

Summary
Transposable elements (TEs) are the main reason for the high plasticity of plant genomes, where they occur as communities of diverse evolutionary lineages. Because research has typically focused on single abundant families or summarized TEs at a coarse taxonomic level, our knowledge about how these lineages differ in their effects on genome evolution is still rudimentary.
Here we investigate the community composition and dynamics of 32 long terminal repeat retrotransposon (LTR‐RT) families in the 272‐Mb genome of the Mediterranean grass Brachypodium distachyon.
We find that much of the recent transpositional activity in the B. distachyon genome is due to centromeric Gypsy families and Copia elements belonging to the Angela lineage. With a half‐life as low as 66 kyr, the latter are the most dynamic part of the genome and an important source of within‐species polymorphisms. Second, GC‐rich Gypsy elements of the Retand lineage are the most abundant TEs in the genome. Their presence explains > 20% of the genome‐wide variation in GC content and is associated with higher methylation levels.
Our study shows how individual TE lineages change the genetic and epigenetic constitution of the host beyond simple changes in genome size.
Keywords: GC content, genome evolution, methylation, random forest, retrotransposons, transposable elements (TEs)
Short abstract
See also the Commentary on this article by Catalán & Vogel, 227: 1587–1590.
Introduction
Transposable elements (TEs) are stretches of DNA which can replicate within genomes (Burt & Trivers, 2006). Since their discovery in the 1950s by Barbara McClintock, their fuzzy status between selfish parasite and integral part of the host genome has puzzled biologists. In contrast to viruses, TEs do not routinely leave the host; instead, their evolutionary history is largely one of vertical transmission and co‐evolution in a host genome which has evolved epigenetic mechanisms to suppress their activity (Lisch, 2009). As suggested by the omnipresence of TEs in the eukaryote domain, this co‐evolution is ancient and has shaped eukaryote genome evolution from the very beginning (Eickbush & Malik, 2002).
Numerous comparative and functional studies have shown how TEs can inflate genome size (Bennetzen & Kellog, 1997; Ma & Bennetzen, 2004; Hawkins et al., 2006; Piegu et al., 2006) and produce novel host phenotypes by inserting into genic regions (McClintock, 1956; Bhattacharyya et al., 1990; van't Hof et al., 2016; Niu et al., 2019). Although these examples have added to the sense that TEs play an important role in evolution (Casacuberta & González, 2013; Belyayev, 2014), it also has become clear that TEs are immensely diverse, and that what is found in one species does not necessarily hold in others. This is particularly true for plants, whose genomes are subject to fewer evolutionary constraints than those of animals (Kejnovsky et al., 2009) and harbour diverse TE lineages with different structures and replication strategies (Wicker & Keller, 2007; Du et al., 2010; Neumann et al., 2019). Transposable element landscapes can diverge rapidly because TE activity depends on multiple interacting factors whose direction and strength can differ within a single species. These factors include environmental triggers (Horváth et al., 2017), the demographic history of the host population (Lynch, 2007) and horizontal transfers (El Baidouri et al., 2014).
Such complexity and historical contingency is the hallmark of ecological systems, and it has therefore been proposed that concepts from ecology are borrowed, conceiving genomes to be ecosystems inhabited by various ‘species’ of TEs differing in behaviour and genomic niches (Kidwell & Lisch, 2001). Although the scope of TE ecology is not very clear‐cut (Brookfield, 2005; Venner et al., 2009; Linquist et al., 2013), one important intuition this metaphor conveys is that our understanding of genome evolution can be improved by taking the diversity of TEs into account rather than lumping them together into coarse taxonomic units (Stitzer et al., 2019). Most studies with an interest in mobile elements consider TEs at the class (DNA transposons vs retrotransposons) or superfamily (Copia vs Gypsy) level. Such comparisons provide valuable overviews of TE communities and have revealed some general patterns, for example DNA transposons tend to occur closer to genes than retrotransposons (Feschotte & Pritham, 2007). Yet, considering that, for example, the Gypsy superfamily consists of more than a dozen of lineages which may be as old as the major divisions of plants (Neumann et al., 2019), generalizations about ‘repeats’, ‘retrotransposons’ or ‘Gypsy elements’ are liable to level out biologically important differences between TE lineages.
With the increasing amount of information available for some model organisms, it has become possible to investigate a single genome ecosystem in detail by relating TEs to recombination rate, methylation levels and other properties of the genomic context (Stitzer et al., 2019). In the present study we use the excellent genomic resources available for the wild Mediterranean grass Brachypodium distachyon in order to investigate the long terminal repeat retrotransposons (LTR‐RT) in this species. In terms of contribution to genome size, LTR‐RTs are the most abundant TEs in plants, are typically between 5 and 15 kb long, and multiply through a copy‐and‐paste mechanism involving an RNA intermediate (Eickbush & Malik, 2002). Apart from the reverse transcription step, which is shared with retroviruses, the two flanking LTRs are the most characteristic feature of LTR‐RT insertions. These sequences not only allow a comparatively easy identification of LTR‐RTs in the genome, but also play a vital role in LTR‐RT life history as they contain regulatory motifs (Schulman, 2013) and are prone to ectopic recombination. In B. distachyon, LTR‐RTs make up 20% of the 272‐Mb genome and are widely dispersed along the five chromosomes (International Brachypodium Initiative, 2010; Schulman, 2015). This modest amount of repeats, compared to the TE ‘jungles’ of wheat, maize and other large‐genome plants, allows the LTR‐RT community to be studied without losing sight of its constituent ‘species’.
The goal of our study is to provide an overview of the LTR‐RT community in B. distachyon, and to characterize its dominant lineages and their effect on genome composition and dynamics. In particular, we address the following questions: Which major plant LTR‐RT lineages are present in B. distachyon, and what is their relative abundance and age? How are these different lineages related to important genomic features such as genes, recombination rate, methylation, GC content and genetic diversity?
Material and Methods
Genome assemblies
Two genome assemblies for Brachypodium distachyon are considered in this study. Although the focus is on the reference accession Bd21 for which most information is available, a new assembly for the Turkish accession BdTR7a is included in order to investigate within‐species differences in long terminal repeat retrotransposons (LTR‐RT) communities. The assembly for the reference accession Bd21 (v.3.0) was downloaded from phytozome 12; it is based on BAC libraries and has well‐assembled repetitive regions (International Brachypodium Initiative, 2010; VanBuren & Mockler, 2016). We chose BdTR7a to create a second assembly because among the 54 recently sequenced accessions it has the highest number of nonreference transposable element insertions (Stritt et al., 2018). The BdTR7a assembly was created by combining PacBio sequencing with Bionano optical mapping (Supporting Information Methods S1, assembly available at https://datadryad.org/stash/dataset/doi:10.5061/dryad.bg79cnp70).
Annotation of LTR retrotransposons and reverse transcriptase phylogeny
Because a consensus library for the different TE families of B. distachyon is available on the TREP database (http://botserv2.uzh.ch/kelldata/trep-db/index.html), we used these sequences as a starting point to annotate LTR‐RTs. For the sake of consistency, we used the same approach to annotate transposable elements (TEs) in the new assembly and to re‐annotate them in the reference genome. The LTR sequence of each of the 21 Copia and 19 Gypsy consensus sequences was blasted against the assemblies. Hits which covered ≥ 80% of the LTR were retained and sorted according to their position on the chromosome. We then traversed the sorted hits and compared adjacent LTR pairs. A hit pair was denoted intact if the two hits belonged to the same family, were on the same strand, and the distance between them corresponded to the distance expected from the consensus sequence, with an error margin of 20% to account for indels. Otherwise the hit was denoted a single LTR. A single LTR was classified as solo LTR if it lacked internal TE sequence in its 500‐bp flanking regions and was flanked by identical 4‐mers, being evidence for the 4–6‐bp‐long target site duplication (TSD) created upon the insertion of LTR retrotransposons (Wicker et al., 2007). For the comparison of intact and solo elements, we included only intact elements satisfying the same stringent criteria, in this case requiring TSDs and the presence of internal TE sequence 500 bp up‐ or downstream of the LTRs. The Python script implementing this annotation method is available at http://www.github.com/cstritt/tes.
In order to determine the evolutionary relatedness of the annotated TE families and their place in the larger phylogeny of plant LTR retrotransposons, we searched the six‐frame translated TE consensus sequences against the Pfam database (pfam.xfam.org) with the Hmmer tool hmmscan (Finn et al., 2011) and extracted the sequences aligning to the reverse transcriptase profiles RVT_1 (Gypsy) and RVT_2 (Copia). Amino acid sequences of the major Copia and Gypsy lineages in plants were obtained from the RepeatExplorer database (Neumann et al., 2019). A reverse transcriptase consensus sequence for each lineage was constructed after aligning the individual RT copies with mafft v.7.402 (Katoh & Standley, 2013). Copia and Gypsy consensus sequences were then merged with the respective B. distachyon RT sequences, aligned with mafft (‐‐auto), and trees were estimated with mrbayes 3.2.2 (Ronquist et al., 2012) by sampling over different amino acid models (aamodelpr = mixed) and running two chains for 500 000 generations. Tracer (Rambaut et al., 2018) was used to assess the convergence and mixing of the MCMC runs. Trees were visualized with figtree (tree.bio.ed.ac.uk/software/figtree).
Estimation of insertion age and family survival functions
In order to estimate the age of intact and solo insertions, we used the LTR sequences to construct LTR genealogies. LTRs from single and intact elements for each family were aligned with Mafft (‐‐auto) and trimmed with trimal (Capella‐Gutiérrez et al., 2009) to remove sites with > 5% gaps in the alignment. Trees were estimated with the uniform clock model in mrbayes, with a HKY substitution model (nst = 2) and an inverse gamma prior on the clock rate (rates = invgamma). From the LTR trees we extracted the terminal branch lengths as a proxy of insertion age. In the case of intact elements, this corresponds to well‐known method of age estimation from LTR divergence (SanMiguel et al., 1998); for solo LTRs, the terminal branch length represents an upper‐limit age estimate as it represents the time to the most recent common ancestor of the solo LTR and another copy rather than the two solo LTRs of a single copy.
Survival curves provide a useful summary of the turnover of LTR‐RT families (Wicker & Keller 2007). They can be obtained by fitting an exponential decay function N t = N 0 e−λt to the observed age distribution of full‐length copies. The main assumptions made here are: (1) that the age distribution of TEs can indeed be approximated by this function; and (2) that the family death rate λ is constant through time. To estimate λ for the families with at least 20 full‐length copies and with age distributions shifted towards young insertions, such that they meet the first assumption, exponential decay functions were fitted to the age distributions using the fitdistr function of R/mass, and the exponential rate was recovered. In addition, we estimated λS, an alternative death rate estimate which includes information on the age of solo LTRs, using the maximum‐likelihood function of Dai et al. (2018). Confidence intervals for λS were obtained from 100 bootstraps. Both procedures were implemented in R and are available on github.com/cstritt/tes.
Genomic niche features
In order to characterize the genomic niches of LTR‐RT lineages, we compiled a dataset of diverse genomic features which might affect or be affected by the presence of LTR‐RTs. Recombination rates were obtained from the linkage map of Huo et al. (2011). Distance to the closest gene was calculated based on v.3.1 of the reference genome annotation, downloaded from phytozome 12. Copy methylation levels were obtained through whole‐genome bisulfite sequencing (Methods S2). Finally, we estimated three population genetic statistics in 10‐kb windows around the annotated TEs, 5 kb on each side: Kelly's Z nS, a measure of multilocus linkage disequilibrium and thus a proxy for local recombination rates; Tajima's D, a statistic indicating deviations from neutrality; and the number of segregating sites S and nucleotide diversity π. Variants for 25 Turkish and Iraqi accessions were extracted from the phytozome 12 variant set, and R/popgenome (Pfeifer et al., 2014) was used to estimate the statistics.
Statistical inference
In order to test whether the ratio of solo‐ to full‐length elements can be predicted by the length of the LTR and the internal TE sequence, we fitted a generalized linear model with a binomial error distribution, using the glm function in R. Because TE families are phylogenetically related and not independent observations, we used only one randomly chosen family per lineage, resulting in a total of 12 observations. The Tekay family RLG_BdisC004 was excluded because of its unusually long LTR.
A random forest approach (Breiman, 2001) was used to discover features of the genomic context associated with the occurrence of specific TE lineages. We preferred this method over a parametric modelling approach because model specification proved impracticable given the high level of variable collinearity, phylogenetic signal and chromosomal autocorrelation in our genome‐wide dataset. A total of 200 decision trees were grown using R/randomforest (Liaw & Wiener, 2002) to predict which TE lineage is present at an insertion site as a function of the genomic niche features described above. Predictor variables were ranked according to the mean decrease in accuracy, a statistic which describes how much worse the model performs when the values of each variable are permuted.
In order to investigate the relationship between TE copy age and GC content, a linear mixed model with the TE lineage as random effect was fitted with the lmer function of R/lme4. All statistical analyses were done with R v.3.4.4.
TE copy synteny and polymorphisms
In order to test whether TE dynamics differ within the species, we compared annotated TEs in Bd21 and BdTR7a, two eastern Mediterranean accessions whose ancestors diverged between 0.5 and 1.5 Myr ago (Ma) (Sancho et al., 2017). The discordant read‐pair approach implemented in detettore (http://github/cstritt/detettore) was used to test whether a copy annotated in one accession is present in the other accession or not. Illumina paired‐end reads for the two accessions were downloaded from the NCBI database (SRA samples SRS1615350 and SRS360854) and aligned to the two respective genome assemblies with BWA MEM (Li, 2013). Detettore was then used to detect clusters of read pairs with abnormal insert sizes which span annotated TEs. A table containing annotated TEs, the values for their genomic niche features, as well as their conservation state between Bd21 and BdTR7a is provided in the supplement (Tables S1 and S2).
Results
Major lineages of LTR retrotransposons in B. distachyon
Dotplots and pairwise alignments of the 40 LTR retrotransposon consensus sequences from the TREP database revealed the presence of elements with sequence similarities > 80% and thus not classifying as families sensu Wicker et al. (2007; Fig. S1). Such elements were merged and given the name of the most abundant subfamily, resulting in a total of 32 families (17 Copia and 15 Gypsy) which were analysed in this study.
Phylogenetic trees based on the reverse transcriptase (RT) sequences show that 11 major lineages of LTR‐RTs are present in B. distachyon (Fig. 1). Some lineages are present as a single family, namely the Copia lineages Bianca, SIRE, Alesia and Ikeros, as well as the Gypsy lineages Reina and Tekay; others comprise multiple families, notably the six Copia families of the Ivana lineage and the three families of the Angela lineage, as well as the nine Gypsy families of the Retand lineage and the three families of the CRM lineage (Fig. 1). The Retand families are further divided into three subclades of which subclade C, with four families and 139 full‐length elements, is the most abundant and diverse. Interestingly, the Gypsy element with most full‐length copies, RLG_BdisC152, could not be classified based on reverse transcriptase as no sequence homology was found. Indeed, apart from a gag fragment, this family lacks internal retrotransposon coding domains altogether. A hint to its origin comes from a stretch of 2000 bp including the 3' half of the LTR and the adjacent region, which has high similarity to the CRM families (Fig. S2).
Figure 1.
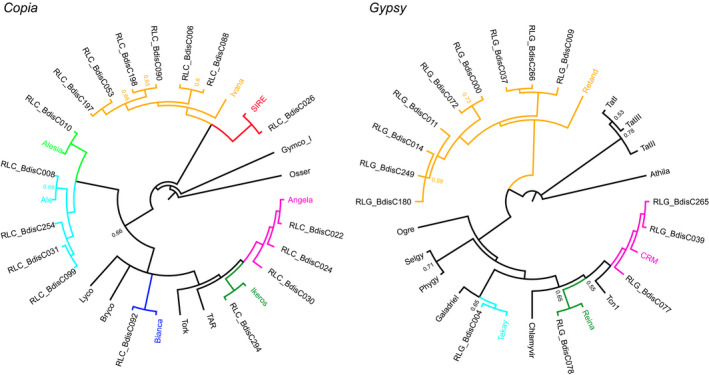
Phylogeny of long terminal repeat (LTR) retrotransposons based on reverse transcriptase sequences. Tip labels not beginning with RL represent consensus sequences of major plant transposable element (TE) lineages; the coloured clades show the lineages present in Brachypodium distachyon. Posterior probabilities for splits are only indicated when they are lower than 0.8. Absent from this figure is RLG_BdisC152, which lacks a reverse transcriptase domain.
Community composition
The abundance of the 32 LTR‐RT families in the reference accession Bd21 varies from 0 (RLG_BdisC037) to 58 full‐length elements (RLG_BdisC152; Table 1; Fig. 2). The three most abundant Copia families, all of which belong to the Angela lineage, have between 27 and 45 full‐length elements. The Alesia family RLC_BdisC010 is dataset‐abundant (24 intact copies), whereas most other Copia families are present in low copy numbers (< 10). Gypsy elements are generally more abundant than Copia elements, the largest contribution coming from families of the Retand lineage, the CRM family RLG_BdisC039 and the unclassified RLG_BdisC152.
Table 1.
Overview of annotated long terminal repeat‐retrotransposon (LTR‐RT) families in the reference accession Bd21.
| Lineage | TE family | Length | LTR length | GC | Intacta | Solo | Unclassified | S/Ib |
|---|---|---|---|---|---|---|---|---|
| Copia | ||||||||
| Angela | RLC_BdisC024 | 7917 | 1355 | 47 | 27 | 20 | 160 | 71 | 8 |
| RLC_BdisC022 | 8676 | 1712 | 44 | 45 | 32 | 95 | 46 | 2.97 | |
| RLC_BdisC030 | 7955 | 1328 | 47 | 28 | 22 | 176 | 133 | 8 | |
| Ikeros | RLC_BdisC294 | 6219 | 420 | 45 | 7 | 0 | 3 | 65 | NAc |
| Bianca | RLC_BdisC092 | 5254 | 228 | 38 | 3 | 3 | 1 | 15 | 0.33 |
| Ivana | RLC_BdisC197 | 5239 | 375 | 49 | 6 | 6 | 0 | 1 | 0 |
| RLC_BdisC053 | 5350 | 409 | 50 | 8 | 8 | 1 | 5 | 0.13 | |
| RLC_BdisC198 | 5210 | 417 | 53 | 4 | 4 | 0 | 0 | 0 | |
| RLC_BdisC090 | 5200 | 366 | 60 | 6 | 5 | 0 | 0 | 0 | |
| RLC_BdisC006 | 5407 | 498 | 50 | 2 | 2 | 0 | 2 | 0 | |
| RLC_BdisC088 | 5161 | 338 | 54 | 4 | 3 | 1 | 1 | 0.33 | |
| SIRE | RLC_BdisC026 | 9276 | 1345 | 39 | 10 | 9 | 131 | 138 | 14.56 |
| Ale | RLC_BdisC099 | 4796 | 200 | 48 | 4 | 3 | 0 | 3 | 0 |
| RLC_BdisC031 | 4846 | 223 | 50 | 4 | 4 | 0 | 2 | 0 | |
| RLC_BdisC254 | 4915 | 208 | 57 | 3 | 3 | 0 | 3 | 0 | |
| RLC_BdisC008 | 5170 | 132 | 52 | 5 | 4 | 2 | 7 | 0.5 | |
| Alesia | RLC_BdisC010 | 5894 | 720 | 45 | 24 | 16 | 30 | 10 | 1.88 |
| Gypsy | ||||||||
| Retand A | RLG_BdisC009 | 13 537 | 821 | 59 | 22 | 9 | 31 | 87 | 3.44 |
| RLG_BdisC266 | 13 197 | 915 | 56 | 45 | 26 | 116 | 230 | 4.46 | |
| RLG_BdisC037 | 15 485 | 854 | 55 | 0 | 0 | 2 | 2 | NA | |
| Retand B | RLG_BdisC000 | 13 500 | 808 | 55 | 57 | 31 | 81 | 349 | 2.61 |
| RLG_BdisC072 | 14 246 | 478 | 53 | 10 | 5 | 7 | 69 | 1.4 | |
| Retand C | RLG_BdisC011 | 14 104 | 972 | 66 | 47 | 29 | 75 | 316 | 2.59 |
| RLG_BdisC014 | 12 887 | 838 | 63 | 26 | 14 | 41 | 260 | 2.93 | |
| RLG_BdisC249 | 13 155 | 915 | 62 | 17 | 4 | 37 | 203 | 9.25 | |
| RLG_BdisC180 | 13 250 | 907 | 64 | 49 | 11 | 79 | 371 | 7.18 | |
| Tekay | RLG_BdisC004 | 12 395 | 3109 | 47 | 8 | 5 | 9 | 19 | 1.8 |
| CRM | RLG_BdisC039 | 8274 | 955 | 43 | 34 | 22 | 40 | 97 | 1.82 |
| RLG_BdisC265 | 7766 | 916 | 45 | 4 | 3 | 13 | 25 | 4.33 | |
| RLG_BdisC077 | 7791 | 939 | 46 | 5 | 3 | 10 | 32 | 3.33 | |
| Reina | RLG_BdisC078 | 5192 | 399 | 58 | 4 | 4 | 11 | 14 | 2.75 |
| ? | RLG_BdisC152 | 5284 | 1012 | 40 | 58 | 28 | 109 | 199 | 3.89 |
TE, transposable element.
The two numbers indicate the total number of annotated paired LTRs and the subset of these which have recognizable target site duplications and the expected 500‐bp flanking regions (see Material and Methods).
Solo‐to‐intact ratio.
Non applicable.
Figure 2.
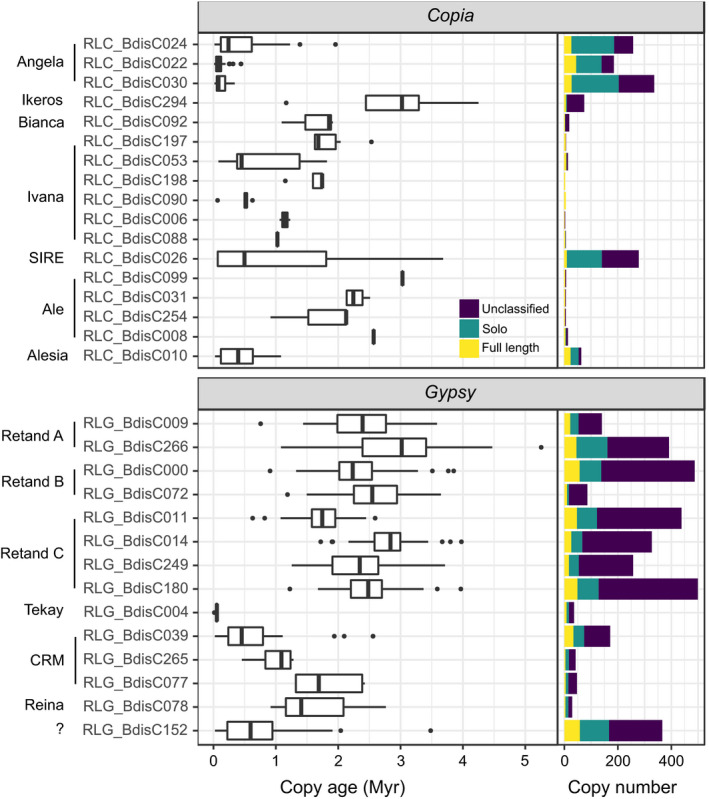
Age distributions of full‐length elements and copy numbers of long terminal repeat‐retrotransposon (LTR‐RT) families in the Brachypodium distachyon reference accession Bd21. The left panel displays, for each family, the age of full‐length elements as estimated from the divergence of their LTRs. The right panel shows the number of full‐length (yellow), solo‐LTR (green), and unclassified copies (purple) in the genome. Families are grouped according to the transposable element (TE) lineage to which they belong. The Retand family RLG_BdisC037 is missing on this figure because it has no full‐length copies.
In most LTR‐RT families, the number of solo LTRs exceeds the number of intact elements (Table 1, Fig. 2). An exception are several low copy‐number Copia families for which we detected only few or no solo LTRs. High solo‐to‐intact (S/I) ratios are associated with long LTRs (r = 0.73, P < 0.001) and high ratios of LTR vs internal sequence length (r = 0.47, P = 0.01). The significance of the latter association is supported by a binomial generalized linear model which includes LTR and internal sequence length as well as their interaction term as predictor variables: although LTR length itself is not significant in this model (P = 0.55), the interaction term is (P = 0.04), indicating that high rates of solo LTR formation are favoured by a combination of long LTRs and short internal sequences separating them.
Finally, a considerable number of annotated LTRs could not be classified as being part of a full‐length or a solo element (Table 1). Annotated Retand LTRs frequently lack target site duplications and/or the expected sequence context that would reveal them as solo LTRs. Among the Copia families, a majority of the elements still have these signatures, although the number of unclassified elements surpasses 100 for RLG_BdisC030 and RLC_BdisC026. As shown in the next paragraph, the lack of signatures is associated with the age of the families.
Age structure of the LTR‐RT community and family half‐lives
Insertion age estimates for full‐length elements based on LTR divergence reveal a multi‐layered age structure of the LTR‐RT community in B. distachyon (Fig. 2). The Angela families are characterized by low LTR divergence (median copy age = 0.4 Ma). For many Angela copies, the 95% credibility interval of the age estimates reaches into the present, illustrating the impossibility to tell whether an element jumped yesterday or a few thousand years ago. The abundant Retand elements, on the other hand, are comparatively old, with a median age of 2.4 my, although this varies for the different families. Ivana and CRM, two other multi‐family lineages, show a stepwise age pattern with some old and apparently inactive families and potentially active families with young copies (RLC_BdisC053, RLG_BdisC039).
Not only average ages, but also the range of age estimates vary between lineages and families. Narrow ranges are due partly to polytomies in the LTR genealogies (Fig. S3): the lack of information in highly similar (Angela) or short (RLC_BdisC008, RLC_BdisC088, RLC_BdisC099) LTRs leads to unresolved nodes in the LTR tree and identical branch lengths for multiple copies. Other families comprise copies of very different ages. Most strikingly, the age distribution of the 10 intact SIRE copies spans 2 Myr, and judging from the presence of one young insertion with identical LTRs (RLC_BdisC026_Bd4_6010841), the family is still active.
Age distributions inferred from LTR divergence only provide an indirect picture of TE activity because their shapes depend not only on TE insertion rates, but also on how rapidly full‐length copies disappear from the genome (Dai et al., 2018). Focusing on the five TE families with > 20 intact elements whose age distribution can be approximated by an exponential decay function, survival curves reveal considerable differences among families (Table 2; Fig. 3). The half‐lives of the three abundant Angela families are the lowest and range from 66 kyr (CI = 53–91) for RLC_BdisC030 to 152 ky (CI = 98–201) for RLC_BdisC024. The two centromeric Gypsy families RLG_BdisC039 and RLG_BdisC152 are more persistent, with half‐lives of 495 kyr (CI = 407–854) and 341 kyr (CI = 298–511).
Table 2.
Half‐life times for nine high copy‐number families.
| Family | t 1/2 (λ) | t 1/2 (λS) | 95% CI |
|---|---|---|---|
| RLC_BdisC022 | 91 | 79 | 75–118 |
| RLC_BdisC030 | 100 | 66 | 53–91 |
| RLC_BdisC024 | 246 | 152 | 98–201 |
| RLG_BdisC152 | 447 | 341 | 298–511 |
| RLG_BdisC039 | 552 | 495 | 407–854 |
Confidence intervals for the λS half‐lives were obtained from 100 bootstrap replicates.
Figure 3.
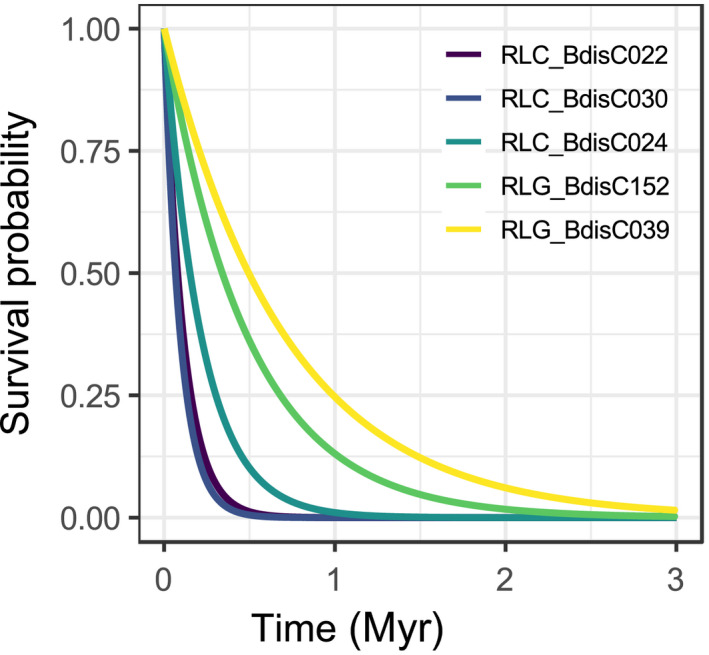
Survival curves of five high copy‐number families in the Brachypodium distachyon reference accession Bd21. For each family, depicted in different colours, these graphs shows the probability (y‐axis) that a new insertion survives up to a specific age (x‐axis). The half‐life of a family, as discussed in the text, is the age up to which 50% of the insertions survive.
TE copy synteny between Bd21 and BdTR7a
In order to find out whether recent TE activity has led to within‐species differences in the LTR‐RT community, we annotated LTR‐RTs in the Turkish accession BdTR7a. The LTR‐RT communities of Bd21 and BdTR7a are highly similar, as can be seen in the strong correlation of family abundances between the two accessions (r = 0.99, P < 0.001; Fig. S4). The largest differences are due to the two Angela families RLC_BdisC022 and RLC_BdisC030, for which 77 and 59 intact elements were annotated in BdTR7a compared to 45 and 26 in Bd21.
In total, 456 of 4612 (9.9%) LTR‐RT copies present in Bd21 have no homolog in BdTR7a, 100 of them full‐length copies, 350 solo LTRs and six unclassified. Strikingly, 235 of these polymorphisms are Angela solo LTRs, compared to 44 intact Angela copies. The other way around, 495 of 4493 (11%) TEs present in BdTR7a have no homolog in Bd21, 126 of them full‐length, 357 solo LTRs and 12 unclassified. Also here more than half of the TE polymorphisms (251) are Angela solo LTRs. Other lineages also contribute to TE polymorphisms: of the annotated Alesia elements, 34% and 42% had no homologs in Bd21 and BdTR7a, respectively. Similar numbers were found for the Tekay family (24% and 42%), whereas the CRM lineage (7% and 10%) and the SIRE lineage (13% and 12%) were more conserved. Almost all annotated elements were conserved between the two accessions in the Ale, Bianca, Ikeros, Reina and Retand lineages, consistent with their older age (Fig. 2).
Nonconserved elements have a median age of 0.43 Myr, whereas for conserved elements this value is 2.5 Myr. Ninety percent of the nonconserved elements are younger than 1.5 Myr, and 90% of the conserved elements older than 0.7 Myr, which agrees well with a divergence time of the ancestral lineages of the two accessions between 0.5 and 1.5 Ma (Sancho et al., 2017). The mean distance to genes of the 456 nonconserved elements in Bd21 is 7897 bases (SD = 11 694), whereas for conserved elements the mean is 11 200 bases (SD = 16 483), a statistically significant difference (Welch's t‐test, P = 6.7e−8).
Niche characteristics of LTR retrotransposon lineages
The LTR retrotransposons analyzed in the present study have a nonhomogenous distribution along chromosomes (Figs 4a, S5). Among the nine lineages with > 50 annotated elements, three ‘macroecological’ distribution patterns can be distinguished: centromeric (CRM, RLG_BdisC152), centrophobic (Alesia) and centrophilic (Angela, Ikeros, SIRE, Retand). Of these, the close association of the CRM families to the centromere is the strongest pattern and suggests an active targeting mechanism rather than a passive aggregation due to selective constraints. Indeed, in all three CRM families we found the Putative Targeting Domain (PTD) at the integrase C‐terminus which is believed to mediate the targeted integration of these elements into the centromere (Fig. S6, Neumann et al., 2011). No such domain was found in the nonautonomous centromere‐specific family RLG_BdisC152.
Figure 4.
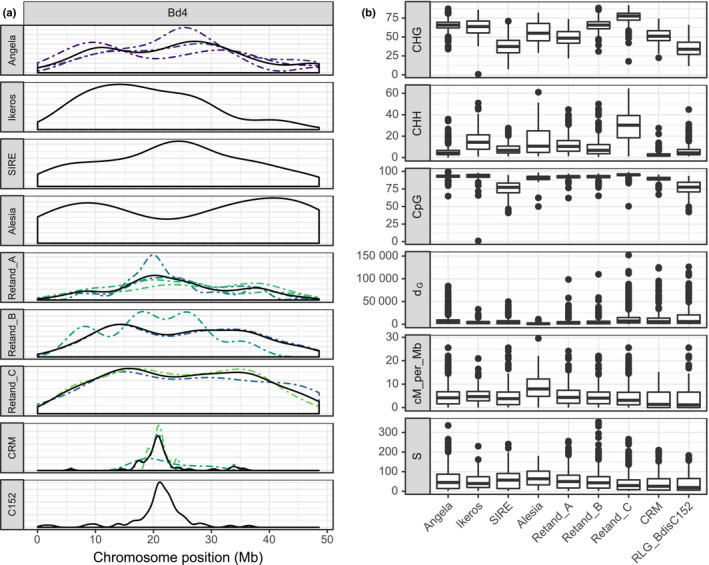
Genomic niches of long terminal repeat‐retrotransposons (LTR‐RTs). (a) Density of the 32 LTR‐RT families along chromosome Bd4. Dashed lines indicate the different families within a lineage, the solid line is their average. (b) Genomic niche features which best predict the different lineages, ranked according to their importance in the random forest model. CHH, CHG and CpG are the three methylation contexts given as percentages, d G is the distance to the closest gene in base pairs, cM_per_Mb is the recombination rate, and S is the number of segregating sites in 10‐kb windows surrounding the annotated transposable elements (TEs).
To get a more precise idea about the genomic niches of different TE lineages, we used a random forest model to identify genomic features associated with the occurrence of individual lineages (see Material and Methods). It correctly predicted two thirds of the observations (OOB error rate 31.58%; Tables S3, S4). The three methylation contexts were by a large margin the best features to distinguish TE lineages, followed by distance to the next gene, recombination rate and number of segregating sites S (Fig. 4b; Table S4).
Some lineages, including the Alesia (SD = 14.6) and the Ikeros elements (SD = 12.9), have copies with a wide range of CHG methylation levels (Fig. 4b); accordingly, they cannot be predicted by their CHG methylation. For other lineages, however, the methylation range is much narrower and quite characteristic: the mean CHG methylation is 78% (SD = 7.0) for the Retand clade C and 65% for Angela (SD = 6.7), whereas it is only 35% (SD = 11.7) for RLG_BdisC152. The same holds for CHH methylation, which correlates with CHG (r = 0.57, P < 0.001) and distinguishes the same lineages, in particular Retand clade C with 29% (SD = 13.1) compared to an overall mean of 15%. Methylation at CpG sites is generally high (mean 91%) and less variable than the other two contexts. Yet also here two lineages stand out with lower means and variability: RLG_BdisC152 and SIRE with means of 76 (SD = 9.8) and 75 (SD = 10.8), respectively.
Distance to the next gene (d G) allows us to distinguish the centromere‐associated lineages Retand C, CRM and RLG_BdisC152, which have mean d G values of 14 026, 18 249 and 20 508 bp, respectively, from the centrophobic Alesia elements with a mean d G of 1870 and the other lineages whose d Gs range from 4687 (Ikeros) to 10 277 (Angela). Recombination rate correlates negatively with d G (r = −0.15, P < 0.001) and distinguishes the same lineages: the genomic neighbourhood of Alesia elements has a median recombination rate of 7.6 cM per Mb compared to 1.1 for RLG_BdisC152 and 3.8 for Angela. The number of segregating sites in the 10‐kb flanking region, finally, also captures this fundamental distinction between LTR‐RTs located mainly in low‐recombination, gene‐poor regions and those also occurring in more frequently recombining, gene‐dense regions: the flanking regions of the former (i.e. CRM, RLG_BdisC152, Retand C) harbour fewer single nucleotide polymorphisms than those of more dispersed lineages like Angela, Alesia or SIRE (Fig. 4b).
TE lineages show great differences in GC content
A possible reason why methylation is a good predictor of the different TE lineages is that it reflects properties of the TE lineages themselves rather than being part of the 'external' niche of the TEs. The 32 LTR‐RT lineages differ substantially in their GC content, which in turn is associated with CHG (r = 0.72, P < 0.001) and CpG (r = 0.57, P < 0.001) methylation levels. The Retand elements are particularly interesting in this regard: compared to the genome‐wide median GC content of 45.6 %, the elements of the three Retand clades have GC contents of 50.3, 52.4 and 60.2 %, respectively (Table 1; Fig. 5a), and differ accordingly in methylation levels (Fig. 4b). At the other end of the spectrum, Bianca and SIRE elements have GC contents way beyond genome‐wide equilibrium level, of 38% and 39%, respectively.
Figure 5.
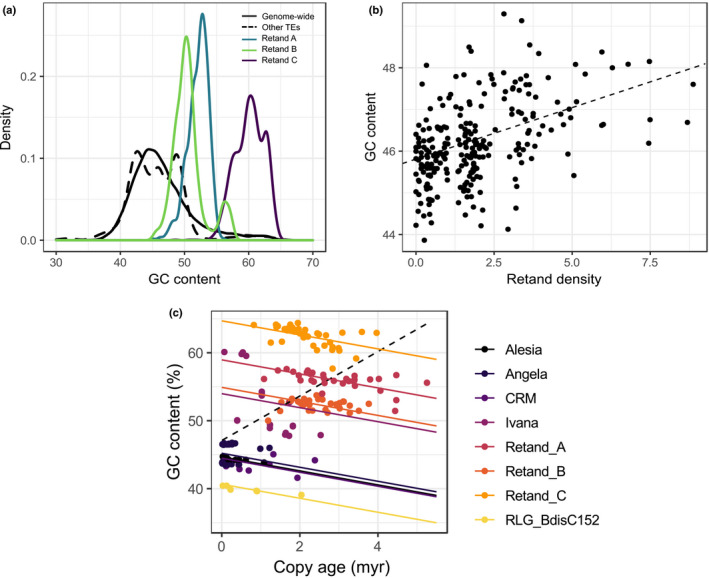
Long terminal repeat‐retrotransposons (LTR‐RTs) and GC content in Brachypodium distachyon. (a) GC content of Retand elements compared to other LTR‐RTs and genome‐wide GC content in in 1‐Mb bins. (b) Association between GC content and the density of Retand copies in 1‐Mb bins across the genome. (c) GC content vs copy age for full‐length elements for eight abundant lineages. The dashed line shows the slope of a simple linear model, the solid lines of the mixed effects model with the lineage as random effect.
Because Retand elements are the most abundant TEs in B. distachyon, their high GC content explains GC‐bias on a genome‐wide scale: the density of Retand elements explains more than a fifth of the variation in GC content in 1‐Mb genomic windows (linear regression, r 2 = 0.21, P < 0.001; Fig. 5b). Recombination rate, however, which explains much of the GC bias in animal genomes (Duret & Galtier, 2009), is a much poorer predictor and has a negative effect size (linear regression, r 2 = 0.08, P < 0.001), which might be due to the negative correlation between Retand elements and recombination rate (r = −0.60, P < 0.001).
The high GC content of these lineages can either be an intrinsic property of the TEs or might be due to GC‐biased gene conversion (gBGC) during ectopic recombination between the numerous homologous copies (Kejnovsky et al., 2007). To test which explanation is more compatible with our data, we looked at the association between GC content and copy age, reflecting that gBGC would cause copies to become more GC‐rich with time, whereas the opposite is expected when initially GC‐rich copies evolve towards equilibrium GC values under the general transition/transversion bias driven by the deamination of methylated cytosines (Ossowski et al., 2010). Indeed we found evidence for the prevalence of the second process, as within each lineage GC content declines with copy age at a rate of −1.03% Myr–1 (standard error = 0.20; Fig. 5c).
Discussion
In this study we investigated the long terminal repeat (LTR) retrotransposon community in the Mediterranean grass Brachypodium distachyon in order to understand how individual transposable element (TE) lineages contribute to genome evolution in this species. The cast of LTR retrotransposons in B. distachyon includes seven major Copia lineages and four major Gypsy lineages (Fig. 1; Table 1). By comparing the properties of these lineages, we identified three ‘foundation species’ in the B. distachyon genome with profound effects on genome composition and dynamics: three active Angela families with an extremely fast turnover, making them an important source of within‐species polymorphisms; an active nonautonomous centromeric TE family, testifying to the dynamic nature of the B. distachyon centromeres; and finally the abundant, old and heavily methylated Retand copies whose presence explains much of the GC bias across the genome.
Live fast, die young: the high‐turnover Angela families
As evident in their high copy number, young age and short half‐lives, the most active components of the B. distachyon genome are the three Copia families belonging to the Angela lineage: RLC_BdisC022, RLC_BdisC030 and RLC_BdisC024. This is the same lineage which has proliferated massively in other pooids, notably in the huge 16 Gb wheat and 5.1 Gb barley genomes. The Angela family itself was first described in wheat, where together with the closely related WIS family it is the most abundant TE in the genome (Wicker et al., 2018). Likewise, the Angela family BARE1 makes up 14% of the diploid barley genome (Wicker et al., 2017). By contrast to wheat and barley, where old intact copies are common and tend to cluster in large heterochromatic regions, Angela copies in B. distachyon are young and consist mostly of dispersed solo LTRs, whereas old intact copies are absent (Fig. 2).
This high number of young solo LTRs, many of which are polymorphic between Bd21 and BdTR7a, indicates that solo LTR formation is frequent in Angela families and can occur shortly after the insertion of the copy. Our comparison of solo‐to‐intact ratios with LTR‐RT structures suggests that fast solo LTR formation in Angela families is helped by long LTRs and a comparatively short internal sequence separating them, which might facilitate ectopic within‐element recombination. Such a mechanism has been suggested in a survey of LTR‐RTs in eight angiosperm species (El Baidouri & Panaud, 2013). In B. distachyon, it would not only explain the lack of old full‐length Angela copies, but also the abundance of old full‐length Retand copies, because these elements have much longer internal sequences and shorter LTRs (Table 1).
More generally, and similar to what was suggested in rice (Vitte & Panaud, 2003; Ma et al., 2004; Tian et al., 2009), it appears that the genome of B. distachyon has remained small not because its TEs are idle, but because the rapid removal of its most active elements prevents a build‐up of TE islands which would provide a ‘safe haven’ for TE insertion (Werren, 2011).
Rare TEs in the genome ecosystem
An important role of LTR length in LTR‐RT life history is not only suggested by high‐turnover families with their long LTRs, but also by their counterparts in the community: the rare, inconspicuous TEs in the genome ecosystem. Within both superfamilies, the rarest TEs (RLG_BdisC078, Ale, Bianca, Ivana) have the shortest LTRs. The observation that all of these families have few or no solo LTRs indicates that the survival of these copies in the genome might be favoured by small LTRs with a low tendency to solo LTR formation.
Lineages which are rare in B. distachyon also are rare in other species, suggesting that their low abundance is not due to an accidental failure to proliferate, but instead the outcome of an evolutionary strategy based on inconspicuousness rather than aggressive proliferation. Also in rice and soybean, Ivana is a diverse clade with low copy numbers (Du et al., 2010); the Ale lineage has been noted for its tendency to evolve a wide variety of low‐copy families, whereas Bianca was found to be present as a low‐copy single family in multiple species (Wicker & Keller, 2007). How these rare lineages manage to persist in the face of the various mutational hazards in the genome is an intriguing question. The answers to it might well modify the current picture of TE evolution, which is based largely on a few highly prolific lineages (e.g. Hawkins et al., 2006; Piegu et al., 2006).
TEs as a source of genetic variation: burst or business as usual?
The survival curves for the three Angela families are steep (Fig. 3): half‐lives are as low as 66 kyr, to our knowledge the fastest rate described so far among plant TEs. Because other causes of TE ‘death’ such as purifying selection are not considered and solo LTR ages are upper‐limit estimates (see Material and Methods), actual death rates might be even higher. Previous estimates at the superfamily level found half‐lives of 1265 kyr for Gypsy and 859 kyr for Copia elements in B. distachyon (International Brachypodium Initiative, 2010). Our analysis shows that these numbers misrepresent the turnover of highly active families by an order of magnitude because they average over active and inactive lineages. The same might be true for half‐life estimates in other species, notably the Copia half‐life estimates of 790 kyr in rice (Wicker & Keller, 2007), 472 kyr in Arabidopsis (Pereira, 2004), and 260 kyr in Medicago trunculata (Wang & Liu, 2008).
Beyond providing an explanation for the small genome size of B. distachyon, the high sequence turnover driven by solo LTR formation advises caution in interpreting skewed age distributions as evidence for a recent activation or ‘burst’ of transposition. A left‐biased age distribution is compatible with both an increased recent activity and a constant transposition rate combined with rapid removal (Dai et al., 2018). Indeed, patterns of TE polymorphisms in 53 diverse natural accessions of B. distachyon are difficult to reconcile with transpositional bursts (Stritt et al., 2018). In this previous study we found 3627 nonreference TE insertions, mainly Angela, SIRE and RLG_BdisC152 elements segregating at low population frequencies, but no evidence for lineage‐specific amplifications of single families, as might be expected if local stress had activated specific TEs (Makarevitch et al., 2015).
Because population genetic surveys of TE polymorphisms rely on discordant read‐pair and splitread methods (Ewing, 2015), they treat TE insertions as presence/absence data and cannot distinguish between intact and truncated copies. By comparing the genomes of Bd21 and BdTR7a, we found that three quarters of the LTR‐RTs which are not conserved between Bd21 and BdTR7a are solo LTRs, most of them belonging to the Angela lineage. This further illustrates the rapidity of solo LTR formation and suggests that a large proportion of the polymorphisms previously identified also are solo LTRs.
Finally, we observed that nonconserved TEs occur closer to genes than conserved insertions. This is consistent with purifying selection shaping the distribution of TEs in the genome, a process important in small, gene‐dense genomes where the insertion of large TEs is likely to disrupt coding or regulatory sequences (Stritt et al., 2018). In this light, conserved, old TEs are conserved and old because they occupy places in the genome where they interfere little with host fitness, whereas nonconserved TEs are primarily recent, low‐frequency variants which are less likely to reach high frequencies the closer they are to genes. Stronger selection against long TEs could explain, together with the centromere specificity of some Gypsy families, why Copia elements tend to occur closer to genes than the typically much larger Gypsy elements, as observed in B. distachyon as well as other plant species (Pereira 2004; Carpentier et al., 2019).
High TE activity in centromeres involves a nonautonomous family
Second to the Angela lineage, centromere‐specific Gypsy elements are the most active part of the B. distachyon genome. Two families stand out with numerous young copies: RLG_BdisC039 and RLG_BdisC152. Interestingly, the latter is a nonautonomous element: it is smaller than the other CRM elements and its 3260‐bp‐long internal sequence lacks retrotransposon open reading frames (ORFs), as well as a chromatin‐targeting domain. It does contain, however, a part with high homology to the CRM families: 2000 bp including most of the LTR and its 3' flanking sequence. This is the region usually containing the polymerase II promoter as well as the primer binding site for reverse transcription (Schulman, 2013). These shared regulatory motifs possibly allow the transcription and reverse transcription of RLG_BdisC152 copies. The insertion into the centromere might then be achieved by scrounging the integration complex of the autonomous CRM families.
GC content as a distinctive feature of TE lineages
The largest contribution to genome size in B. distachyon comes from families of the Retand lineage. The main characteristics of these families are the high number of old full‐length elements, implying a low rate of solo LTR formation, and their high GC content. In Retand C, the clade with the highest copy numbers, median GC content (60.2%) is remarkably higher than the genome‐wide median of 45.6 (Fig. 5). The GC content in Retand copies is not only high, but also shows a phylogenetic pattern: clade A has a median content of 52.4, clade B of 50.3.
The high GC content of Retand elements has two effects: on the one hand, it implies an increased number of methylatable cytosines and higher copy methylation levels (Fig. 4b); on the other, because Retand elements are abundant, they alter the base composition on a genome‐wide scale. Variation in GC content has been studied intensively in animals, where GC‐biased gene conversion (gBGC) has emerged as the favoured explanation because it accounts for the positive association between recombination rate and GC content observed in animal genomes (Duret & Galtier, 2009). In plants, no such general pattern has emerged, possibly because the turnover of intergenic sequence is too fast for broad karyotypic patterns to emerge (Glémin et al., 2014).
Transposable elements previously have been invoked to explain increased GC contents in large plant genomes, in particular of Poaceae species (Smarda et al., 2014). Here we show that individual TE lineages can indeed change the GC content of a genome. More than 20% of the genome‐wide variation in GC content in B. distachyon can be explained by the presence of Retand copies. 1.74% of the genome (4.738 Mb) were annotated as Retand copies, but because Retand elements are old and we ignored the internal sequence of truncated copies in this study, the true percentage must be much higher. As Retand elements are enriched in pericentromeric regions with low recombination rates, the presence of these elements also explains why recombination rate is negatively correlated with GC content in B. distachyon.
Although the presence of GC‐rich transposons explains particularities of the base composition and methylation landscape of B. distachyon, it is an open question why the base composition of these elements is so different from the rest of the genome as well as from other TE lineages. The negative association between GC content and copy age (Fig. 5c) indicates that of the two major and opposing processes affecting GC content, gBGC and the deamination of methylated cytosines (Ossowski et al., 2010), the latter predominates and reduces GC content over time. It thus appears that the differing GC contents are ‘traits’ of the TE lineages themselves. An intriguing possibility is that some TE lineages have evolved elevated GC contents as a means of self‐regulation (Charlesworth & Langley, 1986). The evolution of GC content as an adaptive trait would require fitness differences between TE copies differing in few base pairs. How quantitative variation in GC and methylation affects processes important for TE survival and proliferation such as transcriptional silencing, chromatin formation and ectopic recombination rates is unclear. To clarify these issues, investigating how GC content and methylation levels vary along TE copies and how they relate to transcription factor binding sites and other functional parts of the TE could prove fruitful.
In the present study we have shown that investigating LTR retrotransposon communities at the evolutionary meaningful level of lineages reveals surprising patterns which are missed when averaging over superfamilies or classes. Increasing the resolution at which TE communities are studied led us to re‐appreciate the timescale of TE dynamics: as illustrated by the Angela families, LTR‐RTs can promote genome plasticity in microevolutionary time, with rapid solo LTR formation – and possibly inter‐element recombination – keeping the genome small despite ongoing activity. Applying a similar approach to DNA transposons would be a logical next step to a detailed understanding of the TEs in the model grass B. distachyon, as CACTA, Stowaway and Helitron elements are no less diverse than LTR‐RTs.
Author Contributions
CS conceived the experiment, performed the analyses and wrote the manuscript; MW performed the bisulphite sequencing and subsequent analyses; ELG performed the Bionano experiment; MP assembled the genome of BdTR7a; and ACR conceived and supervised the study and wrote the manuscript. All of the authors read and approved the manuscript.
Supporting information
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Fig. S1 Dotplots of the 40 LTR‐RT families.
Fig. S2 Comparison of the nonautonomous RLG_BdisC152 with other centromeric families.
Fig. S3 Four exemplary LTR genealogies.
Fig. S4 Number of annotated LTR‐RTs in Bd21 and BdTR7a.
Fig. S5 Genomic distribution of the LTR‐RT lineages.
Fig. S6 Putative chromatin‐targeting domain of the centromeric families.
Methods S1 BdTR7a genome assembly.
Methods S2 Whole‐genome bisulfite sequencing.
Table S1 LTR‐RTs annotated in Bd21.
Table S2 LTR‐RTs annotated in BdTR7a.
Table S3 Random forest confusion matrix.
Table S4 Variable importance of the random forest model.
Acknowledgements
This work was supported by the Swiss National Science Foundation (PZ00P3_154724) and the University Research Priority Programs (URPP) Evolution in Action. The authors would like to thank the Genetic Diversity Center at ETH Zurich for providing access to their computing resources, as well as Lucy Poveda and Giancarlo Russo from the Functional Genomic Center Zurich for their support with the Bionano technology. We also thank Michael Thieme, Christian Parisod and the two reviewers for their valuable comments on the manuscript.
See also the Commentary on this article by Catalán & Vogel, 227: 1587–1590.
References
- Belyayev A. 2014. Bursts of transposable elements as an evolutionary driving force. Journal of Evolutionary Biology 27: 2573–2584. [DOI] [PubMed] [Google Scholar]
- Bennetzen JL, Kellog EA. 1997. Do plants have a one‐way ticket to genomic obesity? Plant Cell 9: 1509–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya MK, Smith AM, Ellis N, Hedley C, Martin C. 1990. The wrinkled‐seed character of pea described by Mendel is caused by a transposon‐like insertion in a gene encoding starch‐branching enzyme. Cell 60: 115–122. [DOI] [PubMed] [Google Scholar]
- Breiman L. 2001. Random forests. Machine Learning 45: 5–32. [Google Scholar]
- Brookfield JFY. 2005. The ecology of the genome – mobile DNA elements and their hosts. Nature Reviews Genetics 6: 128–136. [DOI] [PubMed] [Google Scholar]
- Burt A, Trivers R. 2006. Genes in Conflict. London, UK: Harvard University Press. [Google Scholar]
- Capella‐Gutiérrez S, Silla‐Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large‐scale phylogenetic analyses. Bioinformatics 25: 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier M‐C, Manfroi E, Wei F‐J, Wu H‐P, Lasserre E, Llauro C, Debladis E, Akakpo R, Hsing Y‐I, Panaud O. 2019. Retrotranspositional landscape of Asian rice revealed by 3000 genomes. Nature Communications 10: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casacuberta E, González J. 2013. The impact of transposable elements in environmental adaptation. Molecular Ecology 22: 1503–1517. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Langley CH. 1986. The evolution of self‐regulated transposition of transposable elements. Genetics 112: 359–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Wang H, Zhou H, Wang L, Dvořák J, Bennetzen JL, Müller H‐G. 2018. Birth and death of LTR‐retrotransposons in Aegilops tauschii . Genetics 210: 1039–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Tian Z, Hans CS, Laten HM, Cannon SB, Jackson SA, Shoemaker RC, Ma J. 2010. Evolutionary conservation, diversity and specificity of LTR‐retrotransposons in flowering plants: Insights from genome‐wide analysis and multi‐specific comparison. The Plant Journal 63: 584–598. [DOI] [PubMed] [Google Scholar]
- Duret L, Galtier N. 2009. Biased gene conversion and the evolution of mammalian genomic landscapes. Annual Review of Genomics and Human Genetics 10: 285–311. [DOI] [PubMed] [Google Scholar]
- Eickbush TH, Malik HS. 2002. Origins and evolution of retrotransposons In: Craig N, Craigie R, Gellert M, Lambowitz A, eds. Mobile DNA II. Washington, DC, USA: ASM Press. [Google Scholar]
- El Baidouri M, Carpentier MC, Cooke R, Gao D, Lasserre E, Llauro C, Mirouze M, Picault N, Jackson SA, Panaud O. 2014. Widespread and frequent horizontal transfers of transposable elements in plants. Genome Research 24: 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Baidouri M, Panaud O. 2013. Comparative genomic paleontology across plant kingdom reveals the dynamics of TE‐driven genome evolution. Genome Biology and Evolution 5: 954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing AD. 2015. Transposable element detection from whole genome sequence data. Mobile DNA 6: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C, Pritham EJ. 2007. DNA transposons and the evolution of eukaryotic genomes. Annual Review of Genetics 41: 331–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Research 39: 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glémin S, Clément Y, David J, Ressayre A. 2014. GC content evolution in coding regions of angiosperm genomes: a unifying hypothesis. Trends in Genetics 30: 263–270. [DOI] [PubMed] [Google Scholar]
- Hawkins JS, Kim H, Nason JD, Hawkins JS, Kim H, Nason JD, Wing RA, Wendel JF. 2006. Differential lineage‐specific amplification of transposable elements is responsible for genome size variation in Gossypium . Genome Research 16: 1252–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van't Hof AE, Campagne P, Rigden DJ, Yung CJ, Lingley J, Quail MA, Hall N, Darby AC, Saccheri IJ. 2016. The industrial melanism mutation in British peppered moths is a transposable element. Nature 534: 102–105. [DOI] [PubMed] [Google Scholar]
- Horváth V, Merenciano M, González J. 2017. Revisiting the relationship between transposable elements and the eukaryotic stress response. Trends in Genetics 33: 832–841. [DOI] [PubMed] [Google Scholar]
- Huo N, Garvin DF, You FM, McMahon S, Luo MC, Gu YQ, Lazo GR, Vogel JP. 2011. Comparison of a high‐density genetic linkage map to genome features in the model grass Brachypodium distachyon . Theoretical and Applied Genetics 123: 455–464. [DOI] [PubMed] [Google Scholar]
- International Brachypodium Initiative . 2010. Genome sequencing and analysis of the model grass Brachypodium distachyon . Nature 463: 763–768. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kejnovsky E, Hobza R, Kubat Z, Widmer A, Marais GAB, Vyskot B. 2007. High intrachromosomal similarity of retrotransposon long terminal repeats: evidence for homogenization by gene conversion on plant sex chromosomes? Gene 390: 92–97. [DOI] [PubMed] [Google Scholar]
- Kejnovsky E, Leitch IJ, Leitch AR. 2009. Contrasting evolutionary dynamics between angiosperm and mammalian genomes. Trends in Ecology and Evolution 24: 572–582. [DOI] [PubMed] [Google Scholar]
- Kidwell MG, Lisch DR. 2001. Perspective: transposable elements, parasitic DNA, and genome evolution. Evolution 55: 1–24. [DOI] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv: 1303.3997.
- Liaw A, Wiener M. 2002. Classification and regression by random forest. R News 2: 18–22. [Google Scholar]
- Linquist S, Saylor B, Cottenie K, Elliott TA, Kremer SC, Gregory TR. 2013. Distinguishing ecological from evolutionary approaches to transposable elements. Biological Reviews of the Cambridge Philosophical Society 88: 573–584. [DOI] [PubMed] [Google Scholar]
- Lisch D. 2009. Epigenetic regulation of transposable elements in plants. Annual Review of Plant Biology 60: 43–66. [DOI] [PubMed] [Google Scholar]
- Lynch M. 2007. The origins of genome architecture. Sunderland, MA, USA: Sinauer Associates. [Google Scholar]
- Ma J, Bennetzen JL. 2004. Rapid recent growth and divergence of rice nuclear genomes. Proceedings of the National Academy of Sciences, USA 101: 12404–12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Devos KM, Bennetzen JL. 2004. Analyses of LTR‐retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Research 14: 860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarevitch I, Waters AJ, West PT, Stitzer M, Hirsch CN, Ross-Ibarra J, Springer NM. 2015. Transposable elements contribute to activation of maize genes in response to abiotic stress. PLoS Genetics 11: e1004915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClintock B. 1956. Controlling elements and the gene. Cold Spring Harbor Symposia on Quantitative Biology 21: 197–216. [DOI] [PubMed] [Google Scholar]
- Neumann P, Navrátilová A, Koblížková A, Kejnovsk E, Hřibová E, Hobza R, Widmer A, Doležel J, MacAs J. 2011. Plant centromeric retrotransposons: a structural and cytogenetic perspective. Mobile DNA 2: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann P, Novák P, Ho N. 2019. Systematic survey of plant LTR‐ retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mobile DNA 10: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu X, Xu Y, Li Z, Bian Y, Hou X, Chen J, Zou Y. 2019. Transposable elements drive rapid phenotypic variation in Capsella rubella . Proceedings of the National Academy of Sciences, USA 14: 6908–6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, Schneeberger K, Lucas‐Lledó JI, Warthmann N, Clark RM, Shaw RG, Weigel D, Lynch M. 2010. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana . Science 327: 92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira V. 2004. Insertion bias and purifying selection of retrotransposons in the Arabidopsis thaliana genome. Genome Biology 5: R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer B, Wittelsbürger U, Ramos‐Onsins SE, Lercher MJ. 2014. PopGenome: an efficient Swiss army knife for population genomic analyses in R. Molecular Biology and Evolution 31: 1929–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piegu B, Guyot R, Picault N, Roulin A, Saniyal A, Kim H, Collura K, Brar DS, Jackson S, Wing RA et al 2006. Doubling genome size without polyploidization: dynamics of retrotransposition‐driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Research 16: 1262–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. 2018. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Systematic Biology 67: 901–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61: 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho R, Cantalapiedra CP, López‐Alvarez D, Gordon SP, Vogel JP, Catalán P, Contreras‐Moreira B. 2017. Comparative plastome genomics and phylogenomics of Brachypodium: flowering time signatures, introgression and recombination in recently diverged ecotypes. New Phytologist 218: 1631–1644. [DOI] [PubMed] [Google Scholar]
- SanMiguel P, Gaut BS, Tikhonov A, Nakajima Y, Bennetzen JL. 1998. The paleontology of intergene retrotransposons of maize. Nature Genetics 20: 43–45. [DOI] [PubMed] [Google Scholar]
- Schulman AH. 2013. Retrotransposon replication in plants. Current Opinion in Virology 3: 604–614. [DOI] [PubMed] [Google Scholar]
- Schulman A. 2015. Genome size and the role of transposable elements In: Vogel JP, ed. Genetics and genomics of brachypodium. Cham, Switzerland: Springer International Publishing. [Google Scholar]
- Smarda P, Bures P, Horova L, Leitch IJ, Mucina L, Pacini E, Tichy L, Grulich V, Rotreklova O. 2014. Ecological and evolutionary significance of genomic GC content diversity in monocots. Proceedings of the National Academy of Sciences, USA 111: E4096–E4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitzer MC, Anderson SN, Springer NM, Ross‐Ibarra J. 2019. The genomic ecosystem of transposable elements in maize. bioRxiv: doi: 10.1101/559922. [DOI] [PMC free article] [PubMed]
- Stritt C, Gordon SP, Wicker T, Vogel JP, Roulin AC. 2018. Recent activity in expanding populations and purifying selection have shaped transposable element landscapes across natural accessions of the Mediterranean grass Brachypodium distachyon . Genome Biology and Evolution 10: 304–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Z, Rizzon C, Du J, Zhu L, Bennetzen JL, Jackson SA, Gaut BS, Ma J. 2009. Do genetic recombination and gene density shape the pattern of DNA elimination in rice long terminal repeat retrotransposons? Genome Research 19: 2221–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanBuren R, Mockler TC. 2016. The Brachypodium distachyon reference genome In: Vogel JP, ed. Genetics and Genomics of Brachypodium. Cham, Switzerland: Springer International Publishing. [Google Scholar]
- Venner S, Feschotte C, Biémont C. 2009. Dynamics of transposable elements: towards a community ecology of the genome. Trends in Genetics 25: 317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitte C, Panaud O. 2003. Formation of solo‐LTRs through unequal homologous recombination counterbalances amplifications of LTR retrotransposons in rice Oryza sativa L. Molecular Biology and Evolution 20: 528–540. [DOI] [PubMed] [Google Scholar]
- Wang H, Liu JS. 2008. LTR retrotransposon landscape in Medicago truncatula: More rapid removal than in rice. BMC Genomics 9: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH. 2011. Selfish genetic elements, genetic conflict, and evolutionary innovation. Proceedings of the National Academy of Sciences, USA 108: 10863–10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Gundlach H, Spannagl M, Uauy C, Borrill P, Ramírez‐González RH, De Oliveira R, Mayer KFX, Paux E, Choulet F. 2018. Impact of transposable elements on genome structure and evolution in bread wheat. Genome Biology 19: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Keller B. 2007. Genome‐wide comparative analysis of Copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual Copia families. Genome Research 17: 1072–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua‐Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O et al 2007. A unified classification system for eukaryotic transposable elements. Nature Reviews Genetics 8: 973–982. [DOI] [PubMed] [Google Scholar]
- Wicker T, Schulman AH, Tanskanen J, Spannagl M, Twardziok S, Mascher M, Springer NM, Li Q, Waugh R, Li C et al 2017. The repetitive landscape of the 5100 Mbp barley genome. Mobile DNA 8: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Fig. S1 Dotplots of the 40 LTR‐RT families.
Fig. S2 Comparison of the nonautonomous RLG_BdisC152 with other centromeric families.
Fig. S3 Four exemplary LTR genealogies.
Fig. S4 Number of annotated LTR‐RTs in Bd21 and BdTR7a.
Fig. S5 Genomic distribution of the LTR‐RT lineages.
Fig. S6 Putative chromatin‐targeting domain of the centromeric families.
Methods S1 BdTR7a genome assembly.
Methods S2 Whole‐genome bisulfite sequencing.
Table S1 LTR‐RTs annotated in Bd21.
Table S2 LTR‐RTs annotated in BdTR7a.
Table S3 Random forest confusion matrix.
Table S4 Variable importance of the random forest model.