

Abstract
Objective
Depressive disorders are common among about 50% of the patients with drug‐resistant temporal lobe epilepsy (TLE). The underlying etiology remains elusive, but hypothalamus‐pituitary‐adrenal (HPA) axis activation due to changes in glucocorticoid receptor (GR) protein expression could play an important role. Therefore, we set out to investigate expression of the GR in the hippocampus, an important brain region for HPA axis feedback, of patients with drug‐resistant TLE, with and without comorbid depression.
Methods
GR expression was studied using immunohistochemistry on hippocampal sections from well‐characterized TLE patients with depression (TLE + D, n = 14) and without depression (TLE − D, n = 12) who underwent surgery for drug‐resistant epilepsy, as well as on hippocampal sections from autopsy control cases (n = 9). Video–electroencephalography (EEG), magnetic resonance imaging (MRI), and psychiatric and memory assessments were performed prior to surgery.
Results
Abundant GR immunoreactivity was present in dentate gyrus granule cells and CA1 pyramidal cells of controls. In contrast, neuronal GR expression was lower in patients with TLE, particularly in the TLE + D group. Quantitative analysis showed a smaller GR+ area in TLE + D as compared to TLE − D patients and controls. Furthermore, the ratio between the number of GR+/NeuN+ cells was lower in patients with TLE + D as compared to TLE − D and correlated negatively with the depression severity based on psychiatric history. The expression of the GR was also lower in glial cells of TLE + D compared to TLE − D patients and correlated negatively to the severity of depression.
Significance
Reduced hippocampal GR expression may be involved in the etiology of depression in patients with TLE and could constitute a biological marker of depression in these patients.
Keywords: chronic stress, dysthymia, hypothalamus‐pituitary‐adrenal axis, major depression
Key Points.
Depression is the most common psychiatric comorbidity in patients with drug‐resistant temporal lobe epilepsy (TLE).
Lower expression of the glucocorticoid receptor (GR) was found in neurons and glia within the hippocampus of patients with TLE, particularly with comorbid depression.
GR expression correlated negatively with the severity of depression score, which was based on psychiatric history.
Lower GR expression in depressed patients with epilepsy is consistent with possible alterations in hypothalamus‐pituitary‐adrenal (HPA) activity in these patients.
GR expression may be involved in the pathogenesis of comorbid depression in TLE and could constitute a potential biological marker of depression in patients with TLE.
1. INTRODUCTION
Depression disorders are among the most common psychiatric comorbid conditions in patients with drug‐resistant temporal lobe epilepsy (TLE). Its prevalence ranges between 30% and 35%, reaching the highest prevalence (50%) at specialized epilepsy centers. 1 , 2 , 3 Furthermore, a history of depression is frequently found in patients with epilepsy, 4 and a positive correlation between the development of seizures and depressive‐like symptoms has been demonstrated in various animal models of epilepsy, suggesting a bidirectional relationship between depression and epilepsy. 3 , 5 , 6 Comorbid depression is further associated with a poor quality of life, increased suicidal risk, higher medical costs, and an increased risk of developing drug‐resistant epilepsy. 3 , 7
The underlying pathogenic mechanisms remains unknown, but alterations in hippocampal neuroplasticity due to an increased activity of the hypothalamus‐pituitary‐adrenal (HPA) axis is frequently observed in major depression and likely constitutes a common pathway that may also be disturbed in the combination of TLE and depression. Hence, disturbances in HPA axis activity have been implicated as a possible pathogenic mechanism underlying the association between both pathologies. 5 , 8 , 9
In clinical practice, epilepsy surgery is performed on patients with severe, drug‐resistant TLE. The most common histopathological alteration found in these patients is hippocampal sclerosis. 10 , 11 The hippocampus is further particularly sensitive to glucocorticoids (GCs), important steroid hormones released from the adrenal gland after stress. Both, mineralocorticoid receptors (MRs) and glucocorticoid receptors (GRs) are highly expressed in various subregions of the hippocampus, 12 , 13 and their activation following GC binding exerts negative feedback inhibition of HPA axis activity. 14 , 15 Hippocampal GRs further regulate neuronal excitability, 16 , 17 , 18 and particularly in the dentate gyrus, they are involved in neuroplasticity and neurogenesis. 14
It has been reported that high glucocorticoids levels induced by stress can affect epilepsy and may increase seizures. 15 , 16 For example, corticosterone hypersecretion has been found after status epilepticus in rodents. 17 In addition, corticosterone administration and experimental stressors enhance neuronal excitability in the hippocampus, 16 , 18 changes that may be reversed with GR and MR antagonists. 19 Furthermore, patients with TLE who are exposed to a psychosocial stress challenge have higher levels of cortisol, 20 whereas stressful events and particularly early life stressors increase the risk for seizures. 19 Furthermore, a persistent HPA axis hyperactivity has been observed after seizures in patients with epilepsy, suggesting an impairment of the inhibitory control of the HPA system. 9
Several groups have investigated the expression of GRs in animal models with depression and/or chronic stress, 12 , 21 , 22 , 23 , 24 , 25 and a few studies have been done on GRs in postmortem hippocampal tissues from patients with depression. 13 , 25 , 26 , 27 , 28 , 29 So far, only a few addressed hippocampal GR expression in experimental models of epilepsy 29 and/or (resected) brain tissue from patients with epilepsy. 31 , 32 To the best of our knowledge, the GR has not been studied in the hippocampus of patients with epilepsy and depression; we therefore set out to study for the first time hippocampal expression of GRs in a well‐characterized cohort of patients with drug‐resistant TLE, with and without comorbid depression, as well as in control subjects.
2. METHODS
2.1. Study design and patient selection
Hippocampal samples obtained from patients who underwent surgery for drug‐resistant TLE according to the criteria of Kwan et al (2010) 33 were selected within the period from 2006 to 2016. All patients underwent surgery at the epilepsy center of the Ramos Mejía Hospital and/or El Cruce Hospital, Buenos Aires, Argentina. The immunohistochemical procedures were performed at the department of neuropathology of the Amsterdam UMC, The Netherlands.
We included samples from patients who had completed the routine psychiatric assessment protocol before surgery and who had signed the approved informed consent for participation. The psychiatric assessment protocol started at the epilepsy center in 2001 as part of a clinical research project and is now considered a routine measure in all patients before epilepsy surgery. 34 All patients were receiving their habitual medication at the moment of surgery and no one had received glucocorticoids anywhere before surgery.
Samples included in this study were grouped according to the following criteria for depression. Depression was considered positive when patients had experienced at least one current or past interictal episode of major depression and/or other depressive disorder, according to the Axis I of the fourth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM) IV classification using Structured Clinical Interview for DSM Disorders (SCID) I (dysthymia, major depression with or without psychotic symptoms and/or recurrent depression, bipolar disorder). 35 , 36 Patients with primary chronic interictal psychiatric disorders according to other sections in Axis I of DSM IV (ie, chronic psychosis, current posttraumatic stress disorder, and severe anxiety disorder) were excluded, as were patients with mental retardation (IQ < 70 and/ or attendance at a special school). The study was approved by the ethics committee of the Ramos Mejía and El Cruce Hospitals, in accordance with the Ethical Standards laid down in the 1964 Declaration of Helsinki, and full informed consent procedures for participation.
2.2. Diagnosis of drug‐resistant TLE, video‐EEG evaluation, and magnetic resonance imaging
(See Appendix S1.)
2.3. Neuropsychological assessment
All patients underwent a neuropsychological assessment before surgery, which was undertaken by trained specialists. Verbal memory was determined using the Rey Auditory Verbal Learning Test (RAVLT), Spanish Version, 37 consisting of reading a list of words in five different trials and then recovering the immediate memory, differed memory, and recognition in each trial. For visual memory the Rey‐Osterrieth Complex Figure Test (RCFT) was used. This nonverbal test consists of a visual design that is presented to patients who have to copy and then reproduce immediately after the visual presentation (immediate recall) and after 30 minutes (delayed recall). Because there are no regional normative data for these tests for the Argentinian population, international data were used to compare our results. 38 To measure the cognitive status, z‐scores were obtained by comparing each individual result of each test with the normal data corrected by age and sex. 38 According to the high correlation observed between the type of memory and the hippocampal sclerosis laterality, 39 we considered the z‐score for visuospatial memory in patients with a right focus, and the z‐score for verbal memory in patients with a left focus.
2.4. Psychiatric assessment
All patients included in this study also underwent a complete psychiatric assessment prior to surgery. Psychiatric assessment was performed by trained psychiatrists according to a standardized protocol especially designed for patients with drug‐resistant epilepsy. 6 , 34 Psychiatric history was obtained from each patient and relatives, complemented by information from families. The psychiatric semiology of the witnessed examination was supplemented with the Structured Clinical Interview (SCID) Spanish version for DSM IV Axis I diagnoses, and with SCID I and SCID II for personality disorders. 35 Diagnosis of depression was based on DSM IV classification and SCID results. In addition, all patients were assessed according to the Global Assessment of Functioning (GAF) of the DSM IV and to the Beck Depression scale. The GAF is a 100‐point tool that rates overall psychological, social, and occupational functioning in relation to psychiatric symptoms and is included in the DSM IV in the section on multiaxial assessments (Axis V of DSM IV). 36 The interviews were carried out in approximately 2 to 3 hours. To determine depression severity, the Beck Depression Inventory II (BDI II), Spanish version, was also administered to quantify depression symptoms at the moment of psychiatric assessment. The BDI II was added to the protocol in 2010. 40 Depression severity was also determined using an ad hoc composite score based on psychiatric history, using factors 1‐8 for the diagnosis of depression according to the SCID I criteria of DSM‐IV. One point was added for each positive factor: 1, the presence of one episode of an affective disorder codified in Axis I of DSM‐IV; 2, comorbid psychiatric disorders in Axis I or II, present or past (one point for each comorbid disorder); 3, suicide attempts; 4, psychiatric hospitalization; 5, antidepressant treatment (one point was given for patients who had received antidepressants); 6, GAF ≤60; 7, psychotic symptoms associated to depression; and 8, experienced more than one episode of an affective disorder (ie, major depression and dysthymia or recurrent major depression).
2.5. Neuropathological diagnosis and immunohistochemistry
Resected hippocampi were fixed in 10% buffered formalin for >1 week and embedded in paraffin. Coronal hippocampal sections at the anterior‐medial region of hippocampal body were sectioned at 5 µm, mounted on pre‐coated glass slides (Star Frost, Waldemar Knittel, Braunschweig, Germany) and processed. Trained neuropathologists made the neuropathological diagnosis. Archival material of postmortem control hippocampus (postmortem delay was maximum 8 hours) was simultaneously processed. Samples were selected matched by gender and were otherwise free from known neurological injury, drug, and/or alcohol abuse and suicidal events.
Immunohistochemistry was performed to study GR protein expression to protocols described before. 13 , 28 Furthermore, to assess cell type–specific effects, double‐labeling was performed using markers for astrocytes as well as for microglia and for NeuN. Finally, quantitative analysis was performed. (For details see supplementary methods.)
2.6. Statistical analysis
Descriptive statistics was performed, and the chi‐square test was used to analyze qualitative variables. The normal distribution of data was determined using the Shapiro‐Wilk test. The Student's t test, the one‐way analysis of variance (ANOVA), and Pearson correlations were applied when a normal distribution was found and non‐parametric tests (Mann Whitney) and Spearman correlations were applied when the data were not normally distributed (Shapiro‐Wilk test < 0.05). IBM SPSS Statistics 22 was used to perform statistical analysis. A P‐value < 0.05 was assumed to indicate a significant difference.
3. RESULTS
3.1. Clinical data
Hippocampal samples from 12 patients with TLE and depression (TLE + D; age = 31±12 years; 4 men and 8 women), 14 patients with TLE without depression (TLE − D; age = 34± 8 years; 8 men and 6 women), and 9 postmortem controls (age = 52.9 ± 20.6; 6 men and 3 women, cause of death: heart failure = 5, aortic dissection n = 2, pulmonary embolism n = 1, pneumonia n = 1), were included. Postmortem samples were selected matched by gender and free from neurological injury, drug and/or alcohol dependency, and suicidal evidences.
The epilepsy duration, the age at epilepsy onset (both P > .05, Student's t test), and seizure frequency (P > .05, Mann‐Whitney) did not differ between TLE − D and TLE + D. Clinical and neuropathological data of TLE − D and TLE + D cases are summarized in Table 1.
Table 1.
Clinical characteristics of patients
| Case number | Age of epilepsy onset (years) | No seizures per month | Age at surgery (years) | Time of epilepsy duration (years) | Medical history | Depression | Antidepressants | Antiepileptic drugs | Psychiatric Institutionalization | Suicide attempts | GAF score | Beck Depression Inventory | Severity of depression | ILAE HS type | Verbal Memory | z‐score | Visual memory | z‐score | Laterality of epileptic foci and resection | z score ipsilateral to epileptic focus |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 18 | 4 | 24 | 6 | 1 | No | No | DPH, TPM | No | No | 70 | n/a | 0 | 1 | 7 | ‐1.71 | 11.5 | ‐3.89 | R | ‐3.89 |
| 2 | 2 | 10 | 25 | 23 | 5 | MD + P | FLX | CBZ, VPA, CL | No | No | 55 | n/a | 6 | 1 | 10 | ‐0.40 | 8 | ‐4.58 | R | ‐4.58 |
| 3 | 8 | 10 | 22 | 14 | 0 | MD + P | CIT | CBZ, LMT | Yes | Yes | 70 | n/a | 7 | 1 | 8 | ‐1.14 | 9 | ‐4.30 | L | ‐1.14 |
| 4 | 7 | 4 | 45 | 38 | 1 | No | No | CBZ, LMT | No | No | 70 | n/a | 0 | 1 | 4 | ‐1.64 | 17 | ‐0.54 | R | ‐0.54 |
| 5 | 11 | 4 | 28 | 17 | 3 | No | No | CBZ, LMT | No | No | 80 | n/a | 0 | 2 | 10 | ‐0.46 | 21 | ‐1.20 | R | ‐1.20 |
| 6 | 7 | 4 | 21 | 14 | 0 | No | No | CBZ, TPM | No | No | 70 | n/a | 0 | 1 | 8 | ‐1.15 | 22 | ‐0.97 | R | ‐0.97 |
| 7 | 12 | 2 | 22 | 10 | 3 | No | No | DPH, LMT, CL | No | No | 60 | 11 | 1 | 1 | 11 | ‐0.04 | 16 | ‐2.48 | R | ‐2.48 |
| 8 | 1 | 3 | 40 | 39 | 0 | No | No | VPA, CL | No | No | 70 | 6 | 0 | 1 | 9 | ‐0.12 | 14 | ‐1.14 | L | ‐0.12 |
| 9 | 13 | 9 | 34 | 21 | 3 | No | No | VPA, TPM | No | No | 75 | 0 | 0 | 1 | 11 | ‐0.07 | 15 | ‐1.93 | R | ‐1.93 |
| 10 | 11 | 4 | 29 | 18 | 0 | No | No | CBZ, CL | No | No | 70 | 6 | 0 | 1 | 4 | ‐2.96 | 18 | ‐2.01 | L | ‐2.96 |
| 11 | 3 | 10 | 29 | 26 | 4 | MD + P | PARX | TPM, VPA, CL | Yes | Yes | 55 | 25 | 8 | 1 | 4 | ‐2.96 | 10 | 3.50 | L | ‐2.96 |
| 12 | 25 | 4 | 37 | 12 | 3 | No | No | CBZ, TPM, CL | No | No | 85 | 8 | 0 | 2 | 4 | ‐1.76 | 18.5 | ‐0.69 | L | ‐1.76 |
| 13 | 5 | 6 | 35 | 30 | 2 | No | No | LMT, VPA | No | No | 70 | 1 | 0 | 1 | 7 | ‐0.94 | 5 | ‐4.50 | R | ‐4.50 |
| 14 | 3 | 2 | 41 | 38 | 0 | No | No | CBZ, LVT | No | No | 85 | 6 | 0 | 1 | 4 | ‐1.64 | 18 | ‐0.34 | R | ‐0.34 |
| 15 | 10 | 6 | 24 | 14 | 0 | MD | SER | LMT, CBZ | No | No | 55 | 21 | 5 | 1 | 3 | ‐3.00 | 8 | ‐4.00 | L | ‐3.00 |
| 16 | 18 | 2 | 34 | 16 | 2 | No | No | LVT, VPA | No | No | 60 | 8 | 1 | 1 | 8 | ‐1.88 | 7.5 | ‐3.57 | R | ‐3.57 |
| 17 | 6 | 3 | 17 | 11 | 0 | MD | No | OXC, LVT, CL | No | No | 70 | 29 | 3 | 1 | 8 | ‐1.94 | 14 | ‐2.37 | R | ‐2.37 |
| 18 | 21 | 2 | 38 | 17 | 3 | D | No | LVT, LCS | No | No | 65 | 5 | 2 | 1 | 6 | ‐2.00 | 22 | ‐0.40 | L | ‐2.00 |
| 19 | 17 | 3 | 53 | 36 | 1 | MD | FLX | LMT, LVT, CL | No | No | 55 | 24 | 4 | 1 | 10 | 0.00 | 19 | ‐0.36 | L | 0.00 |
| 20 | 8 | 4 | 31 | 23 | 3 | MD | 1 | LVT, LCS | No | No | 65 | 18 | 3 | 1 | 13 | 0.64 | 27 | 0.69 | L | 0.64 |
| 21 | 5 | 3 | 18 | 13 | 0 | D | No | OXC, CL | No | No | 65 | n/a | 2 | NHS | 11 | ‐0.32 | 14 | ‐2.36 | L | ‐0.32 |
| 22 | 8 | 10 | 25 | 17 | 0 | MD | No | TPM, CBZ, DPH | No | No | 55 | 20 | 4 | 1 | 0 | ‐4.63 | 13 | ‐3.29 | L | ‐4.63 |
| 23 | 20 | 8 | 50 | 30 | 0 | MD | SER | CBZ | No | No | 55 | 17 | 5 | 1 | 1 | ‐2.57 | 11 | ‐2.34 | L | ‐2.57 |
| 24 | 2 | 4 | 38 | 36 | 3 | MD + P | CIT | VPA, PB | Yes | Yes | 50 | 22 | 9 | 1 | 5 | ‐2.21 | 22 | ‐0.41 | L | ‐2.21 |
| 25 | 7 | 3 | 33 | 26 | 4 | No | No | LVT, CBZ, CL | No | No | 85 | 2 | 0 | 1 | 6 | ‐1.86 | 11.5 | ‐2.70 | R | ‐2.70 |
| 26 | 25 | 3 | 47 | 22 | 0 | No | No | CBZ, LVT, CL | No | No | 75 | 8 | 0 | NHS | 12 | 0.56 | 21 | 0.08 | R | 0.08 |
Medical history: 0. No history 1. Perinatal hypoxia. 2. Encephalic trauma 3. Febrile seizures 4. Central Nervous System Infection. 5. Status Epilepticus. GAF: Global Assessment of Functioning (DSM IV), Depression: MD, Major Depression; MD + P, Major Depression with psychotic symptoms; D, Dysthymia; n/a, not available. Antidepressants: FLX, Fluoxetine; PARX, Paroxetine; CIT, Citalopram; SER, Sertraline. Antiepileptic drugs: CBZ, Carbamazepine; CL, Clonazepam; DPH, Diphenylhydantoin; LCS, Lacosamide; LMT, Lamotrigine; LVT, Levetiracetam; PB, Phenobarbital; TPM, Topiramate; VPA, Valproate. Severity of Depression score according clinical factors: 0, Absence of depression; 9, Maximum score. HS, Hippocampal Sclerosis; NHS, No hippocampal sclerosis. Laterality of epileptic focus: R, Right; L, Left. Z score ipsilateral to the epileptic focus; R, Visual memory; L, Verbal memory.
3.2. GR expression in neurons
Abundant GR immunoreactivity was present in nuclei of granule cells within the dentate gyrus of controls (Figures 1A and 2A) and pyramidal cells of CA1 (Figures 1A and 3A). A predominant nuclear staining was also found within the granule cell layer of TLE − D (Figures 1B and 2B) and TLE + D samples (Figures 1C and 2C). However, GR immunoreactivity was lower in granule cells in TLE − D as compared to controls, and the lowest expression was found in TLE + D (Figures 1C and 2C). GR immunoreactivity was also lower in CA1 pyramidal cells in TLE − D (Figures 1B and 3B) as compared to controls (Figures 1A and 3A), and the lowest expression was again found in TLE + D samples (Figures 1C and 3C).
Figure 1.
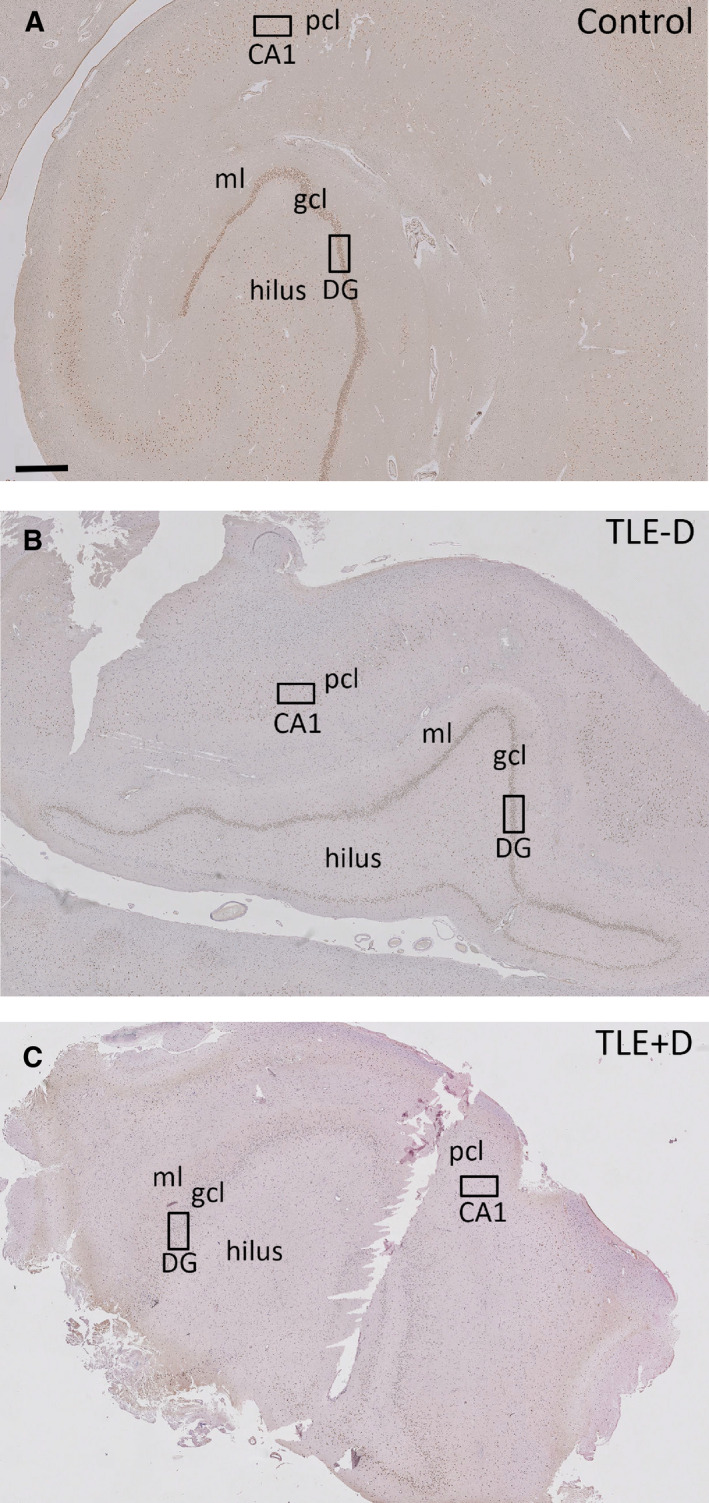
GR expression in the hippocampus. GR expression in postmortem control hippocampus (A), in the resected hippocampus of patients with temporal lobe epilepsy without depression (TLE −D) (B), and in patients with temporal lobe epilepsy with depression (TLE + D) (C). The black rectangles in the dentate gyrus (DG) and CA1 indicate the areas that are shown at high magnification in Figures 2 and 3. Scale bar = 740 µm. ml = molecular layer, gcl = granule cell layer, pcl = pyramidal cell layer.
Figure 2.
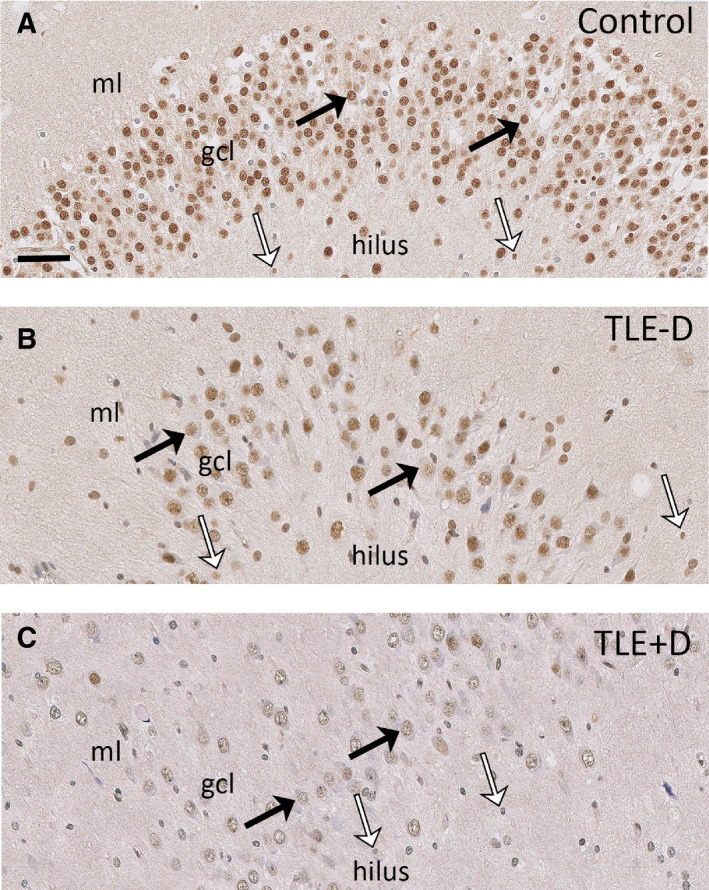
GR expression in the dentate gyrus. GR expression was found in neuronal nuclei (black arrows) and cells with glial morphology (white arrows) in the dentate gyrus of postmortem control hippocampus (A). Lower expression of the GR was evident in neurons (black arrows) and glial cells (white arrows) within the dentate gyrus of patients with TLE − D (B), and this was most evident in patients with TLE + D (C). Scale bar = 50 µm. ml = molecular layer, gcl = granule cell layer, TLE − D = temporal lobe epilepsy without depression, TLE + D = temporal lobe epilepsy with depression.
Figure 3.
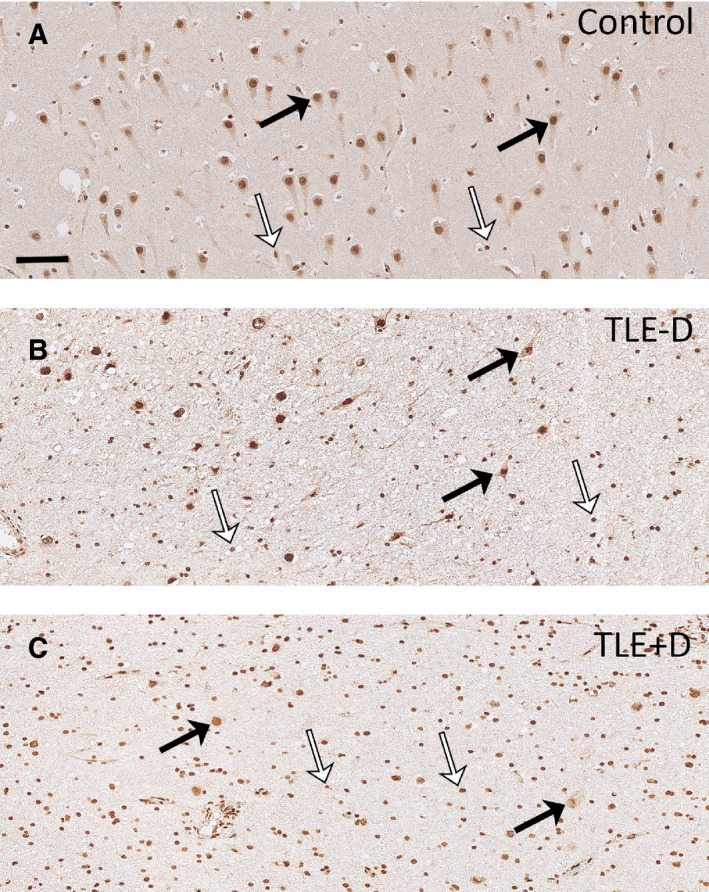
GR expression in CA1. Abundant GR expression was found in neuronal nuclei (black arrows) and cells with glial morphology (white arrows) in the pyramidal cell layer of CA1 of controls (A). A major loss of pyramidal neurons was observed in the CA1 of TLE patients (B and C), and the remaining pyramidal cells showed lower GR expression (black arrows). Lower GR expression was also observed in glial cells (white arrows) of TLE patients. Reduced GR expression was most evident in patients with TLE + D (C). Scale bar = 50 µm. TLE − D, temporal lobe epilepsy without depression; TLE + D, temporal lobe epilepsy with depression.
Quantitative analysis of GR expression in the dentate gyrus showed that the total GR+ area (Figure 4) was different between the three groups (controls, TLE − D, and TLE + D). A smaller GR+ area was found in the dentate gyrus of patients with TLE + D (x = 535.12, SD = 206.9) as compared to TLE − D (x = 787.01, SD = 264; F = 50.3 (2‐33); P = .048; One‐Way ANOVA‐Bonferroni) and as compared to controls (x = 1616.57, SD = 284.26; F = 50.3 (2‐33); P = .0001; one‐way ANOVA‐Bonferroni). Furthermore, the ratio between the number of GR+/NeuN + cells was lower in patients with TLE + D (x = 0.7, SD = 0.27, rank = 17.1) as compared to TLE − D (x = 1.03, SD = 0.24, rank = 9.29; U = 33.50, P = .008; Mann‐Whitney).
Figure 4.
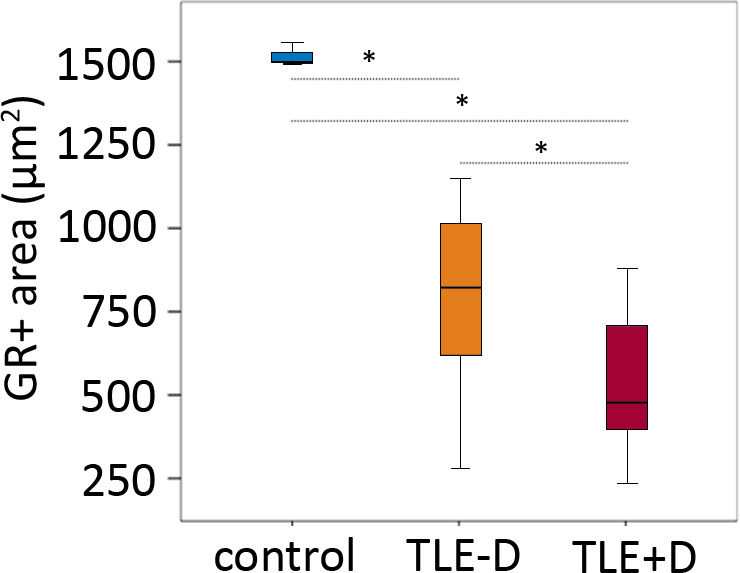
Quantitative analysis of GR expression in the dentate gyrus showed that GR+ area was different between the three groups (controls n = 9, TLE − D n = 14 and TLE + D n = 12; three replicates per case). A smaller GR+ area was found in the dentate gyrus of patients with TLE + D as compared to TLE – D, and as compared to controls. TLE − D, temporal lobe epilepsy without depression; TLE + D, temporal lobe epilepsy with depression. *One‐way ANOVA/Bonferroni P < .05.
3.3. GR expression in glial cells
Consistent with earlier reports, 13 GR immunoreactivity was also found in the nuclei of glia cells (white arrows in Figures 2 and 3). Double labeling confirmed that the GR was expressed predominantly in the nucleus and co‐localized with the microglia marker CR3/43 and the astrocyte marker GFAP (Figure 5). GR expression was observed in 37.9% of the CR3/43+ cells in TLE − D cases vs 40.0% in TLE + D cases. GR expression was found in 50.9% of the GFAP+ cells in TLE − D vs 48.0% in TLE + D cases.
Figure 5.
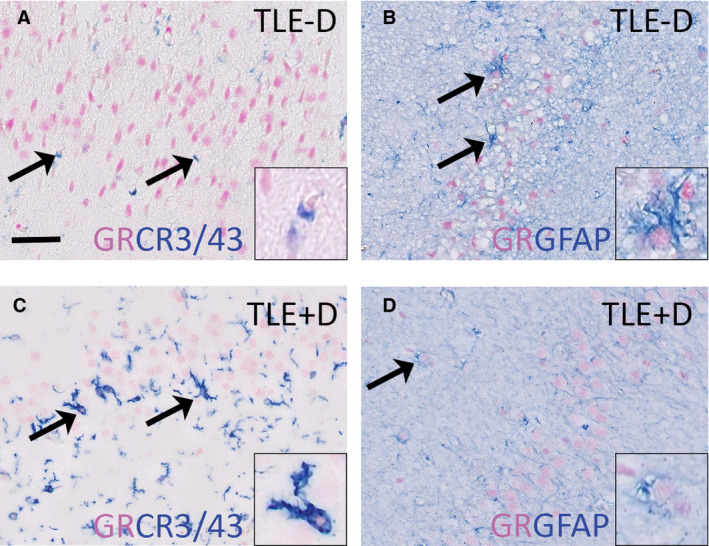
GR expression in glial cells. GR (purple) colocalized with the microglia marker CR3/43(blue) in the dentate gyrus of patients with TLE − D (A) as well as in TLE + D (C) and the GR (purple) also colocalized with the astrocyte marker GFAP (blue) in TLE‐D (B) as well as in TLE + D (D). Insets show higher magnification of glial cells. TLE − D, temporal lobe epilepsy without depression; TLE + D, temporal lobe epilepsy with depression. Scale bar = 50 µm
Quantitative analysis of the GR in glial cells showed that the mean optical density among individual GR+ glia nuclei was lower in TLE + D patients as compared to TLE − D in the granule cell layer/molecular layer (TLE + D x = 83.10, SD = 12.61; TLE‐D x = 95.69, SD = 15.23; t = 2.27; P = .032; Student t test) and in the hilus (TLE + D, x = 80.40, SD = 11.63; TLE‐D x = 97.34, SD = 16.54; t = 2.96; P = .007; Student t test).
3.4. Glucocorticoid receptor expression in relation to clinical parameters
Regarding the psychiatric parameters related to depression, the ratio of GR+/NeuN+ cells correlated negatively with depression severity, based on psychiatric history (ad hoc composite score; r = −0.528; P = .006; Spearman correlation), but not with the Beck Inventory (r = −0.359; P = .131; Spearman correlation). A tendency toward a positive correlation was found between the ratio of GR+/NeuN+ cells and the GAF scores (r = 0.297; P = .141; Spearman correlation). In addition, the expression of the GR in glia within the hilus correlated negatively with depression severity (ad hoc composite score; r = −0.42; P = .032; Spearman correlation).
With regard to the cognitive variables, memory scores (z‐score ipsilateral for the epileptic focus) did not differ between TLE − D and TLE + D cases (P > .05, Student t test) and did not correlate with GR expression in neurons or glial cells (P > .05; Pearson correlation).
With respect to the clinical aspects of epilepsy, the epilepsy duration, age at epilepsy onset, and seizure frequency did not correlate with the GR expression in neurons or glial cells (Pearson correlation, P > .05).
The distribution of male and female patients in TLE − D and TLE + D groups was similar (P > .05, chi‐square test). Regarding sex and GR expression parameters, the expression of the GR in glia was lower in women with TLE‐ D (x = 82.67, SD = 6.23) as compared to men with TLE‐ D (x = 105.46, SD = 12.21; t = 4.15, P = .001; Student t test). These differences were not observed in patients with TLE + D (women x = 86.29, SD = 14.60; men x = 76.71, SD = 1.95; t = −1.27; P = .23; Student t test). The other GR expression parameters did not differ between males and females. GR expression did not correlate with age.
4. DISCUSSION
Comorbid depression in TLE occurs frequently among drug‐resistant epilepsy patients and strongly affects their quality of life. 7 , 17 As such, it forms an important concern for both psychiatrists and neurologists. Here, hippocampal samples from a cohort of well‐characterized TLE patients were studied, focusing on the GR because it has been proposed to be involved in the pathogenesis of depression. 5 , 8 , 9 , 15 , 16
We found lower expression of GRs in the hippocampus of TLE patients as compared to controls. Furthermore, TLE patients with comorbid depression had lower GR immunoreactivity in the hippocampus as compared to TLE patients without depression. Watzka et al investigated expression of the GR in the brain of patients with TLE and showed that MR and GR messenger RNA (mRNA) expression was lower in hippocampal tissue than in frontal and temporal lobe cortical tissue of women with epilepsy, 32 but in this study they did not make a comparison to control brain tissue or between patients with and without depression. In another study, higher GR gene expression was found in the cortex of drug‐resistant TLE patients as compared to controls. 31 However, in that study the hippocampus was not studied, and the authors did not compare TLE patients with and without depression. Furthermore, they discussed that no clear picture has emerged from GR studies using animal models of epilepsy, since in three studies a decrease in GR was observed in the cortex or hippocampus after induced seizures, whereas the expression increased in the hippocampus in another study. 31
In our previous study (using a different cohort), we have shown lower calbindin expression (in the basal part of the granule cell layer), but higher expression in granule cells that were dispersed in the molecular layer of dentate gyrus, in patients with TLE + D vs TLE − D. 6 We discussed that this pattern of calbindin expression may contribute to both hyperexcitability and neuropsychiatric illness, favoring behavioral and cognitive alterations. Liu et al investigated a large group of epilepsy patients (n = 276) and showed indeed that seizure frequency was positively associated with depression severity. 41 We did not observe such differences between epilepsy patients with and without depression in the present study, which may be explained by our smaller sample size compared to Liu et al; however, there was a tendency for a higher seizure frequency in our group of patients with epilepsy and depression.
Our findings about reduced GR expression in the hippocampus, particularly in TLE patients with depression, are comparable to those of other studies on chronic stress and depression models. A downregulation of GR mRNA expression was reported in the rodent hippocampus after chronic stress 21 , 22 , 25 and after chronic corticosterone exposure. 23 Mizoguchi et al further found reduced GR expression in the prefrontal cortex after chronic stress 24 and also in primates lower expression of GR mRNA was reported in the prefrontal cortex 42 following stress exposure. Similarly, López et al reported a decrease in hippocampal MR and GR expression in suicide victims with a history of depression. In addition, similar findings were shown by Klok et al 27 for the GR and MR in major depression in various brain regions including the hippocampus, whereas the GR beta isoform that was thought to be implicated in GR resistance was found to be very rare in the human brain. 29 Furthermore, decreased GR mRNA expression in patients with depression was found exclusively in the dentate gyrus. 26 A reduction in the MR/GR ratio has been reported in the anterior hippocampus from patients with major depression. 12 Overall, these results suggest that alterations in hippocampal GR (and MR) expression are associated with depression. These results appear region‐ and condition‐specific, as GR protein level as well as the percentage of GR‐containing astrocytes were found before to be significantly higher in the amygdala in major depression than in bipolar depressed patients or in control subjects, but these authors focused on older depressed patients. 28 Furthermore, we showed that the ratio of GR+/NeuN+ cells as well as the expression of the GR in glia within the hilus correlated negatively with the severity of depression as based on psychiatric history (ad hoc composite score), but not with the Beck Inventory, which provides a measure of the severity of depression at the moment of assessment. This indicates the importance of taking into account the psychiatric history and highlights that the most recent status may not always reflect the observed GR changes in the brain.
Regarding models of epilepsy, abundant evidence has demonstrated that HPA activity is enhanced during epileptic seizures, 9 but only a few studies identified hippocampal GR expression in epileptogenic brain tissues. In experimental models with rodents, epileptic discharges and ischemic insults were shown to reduce GR expression in the hippocampal neurons of CA1 and the dentate gyrus neurons. 30 , 43
Sex differences in hippocampal GR expression have also been described. Lower GR expression was found in the epileptogenic cortex of men compared to women; however, women showed lower GR expression in the hippocampus. 32 GR expression is generally not altered during aging, although an age‐associated GR decline was reported in the dentate gyrus of females. 13 , 44 In the current study, we observed a lower glial GR content in women with TLE ‐ D as compared to men, but these differences were not observed in TLE + D patients. No other differences in GR expression were observed between female and male patients. Most likely, the sample size of this study was too small to find further differences.
The GR constitutes a key factor in understanding the mechanisms involved in the pathogenesis of TLE and depression, with potential therapeutic implications. Activation of the GR has genomic and, also, rapid nongenomic actions, each of which may affect hippocampal excitability. 16 The MR has a 10‐fold higher affinity than the GR for cortisol, so although MR is almost always occupied, the GR becomes activated only when circulating levels of cortisol increase (ie, under stressful conditions) 12 , 16 and under conditions of epileptic seizures. 9 , 16 The hippocampus and particularly the dentate gyrus cells further exert an inhibitory role on the activity of the HPA axis. This negative feedback is involved in termination of the stress response. 45 GR dysfunction may contribute to impair the negative feedback of the HPA axis, which, in turn, could lead to a feed‐forward activation of the HPA axis. 9 Furthermore, dysregulation of the HPA axis and glucocorticoids may affect local and systemic inflammatory mechanisms, which have been found to be altered in both TLE and depression models. 46 An increase in expression of inflammatory markers has been described extensively in various epilepsy models 47 , 48 , in epilepsy models with depression, 46 and in depression models. 49 In our current study, patients with TLE had hippocampal sclerosis, which is characterized by neuronal loss and gliosis. In addition, we found lower expression of the GR in glial cells from patients with TLE + D. Thus the reduced GR expression in both neurons and glia may indicate that TLE + D patients have been more exposed to glucocorticoids during life, which may lead to downregulation of GRs. As a result of the prolonged downregulation of hippocampal GR, the inhibitory influence on the HPA axis could have become chronically reduced, thereby stimulating HPA activity even further and creating a vicious cycle.
5. CONCLUSION
Reduced hippocampal GR expression may be involved in the etiology of depression in patients with TLE and could constitute a biological marker of depression in these patients.
CONFLICTS OF INTEREST
None of the authors has any conflict of interest to disclose.
Supporting information
Fig S1
ACKNOWLEDGMENTS
We thank all the participants and collaborators of this study, all the team of the Epilepsy Center of Ramos Mejía Hospital and El Cruce Hospital. We also thank Professor Ron de Kloet (Leiden) and Professor Marian Joëls (UMC Groningen) for their help in the collaboration between the Epilepsy Center of Buenos Aires and the Amsterdam UMC, location Academic Medical Center, in Amsterdam. The research leading to these results has received funding from the European Union's Seventh Framework Program (FP7/2007‐2013) under grant agreement 602102 (EPITARGET; EAvV, EA), the Dutch Epilepsy Foundation, project 16‐05 (EAvV), Hersenstichting Nederland (PJL), Alzheimer Nederland (PJL), the UvA research priority area Urban Mental Health (PJL) and the KNAW China Exchange program, grant 05CDP030 (PJL). The authors have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
D’Alessio L, Mesarosova L, Anink JJ, et al. Reduced expression of the glucocorticoid receptor in the hippocampus of patients with drug-resistant temporal lobe epilepsy and comorbid depression. Epilepsia. 2020;61:1595–1605. 10.1111/epi.16598
Aronica and van Vliet shared senior authorship
REFERENCES
- 1. Kanner AM. Is depression associated with an increased risk of treatment‐resistant epilepsy? Research strategies to investigate this question. Epilepsy Behav. 2014;38:3–7. [DOI] [PubMed] [Google Scholar]
- 2. Tellez‐Zenteno JF, Patten SB, Jetté N, Williams J, Wiebe S. Psychiatric comorbidity in epilepsy: a population‐based analysis. Epilepsia. 2007;48(12):2336–44. [DOI] [PubMed] [Google Scholar]
- 3. Kanner AM, Schachter SC, Barry JJ, Hesdorffer DC, Mula M, Trimble M, et al. Depression and epilepsy: epidemiologic and neurobiologic perspectives that may explain their high comorbid occurrence. Epilepsy Behav. 2012;24(2):156–68. [DOI] [PubMed] [Google Scholar]
- 4. Hesdorffer DC, Ishihara L, Mynepalli L, Webb DJ, Weil J, Hauser WA. Epilepsy, suicidality, and psychiatric disorders: a bidirectional association. Ann Neurol. 2012;72(2):184–91. [DOI] [PubMed] [Google Scholar]
- 5. Hester MS, Danzer SC. Hippocampal granule cell pathology in epilepsy ‐ A possible structural basis for comorbidities of epilepsy? Epilepsy Behav. 2014;38:105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. D’Alessio L, Konopka H, Solís P, Scévola L, Fernandez Lima M, Nuñez C, et al. Depression and Temporal Lobe Epilepsy: Expression Pattern of Calbindin Immunoreactivity in Hippocampal Dentate Gyrus of Patients Who Underwent Epilepsy Surgery with and without Comorbid Depression. Behav Neurol. 2019;2019:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scévola L, Sarudiansky M, Lanzillotti A, Oddo S, Kochen S, D’Alessio L. To what extent does depression influence quality of life of people with pharmacoresistant epilepsy in Argentina? Epilepsy Behav. 2017;69:133–8. [DOI] [PubMed] [Google Scholar]
- 8. Hattiangady B, Rao MS, Shetty AK. Grafting of striatal precursor cells into hippocampus shortly after status epilepticus restrains chronic temporal lobe epilepsy. Exp Neurol. 2008;212(2):468–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wulsin AC, Solomon MB, Privitera MD, Danzer SC, Herman JP. Hypothalamic‐pituitary‐adrenocortical axis dysfunction in epilepsy. Physiol Behav. 2016;166:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thom M. Hippocampal Sclerosis in Epilepsy: A neuropathology review. Neuropathol Appl Neurobiol. 2014;40(5):520–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Blümcke I, Thom M, Aronica E, Armstrong DD, Bartolomei F, Bernasconi A, et al. International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a Task Force report from the ILAE Commission on Diagnostic Methods. Epilepsia. 2013;54(7):1315–29. [DOI] [PubMed] [Google Scholar]
- 12. Medina A, Seasholtz AF, Sharma V, Burke S, Bunney W Jr, Myers RM, et al. Glucocorticoid and mineralocorticoid receptor expression in the human hippocampus in major depressive disorder. J Psychiatr Res. 2013;47(3):307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Q, van Heerikhuize J, Aronica E, Kawata M, Seress L, Joëls M, et al. Glucocorticoid receptor protein expression in human hippocampus; stability with age. Neurobiol Aging. 2013;34(6):1662–73. [DOI] [PubMed] [Google Scholar]
- 14. Anacker C, Cattaneo A, Musaelyan K, Zunszain PA, Horowitz M, Molteni R, et al. Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc Natl Acad Sci. 2013;110(21):8708–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Kloet ER, Joëls M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6(6):463–75. [DOI] [PubMed] [Google Scholar]
- 16. Joëls M. Stress, the hippocampus, and epilepsy. Epilepsia. 2009;50(4):586–97. [DOI] [PubMed] [Google Scholar]
- 17. Reagan LP, McEwen BS. Controversies surrounding glucocorticoid‐mediated cell death in the hippocampus. J Chem Neuroanat. 1997;13(3):149–67. [DOI] [PubMed] [Google Scholar]
- 18. Joëls M, de Kloet ER. Effects of Glucocorticoids and Norepinephrine on the Excitability in the Hippocampus. Science. 1989;245(4925):1502–5. [DOI] [PubMed] [Google Scholar]
- 19. Kumar G, Jones NC, Morris MJ, Rees S, O'Brien TJ, Salzberg MR. Early life stress enhancement of limbic epileptogenesis in adult rats: mechanistic insights. PLoS One. 2011;6(9):e24033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Allendorfer JB, Heyse H, Mendoza L, Nelson EB, Eliassen JC, Storrs JM, et al. Physiologic and cortical response to acute psychosocial stress in left temporal lobe epilepsy — A pilot cross‐sectional fMRI study. Epilepsy Behav. 2014;36:115–23. [DOI] [PubMed] [Google Scholar]
- 21. Herman JP, Adams D, Prewitt C. Regulatory changes in neuroendocrine stress‐integrative circuitry produced by a variable stress paradigm. Neuroendocrinology. 1995;61 (2):180–190. [DOI] [PubMed] [Google Scholar]
- 22. Meyer U, van Kampen M, Isovich E, Flügge G, Fuchs E. Chronic psychosocial stress regulates the expression of both GR and MR mRNA in the hippocampal formation of tree shrews. Hippocampus. 2001;11(3):329–36. [DOI] [PubMed] [Google Scholar]
- 23. Hügin‐flores ME, Steimer T, Aubert L, Schulz P. Mineralo‐ and glucocorticoid receptor mRNAs are differently regulated by corticosterone in the rat hippocampus and anterior pituitary. Neuroendocrinology. 2004;79(4):174–84. [DOI] [PubMed] [Google Scholar]
- 24. Mizoguchi K, Ishige A, Aburada M, Tabira T. Chronic stress attenuates glucocorticoid negative feedback: Involvement of the prefrontal cortex and hippocampus. Neuroscience. 2003;119(3):887–97. [DOI] [PubMed] [Google Scholar]
- 25. López JF, Chalmers DT, Little KY, Watson SJ. Regulation of serotonin1A, glucocorticoid, and mineralocorticoid receptor in rat and human hippocampus: implications for the neurobiology of depression. Biol Psychiatry. 1998;43(8):547–73. [DOI] [PubMed] [Google Scholar]
- 26. Webster MJ, Knable MB, O’Grady J, Orthmann J, Weickert CS. Regional specificity of brain glucocorticoid receptor mRNA alterations in subjects with schizophrenia and mood disorders. Mol Psychiatry. 2002;7(9):985–94. [DOI] [PubMed] [Google Scholar]
- 27. Klok MD, Alt SR, Irurzun AJM, Turner JD, Lakke EA, Huitinga I, et al. Decreased expression of mineralocorticoid receptor mRNA and its splice variants in postmortem brain regions of patients with major depressive disorder. J Psychiatr Res. 2011;45(7):871–8. [DOI] [PubMed] [Google Scholar]
- 28. Wang Q, Verweij EWE, Krugers HJ, Joëls M, Swaab DF, Lucassen PJ. Distribution of the glucocorticoid receptor in the human amygdala; changes in mood disorder patients. Brain Struct Funct. 2014;219(5):1615–26. [DOI] [PubMed] [Google Scholar]
- 29. DeRijk RH, Schaaf M, Stam FJ, de Jong IE, Swaab DF, Ravid R, et al. Very low levels of the glucocorticoid receptor beta isoform in the human hippocampus as shown by Taqman RT‐PCR and immunocytochemistry. Brain Res Mol Brain Res. 2003;116(1–2):17–26. [DOI] [PubMed] [Google Scholar]
- 30. Hwang IK, Lee YB, Yoo KY, Kang TC, Kim DW, Moon WK, et al. Seizure‐induced changes of mineralocorticoid and glucocorticoid receptors in the hippocampus in seizure sensitive gerbils. Neurosci Res. 2005;53(1):14–24. [DOI] [PubMed] [Google Scholar]
- 31. Martinez‐Levy GA, Rocha L, Rodriguez‐Pineda F, Alonso‐Vanegas MA, Nani A, Buentello‐García RM, et al. Increased expression of brain‐derived neurotrophic factor transcripts I and VI, cAMP response element binding, and glucocorticoid receptor in the cortex of patients with temporal lobe epilepsy. Mol Neurobiol. 2018;55(5):3698–708. [DOI] [PubMed] [Google Scholar]
- 32. Watzka M, Bidlingmaier F, Beyenburg S, Henke RT, Clusmann H, Elger CE, et al. Corticosteroid receptor mRNA expression in the brains of patients with epilepsy. Steroids. 2000;65(12):895–901. [DOI] [PubMed] [Google Scholar]
- 33. Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51(6):1069–77. [DOI] [PubMed] [Google Scholar]
- 34. D’Alessio L, Giagante B, Papayannis C, Oddo S, Silva W, Solís P, et al. Psychotic disorders in Argentine patients with refractory temporal lobe epilepsy: a case‐control study. Epilepsy Behav. 2009;14(4):604–9. [DOI] [PubMed] [Google Scholar]
- 35. First M, Spitzer R, Gibbon M, Williams J. Entrevista Clínica Estructurada para los Trastornos del Eje I y II del DSM‐IV: SCID‐I y II. Versión Clínica. Barcelona: Masson; 1999. [Google Scholar]
- 36. Diagnostic and statistical manual of mental disorders . DSM IV, fourth edition Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- 37. Campo P, Morales M. Normative data and reliability for a Spanish version of the verbal Selective Reminding Test. Arch Clin Neuropsychol. 2004;19:421–35. [DOI] [PubMed] [Google Scholar]
- 38. Lezak M. Neuropschychogical Assessment. third edition. New York: Oxford University Press; 1995. [Google Scholar]
- 39. Oddo S, Solis P, Consalvo D, Seoane E, Giagante B, D’Alessio L, et al. Postoperative neuropsychological outcome in patients with mesial temporal lobe epilepsy in Argentina. Epilepsy Res Treat. 2012;2012:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Beck AT, Steer RA, Brown GK. Manual for the Beck Depression Inventory–Second Edition (BDI‐II). San Antonio: Psychological Corporation; 1996. [Google Scholar]
- 41. Liu X, Chen H, Zheng X. Effects of seizure frequency, depression and generalized anxiety on suicidal tendency in people with epilepsy. Epilepsy Res. 2020;160:106265. [DOI] [PubMed] [Google Scholar]
- 42. Patel P, Katz M, Karssen AM, Lyons DM, . Stress‐induced changes in corticosteroid receptor expression in primate hippocampus and prefrontal cortex. Psychoneuroendocrinology. 2008;33(3):360–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hwang IK, Yoo K, Nam YS, Choi JH, Lee IS, Kwon YG, et al. Mineralocorticoid and glucocorticoid receptor expressions in astrocytes and microglia in the gerbil hippocampal CA1 region after ischemic insult. Neurosci Res. 2006;54(4):319–27. [DOI] [PubMed] [Google Scholar]
- 44. Wang Q, Joëls M, Swaab DF, Lucassen PJ. Hippocampal GR expression is increased in elderly depressed females. Neuropharmacology. 2012;62(1):527–33. [DOI] [PubMed] [Google Scholar]
- 45. Samuels BA, Leonardo ED, Hen R. Hippocampal subfields and major depressive disorder. Biol Psychiatry. 2015;77(3):210–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kandratavicius L, Peixoto‐santos JE, Monteiro MR, Scandiuzzi RC, Carlotti CG Jr, Assirati JA Jr, et al. Mesial temporal lobe epilepsy with psychiatric comorbidities: a place for differential neuroinflammatory interplay. J Neuroinflammation. 2015;12(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van Vliet EA, Aronica E, Vezzani A, Ravizza T. Review: Neuroinflammatory pathways as treatment targets and biomarker candidates in epilepsy: emerging evidence from preclinical and clinical studies. Neuropathol Appl Neurobiol. 2018;44(1):91–111. [DOI] [PubMed] [Google Scholar]
- 48. Aronica E, Bauer S, Bozzi Y, Caleo M, Dingledine R, Gorter JA, et al. Neuroinflammatory targets and treatments for epilepsy validated in experimental models. Epilepsia. 2017;58(Suppl 3):27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim YK, Na KS, Myint AM, Leonard BE. The role of pro‐inflammatory cytokines in neuroinflammation, neurogenesis and the neuroendocrine system in major depression. Prog Neuropsychopharmacol. Biol Psychiatry. 2016;64:277–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1