

Abstract
Biallelic loss‐of‐function mutations in the centrosomal pericentrin gene (PCNT) cause microcephalic osteodysplastic primordial dwarfism type II (MOPDII), which is characterized by extreme growth retardation, microcephaly, skeletal dysplasia, and dental anomalies. Life expectancy is reduced due to a high risk of cerebral vascular anomalies. Here, we report two siblings with MOPDII and attenuated growth restriction, and pachygyria. Compound heterozygosity for two novel truncated PCNT variants was identified. Both truncated PCNT proteins were expressed in patient's fibroblasts, with a reduced total protein amount compared to control. Patient's fibroblasts showed impaired cell cycle progression. As a novel finding, 20% of patient's fibroblasts were shown to express PCNT comparable to control. This was associated with normal mitotic morphology and normal co‐localization of mutated PCNT with centrosome‐associated proteins γ‐tubulin and centrin 3, suggesting some residual function of truncated PCNT proteins. These data expand the clinical and molecular spectrum of MOPDII and indicate that residual PCNT function might be associated with attenuated growth restriction in MOPDII.
Keywords: microcephalic osteodysplastic primordial dwarfism type II, MOPDII, normal mitotic morphology, pachygyria, pericentrin
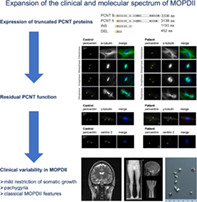
1. INTRODUCTION
Biallelic loss‐of‐function mutations in the centrosomal pericentrin gene (PCNT) cause microcephalic osteodysplastic primordial dwarfism type II (MOPDII). MOPDII is mainly characterized by microcephaly and extreme growth restriction with a final height corresponding to the height of a nearly 4‐year‐old child. Mild skeletal dysplasia, dental anomalies and craniofacial dysmorphism are noted in infancy and insulin resistance and pigmentary abnormalities are common in early adulthood. 1 , 2 , 3 Intellectual development is usually mildly impaired without structural brain malformations. A reduced life expectancy results from a high risk of stroke secondary to cerebral vascular anomalies. 4
PCNT encodes components of the pericentriolar material contributing to the maintenance of centrosomal function, microtubule organization, and cell cycle regulation. 5 Although several patients with MOPDII have been reported, the detailed pathogenic mechanisms underlying the clinical features and contributing to phenotypic variability in MOPDII remain unclear.
Here we report two siblings with MOPDII and an unusual mild restriction of somatic growth, pachygyria, and two novel PCNT variants.
2. MATERIALS AND METHODS
Written informed consent for molecular genetic studies and publication of clinical data was obtained from all participants, and the ethics committees of the Medical Universities of Innsbruck and Vienna approved the study. Whole‐exome sequencing (WES), further genetic and functional studies are described in Supporting Information.
3. RESULTS
3.1. Clinical characteristics
Two siblings from an Austrian family were referred for evaluation of a syndromic form of short stature (Figure 1A‐G), finally diagnosed as MOPDII. Moderate short stature was noted from birth in both siblings, and final adult height was 145.5 cm (> +2 SD on MOPDII‐specific growth charts) in the male patient P1 and 129.5 cm (+1 to 2 SD on MOPDII‐specific growth charts) in the female patient P2 (Figure 1E); At age 26 years P2 weighed 29.9 kg. Microcephaly was noted from age 2 years in both siblings, however, the head circumference was above +1 SD on MOPDII‐specific growth charts. Both patients displayed mild meso‐acromelic shortening of the upper extremities and developed marked scoliosis during childhood. Valgus position of the hip, coxarthroses, jawbone atrophy, and lunar osteonecrosis were seen in P2. In both patients, primary and permanent teeth had been unusually small and opalescent, and all permanent teeth had been poorly rooted and were lost during late adolescence. The following distinctive craniofacial features of MOPDII were present in the patients: small ears, a prominent nasal root and a large, beaked nose with relative hypoplasia of the alae nasi leading to small nostrils, a long‐appearing midface and a small jaw. The palpebral fissures were mildly downslanting, and short with large‐appearing irides. Both patients exhibited skin changes with hyperpigmentation predominantly on sun‐exposed skin, and premature wrinkling of the skin and graying of the hair. P1 died suddenly at the age of 20 years, cerebral hemorrhage was suspected, but autopsy was not performed to confirm the cause of death. Cranial MRI in P2 revealed pachygyria with a posterior to anterior gradient and an arteria basilaris aneurysm at age 26 years; Moyamoya disease was not present. Both patients had completed a school for children with special needs.
FIGURE 1.
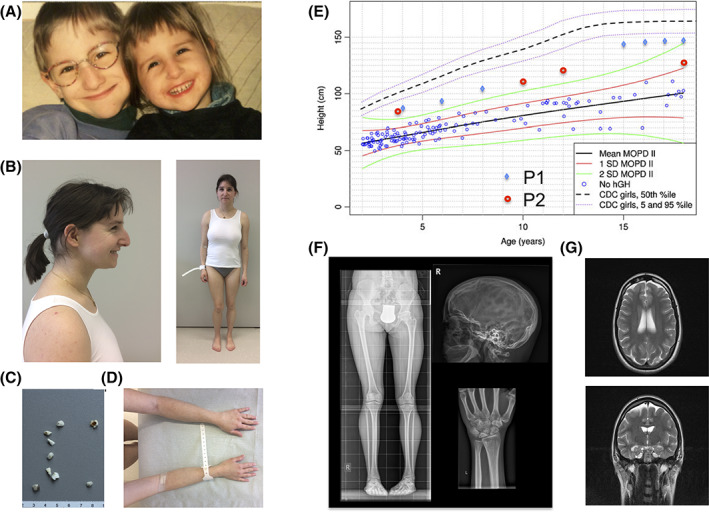
Facial characteristics in both affected siblings, A. P2 at the age of 26 years. A hair dye masks premature graying of the hair, and she wears a dental prosthesis, B. Microdontia, abnormally shaped teeth, rootless molars, and enamel defects of P2's teeth, C. Hyperpigmentation of sun‐exposed skin regions, D. Lengths for age in P1 (blue rhombs) and P2 (red circles) are above + 2 SD on MOPDII‐specific growth charts (modified from Bober et al, 3 with permission) and below the fifth centile of female controls charts (Center for Disease Control [CDC] charts), E. Mild skeletal dysplasia and adontia in P2 at age 26 years, F. Pachygyria, G
3.2. Identification of MOPDII‐causing variants
WES and segregation analysis identified compound heterozygosity for two novel truncating PCNT variants, c.1345_1864del (p.(Leu449Thrfs*5)) and c.3239_3240insCTGG (p.(Gln1081Trpfs*114)) (Figures 2A,B and S1), as the only deleterious variants causing a monogenic disease (Tables S1 and S2). Patient's fibroblasts express a total PCNT mRNA amount of 30% as compared to controls (Figure 2C). Two truncated PCNT transcripts were present in patient's fibroblasts, corresponding to the insertion and deletion mutants (Figure 2D). Truncated proteins were detected in the detergent‐soluble (cytoplasmic) cell fraction, but not in the detergent‐insoluble (cytoskeleton‐associated) cell fraction (Figure 2E) indicating that they display a decreased binding affinity to centrosomal proteins.
FIGURE 2.
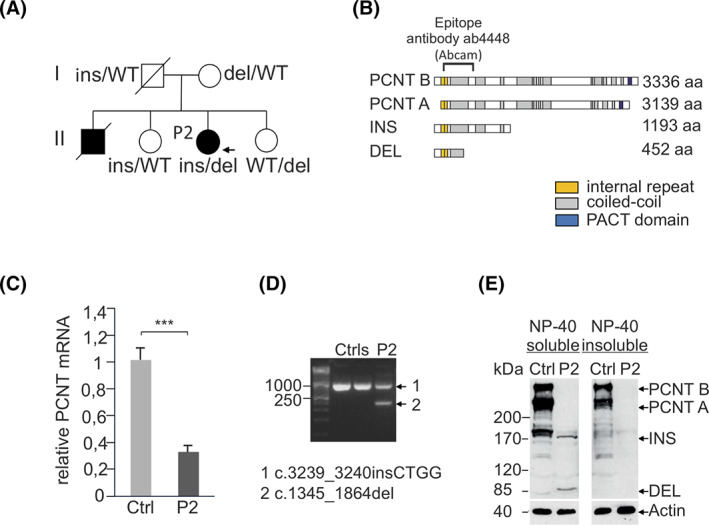
Pedigree and segregation of the PCNT insertion (ins) and deletion (del) variants; father's genotype was reconstructed, A. Schematic of pericentrin A and B isoforms with internal repeats (yellow), coiled‐coil regions (gray) and the PACT domain (blue) and identified PCNT variants. The epitope of the Ab4448 antibody is indicated. B, 30% PCNT mRNA expression in P2's fibroblasts as compared to a healthy control, C. Both mutated PCNT transcripts are expressed in P2's fibroblasts, D. PCNT immunoblot analysis in NP‐40 soluble and insoluble fractions of fibroblast cell lysates from healthy control and P2. In control, pericentrin A and B isoforms of 355 and 378 kDa are detected. Two PCNT‐specific bands were detected in the P2's soluble fraction with apparent molecular weights of ~170 and ~85 kDa, corresponding to the insertion and deletion variants, respectively, E
3.3. Cellular studies of PCNT‐deficient fibroblasts
As a novel finding, we identified 20% of patient's fibroblasts with a regular PCNT expression, associated with normal mitotic morphology (Figure 3A,B) and normal co‐localization of mutated PCNT with centrosome‐associated proteins γ‐tubulin (Figure 3C,D) and centrin 3 (Figure 3E,F). However, PCNT staining was of weaker intensity, suggesting diminished amounts of mutated PCNT or impaired binding to the centrosome. Various abnormal PCNT expression patterns were seen in the remaining 80% of patient's fibroblasts and were associated with abnormal mitotic morphology (Figure S2A) and abnormal co‐localization of PCNT with γ‐tubulin (Figure S2B) and centrin 3 (Figure S2C). As reported in MOPDII, 2 patient's fibroblasts showed decreased proliferation rates and increased numbers of dead and senescent cells (Figure S3A‐C), indicating impaired cell cycle progression.
FIGURE 3.
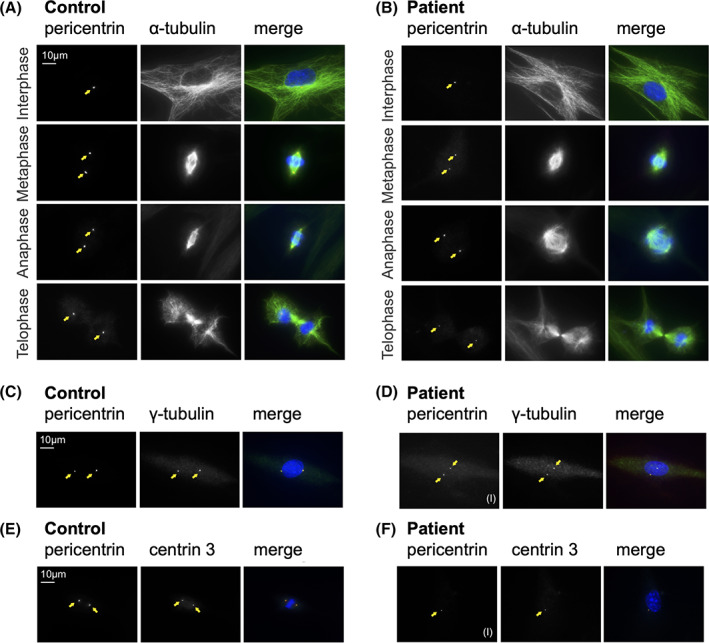
Immunofluorescence images in different mitotic stages of control fibroblasts, A, and patient fibroblasts, B, PCNT, red; α‐tubulin, green; HOECHST, blue). Regular centrosomal localization of PCNT in P2's fibroblasts (yellow arrow) is associated with normal microtubule and spindle organization in all mitotic phases. Co‐localization of PCNT with γ‐tubulin in control fibroblasts, C, and in P2's fibroblasts, D, with a regular (I) PCNT signal (PCNT, red; γ‐tubulin, green; HOECHST, blue). Co‐localization of PCNT with centrin 3 in control fibroblasts, E, and P2's fibroblast, F, with a regular (I) PCNT signal (PCNT, red; centrin 3, green; HOECHST, blue). Irregular (II, III) PCNT signals displayed in Figure S3B,C
4. DISCUSSION
We report here two siblings with an unusually mild presentation of MOPDII with regard to the degree of the patients' short stature, microcephaly, skeletal findings, and facial characteristics. 1 , 2 , 3 Although adontia in late adolescence, mild intellectual disability, and cerebrovascular anomalies pointed toward a diagnosis of MOPDII in these siblings, WES was initiated to determine the underlying genetic cause since the mild degree of short stature was considered unlikely to fit MOPDII. Compound heterozygosity for two novel truncating PCNT variants was identified, and expression of both mutated PCNT transcripts and truncated PCNT proteins in patient's fibroblasts was shown. The latter indicated a partial escape of transcripts containing premature stop codons from NMD, as reported in single MOPDII cases. 2 Decreased proliferation rates, and increased rates of dead and senescent cells indicated that the novel PCNT variants confer loss‐of‐function. This is in‐line with the current hypothesis that mitotic centrosome dysfunction and reduction in the total cellularity of the organism are major mechanisms underlying growth restriction in MOPDII. 2 , 5 , 6 , 7 These mechanisms might also explain the premature graying of hair and the skin affection seen in the patients.
Although PCNT encodes a protein with crucial roles in maintaining centrosomal function, cell cycle regulation and microtubule organization, 5 its complete absence was reported in primary cells from three MOPDII patients with severe growth restriction. 2 This suggests that PCNT is not absolutely required for centrosomal function. We hypothesize that expression of the truncating PCNT proteins identified in this study might confer residual function, explaining the attenuated growth restriction in the MOPDII patients reported here. Indeed, we observed a PCNT signal comparable to control in 20% of patient's fibroblasts. Most importantly, normal mitotic morphology in all mitotic stages and normal co‐localization of mutated PCNT with γ‐tubulin and centrin 3 were observed in these cells. While comparable rates of normal mitotic morphology in up to 30% of patient's cells were reported, 2 we show here that such cells are identifiable by a regular PCNT signal, stemming from the expression of truncated PCNT. The majority of patient's fibroblasts, however, showed impaired interaction between truncated PCNT proteins and centrosomes, indicated by absence of PCNT in the detergent‐insoluble fraction of patient cell extracts and consistent with the disrupted nucleation and organization of α‐tubulin. Mislocalization of γ‐tubulin caused by disrupted assembly of mutated PCNT proteins at the centrosome is probably to prevent proper formation of α‐tubulin as previously observed in all cell cycle stages of MOPDII patients' fibroblasts. 8 The absence of a regular PCNT signal coincides with an abnormal mitotic morphology in about 80% of patient's cells suggesting that PCNT‐related cellular functions are responsible for mitotic spindle formation, as reported. 2 This could be explained either by impaired complex formation of PCNT with γ‐tubulin or the inability of mutated PCNT to properly recruit γ‐tubulin to the site of centrosomes.
Pachygyria was diagnosed in P2, where brain imaging was available. Pachygyria has been reported so far only once in a patient with the clinical diagnosis of MOPDII, but without detection of underlying PCNT variants. 9 Pachygyria is frequently associated with intellectual disability, and it appears to be a probably cause for the observed mild intellectual disability in both siblings reported here; severe cerebrovascular events in infancy and childhood represent another cause for intellectual disability in MOPDII. WES in P2 did not reveal variants in known genes for migration disorders indicating PCNT to be causative for pachygyria. Functioning of PCNT in neuronal migration is supported by studies of Pcnt knockout mice that exhibit overall growth retardation, reduced brain size and abnormal migration of olfactory bulb interneurons. 10 Further evidence comes from pachygyria as a common feature in MOPDI, 11 and in cortical dysplasia caused by γ‐tubulin (TUBG1) mutations (OMIM #615412). Unlike α‐tubulin and β‐tubulin, γ‐tubulin is not incorporated in the microtubule lattice but localizes to the centrosome during interphase. Defective microtubule function has been shown to lead to insufficient proliferation and ultimately, microcephaly. 12 , 13 Microtubule nucleation precedes the formation of bipolar spindles and separation of chromosomes in mitosis, steps that are necessary for the progression of the cell cycle. It is then conceivable that the disrupted binding of γ‐tubulin complex to the pericentrin‐deficient centrosome leads to the cell cycle defects observed in MOPDII patients.
This study expands the spectrum of MOPDII to include pachygyria, and mild postnatal growth restriction, and should increase awareness of this condition.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Appendix S1. Supporting Information
Table S3
ACKNOWLEDGEMENTS
Supported by Jubiläumsfonds of the Austrian National Bank (grant number 17627).
Waich S, Janecke AR, Parson W, et al. Novel PCNT variants in MOPDII with attenuated growth restriction and pachygyria. Clinical Genetics. 2020;98:282–287. 10.1111/cge.13797
Funding information Oesterreichische Nationalbank, Grant/Award Number: 17627
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author, (Julia Vodopiutz), upon reasonable request.
REFERENCES
- 1. Hall JG, Flora C, Scott CI, Pauli RM, Tanaka KI. Majewski osteodysplastic primordial dwarfism type II (MOPD II): natural history and clinical findings. Am J Med Genet A. 2004;130A:55‐72. [DOI] [PubMed] [Google Scholar]
- 2. Rauch A, Thiel CT, Schindler D, et al. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319:816‐819. [DOI] [PubMed] [Google Scholar]
- 3. Bober MB, Niiler T, Duker AL, et al. Growth in individuals with Majewski osteodysplastic primordial dwarfism type II caused by pericentrin mutations. Am J Med Genet A. 2012;158A:2719‐2725. [DOI] [PubMed] [Google Scholar]
- 4. Bober MB, Jackson AP. Microcephalic Osteodysplastic primordial dwarfism, type II: a clinical review. Curr Osteoporos Rep. 2017;15:61‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Delaval B, Doxsey SJ. Pericentrin in cellular function and disease. J Cell Biol. 2010;188:181‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Griffith E, Walker S, Martin CA, et al. Mutations in pericentrin cause Seckel syndrome with defective ATR‐dependent DNA damage signaling. Nat Genet. 2008;40:232‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Delaval B, Doxsey S. Genetics. Dwarfism, where pericentrin gains stature. Science. 2008;319:732‐733. [DOI] [PubMed] [Google Scholar]
- 8. Dictenberg JB, Zimmerman W, Sparks CA, et al. Pericentrin and gamma‐tubulin form a protein complex and are organized into a novel lattice at the centrosome. J Cell Biol. 1998;141:163‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ozawa H, Takayama C, Nishida A, Nagai T, Nishimura G, Higurashi M. Pachygyria in a girl with microcephalic osteodysplastic primordial short stature type II. Brain Dev. 2005;27:237‐240. [DOI] [PubMed] [Google Scholar]
- 10. Endoh‐Yamagami S, Karkar KM, May SR, et al. A mutation in the pericentrin gene causes abnormal interneuron migration to the olfactory bulb in mice. Dev Biol. 2010;340:41‐53. [DOI] [PubMed] [Google Scholar]
- 11. Pierce MJ, Morse RP. The neurologic findings in Taybi‐Linder syndrome (MOPD I/III): case report and review of the literature. Am J Med Genet A. 2012;158A:606‐610. [DOI] [PubMed] [Google Scholar]
- 12. Breuss M, Heng JI, Poirier K, et al. Mutations in the beta‐tubulin gene TUBB5 cause microcephaly with structural brain abnormalities. Cell Rep. 2012;2:1554‐1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Breuss MW, Leca I, Gstrein T, Hansen AH, Keays DA. Tubulins and brain development ‐ the origins of functional specification. Mol Cell Neurosci. 2017;84:58‐67. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information
Table S3
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, (Julia Vodopiutz), upon reasonable request.