connor A. J., Hokura A., Kisler J. M., Shimazu S., Stevens G. W., Komatsu Y., Sep. Purif. Technol. 2006, 48, 197–201. [Google Scholar]
- 12.
- 12a. Ikuo A., Katsumi H., Mutsuo K., Bull. Chem. Soc. Jpn. 1982, 55, 687–689; [Google Scholar]
- 12b. Vinu A., Hossain K. Z., Satish Kumar G., Ariga K., Carbon 2006, 44, 530–536; [Google Scholar]
- 12c. Yang J., Han S., Desalin. Water Treat. 2018, 120, 261–271. [Google Scholar]
- 13. Separation and Purification Technologies in Biorefineries (Eds.: S. Ramaswamy, H.-J. Huang, B. V. Ramarao), John Wiley & Sons, Hoboken, 2013. [Google Scholar]
- 14.
- 14a. Yin C. Y., Aroua M. K., Daud W. M. A. W., Sep. Purif. Technol. 2007, 52, 403–415; [Google Scholar]
- 14b. Chingombe P., Saha B., Wakeman R. J., Carbon 2005, 43, 3132–3143. [Google Scholar]
- 15. Cychosz K. A., Guillet-Nicolas R., García-Martínez J., Thommes M., Chem. Soc. Rev. 2017, 46, 389–414. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Figueiredo J. L., Pereira M. F. R., Freitas M. M. A., Órfão J. J. M., Carbon 1999, 37, 1379–1389; [Google Scholar]
- 16b. Shafeeyan M. S., Daud W. M. A. W., Houshmand A., Shamiri A., J. Anal. Appl. Pyrolysis 2010, 89, 143–151; [Google Scholar]
- 16c. Thommes M., Morlay C., Ahmad R., Joly J. P. J. A., Adsorption 2011, 17, 653; [Google Scholar]
- 16d. Shen W., Li Z., Liu Y., Recent Pat. Chem. Eng. 2008, 1, 27–40. [Google Scholar]
- 17. Zhou J.-H., Sui Z.-J., Zhu J., Li P., Chen D., Dai Y.-C., Yuan W.-K., Carbon 2007, 45, 785–796. [Google Scholar]
- 18. Boehm H. P. in Advances in Catalysis, Vol. 16 (Eds.: D. D. Eley, H. Pines, P. B. Weisz), Academic Press, Oxford, 1966, pp. 179–274. [Google Scholar]
- 19.
- 19a. Cossarutto L., Zimny T., Kaczmarczyk J., Siemieniewska T., Bimer J., Weber J. V., Carbon 2001, 39, 2339–2346; [Google Scholar]
- 19b. Mahle J. J., Carbon 2002, 40, 2753–2759. [Google Scholar]
- 20. Gambino G. L., Lombardo G. M., Grassi A., Marletta G., J. Phys. Chem. B 2004, 108, 2600–2607. [Google Scholar]
- 21. Needham T. E., Paruta A. N., Gerraughty R. J., J. Pharm. Sci. 1971, 60, 565–567. [DOI] [PubMed] [Google Scholar]
- 22. Bansal R. C., Goyal M., Activated Carbon Adsorption, CRC Press, Boca Raton, 2005. [Google Scholar]
- 23. Villacañas F., Pereira M. F. R., Órfão J. J. M., Figueiredo J. L., J. Colloid Interface Sci. 2006, 293, 128–136. [DOI] [PubMed] [Google Scholar]
- 24. Langmuir I., J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar]
- 25.
- 25a. Hill V. M., Ledward D. A., Ames J. M., J. Agric. Food Chem. 1996, 44, 594–598; [Google Scholar]
- 25b. van Boekel M. A. J. S., Biotechnol. Adv. 2006, 24, 230–233. [DOI] [PubMed] [Google Scholar]
- 26. Pereira M. F. R., Soares S. F., Órfão J. J. M., Figueiredo J. L., Carbon 2003, 41, 811–821. [Google Scholar]
- 27. Goertzen S. L., Thériault K. D., Oickle A. M., Tarasuk A. C., Andreas H. A., Carbon 2010, 48, 1252–1261. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary