

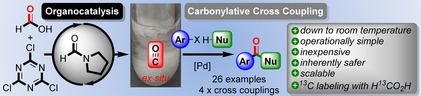
Abstract
Herein, the first organocatalytic method for the transformation of non‐derivatized formic acid into carbon monoxide (CO) is introduced. Formylpyrrolidine (FPyr) and trichlorotriazine (TCT), which is a cost‐efficient commodity chemical, enable this decarbonylation. Utilization of dimethylformamide (DMF) as solvent and catalyst even allows for a rapid CO generation at room temperature. Application towards four different carbonylative cross coupling protocols demonstrates the high synthetic utility and versatility of the new approach. Remarkably, this also comprehends a carbonylative Sonogashira reaction at room temperature employing intrinsically difficult electron‐deficient aryl iodides. Commercial 13C‐enriched formic acid facilitates the production of radiolabeled compounds as exemplified by the pharmaceutical Moclobemide. Finally, comparative experiments verified that the present method is highly superior to other protocols for the activation of carboxylic acids.
Keywords: C1 building blocks, carbon monoxide, carbonylation, CO surrogates, homogenous catalysis, organocatalysis
Double formicable: A simple formamide catalyst enables the rapid transformation of formic acid into carbon monoxide (CO) using inexpensive trichlorotriazine (TCT). Application towards four different carbonylative cross couplings down to room temperature showcases high levels of synthetic utility and versatility. The organocatalytic ex situ formation of CO using commercial H13CO2H allows the synthesis of radiolabeled compounds such as the drug Moclobemide.
Introduction
Carbon monoxide (CO) is one of the most important building blocks in chemistry in both, academia and industry.1, 2 Fundamental applications are, for example, carbonylative cross couplings and hydrocarbonylations of alkenes and alkynes (Scheme 1 A). Since CO is a highly toxic and flammable, odorless gas, its utilization is inevitably associated with severe safety hazards and risks. Therefore, compounds that liberate CO under controlled conditions in stoichiometric quantities have been developed (Scheme 1 B).2 These so‐called CO surrogates are either directly added to the reaction mixture for the in situ CO production, or CO is formed ex situ in a separate reaction vessel. In the latter case, the CO generating and consuming process are spatial separated, for which they do not need to be compatible. Beneficially, this facilitates the substitution of gaseous CO through a CO surrogate significantly. A gas exchange is secured by a connection of the two chambers. Several two chamber gas reactors have been constructed for the ex situ gas synthesis,2, 3a whereby some devices are even commercially available.4
Scheme 1.

Previous and this work. [TM]=transition metal catalyst, dba=dibenzylidene acetone, DIPEA=di‐iso‐propyl ethyl amine, FPyr=N‐formylpyrrolidine, TCT=2,4,6‐trichloro‐1,3,5‐triazine, rt=room temperature.
Examples of common commercial CO surrogates2 are (1) carboxylic acid chlorides like COgen 1 and sila derivatives such as SilaCOgen 2, which have been implemented by Skrydstrup and Lindhardt mainly for the ex situ CO preparation.2c, 2e, 2f, 4 (2) N‐Formyl saccharin (3), the application of which has been pioneered by Manabe (in situ) and Fleischer (ex situ).2b, 5 (3) Aryl formates like 4, which have been discovered by Manabe for the in situ CO production,2b, 2g, 6 and (4) metal carbonyl complexes like 5 as introduced by Larhed for the in and ex situ CO release.2i, 7
In fact, formic acid (6) is an even simpler and significantly cheaper source of CO, which has been initially applied in situ for hydrocarbonylations of alkenes and alkynes and carboxylations of aryl halides, respectively.8, 9 Furthermore, the group of Wu published a range of protocols for carbonylative cross couplings using the mixed anhydride of formic and acetic acid, which was synthesized from Ac2O and HCO2H in prior.10 In addition, the same team discovered that carbodiimides promote the in situ development of CO directly from methanoic acid.11 Finally, de Borggraeve introduced decarbonylations of 6 with methylsulfonyl chloride (MsCl) and NEt3.12
Certain heterogeneous catalysts13 and Brønsted acids like H2SO4 14 facilitate the ex situ decarbonylation of formic acid. Eventually, Cantat and co‐workers recently described a chemical looping strategy for the decarbonylation of formic acid.15 Therein, methyl formate was transformed ex situ to CO and MeOH by means of MeOK as transition metal‐free catalyst.
So far, decarbonylation of methanoic acid typically requires high reaction temperatures of 75–190 °C.8, 9, 10, 11, 13, 14 The only notable exception constitutes the activation with MsCl and NEt3 according to de Borggraeve.12 Alongside lower temperatures were only accomplished in rather specific cases.10a, 11b, 13a Under consideration of the cost‐efficiency of formic acid, the development of approaches for its ex situ decarbonylation under milder conditions (e.g. at room temperature) and with a broader applicability constitutes an indispensable task.
Lately, our group discovered that Lewis bases16 like 1‐formylpyrrolidine (FPyr) and dimethylformamide (DMF) catalyze the transformation of carboxylic acids into the respective acid chlorides by means of trichlorotriazine (TCT), which is also denoted as cyanuric chloride (see Scheme 1 C for structure).17a, 18b Indeed, TCT is the most cost‐efficient reagent for the activation of OH groups like in alcohols and acids besides phosgene (COCl2).17a Since cyanuric chloride contains three Cl atoms, it can be applied in substoichiometric quantities down to 33 mol % with respect to the substrate (100 mol %). In fact, the beneficially low stoichiometry is facilitated by formamide catalysis. Against this background, we envisioned a novel ex situ CO formation approach based on formic acid (Scheme 1 C).
Activation of formic acid should either yield formyl chloride 7 or an intermediate like I, which bears a similarly good leaving group as chloride. Actually, formic acid chloride is known to be labile towards decarbonylation, wherefore a rapid decomposition to CO was predicted.19 Surprisingly, CO evolution from methanoic acid using common chlorination agents has not been reported in the realm of transition metal catalyzed transformations so far.20 Actually, organocatalytic conversion of HCO2H into CO is a challenging task, which has to this end only been accomplished through preceding derivatization to methyl formate.15, 21 Herein, a novel operationally simple method for the transformation of formic acid into CO based on organocatalysis and ex situ application towards four different carbonylative cross couplings is disclosed.
Results and Discussion
At the outset, the reaction of methanoic acid with TCT was investigated by means of gas volumetry (Figure 1, see chp. 2 of the supporting information=SI for details). The use of DMF as solvent and catalyst enabled a rapid CO generation already at room temperature (Figure 1 A, see chp. 2.2 of the Supporting Information). Notably, DMF is a common and inexpensive organic solvent. When TCT was applied in excess (41 mol %=1.2 equiv) with respect to methanoic acid (1.0 equiv), already after 4.5 min (=t2/3) a gas yield n(gas)/n0(HCO2H) of >67 % was noted (green points). Since at room temperature solvent and HCl evaporation can be neglected, the gas yield is equivalent to the yield of CO and consequently also conversion of formic acid. In the present work, at most 1.5 equiv of HCO2H were engaged in the carbonylative transformations (see Scheme 2 and Scheme 3). Hence, 67 % conversion comply to 1 equiv of CO being generated. After 75 min 98 % gas yield were reached, which substantiates an almost quantitative conversion of 6. Such a fast CO generation was not expected, since under catalytic conditions a similarly rapid conversion of carboxylic acids into acid chlorides afforded heating to 80 °C.17a
Figure 1.

Gas volumetry to follow CO formation: A) in DMF, B) solvent screening and C) alteration of catalyst loading. n(gas)=amount of gas as determined by the ideal gas equation, n0(HCO2H)=initial amount of formic acid, THF=tetrahydrofuran, EtOAc=ethyl acetate, 2‐MeTHF=2‐methyltetrahydrofurane.
Scheme 2.

Application of the organocatalytic CO formation (A) towards various carbonylative cross couplings at elevated temperatures (B–G). Yields refer to isolated material after chromatographic purification. For detailed reaction conditions see SI. Bn=benzyl, PMP=para‐methoxyphenyl.
Scheme 3.

A) CO formation and B+C) carbonylative Sonogashira coupling at room temperature. mTol=meta‐tolyl, a. For conditions see Scheme 2 A. b. Prepared from 2.5 equiv of formic acid and 95 mol % TCT. EWG=electron withdrawing group, mTol=meta‐tolyl.
To the best of our knowledge, only SilaCOgen (2),4b which is more expensive than formic acid, and activation of formic acid with MsCl and NEt3 12 allow a similarly fast gas development. Although very limited kinetic data is available, based on the reported reaction temperatures the current approach enables a considerably faster CO formation than almost all other procedures using formic acid.8, 9, 10, 11, 12, 13, 14 With an excess of formic acid (2 equiv) with regard to TCT (33 mol %=1 equiv) the evolution of gas is slower (yellow points).
Nevertheless, after 90 min 80 % yield in terms of CO had been accomplished. This verifies the partial substitution of the third Cl atom of TCT, as also observed in the cases of carboxylic acid and alcohol chlorinations (see mechanism in Scheme 4).17a, 18b From a solvent screening using 10 mol % of FPyr at 70 °C MeCN and THF emerged as optimal (chp. 2.3 in the Supporting Information). Remarkably, the initial rate of gas formation in DMF at room temperature was four times faster than in MeCN at 70 °C.
Scheme 4.

Comparative aminocarbonylations. For detailed reaction conditions see Supporting Information, yield determined with internal standard. [a] isolated yield. NMM=N‐methylmorpholine.
Eventually, a variation of the catalyst loading in MeCN demonstrated the tremendous effect of FPyr (Figure 1 C, chp. 2.4 in the Supporting Information). In these measurements the reaction mixture was heated to 70 °C for 60 min. Both, the gas yield after cooling down to ambient temperature and the initial rate clearly increased from 5 over 10 to 30 mol % of FPyr (green points). Interestingly, a slower development of gas was observed using 10 mol % of DMF (yellow points) than with 5 mol % FPyr, which attests the latter as superior organocatalyst. The blue data points indicate the formation of gas in the absence of TCT, which was similar to heating MeCN to 70 °C (see chp. 2.4, Supporting Information). Therefore, the formed gas consists only of evaporated solvent. As an important aspect, the gas amount was even slightly lower in the absence of FPyr (red points). This highlights the importance of FPyr as catalyst: Without this formamide basically no CO is created at all.
To probe the synthetic utility, in several carbonylative cross couplings either gaseous CO or another CO surrogate was substituted by the present approach (Scheme 2). These reactions were carried out in a commercial two chamber gas reactor named COware from Sytracks, which facilitates the spatially separated ex situ formation of CO (see Scheme 1 C and chp. 3.2 in the Supporting Information).4
In carbonylative transformations at elevated temperatures CO was generated using 10 mol % of FPyr in reaction chamber 1 (Scheme 2 A). As a first example, SilaCOgen (2)4b and Mo(CO)6,7c respectively, were replaced by the present method in the aminocarbonylation of aryl halides 8 (Scheme 2 B). An increase of the NEt3 amount in the chamber 2, in which the amincarbonylation was carried out, by the amount of methanoic acid employed (1.5 equiv) facilitated the neutralization of HCl, which arose from the decarbonylation.
Importantly, production of CO using different solvents furnished amide 10 a in good yields of 81–91 % at 80 °C, which was required for the C‐N cross coupling (Scheme 2 C). Albeit the solvent has a significant impact on the rate of CO evolution (see above), the mitigated effect on the yield of 10 a may be explained by a rather sluggish direct coupling of butyl amine and aryl iodide 8 a. In addition, CO production at room temperature in DMF afforded 10 a in 84 % yield (for conditions see Scheme 3 A). Thus, different solvents can be engaged in the CO generating and consuming process, cross diffusion causes no considerable difficulties. In general, CO formation in DMF is recommended for carbonylative transformations that are carried out at temperatures below 70 °C. At higher temperatures catalytic CO release preferably in MeCN, THF or dioxane is basically equally efficient.
In fact, FPyr could also be substituted by the twofold amount of simpler DMF, which furnished the amide 10 a in 85 % yield. Also aryl bromides 11 could be engaged as starting materials, when the reaction temperature was increased to 100 °C. Worthy of note, in situ CO preparation is in general less suitable for aminocarbonylations, since CO surrogates are usually electrophilic and hence react with amines.7c Indeed, attempted in situ CO release in one reaction vessel furnished 10 a only in minor traces ≤3 %, which testifies the crucial role of two chamber gas reactor. A high synthetic utility is witnessed by the preparation of the insecticide DEET (10 c) and the pharmaceutical Moclobemide 10 b (Scheme 2 D). Actually, the use of commercial H13CO2H as CO surrogate afforded isotopically labeled 10 b. This example also unambiguously accounts for formic acid as CO source. Moreover, a reasonable scalability was demonstrated by the gram synthesis of amide 10 j.
Furthermore, in the alkoxycarbonylation of aryl bromides 11 an atmosphere of CO22a and formic acid decarbonylation at 150 °C using zeolite,13b respectively, were substituted by the current methodology (Scheme 2 E). This process and the aforementioned aminocarbonylation allowed the synthesis of the products 10 d, 12 a and 12 b with acid‐labile functions (Scheme 2 F). These precedents evidence that the simultaneous HCl formation exerts no influence on the functional group compatibility. Thereby, particularly remarkable is example 12 b, which bears a highly acid susceptible acyclic acetal. Furthermore, formamide catalyzed CO release could replace COgen (1) in the preparation of α‐nitro ketone 13 a from nitromethane and the respective iodide 8 a (Scheme 2 G).22b
Ultimately, in a challenging carbonylative Sonogashira coupling at room temperature gaseous CO23a was substituted by the present approach using DMF as solvent (Scheme 3). Albeit in most cases CO was formed at room temperature in DMF as solvent (Scheme 3 A), examples 15 a+b show that catalytic CO release at 80 °C is also feasible. Especially in the case of electron deficient aryl iodides of type 8 the direct Sonogashira coupling without CO incorporation usually is predominant.24
In the past, this problem has been circumvented by exploitation of the expensive and sensitive ligand P(tBu)3.24b In contrast, the present approach allows for the synthesis of the ynones 15 e–h even derived from highly electron poor aryl iodides containing nitro groups using plain PPh3 (Scheme 3 C). Indeed, under standard conditions with 1.5 equiv CO ynone 15 f, which is deduced from electron‐deficient para‐cyanophenyl iodide, was formed as side‐product in 31 % yield besides the respective Sonogashira coupling product in 40 % yield (Scheme 3 C and Supporting Information). An increase of the amount of CO to 2.5 equiv resulted in a minor improvement of the yield to 41 %. However, when CuI was omitted, the desired ketone 15 f could be isolated in 69 % yield. Moreover, even electron poorer 4‐nitrophenyl iodide could be transformed into the corresponding carbonylated product 15 g in 59–62 % yield. As an important aspect, also more reactive aryl alkynes, are suitable substrates, as confirmed by the synthesis of ynone 15 i. Again, without CuI the yield could be essential improved from 55 to 91 %.
Moreover, the amide 10 a was formed in up to 88 % yield when formic acid was engaged as yield‐limiting starting material (1.0 equiv, see chp. 1.3 in the Supporting Information). This outcome proves high levels of CO incorporation. As an important feature, the present method is operationally simple and does not require a reaction setup in a glove box to exclude air like some other CO surrogates (see also graphical procedure in chp. 3.2.2 in the Supporting Information).
To the best of our knowledge, reactions of formic acid with ordinary chlorination agents like oxalyl and thionyl chloride have so far not been harnessed to access CO for carbonylative transformations. Against this background, a rigorous comparative assessment against other protocols for the activation of carboxylic acids was carried out (Scheme 4). Thereby, several experiments for each method were conducted under variation of the amount of base, which is why ranges of yields are stated (see also chp. 1.2 in the Supporting Information).
Indeed, reaction of formic acid with either thionyl or oxalyl chloride (SOCl2 or C2O2Cl2)20 afforded the model amide 10 a in low yields of 5–8 % and 44–54 %, respectively, while the current method provides this amide in 81 % yield. Despite in situ use of the mixed anhydride of formic and acetic acid allows carbonylative cross couplings,10 ex situ preparation from Ac2O and 6 delivered 10 a in synthetically non‐useful yields of 37–42 %. TCT and the amine base NMM have been engaged for the activation of carboxylic acids as mixed anhydrides with cyanuric acid.25 When this protocol was applied to methanoic acid, amide 10 a arose in yields of 51–56 %. Hence, the current organocatalytic approach is indeed superior to other carboxylic acid activation strategies.
The gas volumetric measurements confirmed that basically no decarbonylation takes place without FPyr (Figure 1 C). In contrast, the amide 10 a was still obtained in 35–42 % yield, when FPyr was omitted. A likely explanation for this result is the diffusion of NEt3 from the CO consuming chamber 2 into the CO producing chamber 1, which could effect mixed anhydride activation as in the case of TCT/NMM (see above).
In alignment to our previous work,17a the CO formation should be initiated by a nucleophilic aromatic substitution (SNAr) of a Cl atom of TCT through FPyr (Scheme 5). The emerging intermediate IIa shows structural similarities to the Vilsmeier Haack reagent. Next, substitution with formic acid would deliver salt I and dichlorohydroxytriazine (16). Intermediate I possesses a good leaving group and could undergo decarbonylation directly.
Scheme 5.

Proposed mechanism.
Alternatively, I could also proceed a nucleophilic substitution to yield formyl chloride (7), which is labile and would consequently decompose to CO and HCl.19 As demonstrated through the gas volumetry with an excess of HCO2H (Figure 1 A), 16 passes sequential transformation with FPyr and methanoic acid to generate intermediate I and chlorodihydroxytriazine (17). Finally, the remaining Cl atom of 17 is at least in part replaced by FPyr to again afford carboxy iminium salt I and cyanuric acid. This was proven by gas volumetry (Figure 1 A) and in addition cyanuric acid was confirmed as by‐product in our previous contributions.17a, 18b
Conclusions
The first organocatalytic method for the ex situ formation of CO from non‐derivatized formic acid has been presented. Formamide catalysis enabled the transformation into CO by means of substoichiometric amounts of the bulk chemical TCT. As an important aspect, utilization of DMF as solvent and catalyst facilitates a rapid CO generation at room temperature. Implementation to four different carbonylative cross couplings, which also includes a carbonylative Sonogashira reaction at room temperature with challenging electron poor aryl iodides, proved high synthetic value. High levels of practical relevance were certified by the synthesis of bioactive compounds, namely DEET and Moclobemide.
In addition, isotopically labeled molecules like the drug Moclobemide are amenable using commercial 13C‐enriched formic acid. Although HCl is generated simultaneously, even very acid sensitive functional groups are fully compatible. In order to exchange gaseous CO or other CO surrogates through the current methodology, the amount of base simply has to be adapted according to the amount of formic acid. Finally, comparison experiments witnessed that the present approach is superior to other common carboxylic acid activation procedures.
We are convinced that the high levels of synthetic utility and versatility and the low costs associated will pave the way for a rapid uptake of the current approach. Current efforts are dedicated towards the exploitation of CO and HCl for product incorporation.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We want to thank the German research foundation (DFG) and the Fonds of the Chemical Industry (Liebig fellowship for P.H.) for generous support. In addition, we would like to thank Rudolf Thomes for measuring HR‐MS. Open access funding enabled and organized by Projekt DEAL.
B. Zoller, J. Zapp, P. H. Huy, Chem. Eur. J. 2020, 26, 9632.
References
- 1.Selected reviews on carbonylations:
- 1a. Ojima I., Chem. Rev. 1988, 88, 1011–1030; [Google Scholar]
- 1b. Brennführer A., Neumann H., Beller M., ChemCatChem 2009, 1, 28–41; [Google Scholar]
- 1c. Franke R., Selent D., Börner A., Chem. Rev. 2012, 112, 5675–5732; [DOI] [PubMed] [Google Scholar]
- 1d. Quesnel J. S., Arndtsen B. A., Pure Appl. Chem. 2013, 85, 377–384; [Google Scholar]
- 1e. Wu X.-F., Fang X., Wu L., Jackstell R., Neumann H., Beller M., Acc. Chem. Res. 2014, 47, 1041–1053; [DOI] [PubMed] [Google Scholar]
- 1f. Wu L., Fang X., Liu Q., Jackstell R., Beller M., Wu X.-F., ACS Catal. 2014, 4, 2977–2989; [Google Scholar]
- 1g. Gautam P., Bhanage B. M., Catal. Sci. Technol. 2015, 5, 4663–4702; [Google Scholar]
- 1h. Bai Y., Davis D. C., Dai M., J. Org. Chem. 2017, 82, 2319–2328; [DOI] [PubMed] [Google Scholar]
- 1i. Li Y., Hu Y., Wu X.-F., Chem. Soc. Rev. 2018, 47, 172–194; [DOI] [PubMed] [Google Scholar]
- 1j. Peng J. B., Geng H.-Q., Wu X.-F., Chem. 2019, 5, 526–552; [Google Scholar]
- 1k. Peng J.-B., Wu F.-P., Wu X.-F., Chem. Rev. 2019, 119, 2090–2127. [DOI] [PubMed] [Google Scholar]
- 2.Selected reviews on CO surrogates:
- 2a. Wu L., Liu Q., Jackstell R., Beller M., Angew. Chem. Int. Ed. 2014, 53, 6310–6320; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6426–6436; [Google Scholar]
- 2b. Konishim H., Manabe K., Synlett 2014, 25, 1971–1986; [Google Scholar]
- 2c. Friis S. D., Lindhardt A. T., Skrydstrup T., Acc. Chem. Res. 2016, 49, 594–605; [DOI] [PubMed] [Google Scholar]
- 2d. Cao J., Zheng Z.-J., Xu Z., Xu L.-W., Coord. Chem. Rev. 2017, 336, 43–53; [Google Scholar]
- 2e. Nielsen D. U., Neumann K. T., Lindhardt A. T., Skrydstrup T., J. Label. Compd. Radiopharm. 2018, 61, 949–987; [DOI] [PubMed] [Google Scholar]
- 2f. Nielsen D. U., Neumann K. T., Skrydstrup T., Chimia 2018, 72, 606–609; [DOI] [PubMed] [Google Scholar]
- 2g. Konishi H., Chem. Pharm. Bull. 2018, 66, 1–19; [DOI] [PubMed] [Google Scholar]
- 2h. Mondal K., Halder P., Gopalan G., Sasikumar P., Radhakrishnan K. V., Das P., Org. Biomol. Chem. 2019, 17, 5212–5222; [DOI] [PubMed] [Google Scholar]
- 2i. Åkerbladh L., Odell L. R., Larhed M., Synlett 2019, 30, 141–155. [Google Scholar]
- 3.Review on ex situ gas generation:
- 3a. Demaerel J., Veryser C., De Borggraeve W. M., React. Chem. Eng. 2020, 5, 615–631 10.1039/C9RE00497A; for examples not concerning CO see: [DOI] [Google Scholar]
- 3b. Modvig A., Andersen T. L., Taaning R. H., Lindhardt A. T., Skrydstrup T., J. Org. Chem. 2014, 79, 5861–5868; [DOI] [PubMed] [Google Scholar]
- 3c. Kristensen S. K., Eikeland E. Z., Taarning E., Lindhardt A. T., Skrydstrup T., Chem. Sci. 2017, 8, 8094–8105; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Kristensen S. K., Laursen S. L. R., Taarning E., Skrydstrup T., Angew. Chem. Int. Ed. 2018, 57, 13887–13891; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14083–14087. [Google Scholar]
- 4.
- 4a. Hermange P., Lindhardt A. T., Taaning R. H., Bjerglund K., Lupp D., Skrydstrup T., J. Am. Chem. Soc. 2011, 133, 6061–6071; [DOI] [PubMed] [Google Scholar]
- 4b. Friis S. D., Taaning R. H., Lindhardt A. T., Skrydstrup T., J. Am. Chem. Soc. 2011, 133, 18114–18117; [DOI] [PubMed] [Google Scholar]
- 4c. Collin H. P., Reis W. J., Nielsen D. U., Lindhardt A. T., Valle M. S., Freitas R. P., Skrydstrup T., Org. Lett. 2019, 21, 5775–5778; [DOI] [PubMed] [Google Scholar]; commercial gas reactors (COWare) are available at Sytracks (https://www.sytracks.com/) and via Sigma–Aldrich.
- 5.
- 5a. Ueda T., Konishi H., Manabe K., Angew. Chem. Int. Ed. 2013, 52, 8611–8615; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8773–8777; [Google Scholar]
- 5b. Gehrtz P. H., Hirschbeck V., Fleischer I., Chem. Commun. 2015, 51, 12574–12577; [DOI] [PubMed] [Google Scholar]
- 5c. Hirschbeck V., Gehrtz P. H., Fleischer I., J. Am. Chem. Soc. 2016, 138, 16794–16799. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Ueda T., Konishi H., Manabe K., Org. Lett. 2012, 14, 3100–3103; [DOI] [PubMed] [Google Scholar]
- 6b. Ueda T., Konishi H., Manabe K., Org. Lett. 2012, 14, 5370–5373; [DOI] [PubMed] [Google Scholar]
- 6c. Li H., Neumann H., Beller M., Wu X.-F., Angew. Chem. Int. Ed. 2014, 53, 3183–3186; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3247–3250; [Google Scholar]
- 6d. Konishi H., Matsubara M., Mori K., Tokiwa T., Arulmozhiraja S., Yamamoto Y., Ishikawa Y., Hashimoto H., Shigeta T., Tokiwa H., Manabe K., Adv. Synth. Catal. 2017, 359, 3592–3601. [Google Scholar]
- 7.
- 7a. Kaiser N.-F. K., Hallberg A., Larhed M., J. Comb. Chem. 2002, 4, 109–111; [DOI] [PubMed] [Google Scholar]
- 7b. Wannberg J., Larhed M., J. Org. Chem. 2003, 68, 5750–5753; [DOI] [PubMed] [Google Scholar]
- 7c. Nordeman P., Odell L. R., Larhed M., J. Org. Chem. 2012, 77, 11393–11398; [DOI] [PubMed] [Google Scholar]
- 7d. Schacht M., Mohammadi D., Schützenmeister N., Eur. J. Org. Chem. 2019, 2587–2591. [Google Scholar]
- 8.
- 8a. Simonato J.-P., Walter T., Métivier P., J. Mol. Catal. A 2001, 171, 91–94; [Google Scholar]
- 8b. Simonato J.-P., J. Mol. Catal. A 2003, 197, 61–64; [Google Scholar]
- 8c. Korsager S., Taaning R. H., Skrydstrup T., J. Am. Chem. Soc. 2013, 135, 2891–2894; [DOI] [PubMed] [Google Scholar]
- 8d. Sang R., Kucmierczyk P., Dong K., Franke R., Neumann H., Jackstell R., Beller M., J. Am. Chem. Soc. 2018, 140, 5217–5223. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Hou J., Xie J.-H., Zhou Q.-L., Angew. Chem. Int. Ed. 2015, 54, 6302–6305; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6400–6403; [Google Scholar]
- 9b. Wang Y., Rena W., Shi Y., Org. Biomol. Chem. 2015, 13, 8416–8419; [DOI] [PubMed] [Google Scholar]
- 9c. Fu M.-C., Shang R., Cheng W.-M., Fu Y., ACS Catal. 2016, 6, 2501–2505; [Google Scholar]
- 9d. Hou J., Yuan M.-L., Xiea J.-H., Zhou Q.-L., Green Chem. 2016, 18, 2981–2984; [Google Scholar]
- 9e. Wang Y., Zeng Y., Yanga B., Shi X., Org. Chem. Front. 2016, 3, 1131–1136; [Google Scholar]
- 9f. Liu W., Ren W., Li J., Shi Y., Chang W., Shi Y., Org. Lett. 2017, 19, 1748–1751; [DOI] [PubMed] [Google Scholar]
- 9g. Jiang J., Fu M., Li C., Shang R., Fu Y., Organometallics 2017, 36, 2818–2825; [Google Scholar]
- 9h. Wu F.-P., Peng J.-B., Qi X., Wu X.-F., J. Org. Chem. 2017, 82, 9710–9714. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Qi X., Jiang L.-B., Li C.-L., Li R., Wu X.-F., Chem. Asian J. 2015, 10, 1870–1873; [DOI] [PubMed] [Google Scholar]
- 10b. Qi X., Jiang L.-B., Li H.-P., Wu X.-F., Chem. Eur. J. 2015, 21, 17650–17656; [DOI] [PubMed] [Google Scholar]
- 10c. Qi X., Li C.-L., Wu X.-F., Chem. Eur. J. 2016, 22, 5835–5838; [DOI] [PubMed] [Google Scholar]
- 10d. Qi X., Li H.-P., Wu X.-F., Chem. Asian J. 2016, 11, 2453–2457; [DOI] [PubMed] [Google Scholar]
- 10e. Qi X., Li C.-L., Jiang L.-B., Zhang W.-Q., Wu X.-F., Catal. Sci. Technol. 2016, 6, 3099–3107; [Google Scholar]
- 10f. Li C.-L., Zhang W.-Q., Qi X., Peng J.-B., Wu X.-F., J. Organomet. Chem. 2017, 838, 9–11; [Google Scholar]
- 10g. Qi X., Zhou C., Peng J.-B., Ying J., Wu X.-F., Tetrahedron Lett. 2017, 58, 4153–4155. [Google Scholar]
- 11.
- 11a. Seo Y.-S., Kim D.-S., Jun C.-H., Chem. Asian J. 2016, 11, 3508–3512; [DOI] [PubMed] [Google Scholar]
- 11b. Peng J.-B., Wu F.-P., Li C.-L., Qi X., Wu X.-F., Eur. J. Org. Chem. 2017, 1434–1437; [Google Scholar]
- 11c. Wu F.-P., Peng J.-B., Meng L.-S., Qi X., Wu X.-F., ChemCatChem 2017, 9, 3121–3124; [Google Scholar]
- 11d. Wu F.-P., Peng J.-B., Qia X., Wu X.-F., Catal. Sci. Technol. 2017, 7, 4924–4928; [Google Scholar]
- 11e. Qi X., Ai H.-J., Cai C.-X., Peng J.-B., Ying J., Wu X.-F., Eur. J. Org. Chem. 2017, 7222–7225; [Google Scholar]
- 11f. Wu F.-P., Peng J.-B., Fu L.-Y., Qi X., Wu X.-F., Org. Lett. 2017, 19, 5474–5477; [DOI] [PubMed] [Google Scholar]
- 11g. Wu F.-P., Peng J.-B., Qi X., Wu X.-F., ChemCatChem 2018, 10, 173–177; [Google Scholar]
- 11h. Wu F.-P., Li D., Peng J.-B., Wu X.-F., Org. Lett. 2019, 21, 5699–5703. [DOI] [PubMed] [Google Scholar]
- 12. Veryser C., Van Mileghem S., Egle B., Gilles P., De Borggraeve W. M., React. Chem. Eng. 2016, 1,142–146. [Google Scholar]
- 13.
- 13a. Kuehnel M. F., Wakerley D. W., Orchard K. L., Reisner E., Angew. Chem. Int. Ed. 2015, 54, 9627–9631; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9763–9767; [Google Scholar]
- 13b. Losch P., Felten A.-S., Pale P., Adv. Synth. Catal. 2015, 357, 2931–2938. [Google Scholar]
- 14.
- 14a. Brancour C., Fukuyama T., Mukai Y., Skrydstrup T., Ryu I., Org. Lett. 2013, 15, 2794–2797; [DOI] [PubMed] [Google Scholar]
- 14b. Yin Z., Wu X.-F., Org. Process Res. Dev. 2017, 21, 1869–1871; [Google Scholar]
- 14c. Morgan J. S., J. Chem. Soc. Trans. 1916, 109, 274–283. [Google Scholar]
- 15. Imberdis A., Lefèvre G., Cantat T., Angew. Chem. Int. Ed. 2019, 58, 17215–17219; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 17375–17379. [Google Scholar]
- 16.A recent review on Lewis base catalysis in SN-reactions:
- 16a. An J., Denton R. M., Lambert T. H., Nacsa E. D., Org. Biomol. Chem. 2014, 12, 2993–3003; [DOI] [PubMed] [Google Scholar]
- 16b. Huy P. H., Hauch T., Filbrich I., Synlett 2016, 27, 2631–2636; [Google Scholar]
- 16c. Huy P. H., Eur. J. Org. Chem. 2020, 10–27. [Google Scholar]
- 17.
- 17a. Huy P. H., Mbouhom C., Chem. Sci. 2019, 10, 7399–7406; for a related tropone-catalyzed activation of carboxylic acids by using oxalyl chloride see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Nguyen T. V., Bekensir A., Org. Lett. 2014, 16, 1720–1723; [DOI] [PubMed] [Google Scholar]
- 17c. Nguyen T. V., Lyons D. J. M., Chem. Commun. 2015, 51, 3131–3134. [DOI] [PubMed] [Google Scholar]
- 18.Related formamide-catalyzed chlorination of alcohols and aldehydes:
- 18a. Huy P. H., Motsch S., Kappler S. M., Angew. Chem. Int. Ed. 2016, 55, 10145–10149; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10300–10304; [Google Scholar]
- 18b. Huy P. H., Filbrich I., Chem. Eur. J. 2018, 24, 7410–7416; [DOI] [PubMed] [Google Scholar]
- 18c. Huy P. H., Synthesis 2019, 51, 2474–2483. [Google Scholar]
- 19.Review formylation agents:
- 19a. Olah G. A., Ohannesian L., Arvanaghi M., Chem. Rev. 1987, 87, 671–686; preparation and decarbonylation of formyl chloride: [Google Scholar]
- 19b. Staab H. A., Datta A. P., Angew. Chem. Int. Ed. Engl. 1964, 3, 132; [Google Scholar]; Angew. Chem. 1963, 75, 1203; [Google Scholar]
- 19c. Dowideit P., Mertens R., von Sonntag C., J. Am. Chem. Soc. 1996, 118, 11288–11292; [Google Scholar]
- 19d. Villeneuve G. B., Chan T. H., Tetrahedron Lett. 1997, 38, 6489–6492; [Google Scholar]
- 19e. Bagno A., Kantlehner W., Kress R., Saielli G., Stoyanov E., J. Org. Chem. 2006, 71, 9331–9340; [DOI] [PubMed] [Google Scholar]
- 19f. Bagnoa A., Kantlehner W., Saielli G., J. Phys. Org. Chem. 2008, 21, 682–687. [Google Scholar]
- 20.For ex situ CO formation with oxalyl chloride, a reagent commonly applied for the conversion of acids in acid chlorides, and either NaOH or Zn, see:
- 20a. Hansen S. V. F., Ulven T., Org. Lett. 2015, 17, 2832–2835; [DOI] [PubMed] [Google Scholar]
- 20b. Markovič M., Lopatka P., Koós P., Gracza T., Org. Lett. 2015, 17, 5618–5621. [DOI] [PubMed] [Google Scholar]
- 21.For the organocatalytic conversion of HCO2H to CO2, see: Chauvier C., Tlili A., Das Neves Gomes C., Thuéry P., Cantat T., Chem. Sci. 2015, 6, 2938–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Martinelli J. R., Watson D. A., Freckmann D. M. M., Barder T. E., Buchwald S. L., J. Org. Chem. 2008, 73, 7102–7107; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Lian Z., Friis S. D., Skrydstrup T., Chem. Commun. 2015, 51, 3600–3603; for a recent intriguing photocatalytic strategy overcoming barriers in carbonylative cross couplings allowing amino- and alkoxycarbonylations at room temperature, see: [Google Scholar]
- 22c. Torres G. M., Liu Y., Arndtsen B. A., Science 2020, 368, 318–323. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Karpov A. S., Merkul E., Rominger F., Müller T. J. J., Angew. Chem. Int. Ed. 2005, 44, 6951–6956; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 7112–7117; For a review about ynones see: [Google Scholar]
- 23b. Nájera C., Sydnes L. K., Yus M., Chem. Rev. 2019, 119, 11110–11244. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Liang B., Huang M., You Z., Xiong Z., Lu K., Fathi R., Chen J., Yang Z., J. Org. Chem. 2005, 70, 6097–6100; [DOI] [PubMed] [Google Scholar]
- 24b. Iizuka M., Kondo Y., Eur. J. Org. Chem. 2007, 5180–5182. In addition, ynones derived from aromatic alkynes and (electron-deficient) aryl iodides have been prepared applying PPh3 at 30 °C using the mixed anhydride of formic and acetic acid.[10a, 11b] [Google Scholar]
- 25.
- 25a. Rayle H. L., Fellmeth L., Org. Process Res. Dev. 1999, 3, 172–176; [Google Scholar]
- 25b. Forbes D. C., Barrett E. J., Lewis D. L., Smith M. C., Tetrahedron Lett. 2000, 41, 9943–9947. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary