

Abstract
Objective
To assess the safety and efficacy of lenabasum in diffuse cutaneous systemic sclerosis (dcSSc).
Methods
A randomized, double‐blind, placebo‐controlled, phase II study was conducted at 9 SSc clinics in the US. Adults with dcSSc of ≤6 years’ duration who were receiving stable standard‐of‐care treatment were randomized to receive lenabasum (n = 27) or placebo (n = 15). Lenabasum doses were 5 mg once daily, 20 mg once daily, or 20 mg twice daily for 4 weeks, followed by 20 mg twice daily for 8 weeks. Safety and efficacy were assessed at weeks 4, 8, 12, and 16.
Results
Adverse events (AEs) occurred in 63% of the lenabasum group and 60% of the placebo group, with no serious AEs related to lenabasum. Compared to placebo, lenabasum treatment was associated with greater improvement in the American College of Rheumatology Combined Response Index in diffuse cutaneous Systemic Sclerosis (CRISS) score and other efficacy outcome measures that assessed overall disease, skin involvement, and patient‐reported function. The median CRISS score increased in the lenabasum group during the study, reaching 0.33, versus 0.00 in the placebo group, at week 16 (P = 0.07 by 2‐sided mixed‐effects model repeated‐measures analysis). Gene expression in inflammation and fibrosis pathways was reduced, and inflammation and fibrosis were improved on histologic evaluation of skin biopsy specimens, in the lenabasum group compared to the placebo group (all P ≤ 0.05).
Conclusion
Despite a short trial duration in a small number of patients in this phase II study in dcSSc, our findings indicate that lenabasum improves efficacy outcomes and underlying disease pathology with a favorable safety profile.
INTRODUCTION
Systemic sclerosis (SSc) is a rare, multisystem autoimmune disease of unknown origin characterized by chronic inflammation, fibrosis, and small vessel damage in involved tissues. Activation of the innate immune system is a key component in disease pathogenesis in SSc 1. Immunosuppressive drugs such as mycophenolate, methotrexate, glucocorticoids, cyclophosphamide, and azathioprine are used for overall disease control or treatment of skin or interstitial lung disease in diffuse cutaneous SSc (dcSSc) 2. Immunosuppressive drugs may have only modest efficacy 3, 4 with poor tolerability 5 and cause toxicities such as cytopenias 5 and infection. Furthermore, glucocorticoid use can provoke renal crisis 6. Hematopoietic stem cell transplantation can provide benefit in severe, early dcSSc, but carries substantial treatment‐related morbidity and mortality in the first few years after treatment 7, 8, 9. There remains a major need for treatments that are more effective and safe enough to treat a broad spectrum of individuals with dcSSc 2.
Lenabasum is a synthetic, orally administered agonist of cannabinoid receptor 2 (CB2) 10 that modulates the endocannabinoid system to activate the resolution phase of innate immune responses 11. Lenabasum improves skin and lung inflammation and fibrosis when administered prophylactically or therapeutically in animal models of SSc 12, 13. It reduces transforming growth factor β (TGFβ) and collagen production by isolated SSc fibroblasts and limits fibrosis in an inflammation‐independent animal model of SSc 12, indicating a direct effect on SSc fibroblasts, which express more CB2 than normal fibroblasts 14, in addition to effects on inflammatory cells.
Lenabasum is therefore a rational candidate for treatment of SSc. The hypothesis of this study was that lenabasum would provide clinical benefit by triggering pathways that resolve adverse innate immune responses and ameliorate ongoing fibrotic processes. The primary objectives were to evaluate the safety and tolerability of lenabasum in subjects with active dcSSc and to evaluate the efficacy of lenabasum in SSc using the American College of Rheumatology (ACR) Combined Response Index in diffuse cutaneous Systemic Sclerosis (CRISS) score. To our knowledge, this is the first study in humans to prospectively evaluate an investigational product designed to modulate the endocannabinoid system for treatment of chronic inflammatory and fibrotic diseases such as SSc.
PATIENTS AND METHODS
Study design and subject characteristics
Subjects were 18–69 years of age, met the 2013 ACR/European League Against Rheumatism classification criteria for SSc 15, and had clinically apparent skin involvement proximal to the elbows or knees or on the trunk. Subjects were recruited at 9 SSc clinics in the US. Subjects had a disease duration of ≤3 years since the first non‐Raynaud's sign or symptom. Alternatively, subjects with a disease duration of >3 years and ≤6 years were eligible if they had a C‐reactive protein (CRP) level of >3 mg/liter, interleukin‐6 (IL‐6) level of >5 pg/ml, total modified Rodnan skin thickness score (MRSS) (16) of ≥16, or total MRSS of ≥12 with an increase of ≥5 points in the previous 6 months. The latter option was added during the study based on input from study investigators, to broaden eligibility without anticipated negative effects on safety or efficacy assessments. Subjects could continue most concomitant immunosuppressive medications if doses were stable for ≥3 months before day 1 and remained unchanged during the study.
Cyclophosphamide within 3 months, rituximab within 6 months, and glucocorticoid dosages of >10 mg prednisone or equivalent per day within 28 days prior to day 1 were not allowed. Doses of non‐immunosuppressive medications for SSc were required to be stable for ≥28 days before day 1. Women of childbearing potential could not be pregnant or breastfeeding and must have used ≥1 acceptable method of contraception for ≥28 days before day 1, during dosing, and for ≥28 days after dosing. Concomitant cannabinoid use was not allowed. Subjects were excluded if they were listed for an organ transplant or if they had an organ transplant, pulmonary hypertension requiring active treatment, interstitial lung disease requiring oxygen therapy, gastrointestinal dysmotility requiring total parenteral nutrition or hospitalization in the 6 months prior to day 1 of the study, or renal crisis in the year prior to day 1 of the study.
Each site's institutional review board approved the study. Subjects gave written informed consent for study participation before any study procedures were done. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice.
Randomization and masking
Randomization was computer generated using a random sequence for treatment assignment in an overall 2:1 ratio of lenabasum to placebo, in blocks of 9 subjects. Subject identification numbers, treatment, and investigational product bottle numbers were assigned through a central web‐based randomization system. Lenabasum‐treated subjects received 5 mg once daily and placebo once daily; 20 mg once daily and placebo once daily; or 20 mg twice daily for the first 4 weeks. All subjects in the lenabasum group received 20 mg twice daily for the next 8 weeks. The placebo group received placebo twice daily for all 12 weeks of dosing.
Both lenabasum and placebo (microcrystalline cellulose) were powder‐in‐capsule preparations manufactured by Alcami and presented in identical gelatin capsules in identical bottles with similar labels and handling for masking purposes. All subjects, site staff, and sponsor staff involved in clinical aspects of the study and data analyses were blinded with regard to treatment assignment until after database lock.
Study procedures and safety and efficacy assessments
Investigational product was self‐administered and taken orally twice daily with 8–16 hours between doses. Subjects had study visits on day 1, day 15, and at the ends of week 4, week 8, week 12, and week 16. Safety assessments at all visits included recording adverse events (AEs) and vital signs and performing hematology and chemistry laboratory tests and urinary dipstick for blood, albumin/protein, and glucose. Urine β‐human chorionic gonadotropin tests were performed for female subjects of childbearing potential at all study visits except day 15. Electrocardiograms with QT/corrected QT (QTc) intervals and Addiction Research Center Inventory‐Marijuana scale testing were performed on day 1 and at weeks 4 and 12. Physical examinations were performed on day 1 and at weeks 12 and 16.
An AE was defined as any symptom, sign, illness or experience, or untoward medical occurrence that developed or worsened in severity in a subject who received investigational product. A serious AE was defined as any AE that resulted in death, was life‐threatening, required inpatient hospitalization or prolongation of existing hospitalization, was a congenital anomaly or birth defect, or was an important medical event. The severity (mild, moderate, or severe) and relationship to investigational product (none, unlikely, possible, probably, or definite) of each AE were assigned by the investigator. The Medical Dictionary for Regulatory Activities System Organ Class preferred terms, version 18.0, was used to classify AEs.
Efficacy assessments were done at all visits except day 15 and included total MRSS, Health Assessment Questionnaire (HAQ) disability index (DI) 17, physician global assessment of overall patient health 18, patient global assessment of health 18, and forced vital capacity percent predicted (FVC%) determined using Hankinson's predictive equation 19. For the physician global assessment and the patient global assessment, the subject's overall health related to SSc in the past 7 days was rated on a 10‐cm visual analog scale, where 0 = excellent and 10 = extremely poor 18. Patient‐reported outcomes were obtained before other study visit interactions.
The CRISS score was developed as an outcome measure for 12‐month trials by an international group of experts in SSc. This composite response index for clinical trials in early dcSSc was calculated centrally from changes from baseline in total MRSS, HAQ DI, physician global assessment, patient global assessment, and FVC% 18. A value of 0 was assigned if the subject developed new severe involvement of the kidneys, lungs, heart, or pulmonary vasculature 18.
The Patient‐Reported Outcomes Measurement Information System 29‐item (PROMIS‐29) questionnaire 20, the 5‐D itch scale 21, and an 18‐question patient‐reported SSc skin symptom questionnaire (the Scleroderma Skin Patient‐Reported Outcome [SSPRO] instrument) 22 were administered, and blood biomarkers were measured, on day 1 and at week 4 and week 12. The PROMIS‐29 results are reported as T scores, which rescale the raw score into a standardized score with a mean of 50 and an SD of 10. Study data were captured in an electronic database on case report forms and monitored by Corbus Pharmaceuticals. Scores for calculated efficacy outcome measures were assessed centrally.
Two 3‐mm punch biopsies of involved skin were performed, one on day 1 and one at week 12, at a nearby site to evaluate skin histology and gene transcript expression. Skin biopsy specimens were taken from an active area suitable for repeat biopsy in the opinion of the investigator. The forearm was preferred, and no biopsy specimens were obtained from the face, neck, hands, or feet. The site of the skin biopsy had to have an MRSS score of ≥2, and the repeat biopsy specimen was taken 1–2 cm from the original biopsy site.
Hematoxylin and eosin–stained skin sections were assessed in pairs in a blinded manner (with regard to treatment assignment) for worsening, no change, or improvement in inflammation and fibrosis at week 12 compared to day 1. Inflammation was assessed as the extent of inflammatory infiltrates in the dermis and around adnexal structures. Fibrosis was assessed as the extent of hyalinized extracellular matrix in the skin 23. Gene expression data in skin biopsy specimens were preprocessed and analyzed as previously described 24, with gene expression data generated before unblinding of treatment assignment. Differential expression was determined using Comparative Marker Selection 25, and genes with a q value of <5% were considered significant. Functional profiling for significant genes was performed using g:Profiler 26 and Gene Ontology functional terms. Bonferroni‐corrected P values less than 0.05 were considered significant.
Outcome measures
Safety assessments included AEs, with severity and relatedness to investigational product as judged by the site investigator. Tolerability was assessed as the proportion of subjects who discontinued investigational product due to an AE that was probably or definitely related to the investigational product. A Data Monitoring Committee oversaw safety during the study.
Multiple efficacy outcome measures were used to test the effects of lenabasum on overall disease (CRISS score, change from baseline in physician global assessment and patient global assessment), skin involvement (change from baseline in MRSS, SSPRO score, and 5‐D itch score), and patient‐reported function (change from baseline in HAQ DI score and PROMIS‐29 physical function and social role domains). The primary efficacy outcome measure was the CRISS score. Secondary efficacy outcome measures included change from baseline in MRSS, HAQ DI, patient global assessment, physician global assessment, FVC%, 5‐D itch score, SSPRO score, and PROMIS‐29 domain scores. Biomarker outcome measures included change from baseline in gene expression and in inflammation and fibrosis on histologic evaluation of skin biopsy specimens. This trial was registered with ClinicalTrials.gov (identifier: NCT02465437). An open‐label extension is ongoing.
Statistical analysis
All subjects who received ≥1 dose of investigational product were considered in the safety population. Descriptive statistics were used to compare safety in the lenabasum‐treated and placebo‐treated groups.
The sample size was not formally powered for efficacy and was estimated to be adequate to detect early efficacy signals and feasible to enroll in a limited number of sites. Subjects were analyzed by assigned treatment. The baseline outcomes were measured before treatment was administered. Missing data were imputed using the last observation carried forward (LOCF) method. CRISS scores were calculated in the modified intent‐to‐treat population of subjects who had ≥1 efficacy assessment postbaseline.
The planned primary efficacy outcome measure for statistical analysis was mean CRISS score at week 12. After database lock and unblinding and before issuance of Tables and Listings, analysis by a statistician external to Corbus Pharmaceuticals revealed that CRISS scores were not normally distributed and therefore analysis by t‐test was not an appropriate statistical approach. The CRISS scores then were described using median and interquartile range (IQR), and differences between the lenabasum and placebo groups were analyzed using the Wilcoxon‐Mann‐Whitney test and mixed‐effects model repeated‐measures (MMRM) analysis using rank‐transformed data, with CRISS score as a dependent variable and treatment, visit, and visit × treatment interaction as fixed effects. Visit was a repeated factor, and analyses were done through week 16.
The secondary end points satisfied the assumption of normality for MMRM analysis, and observations were used as‐is and not transformed. An unstructured covariance matrix was utilized for all end points. Differences in change from baseline in other efficacy outcome measures between the lenabasum and placebo groups were also analyzed using MMRM analysis, with change from baseline as a dependent variable, and treatment, visit, and visit × treatment interaction as fixed effects. Because this was a small first‐in‐patient phase II study and improvement in efficacy outcome measures was the expected effect of lenabasum, 1‐sided statistical analyses were done for CRISS scores in addition to 2‐sided analyses (P ≤ 0.05), and 1‐sided and 2‐sided statistical analyses were done for other efficacy outcome measures (P ≤ 0.10). Data were analyzed using SAS version 9.4.
RESULTS
Sixty‐one subjects were assessed for eligibility, and 43 were randomized (28 to receive lenabasum and 15 to receive placebo). One patient was randomized to receive lenabasum but was withdrawn by the investigator before receiving investigational product because of concurrent illness. Forty‐two subjects (27 in the lenabasum group and 15 in the placebo group) received ≥1 dose of investigational product and were included in safety analyses. Forty‐one subjects (26 in the lenabasum group and 15 in the placebo group) had ≥1 efficacy assessment after dosing and were included in efficacy analyses; 38 subjects (24 in the lenabasum group and 14 in the placebo group) completed week 16. Four subjects withdrew after receiving investigational product for the following reasons: physician decision because the subject was taking a prohibited medicine (n = 1 in the lenabasum group; no efficacy assessments done); withdrawal of consent (n = 1 in the lenabasum group and 1 in the placebo group); AE of moderate dizziness probably related to investigational product (n = 1 in the lenabasum group). The date of the first subject's first visit was October 12, 2015, and the date of the last subject's last visit was October 10, 2016.
Demographic and baseline characteristics were similar overall in the lenabasum and placebo groups (Table 1). Approximately 90% of the subjects in each group were receiving background immunosuppressive medications. Mycophenolate mofetil or mycophenolic acid was used alone or in combination with other immunosuppressive medications in 80% of placebo‐treated subjects and 63% of lenabasum‐treated subjects. Other immunosuppressive medications used alone or in combination (in the placebo group versus the lenabasum group) included azathioprine (0% versus 4%), etanercept (0% versus 4%), hydroxychloroquine (13% versus 11%), intravenous immunoglobulin (7% versus 7%), methotrexate (7% versus 19%), and prednisone (13% versus 30%). Mean serum CRP, IL‐6, and CXCL4 levels were comparable between treatment groups and within normal ranges. At baseline, CRP and IL‐6 levels were each elevated in only 7 (17%) of 42 subjects.
Table 1.
Baseline characteristics of the patients with SSc included in the safety populationa
| Characteristic | Placebo (n = 15) | Lenabasum (n = 27) |
|---|---|---|
| Age, years | 47 ± 11.1 | 49 ± 10.4 |
| Sex, no. (%) female | 9 (60) | 23 (85) |
| Race, no. (%) white | 12 (80) | 22 (82) |
| Body mass index, kg/m2 | 24 ± 3.8 | 25 ± 5.6 |
| Duration of SSc, months | 34 ± 18.0 | 34 ± 16.6 |
| Immunosuppressive medications, no. (%) | ||
| Any | 13 (87) | 25 (93) |
| Mycophenolateb | 12 (80) | 17 (63) |
| Prednisone | 2 (13) | 8 (30) |
| Hydroxychloroquine | 2 (13) | 3 (11) |
| Methotrexate | 1 (7) | 5 (19) |
| Intravenous immunoglobulin | 1 (7) | 2 (7) |
| Azathioprine | 0 (0) | 1 (4) |
| Etanercept | 0 (0) | 1 (4) |
| MRSS | 26 ± 11.1 | 24 ± 10.4 |
| Patient global assessment of health | 5 ± 2.8 | 5 ± 2.3 |
| Physician global assessment of health | 5 ± 2.1 | 5 ± 1.8 |
| HAQ DI | 1.5 ± 0.79 | 1.1 ± 0.80 |
| FVC% | 80 ± 10.3 | 86 ± 13.4 |
| CRP, nmoles/liter, median (range) | 19 (2–1,002) | 15 (4–332) |
| IL‐6, pg/ml, median (range) | 3 (1–191) | 3 (1–66) |
| CXCL4 | 9,225 ± 2,694.4 | 8,805 ± 2,704.2 |
Except where indicated otherwise, values are the mean ± SD. SSc = systemic sclerosis; MRSS = modified Rodnan skin thickness score; HAQ DI = Health Assessment Questionnaire disability index; FVC% = forced vital capacity percent predicted; CRP = C‐reactive protein; IL‐6 = interleukin‐6.
Includes mycophenolate mofetil and mycophenolic acid.
Nine (60%) of the placebo‐treated subjects and 17 (63%) of the lenabasum‐treated subjects had AEs (Table 2). There were no deaths. No serious or severe AEs related to lenabasum were observed. Although serum concentrations of lenabasum increased proportionately with dose (data not shown), no dose‐response effect was observed for AEs during weeks 1–4. For total daily doses of 0 mg (placebo), 5 mg, 20 mg, and 40 mg lenabasum, the proportions of subjects with ≥1 AE during weeks 1–4 were 40%, 78%, 56%, and 11%, respectively.
Table 2.
AEs in SSc patients receiving placebo or lenabasum during the 16 weeks of the studya
| Placebo (n = 15) | Lenabasum (n = 27) | |
|---|---|---|
| Subjects with ≥1 AE | 9 (60) | 17 (63) |
| Subjects with ≥1 serious AE | 1 (7) | 1 (4) |
| Abdominal pain | 1 (7) | 0 |
| Vomiting | 0 (0) | 1 (4) |
| Withdrawals because of an AE | 0 (0) | 1 (4) |
| Deaths | 0 (0) | 0 (0) |
| AEs classification by systemb | ||
| Eye disorders | 1 (7) | 0 (0) |
| Gastrointestinal disorder | 3 (20) | 6 (22) |
| General disorders and administration site conditions | 1 (7) | 8 (30) |
| Infections and infestations | 3 (20) | 6 (22) |
| Injury, poisoning, and procedural complications | 2 (13) | 0 (0) |
| Investigations | 3 (20) | 0 (0) |
| Musculoskeletal and connective tissue disorders | 2 (13) | 6 (22) |
| Neoplasms benign, malignant, and unspecified | 0 (0) | 1 (4) |
| Nervous system disorders | 4 (27) | 10 (37) |
| Psychiatric disorders | 2 (13) | 3 (11) |
| Renal and urinary disorders | 0 (0) | 1 (4) |
| Skin and subcutaneous tissue disorders | 2 (13) | 3 (11) |
| Vascular disorders | 0 (0) | 1 (4) |
Values are the number (%) of subjects in the safety population reporting a given event. AEs = adverse events; SSc = systemic sclerosis.
Using Medical Dictionary for Regulatory Activities version 18.0 coding.
AEs that occurred in ≥10% of subjects during the 16 weeks of the study (in the placebo group versus the lenabasum group) were dizziness (13% versus 22%), fatigue (7% versus 19%), headache (7% versus 11%), arthralgia (7% versus 11%), upper respiratory tract infection (0% versus 11%), nausea (13% versus 4%), and decrease in FVC (13% versus 0%). All AEs of dizziness occurred during the first 4 weeks of treatment in both the lenabasum and placebo groups, all were mild except for in 1 subject in each group, who had moderate dizziness, and no dose‐response effect for dizziness was observed in the lenabasum group. One subject (7%) in the placebo group had herpes zoster. There were minimal changes from baseline in both groups for vital signs, hematology tests, liver function tests, renal function tests, electrolytes, electrocardiogram findings, QT/QTc intervals, and Addiction Research Center Inventory‐Marijuana scale scores. There was no evidence of hematologic, hepatic, or renal toxicity. There were no AEs of renal crisis, pulmonary artery hypertension, congestive heart failure, or hospitalizations for infection.
The effect of lenabasum on overall disease was evaluated using the CRISS score and change from baseline in patient global assessment and physician global assessment scores (Figure 1). One subject in the placebo group met step 1 criteria for a CRISS score of 0 due to a decline in FVC% of >15% and FVC% of <80% 18, and none in the lenabasum group did. Improvement in median CRISS scores was observed in the lenabasum group starting at week 8 and increased over time, reaching a maximum of 0.33 (IQR 0.01–0.82), compared to 0.00 (IQR 0.000–0.16) in the placebo group, at week 16 (Figure 1A) (P = 0.04 by 1‐sided MMRM analysis and P = 0.07 by 2‐sided MMRM analysis). Both patient global assessment (Figure 1B) and physician global assessment (Figure 1C) improved with lenabasum treatment. Mean ± SEM treatment differences for patient global assessment were −1.1 ± 0.59 at week 8 (P = 0.04 by 1‐sided MMRM analysis and P = 0.07 by 2‐sided MMRM analysis), −1.2 ± 0.72 at week 12 (P = 0.05 by 1‐sided MMRM analysis and P = 0.11 by 2‐sided MMRM analysis), and −1.2 ± 0.67 at week 16 (P = 0.04 by 1‐sided MMRM analysis and P = 0.08 by 2‐sided MMRM analysis). The mean ± SEM maximum treatment difference for physician global assessment was −0.7 ± 0.4 at week 8 (P = 0.04 by 1‐sided MMRM analysis and P = 0.08 by 2‐sided MMRM analysis).
Figure 1.
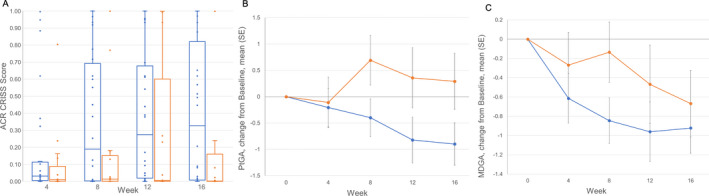
Effects of lenabasum on overall disease in patients with systemic sclerosis (SSc). A, American College of Rheumatology (ACR) Combined Response Index in diffuse cutaneous Systemic Sclerosis (CRISS) score at the indicated weeks in patients with SSc treated with lenabasum (blue) or placebo (orange). CRISS score was not determined at baseline. Data are shown as box plots. Each box represents the 25th to 75th percentiles. Lines inside the boxes represent the median. Lines outside the boxes represent the minimum and maximum values. Circles indicate individual patients (some values overlap). B, Change from baseline in patient global assessment of overall health (PtGA) related to SSc in patients receiving lenabasum (blue) or placebo (orange). Values are the mean ± SEM. The mean ± SD baseline score was 5 ± 2.8 in the placebo group and 5 ± 2.3 in the lenabasum group. C, Change from baseline in physician global assessment of overall health (MDGA) related to SSc in patients receiving lenabasum (blue) or placebo (orange). Values are the mean ± SEM. The mean ± SD baseline score was 5 ± 2.1 in the placebo group and 5 ± 1.8 in the lenabasum group. Missing data were imputed using the last observation carried forward method. For CRISS score at week 16, patient global assessment at week 8, and physician global assessment at week 8, P ≤ 0.05 by 1‐sided mixed‐effects model repeated‐measures (MMRM) analysis and P ≤ 0.10 by 2‐sided MMRM analysis. For CRISS score at week 8 and patient global assessment at week 12, P ≤ 0.10 by 1‐sided MMRM analysis and P ≤ 0.15 by 2‐sided MMRM analysis.
Lenabasum treatment resulted in improvement in 3 measures of skin involvement: change in total MRSS score, change in SSPRO score, and change in 5‐D itch score (Figure 2). The greatest treatment effect was observed at the last time point evaluated for each of these outcome measures: for MRSS, mean ± SEM −2.6 ± 1.9 at week 16 (P = 0.09 by 1‐sided MMRM analysis and P = 0.17 by 2‐sided MMRM analysis); for SSPRO, mean ± SEM −16 ± 6.0 at week 12 (P = 0.005 by 1‐sided MMRM analysis and P = 0.01 by 2‐sided MMRM analysis); and for 5‐D itch score, mean ± SEM −1.8 ± 1.0 at week 12 (P = 0.04 by 1‐sided MMRM analysis and P = 0.08 by 2‐sided MMRM analysis).
Figure 2.
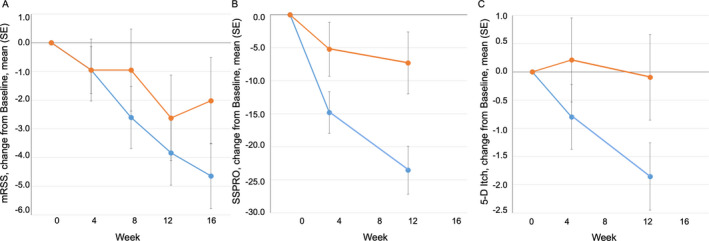
Effects of lenabasum on skin involvement in patients with SSc. A, Change from baseline in modified Rodnan skin thickness score (MRSS) in patients with SSc treated with lenabasum (blue) or placebo (orange). The mean ± SD baseline score was 26 ± 11.1 in the placebo group and 24 ± 10.4 in the lenabasum group. B, Change from baseline in Scleroderma Skin Patient‐Reported Outcome (SSPRO) in patients with SSc treated with lenabasum (blue) or placebo (orange). The mean ± SD baseline score was 83 ± 32.6 in the placebo group and 73 ± 27.3 in the lenabasum group. C, Change from baseline in 5‐D itch score in patients with SSc treated with lenabasum (blue) or placebo (orange). The mean ± SD baseline score was 12.9 ± 5.3 in the placebo group and 10.7 ± 4.4 in the lenabasum group. Values are the mean ± SEM. Missing data were imputed using the last observation carried forward method. For SSPRO at week 12, P ≤ 0.005 by 1‐sided MMRM analysis and P ≤ 0.01 by 2‐sided MMRM analysis. For SSPRO at week 4 and 5‐D itch score at week 12, P ≤ 0.05 by 1‐sided MMRM analysis and P ≤ 0.10 by 2‐sided MMRM analysis. For MRSS at week 16, P ≤ 0.10 by 1‐sided MMRM analysis and P ≤ 0.50 by 2‐sided MMRM analysis. See Figure 1 for other definitions.
The maximal treatment difference for changes in patient‐reported function, as assessed by the HAQ DI and PROMIS‐29 physical function and social role domains, was observed at week 12 (Figure 3). For the HAQ DI, the mean ± SEM treatment difference at 12 weeks was −0.32 ± 0.12 (P = 0.006 by 1‐sided MMRM analysis and P = 0.01 by 2‐sided MMRM analysis). For PROMIS‐29, the mean ± SEM treatment difference at 12 weeks was 3.2 ± 1.9 for physical function (P = 0.05 by 1‐sided MMRM analysis and P = 0.09 by 2‐sided MMRM analysis) and 2.3 ± 1.9 for social role (P not significant).
Figure 3.
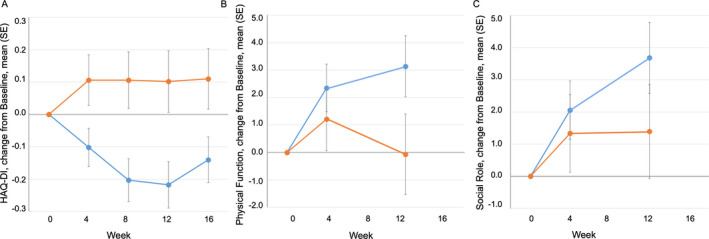
Effects of lenabasum on patient‐reported function in patients with SSc. A, Change from baseline in Health Assessment Questionnaire (HAQ) disability index (DI) in patients with SSc treated with lenabasum (blue) or placebo (orange). The mean ± SD baseline score was 1.5 ± 0.79 in the placebo group and 1.1 ± 0.80 in the lenabasum group. B, Change from baseline in Patient‐Reported Outcomes Measurement Information System 29‐item general health profile (PROMIS‐29) physical function score in patients with SSc treated with lenabasum (blue) or placebo (orange). The mean ± SD baseline score was 38 ± 7.2 in the placebo group and 43 ± 8.5 in the lenabasum group. C, Change from baseline in PROMIS‐29 social role score in patients with SSc treated with lenabasum (blue) or placebo (orange). The mean ± SD baseline score was 41 ± 7.3 in the placebo group and 46 ± 8.59 in the lenabasum group. Values are the mean ± SEM. Missing data were imputed using the last observation carried forward method. For HAQ DI at week 8, P ≤ 0.005 by 1‐sided MMRM analysis and P ≤ 0.01 by 2‐sided MMRM analysis. For HAQ DI at week 12, P ≤ 0.01 by 1‐sided MMRM analysis and P ≤ 0.05 by 2‐sided MMRM analysis. For HAQ DI at week 4 and PROMIS‐29 physical function score at week 12, P ≤ 0.05 by 1‐sided MMRM analysis and P ≤ 0.10 by 2‐sided MMRM analysis. See Figure 1 for other definitions.
FVC% was used as a lung performance measure and improved numerically from baseline in the lenabasum group compared to the placebo group starting at week 8, with a maximal and nonsignificant mean ± SEM treatment difference of 1.7% ± 1.6% observed at week 12.
Gene expression in skin biopsy specimens collected at baseline and at week 12 showed 1,937 genes differentially expressed between treatment groups (data available 12 months after publication at http://www.ncbi.nlm.nih.gov/geo/). Genes with decreased expression after lenabasum treatment included those associated with inflammation and fibrosis: extracellular matrix organization (P = 1.2 × 10−11), collagen metabolism (P = 8.5 × 10−7), inflammatory response (P = 7.3 × 10−7), response to cytokine (P = 2.2 × 10−4), and angiogenesis (P = 1.0 × 10−2) (Figures 4A–D). Histologic examination of skin biopsy specimens showed improvement in the lenabasum group compared to the placebo group in inflammation (P = 0.006 by Fisher's exact test) and fibrosis (P = 0.05 by Fisher's exact test) (Figure 4E).
Figure 4.
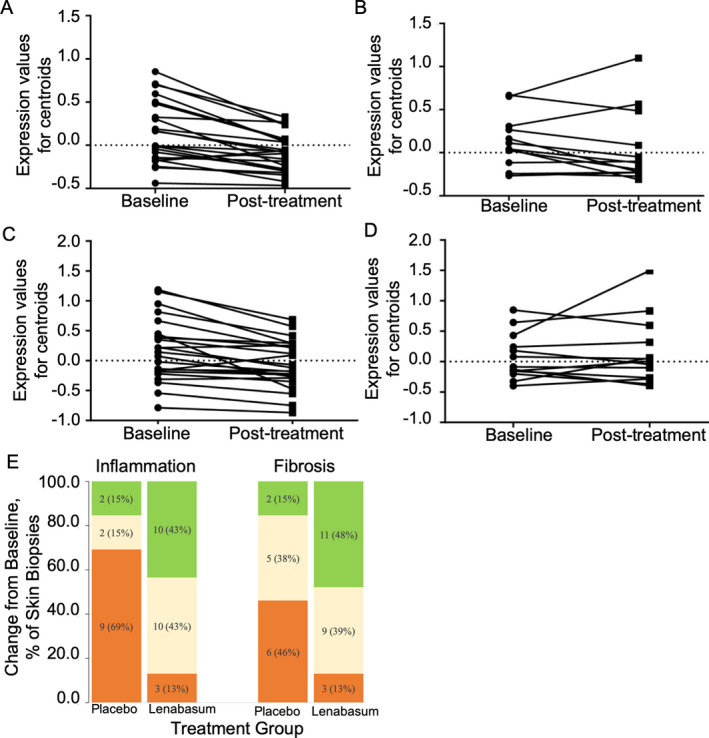
Gene expression, inflammation, and fibrosis in skin biopsy specimens from patients with systemic sclerosis (SSc) treated with lenabasum or placebo. A–D, Gene set centroids for paired biopsy specimens from baseline and week 12. Centroids for each subject were generated by averaging expression values of all genes from the inflammatory response gene set (A and B) and extracellular matrix organization gene set (C and D) separately for baseline and week 12 samples from subjects treated with lenabasum (A and C) or placebo (B and D). P = 0.0002 in A, P = 0.48 in B, P < 0.0001 in C, and P = 0.31 in D, for baseline versus posttreatment, by paired t‐test. E, Change from baseline in inflammation and fibrosis in paired skin biopsy specimens from patients with SSc treated with placebo (n = 13) or lenabasum (n = 23). Values are the number (%) of patients with paired skin biopsy specimens that showed improvement (green), were stable (yellow), or had worsened (orange) at week 12 compared to baseline.
Mean serum levels of CRP, IL‐6, and CXCL4 did not change significantly from baseline in either treatment group and were within normal limits at baseline, week 4, and week 12.
DISCUSSION
This is the first trial to test the impact of modulating the endocannabinoid system through a CB2 agonist in dcSSc patients. In this study, lenabasum treatment was safe, well‐tolerated, and associated with improvement in multiple efficacy assessments of overall disease, skin involvement, and patient‐reported function. These results are encouraging and support the potential for lenabasum to be an effective treatment for dcSSc.
The observed safety profile was consistent with reported effects of endocannabinoid receptor agonists (dizziness, fatigue), clinical manifestations of SSc, and common events in a general population. The most common AE attributed to lenabasum was dizziness, which was associated with study discontinuation in 1 lenabasum‐treated subject. Dizziness also occurred in the placebo group. No severe AEs, infections, or laboratory test abnormalities were attributed to lenabasum treatment.
CB2 is an inducible G protein–coupled receptor that is expressed on activated immune cells 27 and other cell types relevant to SSc, including fibroblasts 14 and endothelial cells 27. CB2 activation inhibits adenylyl cyclase with downstream inhibitory effects on signaling pathways including ERK, JNK, and p38, Smads, and NF‐κB 13, 14, 28, 29. Activation of CB2 leads to engagement of β‐arrestin and changes in ion channels 30. Agonists of CB2 can reduce Toll‐like receptor signaling 31, reduce NLRP3 inflammasome activation 32, reduce the production of multiple inflammatory cytokines 14, 33, 34, 35, 36, increase IL‐10 production 29, 37, induce an M1‐to‐M2 shift in macrophages 31, induce regulatory T cells 37, reduce TGFβ and connective tissue growth factor production 12, 13, 14, reduce myofibroblast accumulation 13, 14, and inhibit collagen production 12, 14. These signaling and biologic effects suggest that activating CB2 might provide benefit in diseases characterized by inflammation and fibrosis.
Indeed, multiple reports suggest that CB2 regulates inflammation and fibrosis in animal models of disease and in humans 11, 12, 13, 14, 31, 32, 33, 34, 35, 36, 37, 38. CB2‐knockout mice develop excessive inflammation and fibrosis in response to different stimuli that activate innate immune responses 32, 38, 39, 40. CB2 agonists including lenabasum lessen inflammation and fibrosis and promote wound healing in animal models 12, 13, 14, 31, 32, 36, 37, 38, 41. Mice exposed to hypochlorite develop an SSc‐like illness with skin and lung inflammation and fibrosis and anti–DNA topoisomerase antibodies 38. This SSc‐like illness improves with treatment with a CB2 agonist 38, and CB2‐knockout mice develop earlier and more skin and lung fibrosis in response to hypochlorite than wild‐type mice 38. Humans with certain CB2 polymorphisms are at an increased risk of developing severe inflammation and fibrosis in response to viral infections and autoimmune diseases 42, 43, 44.
As a CB2 agonist, lenabasum has been shown to activate resolution of an innate immune response in healthy humans, including stimulating production of pro‐resolving lipid mediators and reducing tissue inflammation 11. The results of gene expression analyses and histologic evaluation of skin biopsy specimens in this study are consistent with this mechanism of action and show that lenabasum lessens inflammatory and fibrotic disease pathways in SSc. Although most subjects were taking ≥1 concomitant immunosuppressive drug, they had active inflammation at the site of local skin involvement, as determined by histologic analysis. Worsening of skin inflammation occurred at week 12 in 69% of placebo‐treated subjects but only 13% of lenabasum‐treated subjects. It is unlikely that this difference, as well as differences observed in other outcomes, was due to background immunosuppressive drugs, since background medication was similar in the lenabasum and placebo groups (93% and 87%, respectively).
In exploratory trials of novel therapies in dcSSc, it could be important to identify potential treatment effects in smaller studies of shorter duration. In this first study of lenabasum in SSc patients, potential treatment effect was explored by testing multiple efficacy outcome measures that encompassed overall disease, skin involvement, and patient‐reported function. As a composite outcome measure, CRISS was selected as the primary efficacy outcome measure because it was thought likely to be more sensitive to treatment effect than change in MRSS over a short period of time, acknowledging the challenges of using CRISS in a 16‐week study when it was originally described for use in 52‐week studies 18. In this study, a potential treatment effect of lenabasum was indeed identified using the CRISS score. The physician global assessment and patient global assessment were used as 2 complementary measures of overall health related to SSc and also showed improvement with lenabasum treatment, consistent with the observed improvement in CRISS score. Combined, these 3 outcome measures suggest that lenabasum may provide treatment benefit on overall disease in dcSSc patients.
The mean improvement from baseline in MRSS was −4.6 points at week 16 with lenabasum treatment, reaching a level within the physician‐reported minimally important difference (MID) range of −3.2 to −5.3 in the phase III study of D‐penicillamine 45 and −4.6 to −5.1 at 12 months in the Scleroderma Lung Studies 46. To provide additional evaluation of the effects of lenabasum on skin involvement in the present study, the SSPRO and 5D‐itch scores were assessed. Both showed improvement in the lenabasum group compared to the placebo group. This is the first clinical study to prospectively use the SSPRO 22 to measure patient‐reported symptoms specific to skin involvement in SSc, and its ability to demonstrate a treatment effect suggests its potential usefulness in future trials and as an earlier indicator of improvement in skin in SSc patients than MRSS. Improvement in mean 5D‐itch scores began at 4 weeks in the lenabasum group and is of interest given reports that lenabasum reduces production of IL‐31, which is thought to be a key cytokine driving itch 47.
A potential treatment effect of lenabasum was seen in HAQ DI scores, with improvement in mean HAQ DI score noted within 2 weeks of treatment. This effect was noteworthy, given the short duration of this study and previous finding that mean HAQ DI scores are generally stable to worsening during the course of a clinical study 48. The mean improvement from baseline in HAQ DI reached a level within the estimated physician‐reported MIDs of −0.1 to −0.14 at various times in the phase III study of D‐penicillamine 45 and patient‐reported MIDs (75th percentiles) of −0.125 to −0.250 49. Moderately high correlations have been reported between change in the HAQ DI and change in the PROMIS‐29 physical function score 50. Consistent with those findings, an improvement in PROMIS‐29 physical function score was seen in the lenabasum group in this study.
The limitations of this first‐in‐patient study include a small size and short duration. Missing data were imputed using the LOCF method, which assumes that the outcomes of participants do not change after they have dropped out, which could lead to biased treatment effects when this assumption is not met and could result in biased estimators. However, sensitivity analyses were performed on observed cases (no imputation) for the primary and key secondary end points and CRISS score results were similar, regardless of LOCF or non‐LOCF analysis. It would be important to consider additional sensitivity analyses in a more definitive phase III study. Overall background treatment with immunosuppressive drugs was balanced between the treatment groups, and its use as a covariate in an MMRM model did not substantially alter the results. The use of prednisone and methotrexate was higher, and the use of mycophenolate was lower, in lenabasum‐treated compared to placebo‐treated patients. The impact of these immunosuppressive therapies on observed outcomes will need to be further assessed in a larger future study.
A strength of the present study was the use of the CRISS score as the primary efficacy outcome measure to discern early evidence of a treatment effect of lenabasum compared to placebo. Another strength was the consistency of improvement across multiple physician‐ and patient‐reported outcomes that spanned overall disease, skin involvement, and patient function. By allowing subjects to take stable background immunosuppressive medications, we tested lenabasum under treatment conditions that reflect current standard‐of‐care in clinical practice. This approach likely had a beneficial effect on patient and physician acceptance of the protocol. Overall, the results of this phase II study indicate potential efficacy of lenabasum in dcSSc patients, with consistent improvement in multiple physician‐ and patient‐reported outcomes and apparent improvement in underlying disease mechanisms as shown in skin biopsies. Further evaluation of lenabasum, a CB2 agonist, as a treatment for SSc and other inflammatory and fibrotic diseases is warranted.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Spiera had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Spiera, Constantine, Lee, White.
Acquisition of data
Spiera, Hummers, Chung, Frech, Domsic, Hsu, Furst, Gordon, Mayes, Simms, Lafyatis, Martyanov, Wood, Whitfield.
Analysis and interpretation of data
Spiera, Furst, Lafatis, Martyanov, Whitfield, Dgetluck, White.
ROLE OF THE STUDY SPONSOR
As the study sponsor, Corbus Pharmaceuticals is responsible for the collection of the data. Corbus Pharmaceuticals and the study authors contributed to study design, the analysis and interpretation of the data, the writing of the manuscript, and the decision to submit the manuscript for publication. Publication of this article was not contingent upon approval by Corbus Pharmaceuticals.
ClinicalTrials.gov identifier: NCT02465437.
Supported by Corbus Pharmaceuticals.
1Robert Spiera, MD, Jessica Gordon, MD: Weill Cornell Medicine, New York, New York; 2Laura Hummers, MD: Johns Hopkins University School of Medicine, Baltimore, Maryland; 3Lorinda Chung, MD: Stanford University School of Medicine, Stanford, California, and Palo Alto VA Health Care System, Palo Alto, California; 4Tracy M. Frech, MD: University of Utah and Salt Lake City VA Health Care System; 5Robyn Domsic, MD, Robert Lafyatis, MD: University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania; 6Vivien Hsu, MD: Rutgers Robert Wood Johnson Medical School, New Brunswick, New Jersey; 7Daniel E. Furst, MD: University of California, Los Angeles; 8Maureen Mayes, MD: University of Texas Health Science Center at Houston; 9Robert Simms, MD: Boston University School of Medicine, Boston, Massachusetts; 10Viktor Martyanov, PhD, Tammara Wood, MS, Michael L. Whitfield, PhD: Geisel School of Medicine at Dartmouth, Hanover, New Hampshire; 11Scott Constantine, MS, Elizabeth Lee, BS, Nancy Dgetluck, MS, Barbara White, MD: Corbus Pharmaceuticals, Norwood, Massachusetts.
Dr. Spiera has received consulting fees, speaking fees, and/or honoraria from GlaxoSmithKline, ChemoCentryx, Boehringer Ingelheim, CSL Behring, Sanofi, Janssen, Formation Biologics, Cytori, and Roche/Genentech (less than $10,000 each) and research support from Corbus Pharmaceuticals. Dr. Hummers has received consulting fees, speaking fees, and/or honoraria from CSL Behring, Corbus Pharmaceuticals, Boehringer Ingelheim, Cumberland Pharmaceuticals, Sanofi‐Aventis, and GlaxoSmithKline (less than $10,000 each). Dr. Chung has received consulting fees, speaking fees, and/or honoraria from Bristol‐Myers Squibb and Mitsubishi Tanabe Pharma (less than $10,000 each) and from Eicos Sciences, Boehringer Ingelheim, and Reata (more than $10,000 each). Dr. Domsic has received consulting fees, speaking fees, and/or honoraria from Boehringer Ingelheim and Eicos Sciences (less than $10,000 each). Dr. Hsu has received consulting fees, speaking fees, and/or honoraria from Corbus Pharmaceuticals and Boehringer Ingelheim (less than $10,000 each). Dr. Furst has received research support from Corbus Pharmaceuticals. Dr. Gordon has received consulting fees, speaking fees, and/or honoraria from Corbus Pharmaceuticals and Cumberland Pharmaceuticals (less than $10,000 each) and research support from Corbus Pharmaceuticals. Dr. Mayes has received consulting fees, speaking fees, and/or honoraria from Medtelligence, Actelion Pharma, Astellas Pharma, Mitsubishi Tanabe Pharma, Bayer, Reata, Sanofi, Corbus Pharmaceuticals, Boehringer Ingelheim, and Eicos Sciences (less than $10,000 each) and research support from Corbus Pharmaceuticals. Dr. Lafyatis has received consulting fees, speaking fees, and/or honoraria from Bristol Myers Squibb, Boehringer Mannheim, Merck, and Genentech/Roche (less than $10,000 each) and research support from Formation, Elpidera, and Kiniksa. Dr. Martyanov has received consulting fees, speaking fees, and/or honoraria from Corbus Pharmaceuticals and Celdara Medical (less than $10,000 each). Ms Wood has received consulting fees, speaking fees, and/or honoraria from Corbus Pharmaceuticals (less than $10,000). Dr. Whitfield has received consulting fees, speaking fees, and/or honoraria from Corbus Pharmaceuticals and Celdara Medical (less than $10,000 each). Mr. Constantine, Ms Lee, Ms Dgetluck, and Dr. White own stock or stock options in Corbus Pharmaceuticals. No other disclosures relevant to this article were reported.
References
- 1. Lafyatis R, York M. Innate immunity and inflammation in systemic sclerosis. Curr Opin Rheumatol 2009;21:6–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Frech TM, Shanmugam VK, Shah AA, Assassi S, Gordon JK, Hant FN, et al. Treatment of early diffuse systemic sclerosis skin disease. Clin Exp Rheumatol 2013;31 Suppl 76:166–71. [PMC free article] [PubMed] [Google Scholar]
- 3. Pope JE, Bellamy N, Seibold JR, Baron M, Ellman M, Carette S, et al. A randomized, controlled trial of methotrexate versus placebo in early diffuse scleroderma. Arthritis Rheum 2001;44:1351–8. [DOI] [PubMed] [Google Scholar]
- 4. Tashkin DP, Elashoff R, Clements PJ, Roth MD, Furst DE, Silver RM, et al. Effects of 1‐year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med 2007;176:1026–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D, Kleerup EC, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma‐related interstitial lung disease (SLS II): a randomised controlled, double‐blind, parallel group trial. Lancet Respir Med 2016;4:708–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Steen VD, Medsger TA Jr. Case‐control study of corticosteroids and other drugs that either precipitate or protect from the development of scleroderma renal crisis. Arthritis Rheum 1998;41:1613–9. [DOI] [PubMed] [Google Scholar]
- 7. Burt RK, Shah SJ, Dill K, Grant T, Gheorghiade M, Schroeder J, et al. Autologous non‐myeloablative haemopoietic stem‐cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open‐label, randomised phase 2 trial. Lancet 2011;378:498–506. [DOI] [PubMed] [Google Scholar]
- 8. Van Laar JM, Farge D, Sont JK, Naraghi K, Marjanovic Z, Larghero J, et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA 2014;311:2490–8. [DOI] [PubMed] [Google Scholar]
- 9. Sullivan KM, Goldmuntz EA, Keyes‐Elstein L, McSweeney PA, Pinckney A, Welch B, et al. Myeloablative autologous stem‐cell transplantation for severe scleroderma. N Engl J Med 2018;378:35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tepper MA, Zurier RB, Burstein SH. Ultrapure ajulemic acid has improved CB2 selectivity with reduced CB1 activity. Bioorg Med Chem 2014;22:3245–51. [DOI] [PubMed] [Google Scholar]
- 11. Motwani MP, Bennett F, Norris PC, Maini AA, George MJ, Newson J, et al. Potent anti‐inflammatory and pro‐resolving effects of anabasum in a human model of self‐resolving acute inflammation. Clin Pharmacol Ther 2018;104:675–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gonzalez EG, Selvi E, Balistreri E, Akhmetshina A, Palumbo K, Lorenzini S, et al. Synthetic cannabinoid ajulemic acid exerts potent antifibrotic effects in experimental models of systemic sclerosis. Ann Rheum Dis 2012;71:1545–51. [DOI] [PubMed] [Google Scholar]
- 13. Lucattelli M, Fineschi S, Selvi E, Garcia Gonzalez E, Bartalesi B, de Cunto G, et al. Ajulemic acid exerts potent anti‐fibrotic effect during the fibrogenic phase of bleomycin lung. Respir Res 2016;17:49–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garcia‐Gonzalez E, Selvi E, Balistreri E, Lorenzini S, Maggio R, Natale MR, et al. Cannabinoids inhibit fibrogenesis in diffuse systemic sclerosis fibroblasts. Rheumatology (Oxford) 2009;48:1050–6. [DOI] [PubMed] [Google Scholar]
- 15. Van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum 2013;65:2737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Khanna D, Furst DE, Clements PJ, Allanore Y, Baron M, Czirjak L, et al. Standardization of the modified Rodnan skin score for use in clinical trials of systemic sclerosis. J Scleroderma Relat Disord 2017;2:11–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clements PJ, Wong WK, Hurwitz EL, Furst DE, Mayes M, White B, et al. Correlates of the disability index of the health assessment questionnaire: a measure of functional impairment in systemic sclerosis. Arthritis Rheum 1999;42:2372–80. [DOI] [PubMed] [Google Scholar]
- 18. Khanna D, Berrocal VJ, Giannini EH, Seibold JR, Merkel PA, Mayes MD, et al. The American College of Rheumatology Provisional Composite Response Index for clinical trials in early diffuse cutaneous systemic sclerosis. Arthritis Rheumatol 2016;68:299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general US population. Am J Respir Crit Care Med 1999;59:179–87. [DOI] [PubMed] [Google Scholar]
- 20. Kwakkenbos L, Thombs BD, Khanna D, Carrier ME, Baron M, Furst DE, et al. Performance of the Patient‐Reported Outcomes Measurement Information System‐29 in scleroderma: a Scleroderma Patient‐centered Intervention Network Cohort study. Rheumatology (Oxford) 2017;56:1302–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Elman S, Hynan LS, Gabriel V, Mayo MJ. The 5‐D itch scale: a new measure of pruritus. Br J Dermatol 2010;162:587–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Man A, Correa JK, Ziemek J, Simms RW, Felson DT, Lafyatis R. Development and validation of a patient‐reported outcome instrument for skin involvement in patients with systemic sclerosis. Ann Rheum Dis 2017;76:1374–80. [DOI] [PubMed] [Google Scholar]
- 23. Kissin EY, Merkel PA, Lafyatis R. Myofibroblasts and hyalinized collagen as markers of skin disease in systemic sclerosis. Arthritis Rheum 2006;54:3655–60. [DOI] [PubMed] [Google Scholar]
- 24. Taroni JN, Martyanov V, Huang CC, Mahoney JM, Hirano I, Shetuni B, et al. Molecular characterization of systemic sclerosis esophageal pathology identifies inflammatory and proliferative signatures. Arthritis Res Ther 2015;17:194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gould J, Getz G, Monti S, Reich M, Mesirov JP. Comparative gene marker selection suite. Bioinformatics 2006;22:1924–5. [DOI] [PubMed] [Google Scholar]
- 26. Reimand J, Arak T, Vilo J. g:Profiler–a web server for functional interpretation of gene lists (2011 update). Nucleic Acids Res 2011;39:W307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Galiègue S, Mary S, Marchand J, Dussossoy D, Carrière D, Carayon P, et al. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem 1995;232:54–61. [DOI] [PubMed] [Google Scholar]
- 28. Demuth DG, Molleman A. Cannabinoid signalling. Life Sci 2006;78:549–63. [DOI] [PubMed] [Google Scholar]
- 29. Correa F, Hernangómez M, Mestre L, Loría F, Spagnolo A, Docagne F, et al. Anandamide enhances IL‐10 production in activated microglia by targeting CB2 receptors: roles of ERK1/2, JNK, and NF‐κB. Glia 2010;58:135–47. [DOI] [PubMed] [Google Scholar]
- 30. Franklin JM, Vasiljevik T, Prisinzano TE, Carrasco GA. Cannabinoid agonists increase the interaction between β‐Arrestin 2 and ERK1/2 and upregulate β‐Arrestin 2 and 5‐HT2A receptors. Pharmacol Res 2013;68:46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tomar S, Zumbrun EE, Nagarkatti M, Nagarkatti PS. Protective role of cannabinoid receptor 2 activation in galactosamine/lipopolysaccharide‐induced acute liver failure through regulation of macrophage polarization and microRNAs. J Pharmacol Exp Ther 2015;353:369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ke P, Shao BZ, Xu ZQ, Wei W, Han BZ, Chen XW, et al. Activation of cannabinoid receptor 2 ameliorates DSS‐induced colitis through inhibiting NLRP3 inflammasome in macrophages. PLoS One 2016;11:e0155076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zurier RB, Rossetti RG, Burstein SH, Bidinger B. Suppression of human monocyte interleukin‐1β production by ajulemic acid, a nonpsychoactive cannabinoid. Biochem Pharmacol 2003;65:649–55. [DOI] [PubMed] [Google Scholar]
- 34. Parker J, Atez F, Rossetti RG, Skulas A, Patel R, Zurier RB. Suppression of human macrophage interleukin‐6 by a nonpsychoactive cannabinoid acid. Rheumatol Int 2008;28:631–5. [DOI] [PubMed] [Google Scholar]
- 35. Robinson ES, Bashir M, Alves P, Feng R, Werth VP. Ajulemic acid, a novel cannabinoid, suppresses the secretion of tumor necrosis factor α and interferon α from the peripheral blood mononuclear cells of dermatomyositis patients in vitro. J Investigative Dermatol 2015;135:S10. [Google Scholar]
- 36. Guillot A, Hamdaoui N, Bizy A, Zoltani K, Souktani R, Zafrani ES, et al. Cannabinoid receptor 2 counteracts interleukin‐17‐induced immune and fibrogenic responses in mouse liver. Hepatology 2014;59:296–306. [DOI] [PubMed] [Google Scholar]
- 37. Robinson RH, Meissler JJ, Fan X, Yu D, Adler MW, Eisenstein TK. A CB2‐selective cannabinoid suppresses T cell activities and increases Tregs and IL‐10. J Neuroimmune Pharmacol 2015;10:318–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Servettaz A, Kavian N, Nicco C, Deveaux V, Chéreau C, Wang A, et al. Targeting the cannabinoid pathway limits the development of fibrosis and autoimmunity in a mouse model of systemic sclerosis. Am J Pathol 2010;177:187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Trebicka J, Racz I, Siegmund SV, Cara E, Granzow M, Schierwagen R, et al. Role of cannabinoid receptors in alcoholic hepatic injury: steatosis and fibrogenesis are increased in CB2 receptor‐deficient mice and decreased in CB1 receptor knockouts. Liver Int 2011;31:860–70. [DOI] [PubMed] [Google Scholar]
- 40. Defer N, Wan J, Souktani R, Escoubet B, Perier M, Caramelle P, et al. The cannabinoid receptor type 2 promotes cardiac myocyte and fibroblast survival and protects against ischemia/reperfusion‐induced cardiomyopathy. FASEB J 2009;23:2120–30. [DOI] [PubMed] [Google Scholar]
- 41. Wang LL, Zhao R, Li JY, Li SS, Liu M, Wang M, et al. Pharmacological activation of cannabinoid 2 receptor attenuates inflammation, fibrogenesis, and promotes re‐epithelialization during skin wound healing. Eur J Pharmacol 2016;786:128–36. [DOI] [PubMed] [Google Scholar]
- 42. Coppola N, Zampino R, Bellini G, Stanzione M, Capoluongo N, Marrone A, et al. The impact of the CB2‐63 polymorphism on the histological presentation of chronic hepatitis B. Clin Microbiol Infect 2015;21:609. [DOI] [PubMed] [Google Scholar]
- 43. Sipe JC, Arbour N, Gerber A, Beutler E. Reduced endocannabinoid immune modulation by a common cannabinoid 2 (CB2) receptor gene polymorphism: possible risk for autoimmune disorders. J Leukoc Biol 2005;78:231–8. [DOI] [PubMed] [Google Scholar]
- 44. Rossi F, Bellini G, Tolone C, Luongo L, Mancusi S, Papparella A, et al. The cannabinoid receptor type 2 Q63R variant increases the risk of celiac disease: implication for a novel molecular biomarker and future therapeutic intervention. Pharmacol Res 2012;66:88–94. [DOI] [PubMed] [Google Scholar]
- 45. Khanna D, Furst DE, Hays RD, Park GS, Wong WK, Seibold JR, et al. Minimally important difference in diffuse systemic sclerosis: results from the D‐penicillamine study. Ann Rheum Dis 2006;65:1325–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Khanna D, Clements PJ, Volkmann ER, Wilhalme H, Tseng CH, Furst DE, et al. Minimal clinically important differences for the modified Rodnan Skin Score: results from the Scleroderma Lung Studies (SLS‐I and SLS‐II). Arthritis Res Ther 2019;21:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim HJ, Zeidi M, Bonciani D, Pena SM, Tiao J, Sahu S, et al. Itch in dermatomyositis: the role of increased skin interleukin‐31. Br J Dermatol 2018;179:669–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Merkel PA, Silliman NP, Clements PJ, Denton CP, Furst DE, Mayes MD, et al, for the Scleroderma Clinical Trials Consortium . Patterns and predictors of change in outcome measures in clinical trials in scleroderma: an individual patient meta‐analysis of 629 subjects with diffuse cutaneous systemic sclerosis. Arthritis Rheum 2012;64:3420–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sekhon S, Pope J, Canadian Scleroderma Research Group , Baron M. The minimally important difference in clinical practice for patient‐centered outcomes including health assessment questionnaire, fatigue, pain, sleep, global visual analog scale, and SF‐36 in scleroderma. J Rheumatol 2010;37:591–8. [DOI] [PubMed] [Google Scholar]
- 50. Hinchcliff ME, Beaumont JL, Carns MA, Podlusky S, Thavarajah K, Varga J, et al. Longitudinal evaluation of PROMIS‐29 and FACIT‐dyspnea short forms in systemic sclerosis. J Rheumatol 2015;42:64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]