

Abstract
Thanks to the development of high‐throughput sequencing technologies, target enrichment sequencing of nuclear ultraconserved DNA elements (UCEs) now allows routine inference of phylogenetic relationships from thousands of genomic markers. Recently, it has been shown that mitochondrial DNA (mtDNA) is frequently sequenced alongside the targeted loci in such capture experiments. Despite its broad evolutionary interest, mtDNA is rarely assembled and used in conjunction with nuclear markers in capture‐based studies. Here, we developed MitoFinder, a user‐friendly bioinformatic pipeline, to efficiently assemble and annotate mitogenomic data from hundreds of UCE libraries. As a case study, we used ants (Formicidae) for which 501 UCE libraries have been sequenced whereas only 29 mitogenomes are available. We compared the efficiency of four different assemblers (IDBA‐UD, MEGAHIT, MetaSPAdes, and Trinity) for assembling both UCE and mtDNA loci. Using MitoFinder, we show that metagenomic assemblers, in particular MetaSPAdes, are well suited to assemble both UCEs and mtDNA. Mitogenomic signal was successfully extracted from all 501 UCE libraries, allowing us to confirm species identification using CO1 barcoding. Moreover, our automated procedure retrieved 296 cases in which the mitochondrial genome was assembled in a single contig, thus increasing the number of available ant mitogenomes by an order of magnitude. By utilizing the power of metagenomic assemblers, MitoFinder provides an efficient tool to extract complementary mitogenomic data from UCE libraries, allowing testing for potential mitonuclear discordance. Our approach is potentially applicable to other sequence capture methods, transcriptomic data and whole genome shotgun sequencing in diverse taxa. The MitoFinder software is available from GitHub (https://github.com/RemiAllio/MitoFinder).
Keywords: bioinformatics, DNA barcoding, insects, invertebrates, metagenomics, systematics
1. INTRODUCTION
Next‐generation phylogenomics, in which phylogenetic relationships are inferred from thousands of genomic markers gathered through high‐throughput sequencing (HTS), is on the rise. More specifically, targeted enrichment or DNA sequence capture methods are becoming the gold standard in phylogenetic analyses because they allow subsampling the genome efficiently at reduced cost (Lemmon & Lemmon, 2013; McCormack et al., 2013). The field has witnessed the rapid parallel development of exon capture from transcriptome‐derived baits (Bi et al., 2012), anchored hybrid enrichment techniques (Lemmon, Emme, & Lemmon, 2012), and the capture of ultraconserved DNA elements (UCEs; Faircloth et al., 2012). All hybridization capture methods target a particular portion of the genome corresponding to the defined probes plus flanking regions. Previous knowledge is required to generate sequence capture probes, but ethanol‐preserved tissues, old DNA extractions and museum specimens can be successfully sequenced (Blaimer et al., 2015; Faircloth et al., 2012; Guschanski et al., 2013). The first UCEs were identified by Bejerano et al. (2004) in the human genome and have been shown to be conserved in mammals, birds and even ray‐finned fish (Stephen, Pheasant, Makunin, & Mattick, 2008). Thanks to their large‐scale sequence conservation, UCEs are particularly well suited for sequence capture experiments and have become popular for phylogenomic reconstruction of diverse animal groups (Blaimer et al., 2015; Esselstyn, Oliveros, Swanson, & Faircloth, 2017; Guschanski et al., 2013). Initially restricted to a few vertebrate groups such as mammals (McCormack et al., 2012) and birds (McCormack et al., 2013), new UCE probe sets have been designed to target thousands of loci in arthropods such as hymenopterans (Blaimer et al., 2015; Branstetter, Danforth, et al., 2017; Faircloth, Branstetter, White, & Brady, 2015), coleopterans (Baca, Alexander, Gustafson, & Short, 2017; Faircloth, 2017) and arachnids (Starrett et al., 2017).
It has been shown that complete mitochondrial genomes could be retrieved as by‐products of sequence capture/enrichment experiments such as whole exome capture in humans (Picardi & Pesole, 2012). Indeed, mitogenomes can in most cases be assembled from off‐target sequences of UCE capture libraries in amniotes (do Amaral et al., 2015). Despite its well‐acknowledged limitations (Galtier, Nabholz, Glémin, & Hurst, 2009), mitochondrial DNA (mtDNA) remains a marker of choice for phylogenetic inference (e.g., Hassanin et al., 2012), for species identification or delimitation through barcoding (e.g., Coissac, Hollingsworth, Lavergne, & Taberlet, 2016), and to reveal potential cases of mitonuclear discordance resulting from introgression and/or hybridization events (e.g., Grummer, Morando, Avila, Sites, & Leaché, 2018; Zarza et al., 2016, 2018). mtDNA could also be used to taxonomically validate the specimens sequenced for UCEs using CO1 barcoding (Ratnasingham & Hebert, 2007) and to control for potential cross‐contaminations in HTS experiments (Ballenghien, Faivre, & Galtier, 2017). In practice, the few studies that have extracted mtDNA signal from UCEs (e.g., Meiklejohn et al., 2014; Pie et al., 2017; Wang, Hosner, Liang, Braun, & Kimball, 2017; Zarza et al., 2018) and anchored phylogenomics (Caparroz et al., 2018) have done so manually for only a few taxa. Most studies assembling mitogenomes from UCE libraries have used contigs produced by the trinity RNAseq assembler (Grabherr et al., 2011) as part of the phyluce pipeline (Faircloth, 2016), which was specifically designed to extract UCE loci. Indeed, RNAseq assemblers such as trinity allow to deal with the uneven coverage of target reads in sequence‐capture libraries, but also multicopy genes such as the ribosomal RNA cluster, and organelles (chloroplasts and mitochondria). However, this strategy probably does not scale well to hundreds of taxa because of the high computational demand required by trinity. A potential solution to extract mitochondrial signal from UCE libraries could be the use of iterative mapping against a reference mitogenome using mitobim (Hahn, Bachmann, & Chevreux, 2013). However, this tool requires both closely related reference mitogenomes and good coverage to perform well and also requires UCE and mtDNA assemblies to be conducted separately. Metagenomic assemblers could provide a powerful alternative to assemble both UCE loci and mtDNA simultaneously because they have been designed for efficient de novo assembly of complex read populations by explicitly dealing with uneven read coverage and are computationally and memory efficient. Comparisons based on empirical bulk data sets of known composition (Vollmers, Wiegand, & Kaster, 2017) have identified idba‐ud (Peng, Leung, Yiu, & Chin, 2012), megahit (Li et al., 2016) and metaspades (Nurk, Meleshko, Korobeynikov, & Pevzner, 2017) as the most efficient current metagenomic assemblers.
As a case study, we focused on ants (Hymenoptera: Formicidae) for which only 29 mitogenomes were available on GenBank compared to 501 UCE captured libraries as of March 29, 2018 (Appendix S1). This contrasts sharply with the other most speciose group of social insects, termites (Isoptera), for which almost 500 reference mitogenomes have been produced (Bourguignon et al., 2017) and no UCE study has been conducted so far. Sequencing and assembling difficulties stemming from both the AT‐rich composition (Foster, Jermiin, & Hickey, 1997) and a high rate of mitochondrial genome rearrangements in hymenopterans (Dowton, Castro, & Austin, 2002) might explain the limited number of mitogenomes currently available for ants. It is only recently that a few ant mitogenomes have been assembled from UCE data (Meza‐Lázaro, Poteaux, Bayona‐Vásquez, Branstetter, & Zaldívar‐Riverón, 2018; Ströher et al., 2017; Vieira & Prosdocimi, 2019). Here, we built a pipeline called MitoFinder designed to automatically assemble both UCE and mtDNA from raw UCE capture libraries and to specifically extract and annotate mitogenomic contigs. Using publicly available UCE libraries for 501 ants, we show that complementary mitochondrial phylogenetic signal can be efficiently extracted using metagenome assemblers along with targeted UCE loci.
2. MATERIALS AND METHODS
2.1. Data acquisition
We used UCE raw sequencing data for 501 ants produced in 10 phylogenomic studies (Blaimer et al., 2015; Blaimer et al., 2015; Branstetter, Danforth, et al., 2017; Branstetter, Ješovnik, et al., 2017; Branstetter, Longino, Ward, & Faircloth, 2017; Faircloth et al., 2015; Ješovnik et al., 2017; Pierce, Branstetter, & Longino, 2017; Prebus, 2017; Ward & Branstetter, 2017). This data set includes representatives of 15 of the 16 recognized subfamilies (Ward, 2014) and 30 tribes. Raw sequence reads were downloaded from the NCBI Short Read Archive (SRA) on March 29, 2018 (Appendix S1). For the 501 ant UCE libraries, raw reads were cleaned with trimmomatic version 0.36 (Bolger, Lohse, & Usadel, 2014) using the following parameters: LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50. A reference database with the 29 complete mitochondrial genomes available for ants on GenBank at the time was constructed.
2.2. De novo assembly of mitogenomic and UCE data with MitoFinder
To extract mitogenomic data from UCE libraries, we developed a dedicated bioinformatic pipeline called MitoFinder (Figure 1). This pipeline was designed to assemble sequencing reads from target enrichment libraries, extract, and annotate mitochondrial contigs. To evaluate the impact of assembler choice, contigs were assembled with idba‐ud version 1.1.1, megahit version 1.1.3 and metaspades version 3.13.0 within MitoFinder, and with trinity version 2.1.1 within phyluce using default parameters. Mitochondrial contigs were then identified by similarity search using blastn with e‐value ≥ 1e−06 against our ant reference mitogenomic database. Each detected mitochondrial contig was then annotated with tblastx for protein‐coding genes (CDSs) and blastn for 16S and 12S rRNAs taking advantage of the geneChecker module of mitomaker (Schomaker‐Bastos & Prosdocimi, 2018) that we incorporated into MitoFinder. Finally, we used arwen version 1.2 (Laslett & Canback, 2007) to detect and annotate tRNA genes.
FIGURE 1.
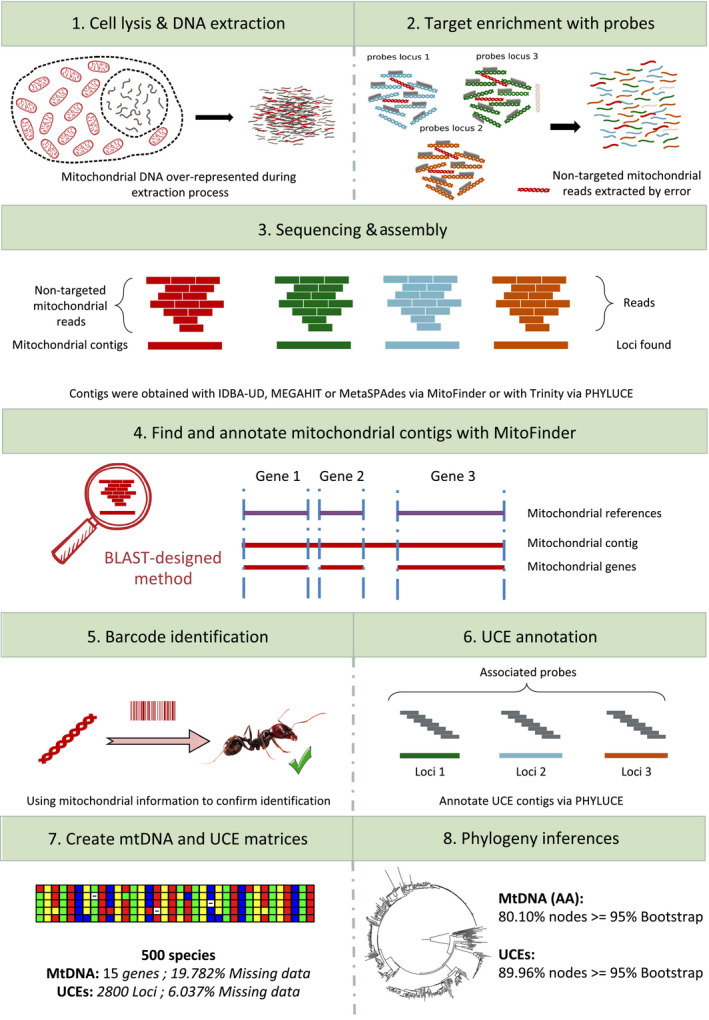
Conceptualization of the pipeline used to assemble and extract UCE and mitochondrial signal from ultraconserved element sequencing data [Colour figure can be viewed at wileyonlinelibrary.com]
Considering possible rearrangements in ant mitogenomes, each annotated mitochondrial CDS was first aligned with mafft version 7.271 (Katoh & Standley, 2013) algorithm FFT‐NS‐2 with option ‐‐adjustdirection. Then, to take into account potential frameshifts and stop codons, mitochondrial CDS alignments were refined with macse version 2.03 (Ranwez, Douzery, Cambon, Chantret, & Delsuc, 2018) with option ‐prog alignSequences, which produces both nucleotide and amino acid alignments. To improve alignment accuracy and reduce calculation time, we used sequences from available ant mitogenomes as references for each CDS (option ‐seq_lr). Sequences with internal stop codons were excluded to remove incorrectly annotated fragments potentially corresponding to nuclear mtDNA segments (NUMTs) in each protein‐coding gene alignment. Then, individual gene alignments were checked by eye to manually remove remaining aberrant sequences. Finally, a nucleotide supermatrix was created by concatenating protein‐coding and ribosomal RNA genes. Because the mitochondrial signal might be saturated for inferring deep phylogenetic relationships, an amino acid supermatrix with the 13 mitochondrial CDSs was also assembled.
2.3. Guided iterative mitogenomic data assembly with mitobim
For comparison purposes, we also ran mitobim (Hahn et al., 2013) to extract mitochondrial sequences from the 501 UCE raw sequencing data. This software is designed to assemble mitochondrial reads using mitochondrial bait such as the CO1 sequence of a related species when available. Then, based on iterative mapping, mitobim extends as much as possible the mitochondrial contig previously obtained. For each library, given the scarcity of closely related complete mitochondrial genomes available for ants, the longest CO1 sequence available for the genus, or the most closely related genus, was used as bait for the initial step of mitobim. As there is no annotation step in mitobim, MitoFinder was used to annotate the resulting mitobim contigs.
2.4. DNA barcoding
To verify species identification of the 501 ant UCE libraries, CO1 sequences extracted by MitoFinder using metaspades (mtDNA recovered for all species) were compared with species‐level barcode records (3,328,881 CO1 sequences including more than 100,000 ants) through the identification server of the Barcode Of Life Data System version 4 (Ratnasingham & Hebert, 2007). The same CO1 sequences were also compared against the NCBI nucleotide database using megablast with default parameters. An identification was considered to be confirmed when the query CO1 sequence had 95% similarity with a reference sequence in BOLD or GenBank with the same identifier.
2.5. Assembly of UCEs
As recommended by Faircloth (2016), we first relied on trinity to assemble UCE contigs using the phyluce_assembly_assemblo_trinity module of phyluce. To assess the impact of assembler choice on UCE loci retrieval, we also used the assemblies obtained with idba‐ud, megahit and metaspades as implemented in MitoFinder. phyluce scripts phyluce_assembly_get_match_counts and phyluce_assembly_get_fastas_from_match_counts were used to match contigs obtained for each sample to the bait set targeting 2,590 UCE loci for Hymenoptera (Branstetter, Ješovnik, et al., 2017). The resulting alignments were then cleaned using gblocks (Castresana, 2000) with the phyluce_align_get_gblocks_trimmed_alignments_from_untrimmed script. Finally, loci found in at least 75% of species were selected to create the four corresponding UCE supermatrices using the phyluce_align_get_only_loci_with_min_taxa script.
2.6. Phylogenetic analyses
Phylogenetic relationships of ants were inferred from a total of 16 different supermatrices corresponding to the four supermatrices constructed from contigs obtained with each of the four assemblers (idba‐ud, megahit, metaspades and trinity). The four supermatrices are as follows: (i) a UCE nucleotide supermatrix built from the concatenation of UCE loci retrieved for at least 75% of species, (ii) a mitochondrial nucleotide supermatrix consisting of the concatenation of the 13 protein‐coding genes and the two rRNA genes, (iii) a mitochondrial amino acid supermatrix of the 13 protein‐coding genes, and (iv) a mixed supermatrix of UCE nucleotides and mitochondrial amino‐acid protein‐coding genes. For all supermatrices, phylogenetic inference was performed with maximum likelihood (ML) as implemented in iq‐tree version 1.6.8 (Nguyen, Schmidt, von Haeseler, & Minh, 2015) using a GTR+Γ4+I model for UCE and mitochondrial nucleotide supermatrices, an mtART+Γ4+I model partitioned by gene for mitochondrial amino acid matrices, and a partitioned model mixing a GTR+Γ4+I model for UCE nucleotides and an mtART+Γ4+I model for mitochondrial amino acids for the mixed supermatrices. Statistical node support was estimated using ultrafast bootstrap (UFBS) with 1,000 replicates (Hoang, Chernomor, von Haeseler, Minh, & Vinh, 2018). Nodes with UFBS values >95% were considered strongly supported. For all supermatrices, the congruence among the different topologies obtained with the four assemblers was evaluated by calculating quartet distances with dquad (Ranwez, Criscuolo, & Douzery, 2010).
3. RESULTS
3.1. Assembly of UCE data sets
De novo assembly of 501 UCE capture sequencing libraries was performed with four different assemblers: idba‐ud, megahit and metaspades via MitoFinder and trinity via phyluce. All assemblers provided different numbers of contigs (Table 1) ranging from 30,544 (idba‐ud) to 114,392 (megahit) on average. The average computational time per assembly was highly variable among assemblers with trinity being by far the slowest (35 CPUs, median time per sample: 1 hr:06 min:22 s, total time for all samples: 26.9 days) and idba‐ud the fastest (5 CPUs, median time per sample: 0 hr:11 min:01 s, total time for all samples: 4.4 days), megahit (5 CPUs, median time per sample: 0 hr:12 min:35 s, total time for all samples: 4.9 days) being slightly slower, and metaspades (5 CPUs, median time per sample: 0 hr:25 min:44 s, total time for all samples: 14.9 days) having a median assembly time about twice as slow as the other two metagenomic assemblers (Table 1; Figure 2a).
TABLE 1.
Summary statistics on assembly results according to the assembler used
| Assembler | Assembly time | UCEs | ||||
|---|---|---|---|---|---|---|
| Number of contigs | Number of loci | Matrix size | % Variable sites | % Missing data | ||
| IDBA‐UD (5 CPU) | 0 hr:11 min:02 s | 30.544 | 2,581 | 132.403 | 43.9 | 6.7 |
| MEGAHIT (5 CPU) | 0 hr:12 min:35 s | 114.392 | 2,579 | 147.589 | 43.2 | 12.5 |
| MetaSPAdes (5 CPU) | 0 hr:25 min:42 s | 113.303 | 2,582 | 156.456 | 44.3 | 6.1 |
| Trinity (35 CPU) | 1 hr:06 min:22 s | 43.481 | 2,579 | 127.803 | 40.5 | 17.8 |
| Assembler | Mitogenomes | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Number of contigs | Number of species | Number of genes | AA matrix size | % Missing data | % Variable sites | NT matrix size | % Missing data | % Variable sites | |
| IDBA‐UD (5 CPU) | 4.2 | 499 | 13.04 | 3,764 | 20.9 | 86.7 | 13.635 | 26.1 | 85.8 |
| MEGAHIT (5 CPU) | 3.9 | 499 | 13.61 | 3,757 | 15.3 | 87.5 | 13.718 | 20.6 | 86.4 |
| MetaSPAdes (5 CPU) | 3.8 | 501 | 13.73 | 3,766 | 14.6 | 88.9 | 13.713 | 19.8 | 87.1 |
| Trinity (35 CPU) | 4.2 | 500 | 13.37 | 3,760 | 18.0 | 86.9 | 13.648 | 26.7 | 86.1 |
The values are averages over the 501 assemblies, except for the assembly time, which is a median value. The two parts of the table report specific statistics for (a) ultraconserved elements data, and (b) mitochondrial data. Note that 35 CPUs were used for trinity whereas 5 CPUs were used for other assemblers.
FIGURE 2.

Comparison of the efficiency of the assemblers in terms of: (a) computational time, (b) number of potential mitochondrial contigs identified, and (c) number of mitochondrial genes annotated. Violin plots reflect the data distribution with a horizontal line indicating the median. Note that for the three metagenomic assemblers, 5 CPUs were used compared to 35 CPUs for trinity. Plots were obtained using plotsofdata (Postma & Goedhart, 2019) [Colour figure can be viewed at wileyonlinelibrary.com]
The UCE supermatrices created by phyluce for each of the four assemblers contained on average 2,580 of the 2,590 UCE loci for Hymenoptera (Table 1). All matrices contained 501 species, but the size of the supermatrix and the percentage of missing data varied depending on the assembler (Table 1). Trinity, which is generally used as the default assembler in phyluce, resulted in the shortest and most incomplete supermatrix with 2,579 loci representing 127,803 sites (40.5% variable) and 17.8% missing data. Among metagenomic assemblers, metaspades provided the largest and most complete supermatrix with 2,582 loci representing 156,456 sites (44.5% variable) and only 6.0% missing data. idba‐ud retrieved 2,581 loci representing 132,403 sites (43.9% variable) with only 6.7% missing data, and megahit resulted in a supermatrix with 2,579 loci representing 147,589 sites (43.2% variable) but with 12.4% missing data. Note that fewer than 30 loci were retrieved for Phalacromyrmex fugax (between four and 27 loci depending on the assembler). This is congruent with the original publication in which this low‐quality library was not included in phylogenetic analyses (Branstetter, Danforth, et al., 2017). Accordingly, we removed the Phalacromyrmex fugax library (SRR5437956) from the data set.
3.2. Extracting mitochondrial sequences from UCE sequencing data
Depending on the assembler used in MitoFinder, mitochondrial reads were recovered in 499, 500 and 501 libraries out of a total of 501 (Table 1; Figure 2b). Overall, mitochondrial signal thus was detected in all libraries but only metaspades retrieved it in all species (Appendix S2). On average, 3.8 contigs per species were identified (Tables 1 and 2; Figure 2b) and 13.7 genes were annotated with MitoFinder (Figure 2c; Table 2). In 296/501 cases, MitoFinder was able to assemble a contig of more than 15,000 bp containing at least 13 annotated genes that probably represents the complete mitochondrial genome. In 52 of these cases, all 15 genes were annotated. In the remaining cases, the putative mitogenome contigs were missing one or two genes, mostly the short and divergent ATP8 (131/296), the 12S rRNA (29/296) and the 16S rRNA (10/296), which were present but not directly annotated by our blast‐based procedure. By comparison, mitobim produced a mitochondrial contig for only 358 libraries for which an average of 3.51 genes were annotated representing 2,840.24 nucleotides on average.
TABLE 2.
Statistical comparison between the performances of the different assemblers
| Number of mtDNA contigs | Number of mtDNA genes | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| IDBA‐UD | MEGAHIT | MetaSPAdes | Trinity | IDBA‐UD | MEGAHIT | MetaSPAdes | Trinity | ||
| IDBA‐UD | ** (+) | *** (+) | NS (–) | IDBA‐UD | *** (–) | *** (–) | *** (–) | ||
| MEGAHIT | ** (–) | * (+) | *** (–) | MEGAHIT | *** (+) | * (–) | *** (+) | ||
| MetaSPAdes | *** (–) | * (–) | *** (–) | MetaSPAdes | *** (+) | * (+) | *** (+) | ||
| Trinity | NS (+) | *** (+) | *** (+) | Trinity | *** (+) | *** (+) | *** (–) | ||
| Number of coding mtDNA nucleotides | Number of UCE nucleotides | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| IDBA‐UD | MEGAHIT | MetaSPAdes | Trinity | IDBA‐UD | MEGAHIT | MetaSPAdes | Trinity | ||
| IDBA‐UD | *** (–) | *** (–) | * (–) | IDBA‐UD | *** (–) | *** (–) | *** (+) | ||
| MEGAHIT | *** (+) | * (–) | *** (–) | MEGAHIT | *** (+) | *** (–) | *** (+) | ||
| MetaSPAdes | *** (+) | * (+) | *** (+) | MetaSPAdes | *** (+) | *** (+) | *** (+) | ||
| Trinity | * (+) | *** (–) | *** (–) | Trinity | *** (–) | *** (–) | *** (–) | ||
Statistical significance was estimated with a paired nonparametric test (paired Wilcoxon test). ***p < .001; **p < .01; *p < .05; NS = p> .05; and (+)/(–) indicates the result of the comparison between the row and the column.
After alignment and cleaning, mitochondrial genes obtained with MitoFinder were used to create nucleotide and amino acid supermatrices. To be consistent with UCE analyses, and despite the recovery of some mitochondrial signal, we ignored Phalacromyrmex fugax in further analyses. In the nucleotide supermatrices (13 protein‐coding + 12S and 16S rRNAs), we obtained 13 genes on average per species, which resulted in supermatrices with 13,679 nucleotide sites (86.4% variable) and 23.3% missing data on average (Table 1). In the amino acid matrices (13 protein‐coding genes), we obtained supermatrices with 3,762 amino acid sites (87.4% variable) and 17.2% missing data on average (Table 1).
3.3. Barcoding analyses
A total of 534 CO1 sequences retrieved from the 501 metaspades assemblies were used to verify species identification of the UCE libraries (Appendix S3). Similarity searches against BOLD and GenBank allowed us to confirm the species identity in only 312 cases probably because of the limited availability of CO1 barcoding data for these ant species. Moreover, in 42 cases, two or three CO1 sequence fragments were retrieved from the same UCE library. In seven of these cases, the slightly overlapping CO1 fragments probably resulted from poor assembly or erroneous annotation. However, in the 35 remaining cases, the genuine complete CO1 sequence overlapped with shorter contigs assembled from a minority of the reads, suggesting either cross‐contaminations, NUMTs, endoparasites or bacterial symbionts. For instance, in Temnothorax sp. mmp11 (SRR5809551), a 391‐bp fragment annotated as CO1 by MitoFinder was found to be 98.2% identical to both the Wolbachia pipientis wAlbB and Wolbachia Pel strain wPip genomes, which are bacterial endosymbionts of the mosquitoes Aedes albopictus and Culex quinquefasciatus, respectively. Also, in Sericomyrmex bondari (SRR5044901) and Sericomyrmex mayri (SRR5044856) short CO1 fragments best matched with nematodes. However, in the 312 cases for which CO1 barcoding allowed us to confirm the species identity of the UCE library, we did not detect any obvious cases of cross‐contaminations where the CO1 extracted from a given library would have been identical to that of another library (Appendix S3).
3.4. Phylogenetic results
The ML topologies inferred from the different UCE supermatrices were very similar with an average quartet distance of 0.005 among assemblers (Appendix S4). However, the percentage of supported nodes (UFBS > 95) differed depending on the assembler: idba‐ud (91.37%), metaspades (89.96%), megahit (89.56%) and trinity (85.85%). In the following, we only discuss the phylogenetic results obtained with MetaSPAdes that provides the most comprehensive assemblies for both UCE and mitochondrial data (Table 1). The following 12 well‐established subfamilies (with several species in the dataset) were retrieved with maximal UFBS support (100%): Amblyoponinae, Apomyrminae, Dolichoderinae, Dorylinae, Ectatomminae, Formicinae, Heteroponerinae, Myrmeciinae, Myrmicinae, Proceratinae, Ponerinae, and Pseudomyrmecinae (Figure 3a). The two supergroups Formicoid and Poneroid were also retrieved with maximal UFBS support, as well as consensual phylogenetic relationships among Formicoid subfamilies (Ward, 2014).
FIGURE 3.
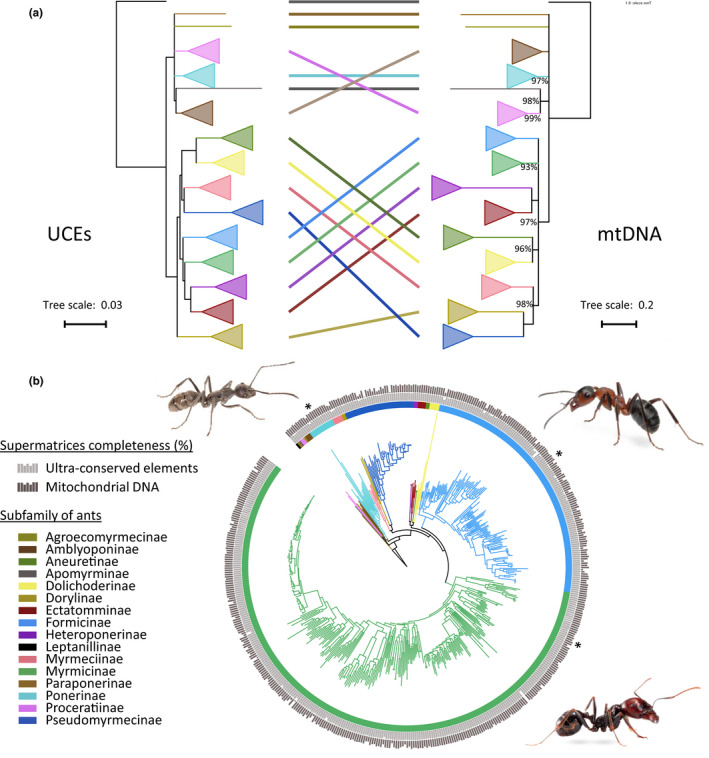
Phylogenomic relationships of ants (Formicidae). (a) Mitonuclear phylogenetic differences among subfamily relationships based on the UCE and mtDNA supermatrices obtained with the MetaSPAdes assembler. Clades corresponding to subfamilies were collapsed. Inter‐subfamily relationships with UFBS <95% were collapsed. Nonmaximal node support values are reported. (b) The topology obtained reflects the results of phylogenetic analyses based on the amino acid mitochondrial supermatrix (using MetaSPAdes as assembler). Histograms reflect the percentage of UCEs (light grey) and mitochondrial genes (dark grey) recovered for each species. Illustrative pictures (*): Diacamma sp. (Ponerinae; top left), Formica sp. (Formicinae; top right) and Messor barbarus (Myrmicinae; bottom right) [Colour figure can be viewed at wileyonlinelibrary.com]
For mitochondrial matrices, the percentage of supported nodes (UFBS > 95) with nucleotides also differed depending on the assembler and was higher than with the amino acids: metaspades (84.5% vs. 80.1%), megahit (84.0% vs. 79.4%), trinity (83.3% vs. 80.4%) and idba‐ud (80.2% vs. 78.0%). However, ML mitogenomic trees inferred from amino acids were more congruent with UCE topologies than those inferred from the mitochondrial nucleotides (average quartet distance = 0.035 vs. 0.063; Appendix S4). Among assemblers, the ML topologies inferred with amino acid matrices were highly congruent with an average quartet distance of 0.007 (Appendix S4). In the ML tree obtained with the metaspades supermatrix (Figure 3b), all ant subfamilies were retrieved with maximal UFBS support values except for Myrmicinae (93%), Ponerinae (97%) and Proceratiinae (99%) (Figure 3a). However, relationships among subfamilies were not congruent with UCE phylogenomic inferences except for Heteroponerinae + Ectatomminae (UFBS = 100) and Dolichoderinae + Aneuretinae (UFBS = 96; Figure 3a).
Finally, phylogenetic inference carried on mixed supermatrices composed of UCEs and mitochondrial amino acids resulted in ML topologies that were also highly similar among assemblers with an average quartet distance of 0.006 (Appendix S4). The percentages of supported nodes (UFBS> 95) were: idba‐ud (91.2%), megahit (92.8%), metaspades (92.2%) and trinity (90.4%). As with UCE matrices, the 12 well‐established subfamilies, the two supergroups Formicoid and Poneroid, and consensus Formicoid inter‐subfamilies relationships (Ward, 2014) were all retrieved with maximal UFBS support.
4. DISCUSSION
4.1. Metagenomic assemblers as powerful tools for assembling UCEs
Currently, genomic and transcriptomic de novo assemblers are commonly used to assemble UCE loci from DNA capture sequencing data (Faircloth, 2016). Because metagenomic assemblers such as idba‐ud, megahit and metaspades have been designed to account for variance in sequencing coverage, they seem to be well adapted for targeted enrichment or DNA sequence capture data. Our results show that metagenomic assemblers are indeed faster at assembling UCE loci than the classically used, but computationally intensive, trinity transcriptomic assembler. Furthermore, they seem more effective and lead to data sets containing more variable sites, less missing data and increased phylogenetic signal (Tables 1 and 2). Indeed, the topologies obtained with the metagenomic assemblers are very similar to the topology obtained with the trinity‐based supermatrix, contain a higher number of supported nodes (UFBS ≥ 95%) and are consistent with previous studies (Ward, 2014). Furthermore, assemblies obtained with the three metagenomic assemblers provide variable numbers of contigs (ranging from 30,544 to 114,392) resulting in differences in the completeness of the matrices (6.0%–17.8% of missing data for UCE matrices and 29.9%–41.3% for mitochondrial matrices) and in numbers of variable sites (for UCE, 40.5%–44.3%; for mtDNA, 77.2%–79.0%). Interestingly, for both UCE matrices and mtDNA matrices, metaspades consistently provides more loci, more variable sites and less missing data. In addition, mitochondrial signal was extracted from all libraries only using metaspades within Mitofinder. Despite a computation time on average twice that of the other two metagenomic assemblers, metaspades was the more effective assembler for ant UCEs. This software therefore provides a much‐needed alternative to trinity for efficiently assembling hundreds of UCE libraries.
4.2. MitoFinder efficiently extracts mitochondrial signal from UCE capture data
Ultraconserved elements are key loci exploited as target capture sequences in an increasing number of phylogenomic studies. DNA sequence capture methods are used to efficiently enrich targeted DNA regions in library preparation prior to sequencing, but nontargeted regions are always sequenced in the process, resulting in so called “off‐target reads.” Interestingly, off‐target reads could represent up to 40% of the sequenced reads in exome capture experiments (Chilamakuri et al., 2014) and many contigs not belonging to targeted UCE loci are typically assembled from UCE capture data (e.g., Faircloth et al., 2015; Smith, Harvey, Faircloth, Glenn, & Brumfield, 2014). Given this high proportion of off‐target reads, we can expect that mtDNA could be found as off‐target sequences in many target enrichment data. Accordingly, several studies have succeeded in extracting mtDNA from UCE libraries (e.g., do Amaral et al., 2015; Smith et al., 2014). The development of MitoFinder allowed the automatic extraction of mitochondrial signal from all 501 ant UCE libraries. This maximum success rate indicates that this approach is highly efficient at least in Formicidae. However, the success in retrieving mitochondrial sequences ultimately depends on the number of mitochondria contained in the tissue used for DNA extraction and library preparation. As expected, mitochondrial off‐target reads are much more common in muscle and heart than in lung tissues in humans (D'Erchia et al., 2015). Similarly, mitochondrial sequences are probably rare or absent in libraries constructed from vertebrate blood, even in birds in which nucleated red blood cells contain mitochondria, but in very low numbers (Reverter et al., 2017). In invertebrates, our case study with a 100% success rate in ant UCEs demonstrates that mitochondrial sequences could probably be easily retrieved for many arthropod taxa as a by‐product of target enrichment sequencing experiments. Finally, the comparison between MitoFinder and mitobim emphasizes that the use of de novo assembly instead of iterative mapping is a suitable solution for recovering mitochondrial signal for groups with limited mitogenomic references.
4.3. The value of complementary mitochondrial signal
Mitochondrial sequences could provide interesting and important complementary information compared to nuclear sequences. First, mtDNA can be used to confirm the identity of the species sequenced for conserved UCE loci. Here, we were able to confirm the identification of 312 ant species out of the 501 UCE libraries using CO1 barcoding without revealing a single case of obvious species misidentification. Given that ant UCE libraries have been constructed from museum specimens, the 501 CO1 sequences we annotated could be used as reference barcoding sequences in future studies. Then, even though we did not detect such cases, the high mutation rate and the absence of heterozygous sites in mtDNA also make it well adapted for cross‐contamination detection analyses (Ballenghien et al., 2017).
Nevertheless, mitochondrial markers also have some well‐identified limitations (Galtier et al., 2009). First, mtDNA could be inserted in the nuclear genome in the form of NUMTs (Bensasson, Zhang, Hartl, & Hewitt, 2001). NUMTs could potentially be assembled as off‐target contigs in DNA capture libraries and we might have indeed extracted some fragments corresponding to NUMTs for the CO1 gene using MitoFinder (Appendix S2). Theoretically, NUMTs could be picked up by analysing the coverage of putative mitochondrial contigs as they are expected to have a coverage comparable to other off‐targets nuclear contigs, whereas genuine mitochondrial contigs should have a higher coverage. A second limitation of mtDNA exists in arthropods where maternally inherited intracellular bacteria are frequent. Among those bacteria, Wolbachia is particularly widespread and could distort the mitochondrial genealogy when a particular strain spreads within the host species, hitchhiking its linked mitochondrial haplotype (Cariou, Duret, & Charlat, 2017). Wolbachia infection is frequent among ants and could therefore be responsible of some mitonuclear discordance (Wenseleers et al., 1998). We indeed discovered such an instance with a Wolbachia CO1 sequence identified in Temnothorax sp. mmp11 (SRR5809551), which was confirmed by several assembled contigs matching to Wolbachia strain genomes in this sample.
Beyond the methodological aspects of species identification and potential cross‐contamination detection, mitochondrial sequences could also be useful to tackle fundamental evolutionary questions. UCEs have also proved to be useful genetic markers for phylogeography and for resolving shallow phylogenetic relationships (Musher & Cracraft, 2018; Smith et al., 2014). In this context, mtDNA could also bring complementary information. In most animals, mtDNA has a maternal inheritance without recombination, which means that all mitochondrial genes behave as a single locus. This simplifies the interpretation of the phylogenetic pattern between closely related species or within subdivided populations of a species. Mitonuclear phylogenetic discordance could also reveal interesting phenomena involving hybridization, sex‐biased dispersal and introgression (Bonnet, Leblois, Rousset, & Crochet, 2017; Toews & Brelsford, 2012). In practice, hybridization events are often identified using mitonuclear discordance (Li et al., 2016) and, in some cases, the mitochondrial introgression events have proven to be adaptive (Seixas, Boursot, & Melo‐Ferreira, 2018). Nevertheless, in our ant case study, a detailed comparison of mitochondrial and UCE phylogenies did not reveal convincing occurrences of such discordances.
4.4. Ant phylogenetic relationships from 500 UCE and mitochondrial data
Both nuclear and mitochondrial data retrieved the most consensual phylogenetic relationships in the ant phylogeny (Borowiec et al., 2019; Branstetter, Ješovnik, et al., 2017; Ward, 2014). Twelve Formicidae subfamilies were recovered as monophyletic in all analyses, with both the nuclear and the mitochondrial data sets, confirming their robustness. However, the well‐defined inter‐subfamily relationships within Formicoids (Borowiec et al., 2019; Branstetter, Ješovnik, et al., 2017; Ward, 2014) were only supported by the UCE data set, but not by the mitochondrial amino acid data set. For example, the army ant subfamily (Dorylinae) was not retrieved as the sister‐group of all other Formicoids, but was the closest relative of Pseudomyrmicinae (UFBS = 100). Similarly, contradicting the classical and well‐defined relationship of Heteroponerinae + Ectatomminae as the sister‐group of Myrmicinae (Borowiec et al., 2019; Branstetter, Ješovnik, et al., 2017; Ward, 2014), the mitochondrial data set supported an alternative relationship with Dolichoderinae + Aneuretinae (UFBS = 96). These differences suggest that mitochondrial data might be not well suited to resolve ancient phylogenetic relationships at the ant inter‐subfamily level diverging about 100 million years ago (Moreau, Bell, Vila, Archibald, & Pierce, 2006), even if they look suitable for more recent nodes such as intra‐subfamily relationships.
Interestingly, these topological incongruences between UCEs and mitochondrial genes also revealed different topologies regarding the existence of the Poneroid taxa, a controversial clade not always retrieved depending on the studies (Ward, 2014), but that tends to be retrieved in the most recent studies (Borowiec et al., 2019; Branstetter, Ješovnik, et al., 2017; UCE data set in this study) and is not recovered by our mitochondrial amino acid data set (Figure 3b). The same applies to the phylogenetic placement of Apomyrminae, a subfamily grouped with either Leptanillinae or Amblyoponinae in previous studies (Ward, 2014), but that was grouped with Proceratiinae in our mitochondrial data set (UFBS = 98; Figure 3b). For such controversial nodes, our study demonstrates that the nature of the phylogenetic markers can provide different results. Such differences between nuclear and mitochondrial data might be due to the substitutional saturation of mitochondrial data even at the amino acid level. This problem may actually be exacerbated in hymenopteran mitochondria that possess high AT content, translating into strongly biased codon usage and potentially leading to phylogenetic reconstruction artefacts (Foster & Hickey, 1999; Foster et al., 1997). Interestingly, such differences between mitochondrial and nuclear inference for ancient phylogenetic relationships are not observed with insects with less AT‐rich mitochondrial genomes such as swallowtail butterflies (Allio et al., 2019; Condamine, Nabholz, Clamens, Dupuis, & Sperling, 2018) or tiger beetles (Vogler & Pearson, 1996). This calls for additional studies on both controversial and consensual ant inter‐subfamily relationships with more comprehensive genome‐wide data sets.
5. CONCLUSIONS
In this study, we developed the MitoFinder tool to automatically extract and annotate mitogenomic data from raw sequencing data in an efficient way. For the assembly step of our pipeline, we tested four different assemblers and showed that metaspades is the most efficient and accurate assembler for both UCE and mitochondrial data. Applying MitoFinder to ants, we were able to extract mitochondrial signal from 501 UCE libraries. This demonstrates that mitochondrial DNA can be found as off‐target sequences in UCE sequencing data. Interestingly, mtDNA extracted from UCE libraries can also be used to: (a) confirm species identification with barcoding methods, (b) highlight potential sample cross‐contamination, and (c) reveal potential cases of mitonuclear discordance caused by hybridization events leading to mitochondrial introgression. Finally, MitoFinder was developed with UCE libraries but our approach should also work with data obtained from other capture methods in which numerous off‐target reads are sequenced, as well as with transcriptomic and whole genome sequencing data, in which mitochondrial reads are over‐represented.
AUTHOR CONTRIBUTIONS
R.A. and F.D. conceived the ideas and designed methodology, analysed the data, and led the writing of the manuscript; R.A. implemented the MitoFinder software in part using code previously written by A.S‐B.; J.R., F.P. and B.N. contributed to the writing of the manuscript. All authors contributed critically to the drafts and gave final approval for publication.
Supporting information
Appendix S1‐S5
ACKNOWLEDGEMENTS
This paper is dedicated to the memory of graduate student Alex Schomaker‐Bastos (1992–2015) who was assassinated by the time he was writing the mitoMaker program on which we built for the annotation module of MitoFinder. We also thank Fabien Condamine and two anonymous reviewers for providing helpful comments on a previous version of the manuscript. This work was supported by grants from the European Research Council (ERC‐2015‐CoG‐683257 ConvergeAnt project) and Investissements d'Avenir of the Agence Nationale de la Recherche (CEBA: ANR‐10‐LABX‐25‐01; CEMEB: ANR‐10‐LABX‐0004). This is contribution ISEM 2020‐071 SUD of the Institut des Sciences de l'Evolution de Montpellier.
Allio R, Schomaker‐Bastos A, Romiguier J, Prosdocimi F, Nabholz B, Delsuc F. MitoFinder: Efficient automated large‐scale extraction of mitogenomic data in target enrichment phylogenomics. Mol Ecol Resour. 2020;20:892–905. 10.1111/1755-0998.13160
DATA AVAILABILITY STATEMENT
The MitoFinder software is available from GitHub (https://github.com/RemiAllio/MitoFinder) and GitLab (https://gitlab.com/RemiAllio/mitofinder). Annotated mitogenomes and partial mitogenomic contigs containing at least two genes and 1,000 bp have been deposited in GenBank and are available in the Third Party Annotation Section of the DDBJ/ENA/GenBank databases under accession numbers TPA: BK012118–BK012857 (Appendix S5). The full analytical pipeline, MitoFinder results including UCE and mtDNA contigs for all assemblers, phylogenetic data sets and corresponding trees can be retrieved from zenodo.org (https://doi.org/10.5281/zenodo.3231390).
We used UCE raw sequencing data for 501 ants produced in 10 phylogenomic studies (Blaimer et al., 2015, Blaimer, LaPolla, Branstetter, Lloyd, & Brady, 2016; Branstetter, Danforth, et al., 2017, Branstetter, Ješovnik, et al., 2017, Branstetter, Longino, et al., 2017; Faircloth et al., 2015; Ješovnik et al., 2017; Pierce et al., 2017; Prebus, 2017; Ward & Branstetter, 2017). (see Appendix S1)
REFERENCES
- Allio, R. , Scornavacca, C. , Nabholz, B. , Clamens, A. L. , Sperling, F. A. , & Condamine, F. (2019). Whole genome shotgun phylogenomics resolves the pattern and timing of swallowtail butterfly evolution. Systematic Biology, 69(1), 38–60. 10.1093/sysbio/syz030 [DOI] [PubMed] [Google Scholar]
- Baca, S. M. , Alexander, A. , Gustafson, G. T. , & Short, A. E. Z. (2017). Ultraconserved elements show utility in phylogenetic inference of Adephaga (Coleoptera) and suggest paraphyly of ‘Hydradephaga’. Systematic Entomology, 42(4), 786–795. 10.1111/syen.12244 [DOI] [Google Scholar]
- Ballenghien, M. , Faivre, N. , & Galtier, N. (2017). Patterns of cross‐contamination in a multispecies population genomic project: Detection, quantification, impact, and solutions. BMC Biology, 15(1), 25 10.1186/s12915-017-0366-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejerano, G. , Pheasant, M. , Makunin, I. , Stephen, S. , Kent, W. J. , Mattick, J. S. , & Haussler, D. (2004). Ultraconserved elements in the human genome. Science, 304(5675), 1321–1325. 10.1126/science.1098119 [DOI] [PubMed] [Google Scholar]
- Bensasson, D. , Zhang, D.‐X. , Hartl, D. L. , & Hewitt, G. M. (2001). Mitochondrial pseudogenes: Evolution’s misplaced witnesses. Trends in Ecology & Evolution, 16(6), 314–321. 10.1016/S0169-5347(01)02151-6 [DOI] [PubMed] [Google Scholar]
- Bi, K. , Vanderpool, D. , Singhal, S. , Linderoth, T. , Moritz, C. , & Good, J. M. (2012). Transcriptome‐based exon capture enables highly cost‐effective comparative genomic data collection at moderate evolutionary scales. BMC Genomics, 13(1), 403 10.1186/1471-2164-13-403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaimer, B. B. , Brady, S. G. , Schultz, T. R. , Lloyd, M. W. , Fisher, B. L. , & Ward, P. S. (2015). Phylogenomic methods outperform traditional multi‐locus approaches in resolving deep evolutionary history: A case study of formicine ants BMC Evolutionary Biology, 15(1), 271 10.1186/s12862-015-0552-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaimer, B. B. , LaPolla, J. S. , Branstetter, M. G. , Lloyd, M. W. , & Brady, S. G. (2016). Phylogenomics, biogeography and diversification of obligate mealybug‐tending ants in the genus Acropyga. Molecular Phylogenetics and Evolution, 102, 20–29. 10.1016/j.ympev.2016.05.030 [DOI] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet, T. , Leblois, R. , Rousset, F. , & Crochet, P. A. (2017). A reassessment of explanations for discordant introgressions of mitochondrial and nuclear genomes. Evolution, 71(9), 2140–2158. 10.1111/evo.13296 [DOI] [PubMed] [Google Scholar]
- Borowiec, M. L. , Rabeling, C. , Brady, S. G. , Fisher, B. L. , Schultz, T. R. , & Ward, P. S. (2019). Compositional heterogeneity and outgroup choice influence the internal phylogeny of the ants. Molecular Phylogenetics and Evolution, 134, 111–121. 10.1016/J.YMPEV.2019.01.024 [DOI] [PubMed] [Google Scholar]
- Bourguignon, T. , Lo, N. , Šobotník, J. , Ho, S. Y. W. , Iqbal, N. , Coissac, E. , … Evans, T. A. (2017). Mitochondrial phylogenomics resolves the global spread of higher termites, ecosystem engineers of the tropics. Molecular Biology and Evolution, 34(3), 589–597. 10.1093/molbev/msw253 [DOI] [PubMed] [Google Scholar]
- Branstetter, M. G. , Danforth, B. N. , Pitts, J. P. , Faircloth, B. C. , Ward, P. S. , Buffington, M. L. , … Brady, S. G. (2017). Phylogenomic insights into the evolution of stinging wasps and the origins of ants and bees Current Biology, 27(7), 1019–1025. 10.1016/J.CUB.2017.03.027 [DOI] [PubMed] [Google Scholar]
- Branstetter, M. G. , Ješovnik, A. , Sosa‐Calvo, J. , Lloyd, M. W. , Faircloth, B. C. , Brady, S. G. , & Schultz, T. R. (2017). Dry habitats were crucibles of domestication in the evolution of agriculture in ants Proceedings of the Royal Society B: Biological Sciences, 284(1852), 20170095 10.1098/rspb.2017.0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branstetter, M. G. , Longino, J. T. , Ward, P. S. , & Faircloth, B. C. (2017). Enriching the ant tree of life: Enhanced UCE bait set for genome‐scale phylogenetics of ants and other Hymenoptera Methods in Ecology and Evolution, 8(6), 768–776. 10.1111/2041-210X.12742 [DOI] [Google Scholar]
- Caparroz, R. , Rocha, A. V. , Cabanne, G. S. , Tubaro, P. , Aleixo, A. , Lemmon, E. M. , & Lemmon, A. R. (2018). Mitogenomes of two neotropical bird species and the multiple independent origin of mitochondrial gene orders in Passeriformes. Molecular Biology Reports, 45(3), 279–285. 10.1007/s11033-018-4160-5 [DOI] [PubMed] [Google Scholar]
- Cariou, M. , Duret, L. , & Charlat, S. (2017). The global impact of Wolbachia on mitochondrial diversity and evolution. Journal of Evolutionary Biology, 30(12), 2204–2210. 10.1111/jeb.13186 [DOI] [PubMed] [Google Scholar]
- Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution, 17(4), 540–552. 10.1093/oxfordjournals.molbev.a026334 [DOI] [PubMed] [Google Scholar]
- Chilamakuri, C. S. , Lorenz, S. , Madoui, M.‐A. , Vodák, D. , Sun, J. , Hovig, E. , … Meza‐Zepeda, L. A. (2014). Performance comparison of four exome capture systems for deep sequencing. BMC Genomics, 15(1), 449 10.1186/1471-2164-15-449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coissac, E. , Hollingsworth, P. M. , Lavergne, S. , & Taberlet, P. (2016). From barcodes to genomes: Extending the concept of DNA barcoding. Molecular Ecology, 25(7), 1423–1428. 10.1111/mec.13549 [DOI] [PubMed] [Google Scholar]
- Condamine, F. L. , Nabholz, B. , Clamens, A.‐L. , Dupuis, J. R. , & Sperling, F. A. (2018). Mitochondrial phylogenomics, the origin of swallowtail butterflies, and the impact of the number of clocks in Bayesian molecular dating. Systematic Entomology, 43(3), 460–480. 10.1111/syen.12284 [DOI] [Google Scholar]
- D'Erchia, A. M. , Atlante, A. , Gadaleta, G. , Pavesi, G. , Chiara, M. , De Virgilio, C. , … Pesole, G. (2015). Tissue‐specific mtDNA abundance from exome data and its correlation with mitochondrial transcription, mass and respiratory activity. Mitochondrion, 20, 13–21. 10.1016/J.MITO.2014.10.005 [DOI] [PubMed] [Google Scholar]
- do Amaral, F. R. , Neves, L. G. , Resende, M. F. R. , Mobili, F. , Miyaki, C. Y. , Pellegrino, K. C. M. , & Biondo, C. (2015). Ultraconserved elements sequencing as a low‐cost source of complete mitochondrial genomes and microsatellite markers in non‐model amniotes. PLoS ONE, 10(9), e0138446 10.1371/journal.pone.0138446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowton, M. , Castro, L. R. , & Austin, A. D. (2002). Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: The examination of genome ‘morphology’. Invertebrate Systematics, 16(3), 345 10.1071/IS02003 [DOI] [Google Scholar]
- Esselstyn, J. A. , Oliveros, C. H. , Swanson, M. T. , & Faircloth, B. C. (2017). Investigating difficult nodes in the placental mammal tree with expanded taxon sampling and thousands of ultraconserved elements. Genome Biology and Evolution, 9(9), 2308–2321. 10.1093/gbe/evx168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faircloth, B. C. (2016). phyluce is a software package for the analysis of conserved genomic loci. Bioinformatics, 32(5), 786–788. 10.1093/bioinformatics/btv646 [DOI] [PubMed] [Google Scholar]
- Faircloth, B. C. (2017). Identifying conserved genomic elements and designing universal bait sets to enrich them. Methods in Ecology and Evolution, 8(9), 1103–1112. 10.1111/2041-210X.12754 [DOI] [Google Scholar]
- Faircloth, B. C. , Branstetter, M. G. , White, N. D. , & Brady, S. G. (2015). Target enrichment of ultraconserved elements from arthropods provides a genomic perspective on relationships among Hymenoptera Molecular Ecology Resources, 15(3), 489–501. 10.1111/1755-0998.12328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faircloth, B. C. , McCormack, J. E. , Crawford, N. G. , Harvey, M. G. , Brumfield, R. T. , & Glenn, T. C. (2012). Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Systematic Biology, 61(5), 717–726. 10.1093/sysbio/sys004 [DOI] [PubMed] [Google Scholar]
- Foster, P. G. , & Hickey, D. A. (1999). Compositional bias may affect both DNA‐based and protein‐based phylogenetic reconstructions. Journal of Molecular Evolution, 48(3), 284–290. 10.1007/PL00006 [DOI] [PubMed] [Google Scholar]
- Foster, P. G. , Jermiin, L. S. , & Hickey, D. A. (1997). Nucleotide composition bias affects amino acid content in proteins coded by animal mitochondria. Journal of Molecular Evolution, 44(3), 282–288. 10.1007/PL00006145 [DOI] [PubMed] [Google Scholar]
- Galtier, N. , Nabholz, B. , Glémin, S. , & Hurst, G. D. D. (2009). Mitochondrial DNA as a marker of molecular diversity: A reappraisal. Molecular Ecology, 18(22), 4541–4550. 10.1111/j.1365-294X.2009.04380.x [DOI] [PubMed] [Google Scholar]
- Grabherr, M. G. , Haas, B. J. , Yassour, M. , Levin, J. Z. , Thompson, D. A. , Amit, I. , … Regev, A. (2011). Full‐length transcriptome assembly from RNA‐Seq data without a reference genome. Nature Biotechnology, 29(7), 644–652. 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grummer, J. A. , Morando, M. M. , Avila, L. J. , Sites, J. W. , & Leaché, A. D. (2018). Phylogenomic evidence for a recent and rapid radiation of lizards in the Patagonian Liolaemus fitzingerii species group. Molecular Phylogenetics and Evolution, 125, 243–254. 10.1016/J.YMPEV.2018.03.023 [DOI] [PubMed] [Google Scholar]
- Guschanski, K. , Krause, J. , Sawyer, S. , Valente, L. M. , Bailey, S. , Finstermeier, K. , … Savolainen, V. (2013). Next‐generation museomics disentangles one of the largest primate radiations. Systematic Biology, 62(4), 539–554. 10.1093/sysbio/syt018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn, C. , Bachmann, L. , & Chevreux, B. (2013). Reconstructing mitochondrial genomes directly from genomic next‐generation sequencing reads a baiting and iterative mapping approach. Nucleic Acids Research, 41(13), 129 10.1093/nar/gkt371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassanin, A. , Delsuc, F. , Ropiquet, A. , Hammer, C. , Jansen van Vuuren, B. , Matthee, C. , … Couloux, A. (2012). Pattern and timing of diversification of Cetartiodactyla (Mammalia, Laurasiatheria), as revealed by a comprehensive analysis of mitochondrial genomes. Comptes Rendus Biologies, 335(1), 32–50. 10.1016/J.CRVI.2011.11.002 [DOI] [PubMed] [Google Scholar]
- Hoang, D. T. , Chernomor, O. , von Haeseler, A. , Minh, B. Q. , & Vinh, L. S. (2018). ufboot2: Improving the Ultrafast Bootstrap Approximation. Molecular Biology and Evolution, 35(2), 518–522. 10.1093/molbev/msx281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ješovnik, A. , Sosa‐Calvo, J. , Lloyd, M. W. , Branstetter, M. G. , Fernández, F. , & Schultz, T. R. (2017). Phylogenomic species delimitation and host‐symbiont coevolution in the fungus‐farming ant genus Sericomyrmex Mayr (Hymenoptera: Formicidae): Ultraconserved elements (UCEs) resolve a recent radiation Systematic Entomology, 42(3), 523–542. 10.1111/syen.12228 [DOI] [Google Scholar]
- Katoh, K. , & Standley, D. M. (2013). mafft Multiple Sequence Alignment Software Version 7: Improvements in performance and usability. Molecular Biology and Evolution, 30(4), 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laslett, D. , & Canback, B. (2007). arwen: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics, 24(2), 172–175. 10.1093/bioinformatics/btm573 [DOI] [PubMed] [Google Scholar]
- Lemmon, A. R. , Emme, S. A. , & Lemmon, E. M. (2012). Anchored hybrid enrichment for massively high‐throughput phylogenomics. Systematic Biology, 61(5), 727–744. 10.1093/sysbio/sys049 [DOI] [PubMed] [Google Scholar]
- Lemmon, E. M. , & Lemmon, A. R. (2013). High‐throughput genomic data in systematics and phylogenetics. Annual Review of Ecology, Evolution, and Systematics, 44(1), 99–121. 10.1146/annurev-ecolsys-110512-135822 [DOI] [Google Scholar]
- Li, D. , Luo, R. , Liu, C.‐M. , Leung, C.‐M. , Ting, H.‐F. , Sadakane, K. , … Lam, T.‐W. (2016). megahit v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods, 102, 3–11. 10.1016/J.YMETH.2016.02.020 [DOI] [PubMed] [Google Scholar]
- McCormack, J. E. , Faircloth, B. C. , Crawford, N. G. , Gowaty, P. A. , Brumfield, R. T. , & Glenn, T. C. (2012). Ultraconserved elements are novel phylogenomic markers that resolve placental mammal phylogeny when combined with species‐tree analysis. Genome Research, 22(4), 746–754. 10.1101/gr.125864.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack, J. E. , Harvey, M. G. , Faircloth, B. C. , Crawford, N. G. , Glenn, T. C. , & Brumfield, R. T. (2013). A phylogeny of birds based on over 1,500 loci collected by target enrichment and high‐throughput sequencing. PLoS ONE, 8(1), e54848 10.1371/journal.pone.0054848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn, K. A. , Danielson, M. J. , Faircloth, B. C. , Glenn, T. C. , Braun, E. L. , & Kimball, R. T. (2014). Incongruence among different mitochondrial regions: A case study using complete mitogenomes. Molecular Phylogenetics and Evolution, 78, 314–323. 10.1016/j.ympev.2014.06.003 [DOI] [PubMed] [Google Scholar]
- Meza‐Lázaro, R. N. , Poteaux, C. , Bayona‐Vásquez, N. J. , Branstetter, M. G. , & Zaldívar‐Riverón, A. (2018). Extensive mitochondrial heteroplasmy in the neotropical ants of the Ectatomma ruidumcomplex (Formicidae: Ectatomminae). Mitochondrial DNA Part A, 29(8), 1203–1214. 10.1080/24701394.2018.1431228 [DOI] [PubMed] [Google Scholar]
- Moreau, C. S. , Bell, C. D. , Vila, R. , Archibald, S. B. , & Pierce, N. E. (2006). Phylogeny of the ants: Diversification in the age of angiosperms. Science, 312(5770), 101–104. 10.1126/science.1124891 [DOI] [PubMed] [Google Scholar]
- Musher, L. J. , & Cracraft, J. (2018). Phylogenomics and species delimitation of a complex radiation of Neotropical suboscine birds (Pachyramphus). Molecular Phylogenetics and Evolution, 118, 204–221. 10.1016/j.ympev.2017.09.013 [DOI] [PubMed] [Google Scholar]
- Nguyen, L.‐T. , Schmidt, H. A. , von Haeseler, A. , & Minh, B. Q. (2015). iq‐tree: A fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular Biology and Evolution, 32(1), 268–274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk, S. , Meleshko, D. , Korobeynikov, A. , & Pevzner, P. A. (2017). metaspades: A new versatile metagenomic assembler. Genome Research, 27(5), 824–834. 10.1101/gr.213959.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, Y. , Leung, H. C. M. , Yiu, S. M. , & Chin, F. Y. L. (2012). idba‐ud: A de novo assembler for single‐cell and metagenomic sequencing data with highly uneven depth. Bioinformatics, 28(11), 1420–1428. 10.1093/bioinformatics/bts174 [DOI] [PubMed] [Google Scholar]
- Picardi, E. , & Pesole, G. (2012). Mitochondrial genomes gleaned from human whole‐exome sequencing. Nature Methods, 9(6), 523–524. 10.1038/nmeth.2029 [DOI] [PubMed] [Google Scholar]
- Pie, M. R. , Ströher, P. R. , Belmonte‐Lopes, R. , Bornschein, M. R. , Ribeiro, L. F. , Faircloth, B. C. , & McCormack, J. E. (2017). Phylogenetic relationships of diurnal, phytotelm‐breeding Melanophryniscus (Anura: Bufonidae) based on mitogenomic data. Gene, 628, 194–199. 10.1016/J.GENE.2017.07.048 [DOI] [PubMed] [Google Scholar]
- Pierce, M. P. , Branstetter, M. G. , & Longino, J. T. (2017). Integrative taxonomy reveals multiple cryptic species within Central American Hylomyrma FOREL, 1912 (Hymenoptera: Formicidae) Myrmecological News, 25, 131–143. 10.25849/myrmecol.news_025:131 [DOI] [Google Scholar]
- Postma, M. , & Goedhart, J. (2019). PlotsOfData—A web app for visualizing data together with their summaries. PLoS Biology, 17(3), e3000202 10.1371/journal.pbio.3000202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prebus, M. (2017). Insights into the evolution, biogeography and natural history of the acorn ants, genus Temnothorax Mayr (hymenoptera: Formicidae) BMC Evolutionary Biology, 17(1), 250 10.1186/s12862-017-1095-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranwez, V. , Criscuolo, A. , & Douzery, E. J. P. (2010). supertriplets: A triplet‐based supertree approach to phylogenomics. Bioinformatics, 26(12), i115–i123. 10.1093/bioinformatics/btq196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranwez, V. , Douzery, E. J. P. , Cambon, C. , Chantret, N. , & Delsuc, F. (2018). macse v2: Toolkit for the alignment of coding sequences accounting for frameshifts and stop codons. Molecular Biology and Evolution, 35(10), 2582–2584. 10.1093/molbev/msy159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnasingham, S. , & Hebert, P. D. N. (2007). bold: The Barcode of Life Data System (http://www.barcodinglife.org). Molecular Ecology Notes, 7(3), 355–364. 10.1111/j.1471-8286.2007.01678.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reverter, A. , Okimoto, R. , Sapp, R. , Bottje, W. G. , Hawken, R. , & Hudson, N. J. (2017). Chicken muscle mitochondrial content appears co‐ordinately regulated and is associated with performance phenotypes. Biology Open, 6(1), 50–58. 10.1242/bio.022772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schomaker‐Bastos, A. , & Prosdocimi, F. (2018). mitoMaker: a pipeline for automatic assembly and annotation of animal mitochondria using raw NGS data. 10.20944/preprints201808.0423.v1 [DOI]
- Seixas, F. A. , Boursot, P. , & Melo‐Ferreira, J. (2018). The genomic impact of historical hybridization with massive mitochondrial DNA introgression. Genome Biology, 19(1), 91 10.1186/s13059-018-1471-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, B. T. , Harvey, M. G. , Faircloth, B. C. , Glenn, T. C. , & Brumfield, R. T. (2014). Target capture and massively parallel sequencing of ultraconserved elements for comparative studies at shallow evolutionary time scales. Systematic Biology, 63(1), 83–95. 10.1093/sysbio/syt061 [DOI] [PubMed] [Google Scholar]
- Starrett, J. , Derkarabetian, S. , Hedin, M. , Bryson, R. W. , McCormack, J. E. , & Faircloth, B. C. (2017). High phylogenetic utility of an ultraconserved element probe set designed for Arachnida. Molecular Ecology Resources, 17(4), 812–823. 10.1111/1755-0998.12621 [DOI] [PubMed] [Google Scholar]
- Stephen, S. , Pheasant, M. , Makunin, I. V. , & Mattick, J. S. (2008). Large‐scale appearance of ultraconserved elements in tetrapod genomes and slowdown of the molecular clock. Molecular Biology and Evolution, 25(2), 402–408. 10.1093/molbev/msm268 [DOI] [PubMed] [Google Scholar]
- Ströher, P. R. , Zarza, E. , Tsai, W. L. E. , McCormack, J. E. , Feitosa, R. M. , & Pie, M. R. (2017). The mitochondrial genome of Octostruma stenognatha and its phylogenetic implications. Insectes Sociaux, 64(1), 149–154. 10.1007/s00040-016-0525-8 [DOI] [Google Scholar]
- Toews, D. P. , & Brelsford, A. (2012). The biogeography of mitochondrial and nuclear discordance in animals. Molecular Ecology, 21(16), 3907–3930. 10.1111/j.1365-294X.2012.05664.x [DOI] [PubMed] [Google Scholar]
- Vieira, G. A. , & Prosdocimi, F. (2019). Accessible molecular phylogenomics at no cost: Obtaining 14 new mitogenomes for the ant subfamily Pseudomyrmecinae from public data. PeerJ, 7, e6271 10.7717/peerj.6271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler, A. P. , & Pearson, D. L. (1996). A molecular phylogeny of the tiger beetles (Cicindelidae): Congruence of mitochondrial and nuclear rDNA data sets. Molecular Phylogenetics and Evolution, 6(3), 321–338. 10.1006/mpev.1996.0083 [DOI] [PubMed] [Google Scholar]
- Vollmers, J. , Wiegand, S. , & Kaster, A.‐K. (2017). Comparing and evaluating metagenome assembly tools from a microbiologist’s perspective – not only size matters!. PLoS ONE, 12(1), e0169662 10.1371/journal.pone.0169662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, N. , Hosner, P. A. , Liang, B. , Braun, E. L. , & Kimball, R. T. (2017). Historical relationships of three enigmatic phasianid genera (Aves: Galliformes) inferred using phylogenomic and mitogenomic data. Molecular Phylogenetics and Evolution, 109, 217–225. 10.1016/J.YMPEV.2017.01.006 [DOI] [PubMed] [Google Scholar]
- Ward, P. S. (2014). The phylogeny and evolution of ants. Annual Review of Ecology, Evolution, and Systematics, 45(1), 23–43. 10.1146/annurev-ecolsys-120213-091824 [DOI] [Google Scholar]
- Ward, P. S. , & Branstetter, M. G. (2017). The acacia ants revisited: Convergent evolution and biogeographic context in an iconic ant/plant mutualism Proceedings of the Royal Society B: Biological Sciences, 284(1850), 20162569 10.1098/rspb.2016.2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenseleers, T. , Ito, F. , Van Borm, S. , Huybrechts, R. , Volckaert, F. , & Billen, J. (1998). Widespread occurrence of the microorganism Wolbachia in ants. Proceedings of the Royal Society B: Biological Sciences, 265(1404), 1447–1452. 10.1098/rspb.1998.0456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarza, E. , Connors, E. M. , Maley, J. M. , Tsai, W. L. , Heimes, P. , Kaplan, M. , & McCormack, J. E. (2018). Combining ultraconserved elements and mtDNA data to uncover lineage diversity in a Mexican highland frog (Sarcohyla; Hylidae). PeerJ, 6, e6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarza, E. , Faircloth, B. C. , Tsai, W. L. E. , Bryson, R. W. , Klicka, J. , & McCormack, J. E. (2016). Hidden histories of gene flow in highland birds revealed with genomic markers. Molecular Ecology, 25(20), 5144–5157. 10.1111/mec.13813 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1‐S5
Data Availability Statement
The MitoFinder software is available from GitHub (https://github.com/RemiAllio/MitoFinder) and GitLab (https://gitlab.com/RemiAllio/mitofinder). Annotated mitogenomes and partial mitogenomic contigs containing at least two genes and 1,000 bp have been deposited in GenBank and are available in the Third Party Annotation Section of the DDBJ/ENA/GenBank databases under accession numbers TPA: BK012118–BK012857 (Appendix S5). The full analytical pipeline, MitoFinder results including UCE and mtDNA contigs for all assemblers, phylogenetic data sets and corresponding trees can be retrieved from zenodo.org (https://doi.org/10.5281/zenodo.3231390).
We used UCE raw sequencing data for 501 ants produced in 10 phylogenomic studies (Blaimer et al., 2015, Blaimer, LaPolla, Branstetter, Lloyd, & Brady, 2016; Branstetter, Danforth, et al., 2017, Branstetter, Ješovnik, et al., 2017, Branstetter, Longino, et al., 2017; Faircloth et al., 2015; Ješovnik et al., 2017; Pierce et al., 2017; Prebus, 2017; Ward & Branstetter, 2017). (see Appendix S1)