

Abstract
Cannabinoid subtype 2 receptors (CB2Rs) are G protein‐coupled receptors (GPCRs) belonging to the endocannabinoid system, a complex network of signalling pathways leading to the regulation of key physiological processes. Interestingly, CB2Rs are strongly up‐regulated in pathological conditions correlated with the onset of inflammatory events like cancer and neurodegenerative diseases. Therefore, CB2Rs represent an important biological target for therapeutic as well as diagnostic purposes. No CB2R‐selective drugs are yet on the market, thus underlining a that deeper comprehension of CB2Rs’ complex activation pathways and their role in the regulation of diseases is needed. Herein, we report an overview of pharmacological and imaging tools such as fluorescent, positron emission tomography (PET), photochromic and covalent selective CB2R ligands. These molecular probes can be used in vitro as well as in vivo to investigate and explore the unravelled role(s) of CB2Rs, and they can help to design suitable CB2R‐targeted drugs.
Keywords: drug design, endocannabinoid system, G protein-coupled receptors, inflammation, pharmacologic tools
Exploring cannabinoid subtype 2 receptor: CB2R is upregulated in pathological conditions involving the onset of inflammatory processes, but no selective drug for CB2R is on the market and there is still a lot of uncertainty about its activation pathway. The development of potent and selective CB2R functionalized probes provides valuable tools to deepen our knowledge of the CB2R universe.

1. Introduction
Cannabinoid subtype 2 receptor (CB2R) belongs to the superfamily of G protein‐coupled receptors (GPCRs) and together with cannabinoid subtype 1 receptor (CB1R), embraced by their respective cascades of signalling pathways, constitute the endocannabinoid system (ECS), a complex and pleiotropic endogenous signalling system.1, 2 Besides CBRs, the ECS includes also a wide variety of biological players like ionotropic cannabinoid receptors (e. g., transient receptor potential channels), nuclear hormone receptors (e. g., PPAR family) and all the catabolic/anabolic enzymes involved in EC metabolism (e. g., FAAH, MAGL), which jointly regulate essential homeostatic processes.3 Since the cloning of the CB2R in 1993, CBRs have basically been distinguished according to their biodistribution: CB1R in the brain with neuromodulatory properties and CB2R peripherically with immunomodulatory properties.4 Nevertheless, nowadays this characterization has been superseded by the discovery of low spotted expression of CB2R in the central nervous system (CNS)5 and widely distribution of CB1R also in the peripherical immune cells.6, 7
Unlike the well‐characterized CB1R's signal transduction pathways, for CB2R different possible activation pathways exist, still not fully elucidated, depending on the respective tissue, agonist applied and pathological conditions involved. For sure, we know that CB2R signalling is mediated via Gi/o proteins, thereby reducing cAMP levels, triggering kinases cascade or intracellular calcium mobilization through phospholipase C (PLC) activation and via β‐arrestins causing receptor desensitization and internalization.2, 8 Due to a remarkable variety and complexity of CB2R's activation pathways, certain CB2R‐selective ligands can cause biased signalling, inducing different pharmacological outputs owed to a preferential activation of a particular effector cascade among others.9, 10, 11 In drug development, such intriguing functional selectivity can cause problems in obtaining a clear and full pharmacological evaluation of CB2R ligands, but also stimulates CB2R biased drugs discovery to evaluate their potential therapeutic approach.12
The CB2R has gained major attention as potential pharmacological target for its peculiarly strong up‐regulation in pathological conditions correlated with the onset of inflammatory processes such as in cancer and in neurodegenerative diseases.13, 14, 15, 16 Furthermore, CB2R activation is related to pharmacological effects without unwanted psychotropic effects related to CB1R.17, 18 In particular, triggering CB2R‐driven cascades leads to strong reduction of inflammatory processes through the switch of microglia into the M2 anti‐inflammatory phenotype, which promotes the release of anti‐inflammatory cytokines.8, 19 For example, it has been found in post‐mortem cortical brain tissues of neuropathologically confirmed Alzheimer's disease (AD) patients that expression of CB2Rs was 40 % higher than in healthy brain and that their activation in AD animal models not only reduced inflammatory response but also seemed able to decrease amyloid‐toxicity, facilitating its clearance, and improve cognitive performance.18, 20 Moreover, in solid tumours, such as renal cell carcinoma,21 breast,15 colon16 or non‐small‐cell lung14 cancers, researchers have demonstrated a correlation between CB2R overexpression and tumour size, aggressiveness or growth and that their activation in vitro inhibits tumour growth and metastasis. The same correlation between overexpression and beneficial effects derived from CB2R activation was found also in other neuroinflammatory disorders like Parkinson's disease (PD),22 Huntington's disease (HD),23 amyotrophic lateral sclerosis (ALS)24 or peripheral pathological conditions such as rheumatoid arthritis, inflammatory bowel disease and atherosclerosis.8 Despite all the promising experimental observations listed above there is still no selective CB2R drug approved and all clinical trials involving CB2R ligands failed until now. They have been clinically evaluated in neuropathic pain, osteoarthritis, neurodegenerative diseases (e. g., AD, PD, multiple sclerosis, amyotrophic lateral sclerosis, HD) resulting in generally safe drugs, but with poor therapeutic efficiencies.25
It has been almost one year now that the crystal structure of hCB2R is known, albeit in an antagonist‐bound structure, whereas still more recently the structure of the agonist‐bound hCB2R‐Gi signalling complex was elucidated by cryo‐EM.26 These findings will facilitate rational structure‐based design of selective and high affinity ligands. However, also with the previous ligand‐based approaches there were numerous highly potent and selective compounds published, but unfortunately not taking the last step from preclinical premises to clinical application.27 This lack of translation may not only suggest possible weaknesses in the predictive utility of the preclinical models and important CBRs interspecies differences, but also highlight that further understanding of the CB2R's pharmacology is needed under pathophysiological as well as physiological conditions.10, 28, 29 First of all, it should be noted that CB2R belongs to GPCR family and ECS is a lipid signalling pathway which are both proved to be promiscuous regulatory systems not leading to an univocal ligand‐binding activation cascade. In addition, the assay's readouts should always be correlated to the employed experimental conditions and not arise as a final verdict, instead of a relative definition. For example, cellular outcomes will vary depending on values like receptor density, analysed downstream signalling molecules, intrinsic efficacy, type of cell lines and all of these parameters should always be reported with the activity rank.28 It seems fundamental also to keep in mind bias correlated to functional selectivity as described above.11, 30 Furthermore, different ligands can be correlated to different CBRs pharmacology only depending on the specific biological assay applied, while in a more complex physiological setting they may act differently. At last, there is the need to deepen our insights about CB2R regulation or expression in physiological and diseased conditions (e. g., chronic activation, desensitization, mechanisms of upregulation), as well as deflections correlated to proven inter‐species divergences.28, 29
Regarding the above issues, researchers have focused their efforts to the development of chemical tools that allow the unsolved questions surrounding CB2R pharmacology to be addressed, and therefore optimise and support the discovery of a successful CB2R drug. Here are reported advances in the field of selective CB2R ligands properly functionalized for positron emission tomography (PET) and fluorescent, photochromic or covalent chemical probes, respectively, used as imaging and pharmacological tools (Figure 1).
Figure 1.
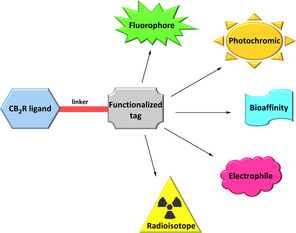
General template for the development of selective CB2R functionalized ligands.
These probes should enable better “visualization” of the actions of the receptor and deepen our knowledge on how it behaves in the regulation of physiological processes or pathophysiological pathways of the various disorders in which is involved. This review will not deal about other CB2R functionalized ligands such as multifunctional and bivalent compounds or pharmacological tools like antibodies which have been discussed elsewhere.7, 31
2. CB2R PET Radiotracers
PET is a powerful non‐invasive imaging technique providing high‐sensitivity and real‐time monitoring of patho/physiological conditions.32 As opposed to fluorescent imaging, PET needs radiation exposure but the radioisotope insertion into the selected ligands is often less structural‐altering than fluorescent moiety introduction and, furthermore, it has an immediate ex vivo and/or in vivo application. Thanks to these properties, development of selective PET radiotracers can provide valuable pharmacological opportunities to quantitatively visualize expression of biological patterns and help evaluation of the pharmacokinetic or pharmacodynamic properties of a drug concerned.33 CB2R's PET imaging has mainly been exploited for studying and deepen knowledge about neuroinflammation and their involvement in it.34 Up to now, no clinically approved CB2R PET radiotracer has been reported, due to the occurrence of poor in vivo specificity/selectivity, unfavourable metabolic fate, low brain uptake associated with poor CB2R expression levels in non‐diseased conditions in the central nervous system (CNS). Despite this, several PET radiotracers for CB2R imaging have been developed and a complete structure–activity relationship (SAR) analysis of them has been discussed in a recently published review by Spinelli et al.35 Herein, selected highlights of most recent advances in CB2R PET radiotracer development are reported, and a glimpse on the more promising in vivo CB2R PET imaging tools is presented.
2.1. Oxoquinolines’ radiotracer journey: from [11C]NE‐40 to [11C]RS‐028
The first high‐affinity CB2R‐selective radiotracer able to pass the blood–brain barrier (BBB) was developed by Evens et al. in 2009 who achieved nanomolar affinity and high selectivity over CB1R with solid in vivo imaging results in mice, e. g. high brain uptake and fast brain washout ([11C]NE40, Figure 2).36 [11C]NE40 was further tested in a rat model with local overexpression of hCB2R confirming in vivo specific binding (e. g., 2.5‐fold uptake compared to control and a successful blocking experiment), and in microPET studies in rhesus monkey and healthy men corroborating valid brain penetration and clearance.37 Starting from these promising results of the 2‐oxoquinoline scaffold, the 3‐carboxamide function was further explored to optimise CB2R affinity and selectivity. [11C]‐KP23 showed the highest affinity and selectivity among all the 2‐oxoquinoline derivatives (Figure 2). It showed promising in vitro and in vivo spleen uptake (well‐described CB2R rich tissue), whereas it lacked distribution to the brain in the in vivo PET experiments, probably due to the high non‐specific binding.38 With the 4‐oxoquinoline [11C]KD2, the same promising experimental outputs in term of specific binding found in the in vitro characterization were not translated to in vivo brain uptake because of its relatively high plasma protein binding and lipophilicity (Figure 2).39 After a SAR campaign on exchanging the N1‐substituent with a more hydrophilic ether chain, [11C]RS‐016 maintained all effective affinity/selectivity [11C]KD2’s properties and added higher specific binding when compared to in vitro autoradiography studies. Rat brain distribution of [11C]RS‐016 was low under healthy conditions, whereas in a neuroinflammation mouse model higher and more specific CNS uptake was demonstrated in blocking studies in vitro and in vivo.40 In order to obtain a radiotracer with longer half‐life, fluorinated derivatives of RS‐016 were evaluated.41 RS‐126 bearing a terminal fluoroethyl chain was selected for further biological evaluation (Figure 2). [18F]RS‐126 exhibited high specific binding in rat spleen (79 %) in vivo but it was missing brain uptake in lipopolysaccharide (LPS)‐treated mice using CB2R expression as inflammatory marker. This is probably due to lower specific activity of the tracer and its fast in vivo defluorination, not providing sufficient BBB crossing.41 Fluorinated RS‐126’s derivatives with longer and different N1‐alkyl chain were correlated to higher metabolic stability at in vitro level, but losing CB2R selectivity.42
Figure 2.
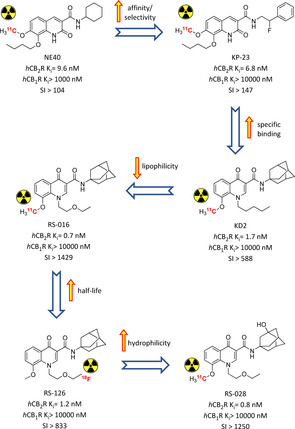
Radiolabelled oxoquinoline derivatives developed in a step‐by‐step optimisation of pharmacokinetic and metabolic properties.36, 37, 38, 39, 40, 41, 42 Affinities reported in the picture are those of the non‐radioactive analogues.
More recently [18F]RS‐126 was used to visualize CB2Rs in amyotrophic lateral sclerosis (ALS) spinal cord tissue, revealing high unspecific binding under the selected experimental conditions. In order to overcome this, more hydrophilic [11C]RS‐028 (ClogP 0.94 vs 2.74 of RS‐126) was evaluated, showing important radioactivity detection in ALS tissue compared to healthy one, which was further reduced in a blocking experiment (Figure 2).42 [11C]RS‐028 demonstrated stability in brain, plasma, spleen in an ex vivo rat study with a moderate spleen uptake correlated with a very fast washout.42 All these findings corroborated the possibility to effectively exploit the oxoquinoline scaffold for in vitro PET studies, but until now also showed strong metabolic and pharmacokinetic down‐sides.
2.2. Oxadiazole derivatives
Around an oxadiazole core a lot of substituents were tested in search of novel cannabinoid ligands suitable for PET‐radiotracer development. Bulky aromatic substituents linked via alkylene chains to the central oxadiazole exhibited the best biological premises for potential imaging applications. Fluorinated carbazole derivatives [18F]1 and [18F]2 represented a starting point for further investigations as they combine high CB2R affinity and selectivity with very fast metabolic degradation (Figure 3).43 Recent in‐depth analysis on these radiotracers yielded a series of cold fluorinated analogues with more promising metabolic and pharmacokinetic fate, but this has still to be translated into development of novel radiotracers.44 [11C]MA2 and [18F]MA3 were developed later, maintaining the central oxadiazole cycle with side 3‐quinoline scaffold. Both of them demonstrated strong brain uptake compared to other CB2R radiotracers, associated to rapid clearance from other organs.45 Based on these results, more potent [18F]MA3 was tested further in vivo.46 In a rat model with brain‐localized hCB2R overexpression it caused high radiation uptake in the interested region when compared to the counterpart, with reduction to non‐significant differences in blocking experiment with NE40. A subsequent evaluation in healthy non‐human primates demonstrated acceptable but not specific brain uptake, highlighting the need of further studies in pathological models.46
Figure 3.
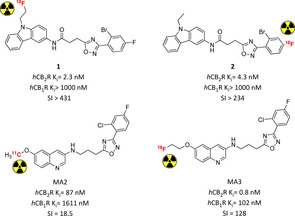
CB2R PET radiotracers bearing an oxadiazole core with in vivo application.43, 45 Affinities reported in the picture are those of the non‐radioactive analogues.
2.3. Thiophene and thiazole derivatives
Starting from the promising results of thiazole derivative A‐836339 biologically characterized by Abbott in 2009, the respective radioactive analogue [11C]A‐836339 was developed to translate its CB2R's binding properties into suitable imaging properties (Figure 4).47, 48 It showed low brain uptake with important specific binding in mouse spleen, whereas it bound almost nonspecifically to other organs. In an inflammation mouse model it exerted strong uptake in all brain regions with a reduction of ∼80 % specific binding in a blocking experiment under these experimental conditions. The same findings were confirmed in PET analysis with an almost fourfold increase in initial whole brain uptake.48 [11C]A‐836339 was also used as imaging biomarker of neuroinflammation in an AD mouse model, confirming high and selective brain uptake under these pathological conditions.49 In order to optimise [11C]A‐836339’s premises, a series of fluorinated analogues were synthesised and analysed through modification of N1‐alkoxy chain.50 PET studies on N1‐fluorobutyl analogue [18F]3 in healthy mice demonstrated identical spleen's specific binding to its parent compound (Figure 4). In LPS‐treated mice micro‐PET scans revealed [18F]3’s higher brain uptake with almost total CB2R specific binding of the accumulation, as verified in blocking analysis.50 Another radiofluorinated thiazole, this time with a N1‐ethoxyethyl side chain, confirmed the promising imaging results of the parent compound with the same high specific binding proved in vivo (4, Figure 4).51 A similar class of thiophene derivatives with high CB2R affinity and selectivity were radiolabelled with 11C to evaluate its in vivo profile. Unfortunately, no traces of specific binding were detected in vitro and in vivo, lowering all the previous biological premises on this new class of compounds for radioimaging.52
Figure 4.
Selective PET radiotracers developed for CB2R imaging bearing a thiophene or thiazole central core.48, 50, 51, 52 Affinities reported in the picture are those of the non‐radioactive analogues.
2.4. Pyridine derivatives
A comprehensive SAR campaign was conducted by Ametamey et al. on the 2,5,6‐substituted pyridine scaffold to develop selective potent CB2R radiotracers. 5‐Methoxyazetidine RSR‐056 was identified as the most suitable derivative of the series exhibiting low nanomolar affinity, high selectivity and pronounced in vitro stability.53 [11C]RSR‐056 demonstrated specific in vivo binding in rat spleen (79 % reduction in a blocking experiment) with fast washout and moderate metabolic stability (21 % intact parent tracer in plasma 20 min after iv injection). In a neuroinflammatory mouse model [11C]RSR‐056 exhibited brain accumulation compared to vehicle or to healthy animals, but unfortunately a similar specific binding was not confirmed in these conditions.53 In order to prolong the tracer's half‐life, more recently a series of fluorinated RSR‐056 analogues were also evaluated.54 Compound 5 (Figure 5) is a potent selective inverse agonist for CB2R and its radiofluorinated analogue was studied in vivo. Radiotracer [18F]5 bound specifically and reversibly to CB2R in rat with a low abundance in the brain. However and interestingly, even after 45 min, only the intact radioligand was detected in the brain, with no metabolites formed. Furthermore, significant upregulation of CB2R was found in ALS spinal cord thanks to [18F]5 specific detection. These last two promising findings suggest the possibility to exploit [18F]5 for neuroinflammatory imaging.54
Figure 5.
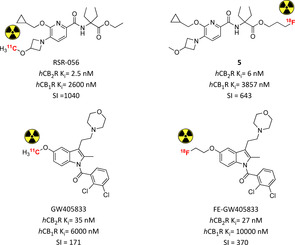
Pyridine‐ and indole‐bearing PET radiotracers for in vivo CB2R imaging.53, 54, 55, 56 Affinities reported in the picture are those of the non‐radioactive analogues.
2.5. Indole derivatives
The first indole CB2R‐selective radiotracer was initially radiosynthesised and biologically evaluated in order to develop a new selective brain reporter gene system (experimental practice used to deliver nucleic acid sequences encoding selected peptides).55 [11C]GW405833 biodistribution analysis in rats and mice showed high initial uptake and stability in brain followed by fast excretion, confirmed in a rhesus monkey PET study (Figure 5).55 A longer‐lasting fluorinated derivative was further explored to evaluate its imaging properties.56 Like its parent compound, it acts as CB2R‐selective low nanomolar inverse agonist. [18F]FE‐GW405833 maintained the same high brain uptake with slower washout and metabolic degradation, but this might be due to false positive radiometabolites being BBB permeable. MicroPET in a rat model with brain‐localized hCB2R overexpression confirmed highly selective radioactivity accumulation with a slower clearance, confirming this compound's potential for neuroinflammation imaging.56 More recently, following the same approach, a series of radiofluorinated benzimidazoles was prepared and tested in vitro. Unfortunately, the radiotracers tested in rat spleen tissue revealed only 48 % CB2R‐specific binding, requiring further biological optimisation.57
3. CB2R Fluorescent Ligands
Fluorescent probes have been widely exploited for the study of receptors at cellular and molecular levels, due to their favourable imaging properties. Compared to the other imaging techniques, fluorescence is a low‐cost method with high sensitivity and resolution that allows a plethora of possible experimental set‐ups using different techniques and respective fluorophores.58 For small‐molecule fluorescent probes, properties of the protein or the ligand itself can be exploited and explored, which makes it a valuable approach also for the drug discovery pipeline.59 On the other hand, the main weakness of fluorescent ligands is limited tissue penetration and possible interferences due to endogenous molecules’ absorbance. This last issue can be addressed by near‐infrared (NIR, 650–900 nm) dyes, because in this wavelength range the tissues of interest exhibit relatively low absorption, autofluorescence and scattering.60
The major challenge in designing new selective fluorescent ligands is to select a known active and selective ligand which is then conjugated, through a suitable linker, with the selected fluorophore avoiding loss of affinity or selectivity (Figure 1).61 This turned out particularly challenging for CB2R, because of the high structural homology between CBRs that often leads to a loss or even switch of selectivity by only minor modifications. Furthermore, CB2R usually prefers lipophilic scaffolds, and combinations with bulky fluorophores can lead to excessive lipophilic tools with high non‐specific binding.
Despite these difficulties, interesting CB2R fluorescent tools have been developed and they are here discussed based on their core structure.
3.1. Biarylpyrazole: development of mbc94 and its functionalized derivatives
Actually, this subclass of fluorescent CB2R ligands is based on several fluorophore optimisations introduced into the biarylpyrazole structure of SR 144528. It was the first selective CB2R antagonist with high affinity (K i CB2R=0.6 nM, Figure 11), later shown to act as inverse agonist, developed in 1998 by Rinaldi‐Carmona et al.62 However, despite its desirable pharmacological profile, it lacks functional groups for easy fluorophore conjugation. In 2008 a suitable analogue of SR 144528 was synthesized and named mbc94. It presents a terminal amino group which could be quickly linked to imaging scaffolds, decreasing affinity only 15‐fold (K i=15 nM).63, 64 Conjugating mbc94 nucleus with the NIR dye IRDye 800CW via its NHS ester, Bai et al. developed NIR‐mbc94, one of the first CB2R's fluorescent ligands with submicromolar affinity (K i=260 nM, Figure 6). In NIR microscopy experiments it demonstrated promising imaging properties (signal to noise ratio S/N=1.6). Furthermore, it exhibited an interesting pharmacological profile thanks to the lack of non‐specific binding and high selectivity in competitive binding experiments using SR 144528 as displacing ligand.64 NIR‐mbc94 was successfully validated as a molecular imaging agent assessing its CB2R selectivity in DBT intact cells, that is, cells heterologously expressing mCB2Rs at levels that are found in native cells, and in two cell lines known to naturally express this target, that is, BV‐2 cells (mouse microglia cell line) and mouse microglia cells in primary culture. Accordingly, it lacked binding in untransfected DBT cells free of endogenous CB2Rs.64 All these experimental outputs corroborated the potential use of NIR‐mbc94 for high‐throughput screening (HTS) of chemical libraries of compounds binding to CB2R. A preliminary screening of compounds was done, validating this hypothesis and suggesting new possible hits.64 However, downsides of NIR‐mbc94 arise from its fluorophore IRDye 800CW, which is a high‐cost dye with relevant stability issues. Bai and co‐workers overcame these problems choosing NIR760 as a new probe to conjugate with mbc‐94.65 NIR760 is a symmetric dye easy to synthesize, more stable in the applied experimental conditions (37 % vs 55 % fluorescence intensity reduction after 2 h of exposure, when compared to IRDye 800CW) and characterized by overall better spectroscopic properties in terms of fluorescence quantum yield and molar extinction coefficient.65 The new compound NIR760‐mbc94 showed nanomolar affinity (K d=26.9 nM) with a moderate specific binding (40 % fluorescence intensity reduction after 30 min pretreatment with 10 μM of SR 144528, Figure 6). More importantly, NIR760‐mbc94 was the first CB2R fluorescent probe tested in vivo in a CB2‐mid DBT mouse tumour model to assess its potential as cancer imaging tool. Tumour uptake reached the maximum after 24 h post iv injection while NIR760‐mbc94 achieved highest tumour area to normal area ratio (T/N) at 72 h increasing gradually over time, providing evidence of long half‐life. Specific binding of the probe was tested also in vivo and ex vivo with a preinjection of SR 144528, confirming the good results shown in vitro (31–35 % vs 40 % respectively).65 NIR760‐mbc94 was also tested as fluorescent CB2R probe for inflammation imaging in vitro and in vivo.66 Specific binding at cellular levels was assessed in regular RAW‐264.7 (mouse macrophages) and LPS‐activated cell lines. SR 144528 was used as blocking agent, causing a reduction of fluorescence signal by 36 % in regular and 23 % in activated cells. In vivo imaging in a mouse inflammation model showed an image contrast (fluorescence detected in inflamed paw vs uninflamed paw) increasing over time, confirming metabolic stability previously detected and highest blockage of 30 % at 36 h. Immunofluorescence staining on frozen paw sections validated previous outcomes but also highlighted once more the moderate specific binding of NIR‐mbc94.66 The authors suggest that it might be due to its net negative charge and to overcome this problem they have further developed a zwitterionic CB2R NIR probe called ZW760‐mbc94 (Figure 6).67 Similar to its ancestor, ZW760‐mbc94 binds to CB2R with nanomolar affinity (K d=53.9 nM). As hoped, the uptake of ZW760‐mbc94 in CB2R‐mid DBT cells was blocked by 4‐quinolone‐3‐carboxamide (4Q3 C, used as selective CB2R blocking agent) by roughly 50 %, higher than previous result obtained with NIR760‐mbc94.67 In an in vivo mice model inoculated with CB2R‐mid DBT tumour cells subcutaneously, ZW760‐mbc94 showed higher fluorescence intensity than in blocked animals (pre‐injection with 4Q3 C) with important uptake in liver and kidneys (metabolizing organs). In an ex vivo biodistribution study, a blocking effect around 47 % was observed only in the cancer cells, indicating higher specific binding of ZW760‐mbc94 than its parent compound NIR760‐mbc94 at both cellular and animal level.67
Figure 11.
Biarylpyrazole hCB2R covalent ligands and its parent compound SR144528.98 An asterisk refers to “apparent K i” because of the plausible covalent linking. nd=not determined.
Figure 6.
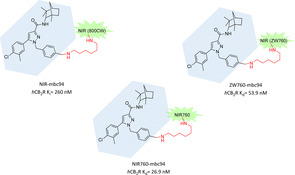
Mbc94 CB2R‐selective fluorescent derivatives with various NIR fluorophores.63, 65, 67
3.2. Oxoquinoline derivatives
Oxoquinolines have been widely exploited to obtain selective and potent CB2R ligands.68 As a consequence, a lot of detailed SARs had been described for this subclass, which allowed the development of oxoquinoline‐based fluorescent tools with high affinity and selectivity. In the first two successful examples, the fluorophore, in this case NIR760, was directly introduced into the aromatic moiety through a proper linker. Pyrazolo [1,5‐a]pyrimidin‐7‐one moiety has been reported to show high CB2R binding affinity and selectivity with the important feature of wide tolerability about chemical modification at the C2 position.69 Through an 4‐aminohexylphenylamide linker, NIR760 was connected to the pyrazolo [1,5‐a]pyrimidin‐7‐one core affording NIR760‐XLP6 (Figure 7).70 It evidenced important CB2R affinity and selectivity (K d=169.1 nM) in the appropriate DBT‐CBR cells with noticeable amount of non‐specific binding, confirmed by in vitro fluorescence imaging. In vivo and ex vivo optical imaging in tumour mouse model corroborated NIR760‐XLP6’s CB2R preference over CB1R with only a moderate CB2R specific binding.70 NIR760‐XLP6 was also evaluated as fluorescence probe to image pancreatic duct adenocarcinoma (PDAC), obtaining high imaging contrast both in a xenograft tumour and PDAC lymph node metastasis models and finding also confirmation of CB2R overexpression in this kind of tumour.71 The same NIR760 fluorophore was further exploited attached to a selective CB2R quinolone structure in position 6 via triazole‐PEGylated linker to obtain NIR760‐Q (Figure 7).72 In this case, its CB2R's binding affinity and targeting specificity were tested in living Jurkat cells, which naturally express CB2R. NIR760‐Q has nanomolar affinity (K d=75.51 nM) and moderate specific binding, confirming the same trend emerged for the charged NIR fluorescent dyes.72
Figure 7.
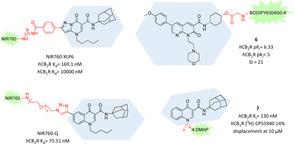
2‐Oxo‐ and 4‐oxoquinoline analogues as highly selective CB2R fluorescent tools.70, 72, 73, 74
More recently, also positions N1 and C3 of the oxoquinoline core were explored as a possible attachment point for fluorescent dyes. In particular, Cooper et al. developed a series of 1,8‐naphthyridin‐2‐(1H)‐one‐3‐carboxamide derivatives with fluorophore BODIPY630/650‐X in that positions.73 In contrast to other CB2R scaffolds until now described, naphthyridine moiety presents higher hydrophilicity, which should be beneficial to avoid unwanted non‐specific binding. Unfortunately, all N1‐attached fluorescent ligands reported resulted in almost no affinity for CB2R whereas the C3‐attached derivative 6, exploiting a glycine bridge, exerted appreciable CB2R affinity and selectivity (pK i=6.33, SI>21), acting as an inverse agonist (Figure 7).73 Interestingly, compound 6 is one of the rare CB2R ligands which increases affinity after fluorophore addition. Despite these good premises, ligand 6 showed poor CB2R imaging properties in cells, likely due to high levels of membrane association and non‐CB2R associated intracellular accumulation.73 Finally, in a recent successful SAR campaign on 4‐oxo‐1,4‐dihydroquinoline analogues, a N1‐attached fluorescent CB2R ligand was obtained.74 A series of 4‐DMAP, NBD and FTU derivatives with different linkers were evaluated, with compound 7, bearing the 6‐hexyl‐4‐DMAP fragment in N1, emerging for the high CB2R affinity and selectivity (Figure 7). The authors proposed derivative 7 as a novel potential tool for fluorescent‐based competition binding assays and detection of CB2Rs in specific cell populations tested via fluorescence‐activated cell sorting (FACS) analysis. Results obtained in a preliminary competitive binding assay to measure IC50 of two reference compounds (WIN55,212‐2 and GW405833) and saturation binding assay on CB2R‐HEK293 cells with GW405833 as reference and [3H]‐CP55,940 as radioligand, using compound 7 at 100 nM with incubation time of 90 min as fluoligand, gave results comparable to K i values reported in literature.75 Saturation and binding assays were also performed in tumour cells physiologically expressing CB2Rs, corroborating results previously reported in engineered cells. Further immunofluorescence experiments with compound 7 at 15 μM used as fluorescent probe to visualize CB2R in cells revealed its important specificity and reliability as potential fluorescent alternative to usual radioligand assays.74
3.3. Indole derivatives
To the indole subclass belongs the first CB2R fluorescent probe reported in 2005 by Yates et al.76 Thanks to the promising affinity, selectivity and easy accessibility of the agonist JWH‐015 (Ki=13.8 nM, SI=28),77 they underwent an in silico modelling campaign to identify better positioning of the fluorescent dye 4‐fluoro‐7‐nitrobenzofurazan (NBD‐F). Through exploitation of binding mode of WIN 55,212‐2, which has a significant structural homology to JWH‐015, a rational binding model of JWH‐015 bound to CB2R was generated.76 In the end, the 3‐position of the naphthyl ring seemed the best to tolerate bulky substituents and was therefore used to insert NBD‐F through a short amide linker, necessary to prevent photoinduced electron transfer (8, Figure 8). Unfortunately, the obtained probe showed a >250‐fold loss in CB2R affinity, preventing its use as selective CB2R fluorescent tool.76
Figure 8.
Indole scaffold exploited to develop selective CB2R fluorescent ligands.76, 79
The 3‐position of indole scaffold was further exploited as conjugation site for fluorophores using more flexible and hydrophilic linkers. Actually, the starting indole scaffold was modified to an isatin acylhydrazone to gain higher CB2R affinity and selectivity.78 In particular, the binding mode of a morpholino derivative with low nanomolar affinity (K i=13.3 nM, SI>700) in which the morpholine moiety fits into a highly lipophilic pocket of a CB2R model was used as source of inspiration for NMP6, a new CB2R fluorescent probe where morpholine is substituted by the NBD fluorophore (Figure 8).79 NMP6 still maintained nanomolar affinity and high selectivity for CB2R and therefore was further tested in CB2R cellular imaging. Both using confocal microscopy in non‐transfected cells and cytometric analysis in B lymphocytes, NMP6 was able to visualize CB2R and its binding was blocked in competition studies or by preincubation with GW842166X, a selective CB2R agonist, evidencing NMP6 CB2R's selectivity.79
More recently also positions 5 and 7 of the indole moiety were exploited for potential linker and fluorophore attachment. After a SAR campaign on the indole scaffold Cooper and co‐workers found CB2R ligands with nanomolar affinity with bulky substituents in position 1 or 3 like ethylmorpholine, methoxyphenyl or methyltetrahydropyran. Interestingly, derivatives with substituents of different length in position 7 lead to strong inverse agonist activity, pointing 7‐propyloxy compound as the best of the series (K i=5.7 nM, SI=89), while C5 analogues were characterized by agonist behaviour in a cAMP assay.80 Unfortunately, conjugating both of these promising indole derivatives with fluorophore BODIPY 630/650‐X in C5 or C7 through alkylene, PEG, or dipeptide linkers, respectively, resulted in fluorescent ligands with no CB2R affinity (K i>10000 nM), showing that only smaller substituents are tolerated in these positions.80
3.4. Other CB2R fluorescent probes
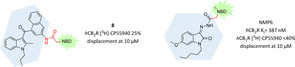
Among all the CB2R functionalized probes it's not so common to find selective ligands bearing the original tetrahydrocannabinol (THC) core or analogues, because this moiety is usually correlated with a lack of CBR subselectivity and interference with other targets of the ECS. Recently, the tricyclic cannabinoid scaffold has gained major attention for its peculiar biological properties and because, thanks to proper substitutions, CBR subtype selectivity can be achieved. Starting from the bioisoster chromenopyrazole nucleus, a series of fluorescent ligands were synthesised and biologically evaluated.81 Among all, the Cy5‐derivative 9 came out for the best affinity and selectivity (131‐fold over CB1R, Figure 9), acting as inverse agonist with high efficacy and potency. In vitro fluorescence microscopy imaging experiments with 9 at 1 μM showed very high selectivity and almost no unspecific binding.81 In 2020 Carreira and co‐workers developed double‐functionalized CB2R‐selective ligands with both electrophile moiety at one side and fluorescent or photochromic moieties, always retaining a bicyclic THC‐derived core. A small series of fluorescent derivatives, labelled with NBD, exhibited high affinity and selectivity, but they were not further tested to evaluate their actual imaging properties (10 and 11, Figure 9).82 Starting from these premises and keeping the terminal azido group, different fluorescent dyes were linked using side amino group as attachment point, obtaining new CB2R fluorescent probes with interesting in vitro application.83 Compounds 12 and 13 pointed out as the best of the series regarding affinity and selectivity and, interestingly, only varying the fluorescent dye moiety led to different efficacy (partial agonism for 13 and agonism for 12) with very high functional selectivity over hCB1R (Figure 9). Both probes were evaluated as fluorescent tools for flow cytometry, time‐resolved confocal microscopy and a time‐resolved fluorescence resonance energy transfer (TR‐FRET) assay available for high throughput screening. Particularly compound 12 exhibited very high in vitro specificity, ability to detect hCB2R human breast cancer cells and monitor hCB2R in live cells through real‐time confocal fluorescence microscopy with minimal internalization over long period with very low levels of nonspecific bindings.83 Recently, for the first time to our knowledge, the pyridine scaffold was exploited for the design of new selective CB2R fluorescent probes. Firstly, docking studies were applied to determine a promising linker and attachment point for the pharmacophore among pyrazine‐ and pyridine‐based selective CB2R ligands.84 A series of fluorescent ligands with dyes connected through a PEG linker to the geminal diethyl group of the properly substituted pyridine moiety were developed and biologically evaluated in vitro (Figure 9). Derivative 14 came out as the best ligand regarding potency, selectivity and agonist functional properties, but only compounds 15 and 16 were further tested in fluorescence imaging thanks to the better imaging properties of their dyes (Figure 9). In a FACS analysis, both derivatives 15 and 16 demonstrated high target specificity with almost no unspecific binding. In addition, probe 16 was successfully employed to visualize hCB2R in living cells by confocal imaging.84
Figure 9.
Selective CB2R fluorescent ligands bearing tricyclic, tricyclic‐derived or pyridine cores.81, 82, 83, 84
4. CB2R Covalent Ligands
Covalently binding molecular probes are compounds which can activate or inactivate – temporarily or permanently – specific biological targets through formation of a covalent bond. Thanks to the strong interaction and the related long‐lasting effects, associated with tuned reactivity toward the target of interest, covalent drugs gained increasing interest and application in recent years.85 Covalent drugs include for example antibiotics,86 antiplatelet clopidogrel,87 aspirin,88 antacid omeprazole89 and anticancer treatments.90 Formation of a covalent adduct can be due to the activation of a photoaffinity fragment or to an intrinsically reactive electrophilic moiety which reacts with nucleophilic amino acid residues present in the binding pocket, usually cysteine or lysine. Examples referring to a photoactivation step will be discussed in the next chapter.
4.1. Covalent CB2R agonists
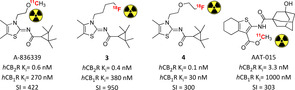
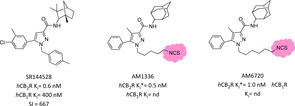
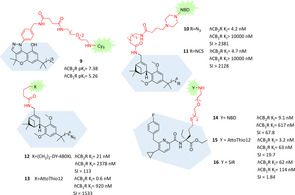
The possibility to selectively and covalently modify receptors represents a valuable tool for elucidating structures and functions of membrane proteins without known crystal structure.91 Regarding the CB2R, series of selective covalent ligands were developed from Makriyannis’ group with the aim to define the structural aspects of ligand recognition in hCB2R, using a direct experimental approach termed ligand‐assisted protein structure (LAPS). This approach lies in rational design and development of high affinity irreversible probes combined with their biological evaluation by means of point‐mutation analysis and mass‐based proteomic analysis. A final computer‐based molecular modelling approach enables to characterize at molecular level the structural details of ligand recognition.92 This successful approach started with the development of AM841, a THC derivative with 7‐isothiocyanate fragment at the end of C3‐terminus alkyl side chain (Table 1).93 In this case, isothiocyanate moiety represents the reactive electrophile core, which undergoes at the electrophilic carbon the nucleophilic attack of amino acid's responsive side groups present in ligand‐binding pocket, usually thiol group of cysteines. Identified by an interactive docking simulation and a point‐mutating affinity and activity evaluation campaign, AM841 is a selective high affinity CB2R agonist that was proved to irreversibly activate hCB1R through formation of a covalent adduct between ‐NCS moiety and cysteine 355 (C355) in the transmembrane helix 6 (TMH6).94 The same LAPS approach was further applied with AM841 to explore the ligand‐binding motif of hCB2R.92 Initially, C257 in TMH6 of hCB2R was identified as homologous of C355 of hCB1R, previously shown to covalently bind AM841, and its role was exploited, among other nucleophilic residues (K109 in TMH3, C288 and C284 in TMH7), with regard to hCB2R‐ligand interactions. All the mutant receptors involving point‐variations at C257 residue after AM841 pretreament or wild‐type (WT) hCB2R after AM4043 or AM4056 (non‐electrophilic analogues of AM841, Table 1) pretreatment lead to no significant decrease at [3H]‐CP55940 B max, whereas WT‐hCB2R and all other mutant isoforms exhibited around 80 % decrease of [3H]CP55940 B max after AM841 pretreament.92 These findings prove that C257 in TMH6 is the only residue involved in covalent adduct formation with ‐NCS moiety of AM841, activating WT‐hCB2R with exceptional potency (IC50=0.079 nM, 12 times greater than at hCB1R).
Table 1.
|
| |||||
|---|---|---|---|---|---|
|
Compound |
R1 |
R2 |
hCB2R K i [nM] |
hCB1R K i [nM] |
SI |
|
AM4056 |
OH |
H |
2.14 |
2.99 |
1.40 |
|
AM4043 |
OH |
Br |
2.64 |
3.99 |
1.51 |
|
AM841 |
OH |
NCS |
1.51* |
9.05* |
5.99 |
|
AM4073 |
NCS |
H |
3.3* |
nd |
nd |
|
AM4099 |
NCS |
NCS |
12.6* |
nd |
nd |
An asterisk refers to “apparent K i” because of the plausible covalent linking. nd: not determined.
Another interesting output concerns lysine residue 109 in hCB2R (K192 in hCB1R), which was demonstrated to play a crucial role in hCB1R‐ligand binding architecture causing a loss of affinity after selective point‐mutation. Notably, in hCB2R its mutation does not affect ligand recognition, suggesting different structural binding rearrangements for the two CBRs.92 Covalent C257‐AM841 interaction was further evaluated and endorsed also in a mass‐based proteomic analysis in N‐terminal FLAG‐tagged/C‐terminal 6His‐tagged hCB2R (FLAG‐hCB2R‐6His) overexpressed in Spodoptera frugiperda cells.95 In a first multiple reaction monitoring (MRM)‐MS method assay all the seven hCB2R TMH peptides were separated and identified and a following treatment with AM841 emphasized how only TMH6‐associated signal decreased by ∼75 % (roughly same result found before with LAPS approach) with respect to the peak area of TMH6 from untreated FLAG‐hCB2R‐6His. A final Q‐TOF MS/MS analysis underlined that AM841 reacts selectively and irreversibly with C257 of TMH6, a critical interaction determinant for AM841 strong agonist activity and hCB2R activation.95
More recently, the same LAPS approach was applied to explore functions of C9/C11 positions at the cyclohexenyl C‐ring of THC in recognition and binding to hCB2R.96 With this purpose, two AM841 analogues have been developed bearing electrophilic −NCS fragment only at C11 (AM4073) and both at C11 and in the terminus of C3 alkyl chain (AM4099, Table 1). Like AM841, these new two derivatives bound to WT‐hCB2R with low‐nanomolar affinities and acted as strong agonists, albeit 100 times less potent than the parent compound.
As expected, AM4073 or AM4099 pretreatment reduced subsequent B max for specific [3H]‐CP55940 binding by 60 % in WT‐hCB2R when compared to non‐pretreated membranes, but – surprisingly – in this case this is the result of a covalent interaction with the receptor at its nucleophilic C89 residue in TMH2. This interesting output suggested that simply adding an electrophilic moiety at C11 completely changed ligand‐binding architecture losing C257 interaction (also in AM4099 which presents ‐NCS in C3 side chain like AM841) and the consequent strong agonist effect of the parent compound (over 100 times less potent) while maintaining high affinity and irreversibility.96 It's worth noting that different electrophilic substitutions at the same core structure lead to completely different binding modes, underlining the driving force of a covalent adduct, and stabilize the ligand‐binding architecture allowing the activation of the same adenylyl cyclase‐mediated signalling with distinct potencies.96
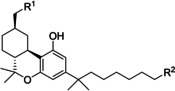
By using the same THC scaffold, different −NCS‐bearing C3 substituents were exploited to further explore their pharmacological properties.97 Bulky and lipophilic groups like adamantyl or norbornyl moieties are well described to correlate with high CBR affinity. A small library of C3‐adamantyl THC derivatives was developed and tested to evaluate their biological behaviour toward CBRs. Among them, two electrophilic tools with isothiocyanate and isothiocyanatemethylene moieties in the 3 position showed a peculiar biological profile, albeit not high hCB2R selectivity (Figure 10).97 They exhibit nanomolar affinity, but only derivative 18 was slightly selective for hCB2R and, surprisingly, they almost showed no efficacy in forskolin‐stimulated cAMP assay, with only compound 17 presenting very low potency as inverse hCB2R agonist, whereas both of them were strong hCB1R agonists. Ligand 17, more than ligand 18, was able to covalently label hCB2R (74 vs 25 % respectively), but further experiments are needed to identify the amino acid residue involved.97
Figure 10.
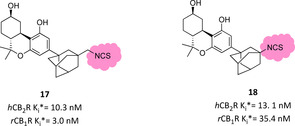
Adamantyl THC derivatives bearing an electrophilic moiety.97 An asterisk refers to “apparent K i” because of the plausible covalent linking.
Recently, a series of double functionalized bicyclic derivatives of HU‐308 were developed bearing a terminal isothiocyanate or alkyne group. They exerted pronounced hCB2R affinity and selectivity but, unfortunately, none of them demonstrated ability to form irreversible binding under two different set of experimental conditions.82
4.2. Covalent antagonist/inverse agonist CB2R ligands
The above described covalent ligands can deepen our knowledge regarding agonist binding modes and associated functional receptor activation pathways. An interesting comparison in the antagonist/inverse agonist ligand‐receptor binding architecture was provided for the first time by Mercier and co‐workers using irreversible SR144528 (biarylpyrazole high affinity inverse agonist) derivatives bearing an isothiocyanate moiety at the end of N1‐aliphatic side chain (AM1336 and AM6720, Figure 11).98 In a saturation binding assay using [3H]CP55940 after AM1335/AM6720 pretreatment, these electrophilic ligands showed a 60 % level of irreversible binding to WT‐hCB2R and all other selective cysteine receptor mutants except for variants with mutations on cysteine residues in TMH7. Regarding AM1336, selective point mutations at C284 or C288 diminished covalent labelling from 25 to 40 % while in double mutants C284‐C288 the covalent interaction was completely lost, suggesting that both TMH7 cysteines were involved in covalent adduct and in ligand‐binding rearrangement. Conversely, AM6720 – carrying a longer NCS terminal side chain – interacted only with C288, which is positioned more in depth of the TMH7 gorge.98 Like parent compound AM1336, it acted as antagonist/inverse agonist and was able to fully activate the receptor nine times stronger than the longer AM6720, underlining the possibility of double TMH7 covalent interaction. Interestingly, AM1336 was not able to covalently bind to the hCB1R, confirming that CBRs ligand‐binding domains differ at amino acid level.98 These experimental findings were further evaluated through the same MS‐based proteomic analysis mentioned above for the study of THC agonist derivatives. Surprisingly, in this case only C284 in TMH7 was confirmed to undergo a covalent interaction with AM1336.99
5. CB2R Photochromic Ligands
As discussed above, one of the major challenges in CBR ligand discovery is selectivity, not only between the two receptor subtypes,2 but also over other parts of the endocannabinoid system like ion channels,100 nuclear receptors,101 and also over distinct GPCRs in general due to their usually high lipophilicity. In recent years “photopharmacology” has emerged as a valid experimental application to remotely and directly control selective and localized biological action by properly functionalized molecules. Furthermore, administration to cells or organs of compounds incorporating photo‐activatable fragments allow the pharmacophore's activation to be controlled in time and space through an external light‐based modality, with the intention to avoid undesired side effects. The same principles can be applied in biological assays for the study of target molecules or selected cellular pathways, their interactions and their involvement in patho/physiological processes after activation/inactivation of the photochromic probe.102, 103 Controlled light‐derived activation of photosensible compounds can lead to different photopharmacological approaches. Herein only CB2R chemistry‐based approach will be discussed, such as phototherapy or molecular photoswitches.104
5.1. Phototherapy with CB2R ligands
Phototherapy exploits selective pharmacologic tools coupled with specific dyes which, upon local irradiation, are able to exert therapeutic activities (usually killing cancer cells) by means of ROS overproduction (photodynamic therapy, PDT) or generating hyperthermia (photothermal therapy, PTT). Regarding CB2R, in 2014 Zhang et al. reported the first CB2R‐targeted photosensitizer called IR700DX‐mbc94. In its structure, the previously described CB2R‐selective ligand mbc94 is functionalized with a phthalocyanine dye (IR700DX) which acts as photosensitizer moiety (Figure 12).105 IR700DX‐mbc94 showed no cytotoxicity in CB2R‐mid DBT cells without light irradiation, whereas destroyed ∼80 % after 20 min irradiation following a 5 μM pretreatment. Particularly, its phototoxicity worked only in a target‐specific manner because there was no activity registered for the unbound ligand, for the dye or light irradiation alone and an important reduction was found in WT‐DBT vs CB2R‐mid DBT cells treated (47 vs 80 % cell death). Furthermore, the IR700DX‐mbc94 induced cell death increased with longer incubation time, higher ligand concentration and increased light irradiation dose.105 These promising results were further evaluated in an in vivo CB2R‐mid DBT tumour‐bearing mouse model.106 Treatment with IR700DX‐mbc94 (10 nM) and light irradiation (45 J/cm2) strongly reduced tumour volume and almost duplicated survival time when compared to untreated mice. At the same time, ligand or irradiation alone, dye treatment with irradiation and IR700DX‐mbc94 administration in tumour‐model lacking CB2R caused no effects, confirming target‐specific action of this photosensitizer and CB2R potential for specific cancer treatment.106
Figure 12.
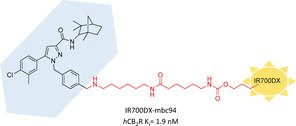
First photoaffinity CB2R probe studied for PDT.105
In a follow‐up study pursuing a multifunctional approach, CB2R photosensitizer IR700DX‐TK‐mbc94 (Figure 13) was developed, which combines a CB2R‐targeted prodrug (mbc94‐SH, K i=47.7 nM), a ROS‐activatable linker (heterobifunctional thioketal linker that releases free thiol groups in an oxidant environment) and a hydrophilic photosensitizer (IR700DX).107 In a fluorescence assay ROS‐caused linker cleavage upon light irradiation was confirmed, as two thiol fragments were released, one of which belonging to the CB2R‐directed mbc94‐SH. In a CB2R+ mouse brain tumour model IR700DX‐TK‐mbc94 strongly induced cell death after 24 h irradiation (78 % at 0.5 μM concentration), whereas no significant effect was observed for IR700DX‐mbc94. The 44 % blocking effect of IR700DX‐TK‐mbc94 cellular uptake by SR144528 (CB2R‐selective ligand) and its apparently safe treatment to non‐CB2R+ cells, firmly strengthened IR700DX‐TK‐mbc94 phototoxicity activity in a target‐specific manner. IR700DX‐TK‐mbc94 produced lower amounts of ROS when compared to IR700DX‐mbc94, whereas it exerted a remarkable therapeutic effect when compared to the parent compound. This could be probably due to the cannabinoid‐derived synergistic effect of mbc94‐SH, which has no relevant therapeutic effect alone, and PDT‐related ROS toxicity.107
Figure 13.
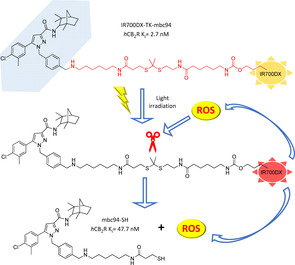
Prodrug IR700DX‐TK‐mbc94 and its proposed activation pathway. As a result of light irradiation, IR700DX promotes cytotoxic ROS production that concurrently cleaves the linker at the thioketal fragment releasing mbc94‐SH and having therapeutic effect.107
5.2. Photolabelling CB2R ligands
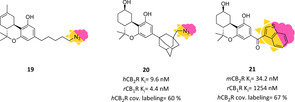
Regarding molecular photoswitches, irradiation by light drives a structural modification and thereby can induce a twist of biological profiles (e. g., affinity, activity, etc.). The light‐derived structural change depends on the photochemical properties of photoaffinity moieties, inserted into the interested hybrid, which, upon irradiation activation, can respond in different ways.103 One example is a photoreactive covalent group (e. g., azide, diazirine), which upon photoactivation converts itself into a reactive fragment able to undergo covalent attachment with amino acid residues present in the active site.108, 109 The first CBR‐targeted photoaffinity probe was CB1R‐directed; it was developed in 1992 by introducing an azido terminal group in the THC side chain, which upon light irradiation was converted into nitrene able to covalently react and completely abolish [3H]CP55940 specific binding (19, Figure 14).110 The same tricyclic cannabinoid moiety core was further functionalized with a bulky adamantyl group in C3 bearing the same terminal azide or methyleneazide (20) fragment leading to nonselective nanomolar photoaffinity probes, which upon light irradiation lead to hCB2R covalent labelling of 26 and 60 %, respectively (Figure 14).97 Bifunctional nonselective photoactivable probes were also reported, but not providing the same interesting results of their isothiocyanate analogues.111 The first photolabelling CB2R‐selective THC analogue was derived from a SAR campaign regarding C3‐heteroyl THC derivatives for exploring CBRs binding sites and exploiting new fragments as photoactivatable moiety.112 The most promising results were observed for bicyclic 3‐heteroaroyl substituted analogues (e. g., benzofuran, indole, etc.), with the 3‐benzothiopheno derivative emerging as the best compound of the series with low‐nanomolar CB2R affinity and good selectivity (21, Figure 14). Furthermore, compound 21 showed the ability to covalently bind mCB2R at 67 % upon light irradiation as compared to its 2 regioisomer (77 %), suggesting the possible harnessing of 3‐arylphenone moieties as a photolabelling fragment for CB2R binding studies.112
Figure 14.
Photoactivatable probes can also be exploited, when coupled to fluorescent or affinity tags to visualize ligand‐protein interaction in cells, to monitor receptor expression or target‐engagement based on a photoaffinity‐based protein profiling (pAfBPP) approach.108 By these means compound LEI121 was developed, the first CB2R‐selective ligand containing both a photoaffinity moiety (diazirine) and a ligation handle (terminal alkyne, red in Figure 15) for introducing a tag (red star in Figure 15) merged in a cannabinoid‐targeted core to study CB2R behaviour (Figure 15).113 LEI121 represents a highly selective hCB2R ligand acting as inverse agonist capable of covalently trap hCB2R (blue in Figure 15) upon irradiation (∼50 %). In a two‐step process, consisting of a first photolabelling and in situ click reaction with the dye, LEI121 allowed CB2Rs endogenously expressed both in a human promyelocytic leukaemia cell line and in primary human immune cells to be visualized by flow cytometry. When coupled to an affinity tag (e. g., biotin), it was also applied to test target engagement of various ligands in living systems, confirming the wide biological applicability of selective photolabelling ligands to map ligand‐protein interactions.113
Figure 15.
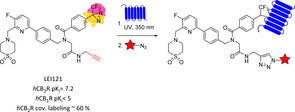
Photoaffinity‐based protein profiling as two‐step process with hCB2R‐selective compound LEI121.113
5.3. Photoswitchable CB2R ligands
Reversibly photoswitchable fragments (e. g., azobenzenes, diarylethenes) connected to or incorporated into pharmacophores can lead to fast and reversible conversion in different photoisomers upon irradiation by light (normally UV), leading to diverse and remotely controlled binding modes to the target of interest, thus allowing a deeper comprehension of complex biological processes.114
Using profound SAR studies performed on a benzimidazole scaffold, Decker and co‐workers spotted in position 5 the best introduction point for a photoswitchable azobenzene moiety with different substituents.115, 116 Most pronounced hCB2R affinity‐switch upon irradiation with UV‐light occurred with m‐substitutions, with an m‐methyl analogue exhibiting the highest difference between the two photoisomers of the series (∼eightfold), while maintaining good selectivity over the hCB1R (22, Figure 16). For this compound series, the affinity switch was not concurrent with any efficacy change, picking out compound 22 as an interesting functionalized tool compound due to its single pharmacological property variation at a time and its affinity boost upon light irradiation.116
Figure 16.
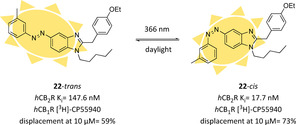
First hCB2R‐selective photoswitchable ligand and changes in its pharmacological properties upon irradiation.116
Very recently, Westphal et al. exploited the scaffold of bicyclic cannabinoids for the development of selective photochromic probes. Particularly, derivatives with the same nucleus of compounds 10–13 reported in Figure 9 were functionalized both with photoactivable groups (e. g., azido, diazirine) and photoswitchable ones (substituted azobenzene), always keeping strong hCB2R affinity and selectivity. For now, none of these derivatives have been tested to evaluate their photoaffinity/photolabelling/photoswitchable properties.82
6. CB2R Bioaffinity Ligands
Biotin has been widely used as affinity tag to enable characterization of receptor–ligand interactions with the isolation and identification of receptor complexes through a simple trapping method. It exploits the high affinity of biotin for avidin conjugates (K d=∼10−14 mol/L) and, furthermore, the fact that such complexes being very resistant to a wide variety of harsh experimental conditions. Because of these advantages, avidin–biotin technology is used to achieve site‐specific delivery, sensing, labelling and imaging of biological targets such as GPCRs.117 The first CB2R biotinylated ligand was developed within a functionalization project of endocannabinoids to study CBR. Derivative 23 originating from functionalization of 2‐arachidonoyl glycerol exhibited CB2R selectivity with nanomolar affinity (Figure 17).118 In the same research group further biotinylated agents with higher CB2R affinity were developed and used for in vitro labelling of hCB2R through direct or indirect streptavidin‐fluorescent imaging.119 The synthesised probes, reported in figure 17, kept the typical tricyclic cannabinoid core or the bicyclic derived moiety and were functionalized with biotinylated linker at free hydroxy groups. For probe 24 the highest affinity with no selectivity for CB2R was reported while the other compounds gained selectivity but lost affinity. Compounds 24 demonstrated high in vitro specificity with the ability to label both CB2R and CB1R and visualize CBRs in a monocytic cell line by flow cytometry. Furthermore, derivatives 24 and 25 were successfully used to image CB2R in microglia in both activated and resting states.119 More recently, the bicyclic structure of derivative 25 was further exploited as starting point for double functionalized selective CB2R ligands. Particularly, a biotinylated derivative was developed keeping nanomolar affinity and good selectivity over CB1R, but no further biological evaluations were reported yet.82
Figure 17.
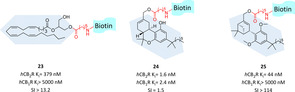
CB2R‐selective biotinylated ligands and their pharmacological properties.118, 119
7. Summary and Outlook
The CB2R represents a very up‐to‐date target for several unmet pharmaceutical and medical needs, such as neuroinflammation. Despite great efforts to develop highly potent and selective ligands, ligand design is challenging, mainly due to the high lipophilicity of the compounds obtained and due to the complex pharmacology of the endocannabinoid system.3 Surprisingly, there are only limited efforts on the very basic problem of high lipophilicity and low solubility. Basic medicinal chemistry endavours to find novel scaffolds could significantly improve the development of functionalized ligands also.68, 120
To unravel CB2R pharmacology and the role of the receptor in health and disease, the above chapters describe how medicinal chemists use the latest techniques available such as PET radiotracer development, covalent ligand, and photochromic as well as fluorescent ligand design to provide versatile and at the same time highly specific molecular tools and probes. These probes help to elucidate CB2R receptor pharmacology and investigate specific processes contributing to receptor dynamics, but of course also enable further drug discovery efforts. These successful approaches are also transferable to other GPCR ligands and therefore of considerable relevance in GPCR research in general.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Filippo Basagni graduated in pharmaceutical chemistry and technology at the University of Bologna in 2017. He spent a research period at King's College London in Prof. Gee's group working on the development of PET radiotracers, before starting his PhD in November 2017 at the University of Bologna under the supervision of Prof. Rosini. He is involved in the development of pharmacologic tools to investigate neuroinflammatory processes characterizing neurodegenerative diseases. He is currently a visiting PhD student in Prof. Decker's group at Würzburg University working on the development of functionalized and multitarget probes.

Biographical Information
Michela Rosini obtained her degree in pharmaceutical chemistry and technology in 1997 followed by a Ph.D. in pharmaceutical sciences in 2001 from the University of Bologna, Italy. In 1998 and 2000, she spent some months at The Royal Danish School of Pharmacy in Copenhagen, Denmark. She is currently Associate Professor of Medicinal Chemistry at the University of Bologna. Her research focuses on the design and synthesis of small molecules as probes for investigating biological processes or as drug candidates for neurodegenerative diseases.

Biographical Information
Michael Decker studied chemistry at Bonn and Cambridge Universities. He received his PhD in pharmaceutical chemistry from Bonn University in 2001 and pursued his postdoctoral research qualification (“Habilitation”) in pharmacy at Jena University. After that, he was a Leopoldina Scholar at the McLean Hospital of Harvard Medical School. He was appointed a Lecturer at Queen's University Belfast, followed by a position at Regensburg University. Since 2012, he has been Professor of Pharmaceutical and Medicinal Chemistry at Würzburg University, where his group works on various GPCRs and enzymes developing hybrid molecules, PET radiotracers, fluorescent and photoswitchable probes.

Acknowledgements
The authors acknowledge the German Research Foundation (DFG DE1546/10‐1 to M.D.) and the University of Bologna (grants from RFO to M.R.).
F. Basagni, M. Rosini, M. Decker, ChemMedChem 2020, 15, 1374.
Dedicated to Professor John L. Neumeyer on the occasion of his 90th birthday.
References
- 1. De Petrocellis L., Di Marzo V., Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 1–15. [DOI] [PubMed] [Google Scholar]
- 2.A. C. Howlett, M. E. Abood in Cannabinoid Pharmacology, Vol. 80 (Eds.: D. Kendall, S. P. H. Alexander), Academic Press, Cambridge, 2017, pp. 169–206.
- 3.D. Kendall, S. Alexander, Cannabinoid Pharmacology, Vol. 80, Academic Press, Cambridge, 2017.
- 4. Munro S., Thomas K., Abushaar M., Nature 1993, 365, 61–65. [DOI] [PubMed] [Google Scholar]
- 5. Van Sickle M. D., Duncan M., Kingsley P. J., Mouihate A., Urbani P., Mackie K., Stella N., Makriyannis A., Piomelli D., Davison J. S., Marnett L. J., Di Marzo V., Pittman Q. J., Patel K. D., Sharkey K. A., Science 2005, 310, 329–332. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Howlett A. C., Barth F., Bonner T. I., Cabral G., Casellas P., Devane W. A., Felder C. C., Herkenham M., Mackie K., Martin B. R., Mechoulam R., Pertwee R. G., Pharmacol. Rev. 2002, 54, 161–202; [DOI] [PubMed] [Google Scholar]
- 6b. Mackie K., J. Neuroendocrinol. 2008, 20, 10–14. [DOI] [PubMed] [Google Scholar]
- 7. Atwood B., Mackie K., Br. J. Pharmacol. 2010, 160, 467–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Turcotte C., Blanchet M. R., Laviolette M., Flamand N., Cell. Mol. Life Sci. 2016, 73, 4449–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wouters E., Walraed J., Banister S. D., Stove C. P., Biochem. Pharmacol. 2019, 169, 113623. [DOI] [PubMed] [Google Scholar]
- 10. Morales P., Jagerovic N., Expert Opin. Drug Discovery 2020, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dhopeshwarkar A., Mackie K., J. Pharmacol. Exp. Ther. 2016, 358, 342–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Oyagawa C. R. M., de la Harpe S. M., Saroz Y., Glass M., Vernall A. J., Grimsey N. L., Front. Pharmacol. 2018, 9, 1202; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Mallipeddi S., Janero D. R., Zvonok N., Makriyannis A., Biochem. Pharmacol. 2017, 128, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Benito C., Tolon R., Pazos M., Nunez E., Castillo A., Romero J., Br. J. Pharmacol. 2008, 153, 277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu S., Ma H., Bo Y., Shao M., Biomed. Pharmacother. 2019, 117. [DOI] [PubMed] [Google Scholar]
- 15. Perez-Gomez E., Andradas C., Blasco-Benito S., Caffarel M., Garcia-Taboada E., Villa-Morales M., Moreno E., Hamann S., Martin-Villar E., Flores J., Wenners A., Alkatout I., Klapper W., Rocken C., Bronsert P., Stickeler E., Staebler A., Bauer M., Arnold N., Soriano J., Perez-Martinez M., Megias D., Moreno-Bueno G., Ortega-Gutierrez S., Artola M., Vazquez-Villa H., Quintanilla M., Fernandez-Piqueras J., Canela E., McCormick P., Guzman M., Sanchez C., J. Natl. Cancer Inst. 2015, 107. [DOI] [PubMed] [Google Scholar]
- 16. Martínez-Martínez E., Gómez I., Martín P., Sánchez A., Román L., Tejerina E., Bonilla F., Merino A. G., de Herreros A. G., Provencio M., García J. M., Oncoscience 2015, 2, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Velasco G., Sanchez C., Guzman M., Nat. Rev. Cancer 2012, 12, 436–444; [DOI] [PubMed] [Google Scholar]
- 17b. Javed H., Azimullah S., Haque M. E., Ojha S. K., Front. Neurosci. 2016, 10, 321; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17c. Bouchard J., Truong J., Bouchard K., Dunkelberger D., Desrayaud S., Moussaoui S., Tabrizi S. J., Stella N., Muchowski P. J., J. Neurosci. 2012, 32, 18259–18268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aso E., Ferrer I., Front. Neurosci. 2016, 10, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Mecha M., Feliú A., Carrillo-Salinas F. J., Rueda-Zubiaurre A., Ortega-Gutiérrez S., de Sola R. G., Guaza C., Brain Behav. Immun. 2015, 49, 233–245; [DOI] [PubMed] [Google Scholar]
- 19b. Mecha M., Carrillo-Salinas F. J., Feliú A., Mestre L., Guaza C., Pharmacol. Ther. 2016, 166, 40–55. [DOI] [PubMed] [Google Scholar]
- 20. Solas M., Francis P. T., Franco R., Ramirez M. J., Neurobiol. Aging 2013, 34, 805–808. [DOI] [PubMed] [Google Scholar]
- 21. Khan M., Sobocinska A. A., Brodaczewska K. K., Zielniok K., Gajewska M., Kieda C., Czarnecka A. M., Szczylik C., BMC Cancer 2018, 18, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gómez-Gálvez Y., Palomo-Garo C., Fernández-Ruiz J., García C., Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 64, 200–208. [DOI] [PubMed] [Google Scholar]
- 23. Palazuelos J., Aguado T., Pazos M., Julien B., Carrasco C., Resel E., Sagredo O., Benito C., Romero J., Azcoitia I., Fernandez-Ruiz J., Guzman M., Galve-Roperh I., Brain 2009, 132, 3152–3164. [DOI] [PubMed] [Google Scholar]
- 24. Shoemaker J., Seely K., Reed R., Crow J., Prather P., J. Neurochem. 2007, 101, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.
- 25a. Morales P., Hernandez-Folgado L., Goya P., Jagerovic N., Expert Opin. Ther. Pat. 2016, 26, 843–856; [DOI] [PubMed] [Google Scholar]
- 25b. Fernández-Ruiz J., Moro M. A., Martínez-Orgado J., Neurotherapeutics 2015, 12, 793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a. Li X., Hua T., Vemuri K., Ho J. H., Wu Y., Wu L., Popov P., Benchama O., Zvonok N., Locke K., Qu L., Han G. W., Iyer M. R., Cinar R., Coffey N. J., Wang J., Wu M., Katritch V., Zhao S., Kunos G., Bohn L. M., Makriyannis A., Stevens R. C., Liu Z. J., Cell 2019, 176, 459–467.e413; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Xing C., Zhuang Y., Xu T. H., Feng Z., Zhou X. E., Chen M., Wang L., Meng X., Xue Y., Wang J., Liu H., McGuire T. F., Zhao G., Melcher K., Zhang C., Xu H. E., Xie X. Q., Cell 2020, 180, 645–654.e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Soethoudt M., Grether U., Fingerle J., Grim T. W., Fezza F., de Petrocellis L., Ullmer C., Rothenhäusler B., Perret C., van Gils N., Finlay D., MacDonald C., Chicca A., Gens M. D., Stuart J., de Vries H., Mastrangelo N., Xia L., Alachouzos G., Baggelaar M. P., Martella A., Mock E. D., Deng H., Heitman L. H., Connor M., Di Marzo V., Gertsch J., Lichtman A. H., Maccarrone M., Pacher P., Glass M., van der Stelt M., Nat. Commun. 2017, 8, 13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Atwood B. K., Straiker A., Mackie K., Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 38, 16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dhopeshwarkar A., Mackie K., Mol. Pharmacol. 2014, 86, 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ibsen M. S., Connor M., Glass M., Cannabis Cannabinoid Res. 2017, 2, 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nimczick M., Decker M., ChemMedChem 2015, 10, 773–786. [DOI] [PubMed] [Google Scholar]
- 32.E. E. Kim, M. Lee, T. Inoue, W. Wong, Clinical PET and PET/CT: Principles and Application, Springer Science, New York, 2013.
- 33. Honer M., Gobbi L., Martarello L., Comley R. A., Drug Discov. Today 2014, 19, 1936–1944. [DOI] [PubMed] [Google Scholar]
- 34. Ni R., Mu L., Ametamey S., Acta Pharmacol. Sin. 2019, 40, 351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spinelli F., Mu L., Ametamey S. M., J. Labelled Compd. Radiopharm. 2017, 61, 299–308. [DOI] [PubMed] [Google Scholar]
- 36. Evens N., Muccioli G., Houbrechts N., Lambert D., Verbruggen A., Van Laere K., Bormans G., Nucl. Med. Biol. 2009, 36, 455–465. [DOI] [PubMed] [Google Scholar]
- 37.
- 37a. Evens N., Vandeputte C., Coolen C., Janssen P., Sciot R., Baekelandt V., Verbruggen A., Debyser Z., Van Laere K., Bormans G., Nucl. Med. Biol. 2012, 39, 389–399; [DOI] [PubMed] [Google Scholar]
- 37b. Ahmad R., Koole M., Evens N., Serdons K., Verbruggen A., Bormans G., Van Laere K., Mol. Imaging Biol. 2013, 15, 384–390. [DOI] [PubMed] [Google Scholar]
- 38. Mu L., Slavik R., Müller A., Popaj K., Cermak S., Weber M., Schibli R., Krämer S. D., Ametamey S. M., Pharmaceuticals 2014, 7, 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mu L., Bieri D., Slavik R., Drandarov K., Muller A., Cermak S., Weber M., Schibli R., Kramer S., Ametamey S., J. Neurochem. 2013, 126, 616–624. [DOI] [PubMed] [Google Scholar]
- 40. Slavik R., Herde A., Bieri D., Weber M., Schibli R., Kramer S., Ametamey S., Mu L., Eur. J. Med. Chem. 2015, 92, 554–564. [DOI] [PubMed] [Google Scholar]
- 41. Slavik R., Herde A., Haider A., Kramer S., Weber M., Schibli R., Ametamey S., Mu L., J. Neurochem. 2016, 138, 874–886. [DOI] [PubMed] [Google Scholar]
- 42. Haider A., Spinelli F., Herde A., Mu B., Keller C., Margelisch M., Weber M., Schibli R., Mu L., Ametamey S., Eur. J. Med. Chem. 2018, 145, 746–759. [DOI] [PubMed] [Google Scholar]
- 43. Teodoro R., Moldovan R. P., Lueg C., Günther R., Donat C. K., Ludwig F. A., Fischer S., Deuther-Conrad W., Wünsch B., Brust P., Org. Med. Chem. Lett. 2013, 3, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.
- 44a. Heimann D., Borgel F., de Vries H., Bachmann K., Rose V. E., Frehland B., Schepmann D., Heitman L. H., Wünsch B., Eur. J. Med. Chem. 2018, 143, 1436–1447; [DOI] [PubMed] [Google Scholar]
- 44b. Heimann D., Borgel F., de Vries H., Patberg M., Jan-Smith E., Frehland B., Schepmann D., Heitman L. H., Wünsch B., Eur. J. Med. Chem. 2018, 146, 409–422. [DOI] [PubMed] [Google Scholar]
- 45. Ahamed M., van Veghel D., Ullmer C., Van Laere K., Verbruggen A., Bormans G., Front. Neurosci. 2016, 10, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Attili B., Celen S., Ahamed M., Koole M., Van Den Haute C., Vanduffel W., Bormans G., Br. J. Pharmacol. 2019, 176, 1481–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yao B. B., Hsieh G., Daza A. V., Fan Y., Grayson G. K., Garrison T. R., El Kouhen O., Hooker B. A., Pai M., Wensink E. J., Salyers A. K., Chandran P., Zhu C. Z., Zhong C., Ryther K., Gallagher M. E., Chin C. L., Tovcimak A. E., Hradil V. P., Fox G. B., Dart M. J., Honore P., Meyer M. D., J. Pharmacol. Exp. Ther. 2009, 328, 141–151. [DOI] [PubMed] [Google Scholar]
- 48. Horti A. G., Gao Y., Ravert H. T., Finley P., Valentine H., Wong D. F., Endres C. J., Savonenko A. V., Dannals R. F., Bioorg. Med. Chem. 2010, 18, 5202–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Savonenko A. V., Melnikova T., Wang Y., Ravert H., Gao Y., Koppel J., Lee D., Pletnikova O., Cho E., Sayyida N., Hiatt A., Troncoso J., Davies P., Dannals R. F., Pomper M. G., Horti A. G., PLoS One 2015, 10, e0129618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moldovan R. P., Teodoro R., Gao Y., Deuther-Conrad W., Kranz M., Wang Y., Kuwabara H., Nakano M., Valentine H., Fischer S., Pomper M. G., Wong D. F., Dannals R. F., Brust P., Horti A. G., J. Med. Chem. 2016, 59, 7840–7855. [DOI] [PubMed] [Google Scholar]
- 51. Caillé F., Cacheux F., Peyronneau M. A., Jego B., Jaumain E., Pottier G., Ullmer C., Grether U., Winkeler A., Dollé F., Damont A., Kuhnast B., Mol. Pharm. 2017, 14, 4064–4078. [DOI] [PubMed] [Google Scholar]
- 52. Haider A., Müller A. H., Slavik R., Weber M., Mugnaini C., Ligresti A., Schibli R., Mu L., Ametamey S. M., Front. Neurosci. 2016, 10, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Slavik R., Grether U., Müller Herde A., Gobbi L., Fingerle J., Ullmer C., Krämer S. D., Schibli R., Mu L., Ametamey S. M., J. Med. Chem. 2015, 58, 4266–4277. [DOI] [PubMed] [Google Scholar]
- 54. Haider A., Kretz J., Gobbi L., Ahmed H., Atz K., Burkler M., Bartelmus C., Fingerle J., Guba W., Ullmer C., Honer M., Knuesel I., Weber M., Brink A., Herde A., Keller C., Schibli R., Mu L., Grether U., Ametamey S., J. Med. Chem. 2019, 62, 11165–11181. [DOI] [PubMed] [Google Scholar]
- 55. Vandeputte C., Evens N., Toelen J., Deroose C., Bosier B., Ibrahimi A., Van der Perren A., Gijsbers R., Janssen P., Lambert D., Verbruggen A., Debyser Z., Bormans G., Baekelandt V., Van Laere K., J. Nucl. Med. 2011, 52, 1102–1109. [DOI] [PubMed] [Google Scholar]
- 56. Evens N., Vandeputte C., Muccioli G., Lambert D., Baekelandt V., Verbruggen A., Debyser Z., Van Laere K., Bormans G., Bioorg. Med. Chem. 2011, 19, 4499–4505. [DOI] [PubMed] [Google Scholar]
- 57. Kallinen A., Boyd R., Lane S., Bhalla R., Mardon K., Stimson D. H. R., Werry E. L., Fulton R., Connor M., Kassiou M., Org. Biomol. Chem. 2019, 17, 5086–5098. [DOI] [PubMed] [Google Scholar]
- 58. McGrath J. C., Arribas S., Daly C. J., Trends Pharmacol. Sci. 1996, 17, 393–399. [DOI] [PubMed] [Google Scholar]
- 59.
- 59a. Ma Z., Du L., Li M., J. Med. Chem. 2014, 57, 8187–8203; [DOI] [PubMed] [Google Scholar]
- 59b. Iliopoulos-Tsoutsouvas C., Kulkarni R. N., Makriyannis A., Nikas S. P., Exp. Opin. Drug Discov. 2018, 13, 933–947. [DOI] [PubMed] [Google Scholar]
- 60. Weissleder R., Nat. Biotechnol. 2001, 19, 316–317. [DOI] [PubMed] [Google Scholar]
- 61. Leopoldo M., Lacivita E., Berardi F., Perrone R., Drug Discov. Today 2009, 14, 706–712. [DOI] [PubMed] [Google Scholar]
- 62.
- 62a. Rinaldi-Carmona M., Barth F., Millan J., Derocq J., Casellas P., Congy C., Oustric D., Sarran M., Bouaboula M., Calandra B., Portier M., Shire D., Breliere J., Le Fur G., J. Pharmacol. Exp. Ther. 1998, 284, 644–650; [PubMed] [Google Scholar]
- 62b. Pertwee R., Ross R., Prostag. Leukotr. Ess. 2002, 66, 101–121. [DOI] [PubMed] [Google Scholar]
- 63. Bai M., Sexton M., Stella N., Bornhop D., Bioconjugate Chem. 2008, 19, 988–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sexton M., Woodruff G., Horne E., Lin Y., Muccioli G., Bai M., Stern E., Bornhop D., Stella N., Chem. Biol. 2011, 18, 563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang S., Shao P., Bai M., Bioconjugate Chem. 2013, 24, 1907–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhang S., Shao P., Ling X., Yang L., Hou W., Thorne S. H., Beaino W., Anderson C. J., Ding Y., Bai M., Am. J. Nucl. Med. Mol. Imaging 2015, 5, 246–258. [PMC free article] [PubMed] [Google Scholar]
- 67. Wu Z., Shao P., Zhang S., Bai M., J. Biomed. Opt. 2014, 19, 36006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Spinelli F., Capparelli E., Abate C., Colabufo N., Contino M., J. Med. Chem. 2017, 60, 9913–9931. [DOI] [PubMed] [Google Scholar]
- 69. Tabrizi M., Baraldi P., Saponaro G., Moorman A., Romagnoli R., Preti D., Baraldi S., Ruggiero E., Tintori C., Tuccinardi T., Vincenzi F., Borea P., Varani K., J. Med. Chem. 2013, 56, 4482–4496. [DOI] [PubMed] [Google Scholar]
- 70. Ling X., Zhang S., Shao P., Li W., Yang L., Ding Y., Xu C., Stella N., Bai M., Biomaterials 2015, 57, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Guo X., Ling X., Du F., Wang Q., Huang W., Wang Z., Ding X., Bai M., Wu Z., Transl. Oncol. 2018, 11, 1065–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wu Z., Shao P., Zhang S., Ling X., Bai M., J. Biomed. Opt. 2014, 19, 76016. [DOI] [PubMed] [Google Scholar]
- 73. Cooper A. G., Oyagawa C. R. M., Manning J. J., Singh S., Hook S., Grimsey N. L., Glass M., Tyndall J. D. A., Vernall A. J., MedChemComm 2018, 9, 2055–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Spinelli F., Giampietro R., Stefanachi A., Riganti C., Kopecka J., Abatematteo F. S., Leonetti F., Colabufo N. A., Mangiatordi G. F., Nicolotti O., Perrone M. G., Brea J., Loza M. I., Infantino V., Abate C., Contino M., Eur. J. Med. Chem. 2020, 188, 112037. [DOI] [PubMed] [Google Scholar]
- 75. Valenzano K. J., Tafesse L., Lee G., Harrison J. E., Boulet J. M., Gottshall S. L., Mark L., Pearson M. S., Miller W., Shan S., Rabadi L., Rotshteyn Y., Chaffer S. M., Turchin P. I., Elsemore D. A., Toth M., Koetzner L., Whiteside G. T., Neuropharmacology 2005, 48, 658–672. [DOI] [PubMed] [Google Scholar]
- 76. Yates A. S., Doughty S. W., Kendall D. A., Kellam B., Bioorg. Med. Chem. Lett. 2005, 15, 3758–3762. [DOI] [PubMed] [Google Scholar]
- 77. Showalter V. M., Compton D. R., Martin B. R., Abood M. E., J. Pharmacol. Exp. Ther. 1996, 278, 989–999. [PubMed] [Google Scholar]
- 78. Diaz P., Phatak S. S., Xu J., Astruc-Diaz F., Cavasotto C. N., Naguib M., J. Med. Chem. 2009, 52, 433–444. [DOI] [PubMed] [Google Scholar]
- 79. Petrov R. R., Ferrini M. E., Jaffar Z., Thompson C. M., Roberts K., Diaz P., Bioorg. Med. Chem. Lett. 2011, 21, 5859–5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cooper A. G., MacDonald C., Glass M., Hook S., Tyndall J. D. A., Vernall A. J., Eur. J. Med. Chem. 2018, 145, 770–789. [DOI] [PubMed] [Google Scholar]
- 81. Singh S., Oyagawa C. R. M., Macdonald C., Grimsey N. L., Glass M., Vernall A. J., ACS Med. Chem. Lett. 2019, 209–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Westphal M. V., Sarott R. C., Zirwes E. A., Osterwald A., Guba W., Ullmer C., Grether U., Carreira E. M., Chem. Eur. J. 2020, 26, 1380–1387. [DOI] [PubMed] [Google Scholar]
- 83.R. C. Sarott, M. V. Westphal, P. Pfaff, C. Korn, D. A. Sykes, T. Gazzi, B. Brennecke, K. Atz, M. Weise, Y. Mostinki, P. Hompluem, T. Miljus, N. J. Roth, H. Asmelash, M. C. Vong, J. Piovesan, W. Guba, A. C. Rufer, E. A. Kusznir, S. Huber, C. Raposo, E. A. Zirwes, A. Osterwald, A. Pavlovic, S. Moes, J. Beck, I. Benito-Cuesta, T. Grande, A. Yeliseev, F. Drawnel, G. Widmer, D. Holzer, T. van der Wel, H. Mandhair, C. Y. Yuan, W. R. Drobyski, Y. Saroz, N. Grimsey, M. Honer, J. Fingerle, K. Gawrisch, J. Romero, C. J. Hillard, Z. V. Varga, M. van der Stelt, P. Pacher, J. Gertsch, P. J. McCormick, C. Ullmer, S. Oddi, M. Maccarrone, D. B. Veprintsev, M. Nazaré, U. Grether, E. M. Carreira, ChemRxiv preprint 2019, https://doi.org/10.26434/chemrxiv.10288547.v10288541.
- 84.T. Gazzi, B. Brennecke, K. Atz, C. Korn, D. Sykes, R. C. Sarott, M. V. Westphal, P. Pfaff, M. Weise, Y. Mostinski, B. L. Hoare, T. Miljus, M. Mexi, W. Guba, A. Alker, A. C. Rufer, E. A. Kusznir, S. Huber, C. Raposo, E. A. Zirwes, A. Osterwald, A. Pavlovic, S. Moes, J. Beck, I. Benito-Cuesta, T. Grande, F. Drawnel, G. Widmer, D. Holzer, T. van der Wel, H. Mandhair, Y. Saroz, N. Grimsey, M. Honer, J. Fingerle, K. Gawrisch, J. Romero, C. J. Hillard, P. J. McCormick, Z. V. Varga, M. van der Stelt, P. Pacher, J. Gertsch, C. Ullmer, S. Oddi, M. Maccarrone, D. B. Veprintsev, E. M. Carreira, U. Grether, M. Nazaré, ChemRxiv preprint 2019, https://doi.org/10.26434/chemrxiv.10283027.v10283021.
- 85.
- 85a. Singh J., Petter R. C., Baillie T. A., Whitty A., Nat. Rev. Drug Discovery 2011, 10, 307–317; [DOI] [PubMed] [Google Scholar]
- 85b. Ghosh A. K., Samanta I., Mondal A., Liu W. R., ChemMedChem 2019, 14, 889–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yocum R. R., Waxman D. J., Rasmussen J. R., Strominger J. L., Proceed. Natl. Acad. Sci. U. S. A. 1979, 76, 2730–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Savi P., Zachayus J., Delesque-Touchard N., Labouret C., Herve C., Uzabiaga M., Pereillo J., Culouscou J., Bono F., Ferrara P., Herbert J., Proc. Natl. Acad. Sci. USA 2006, 103, 11069–11074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Toth L., Muszbek L., Komaromi I., J. Mol. Graphics Modell. 2013, 40, 99–109. [DOI] [PubMed] [Google Scholar]
- 89. Shin J., Kim N., J. Neurogastroenterol. 2013, 19, 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mullard A., Nat. Rev. Drug Discov. 2018, 17, 81. [DOI] [PubMed] [Google Scholar]
- 91. Weichert D., Gmeiner P., ACS Chem. Biol. 2015, 10, 1376–1386. [DOI] [PubMed] [Google Scholar]
- 92. Pei Y., Mercier R., Anday J., Thakur G., Zvonok A., Hurst D., Reggio P., Janero D., Makriyannis A., Chem. Biol. 2008, 15, 1207–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Guo Y., Abadji V., Morse K. L., Fournier D. J., Li X. Y., Makriyannis A., J. Med. Chem. 1994, 37, 3867–3870. [DOI] [PubMed] [Google Scholar]
- 94. Picone R., Khanolkar A., Xu W., Ayotte L., Thakur G., Hurst D., Abood M., Reggio P., Fournier D., Makriyannis A., Mol. Pharmacol. 2005, 68, 1623–1635. [DOI] [PubMed] [Google Scholar]
- 95. Szymanski D., Papanastasiou M., Melchior K., Zvonok N., Mercier R., Janero D., Thakur G., Cha S., Wu B., Karger B., Makriyannis A., J. Prot. Res. 2011, 10, 4789–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhou H., Peng Y., Halikhedkar A., Fan P., Janero D., Thakur G., Mercier R., Sun X., Ma X., Makriyannis A., ACS Chem. Neurosci. 2017, 8, 1338–1347. [DOI] [PubMed] [Google Scholar]
- 97. Ogawa G., Tius M., Zhou H., Nikas S., Halikhedkar A., Mallipeddi S., Makriyannis A., J. Med. Chem. 2015, 58, 3104–3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mercier R., Pei Y., Pandarinathan L., Janero D., Zhang J., Makriyannis A., Chem. Biol. 2010, 17, 1132–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Mallipeddi S., Kreimer S., Zvonok N., Vemuri K., Karger B., Ivanov A., Makriyannis A., J. Prot. Res. 2017, 16, 2419–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.L. De Petrocellis, M. Nabissi, G. Santoni, A. Ligresti in Cannabinoid Pharmacology, Vol. 80 (Eds.: D. Kendall, S. P. H. Alexander), Academic Press, Cambridge, 2017, pp. 249–289. [DOI] [PubMed]
- 101.M. Pistis, S. E. O′Sullivan in Cannabinoid Pharmacology, Vol. 80 (Eds.: D. Kendall, S. P. H. Alexander), Academic Press, Cambridge, 2017, pp. 291–328.
- 102.
- 102a. Lerch M. M., Hansen M. J., van Dam G. M., Szymanski W., Feringa B. L., Angew. Chem. Int. Ed. 2016, 55, 10978–10999; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11140–11163; [Google Scholar]
- 102b. Lerch M. M., Hansen M. J., van Dam G. M., Szymanski W., Feringa B. L., Angew. Chem. 2016, 128, 11140–11163; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2016, 55, 10978–10999. [DOI] [PubMed] [Google Scholar]
- 103. Velema W., Szymanski W., Feringa B., J. Am. Chem. Soc. 2014, 136, 2178–2191. [DOI] [PubMed] [Google Scholar]
- 104.
- 104a. Agostinis P., Berg K., Cengel K., Foster T., Girotti A., Gollnick S., Hahn S., Hamblin M., Juzeniene A., Kessel D., Korbelik M., Moan J., Mroz P., Nowis D., Piette J., Wilson B., Golab J., Ca-Cancer J. Clin. 2011, 61, 250–281; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104b.L. Agnetta, M. Decker in Design of Hybrid Molecules for Drug Development (Ed.: M. Decker), Elsevier, Oxford, 2017, pp. 279–315.
- 105. Zhang S., Jia N., Shao P., Tong Q., Xie X., Bai M., Chem. Biol. 2014, 21, 338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Jia N. Y., Zhang S. J., Shao P., Bagia C., Janjic J. M., Ding Y., Bai M. F., Mol. Pharmaceutics 2014, 11, 1919–1929. [DOI] [PubMed] [Google Scholar]
- 107. Ling X., Zhang S., Liu Y., Bai M., J. Biomed. Opt. 2018, 23, 1–9. [DOI] [PubMed] [Google Scholar]
- 108.P. P. Geurink, L. M. Prely, G. A. van der Marel, R. Bischoff, H. S. Overkleeft in Activity-Based Protein Profiling (Ed.: S. A. Sieber), Springer, Heidelberg, 2011, pp. 85–113. [DOI] [PubMed]
- 109. Szymanski D., Papanastasiou M., Pandarinathan L., Zvonok N., Janero D. R., Pavlopoulos S., Vouros P., Makriyannis A., J. Med. Chem. 2018, 61, 11199–11208. [DOI] [PubMed] [Google Scholar]
- 110. Charalambous A., Guo Y., Houston D. B., Howlett A. C., Compton D. R., Martin B. R., Makriyannis A., J. Med. Chem. 1992, 35, 3076–3079. [DOI] [PubMed] [Google Scholar]
- 111. Khanolkar A. D., Palmer S. L., Makriyannis A., Chem. Phys. Lipids 2000, 108, 37–52. [DOI] [PubMed] [Google Scholar]
- 112. Dixon D., Tius M., Thakur G., Zhou H., Bowman A., Shukla V., Peng Y., Makriyannis A., Bioorg. Med. Chem. Lett. 2012, 22, 5322–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Soethoudt M., Stolze S., Westphal M., van Stralen L., Martella A., van Rooden E., Guba W., Varga Z., Deng H., van Kasteren S., Grether U., IJzerman A., Pacher P., Carreira E., Overkleeft H., Ioan-Facsinay A., Heitman L., van der Stelt M., J. Am. Chem. Soc. 2018, 140, 6067–6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Rodriguez-Soacha D. A., Decker M., Adv. Ther. 2018, 1, 180037. [Google Scholar]
- 115.
- 115a. Page D., Balaux E., Boisvert L., Liu Z., Milburn C., Tremblay M., Wei Z., Woo S., Luo X., Cheng Y., Yang H., Srivastava S., Zhou F., Brown W., Tomaszewski M., Walpole C., Hodzic L., St-Onge S., Godbout C., Salois D., Payza K., Bioorg. Med. Chem. Lett. 2008, 18, 3695–3700; [DOI] [PubMed] [Google Scholar]
- 115b. Dolles D., Nimczick M., Scheiner M., Ramler J., Stadtmüller P., Sawatzky E., Drakopoulos A., Sotriffer C., Wittmann H., Strasser A., Decker M., ChemMedChem 2016, 11, 1270–1283. [DOI] [PubMed] [Google Scholar]
- 116. Dolles D., Strasser A., Wittmann H. J., Marinelli O., Nabissi M., Pertwee R. G., Decker M., Adv. Ther. 2018, 1, 1700032. [Google Scholar]
- 117. Ren W. X., Han J., Uhm S., Jang Y. J., Kang C., Kim J. H., Kim J. S., Chem. Commun. 2015, 51, 10403–10418. [DOI] [PubMed] [Google Scholar]
- 118. Martín-Couce L., Martín-Fontecha M., Capolicchio S., López-Rodríguez M. L., Ortega-Gutiérrez S., J. Med. Chem. 2011, 54, 5265–5269. [DOI] [PubMed] [Google Scholar]
- 119.
- 119a. Martín-Couce L., Martín-Fontecha M., Palomares O., Mestre L., Cordomí A., Hernangomez M., Palma S., Pardo L., Guaza C., López-Rodríguez M. L., Ortega-Gutiérrez S., Angew. Chem. Int. Ed. 2012, 51, 6896–6899; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7002–7005; [Google Scholar]
- 119b. Martín-Couce L., Martín-Fontecha M., Palomares O., Mestre L., Cordomí A., Hernangomez M., Palma S., Pardo L., Guaza C., López-Rodríguez M. L., Ortega-Gutiérrez S., Angew. Chem. 2012, 124, 7002–7005; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012, 51, 6896–6899. [DOI] [PubMed] [Google Scholar]
- 120.
- 120a. Leleu-Chavain N., Baudelet D., Heloire V. M., Rocha D. E., Renault N., Barczyk A., Djouina M., Body-Malapel M., Carato P., Millet R., Eur. J. Med. Chem. 2019, 165, 347–362; [DOI] [PubMed] [Google Scholar]
- 120b. Mugnaini C., Rabbito A., Brizzi A., Palombi N., Petrosino S., Verde R., Di Marzo V., Ligresti A., Corelli F., Eur. J. Med. Chem. 2019, 161, 239–251. [DOI] [PubMed] [Google Scholar]