

Abstract
The link between drug‐metabolizing enzymes and gut microbiota is well established. In particular, hepatic cytochrome P450 (CYP) 3A activities are presumed to be affected by gut microbiota. However, there is no direct evidence that the gut microbiota affects CYP3A metabolism or the clearance of clinically relevant drugs in vivo. Our purpose was to evaluate the effects of gut microbiota on in vitro and in vivo drug metabolism and on the clearance of midazolam, which is a standard CYP3A metabolized drug. Hepatic Cyp3a activity and in vitro midazolam hydroxylase activity were compared using specific pathogen‐free (SPF) and germ‐free (GF) mice. In a pharmacokinetics (PK) study, SPF and GF mice were intraperitoneally injected with 60 mg/kg of midazolam, and plasma and tissue concentrations were measured. Hepatic Cyp3a activity and midazolam hydroxylase activity were significantly lower in GF mice than in SPF mice. Notably, in the PK study, the area under the plasma concentration–time curve from time zero to infinity and the elimination half‐life were approximately four‐fold higher in GF mice compared with SPF mice. Furthermore, the concentration of midazolam in the brain 180 min after administration was about 14‐fold higher in GF mice compared with SPF mice. Together, our results demonstrated that the gut microbiota altered the metabolic ability of Cyp3a and the tissue accumulation of midazolam.
Keywords: CYP3A, germ‐free, gut microbiota, midazolam, pharmacokinetics
1. INTRODUCTION
The gut microbiota plays an important role in maintaining host health (Clemente, Ursell, Parfrey, & Knight, 2012; Lynch & Pedersen, 2016; Valdes, Walter, Segal, & Spector, 2018). Drug metabolism is also considered to be affected by gut microbiota (Collins & Patterson, 2020; Kuno, Hirayama‐Kurogi, Ito, & Ohtsuki, 2016, 2018; Selwyn, Cheng, Klaassen, & Cui, 2016; Toda et al., 2009; Zimmermann, Zimmermann‐Kogadeeva, Wegmann, & Goodman, 2019). Zimmermann et al. (2019) reported that several drugs were directly metabolized by gut microbiota in vitro. However, many drugs administered are first metabolized in the liver, and thus the metabolic functions of the liver are critical for many drugs.
Several studies have reported the relationship between gut microbiota and host hepatic drug‐metabolizing enzymes (Kuno et al., 2016, 2018; Selwyn et al., 2016; Toda et al., 2009). Previous studies using antibiotic‐treated mice demonstrated that the expression of hepatic cytochrome P450 (CYP) 3a was decreased compared with untreated mice (Kuno et al., 2016, 2018). Other studies concluded that the activity of Cyp3a was significantly lower in germ‐free (GF) mice than in specific pathogen‐free (SPF) mice (Selwyn et al., 2016; Toda et al., 2009). As CYP3A is the major isoform of CYP and is involved in the metabolism of more than 50% of drugs on the market (Zuber, Anzenbacherová, & Anzenbacher, 2002), it is possible that the gut microbiota affects the metabolism and bioavailability of several drugs. Thus, it is important to clarify the effects of gut microbiota on drug pharmacokinetics (PK) and to isolate these drug metabolism‐modulating microbiomes for the development and evaluation of therapeutic drugs.
Decreased drug metabolism results in increased accumulation of the drug in the blood and target tissues. Since it is well known that there is a strong correlation between the drug concentration in a target tissue and drug efficacy, increased accumulation could alter the therapeutic effects or the toxicity of a drug. A previous study suggested that purified diet components impacted the intestinal environment, resulting in changes in Cyp3a expression and in the PK of triazolam (Tajima et al., 2013). However, there is no direct evidence that the gut microbiota affects in vivo drug metabolism or the clearance of clinically relevant drugs that are metabolized by Cyp3a. Furthermore, no reports have focused on whether such effects alter the accumulation of drugs in target tissues.
Midazolam, a benzodiazepine sedative, is one of the drugs metabolized by CYP3A. It acts on the central nervous system and is commonly used as an anesthetic or sedative. The drug is also considered to be a standard substrate for human CYP3A and is metabolized to 1′‐OH midazolam and 4‐OH midazolam (Greenblatt, 2003; Perloff et al., 2000). Although midazolam is also used to evaluate Cyp3a activity in mice, several studies have reported that there is a significant contribution of Cyp2c as well as Cyp3a to the metabolism of midazolam into 1′‐OH midazolam in mice (Perloff et al., 2000; Van Waterschoot et al., 2008). Additionally, 1′‐OH and 4‐OH midazolam undergo glucuronidation by glucuronosyltransferase (Ugt; Klieber et al., 2008). Most CYP3A substrates also act as substrates for P‐glycoprotein, a transporter that mediates drug excretion (Kirn et al., 1999). However, midazolam is not a P‐glycoprotein substrate. In view of the above, we conducted an experiment using SPF and GF mice to investigate the effects of gut microbiota on Cyp3a‐mediated drug metabolism and the tissue accumulation of midazolam.
2. MATERIALS AND METHODS
2.1. Chemicals
Midazolam was obtained from Fujifilm Wako Pure Chemical Co. 1′‐OH Midazolam was obtained from Funakoshi Co., Ltd. 4‐OH Midazolam was obtained from Sigma‐Aldrich. Midazolam Injection (Dormicam® Injection 10 mg; Astellas Pharma Inc.) was used for the in vivo study. Phenacetin, disodium hydrogen phosphate 12‐water, sodium dihydrogen phosphate dihydrate, potassium hydrogen phosphate, potassium dihydrogen phosphate, sucrose, Tris, EDTA‐2K, and glycerol were obtained from Fujifilm Wako Pure Chemical Co. All chemicals used for high‐performance liquid chromatography (HPLC) were HPLC grade and obtained from Fujifilm Wako Pure Chemical Co.
2.2. Animals
2.2.1. Experiment 1 (measurement of Cyp activities and midazolam hydroxylase activities)
To analyse microsomal activities of Cyp3a, Cyp2c, and Ugt and midazolam hydroxylase activities, the livers of SPF and GF BALB/cAJcl mice (male, 10 weeks of age, n = 5 in each group) were purchased from CLEA Japan, Inc. the samples were stored in a −80°C freezer until use.
2.2.2. Experiment 2 (midazolam PK test)
For the PK study, SPF and GF BALB/cAJcl mice (male, 9 weeks of age, n = 6 in each group) were purchased from CLEA Japan, Inc. The GF mice were transported to a flexible film isolator. Both mice were housed in polymethylpentene cages. Their diet consisted of radiation‐sterilized chow (FR‐2 50 kGy; Funabashi Farm Co., Ltd), and drinking water was provided ad libitum. The breeding room was maintained at a 12 h light/dark cycle. The room temperature and humidity were maintained within 23 ± 3°C and 50% ± 20%, respectively. The acclimation period was more than 1 week. The body weights of SPF and GF mice after the acclimation period were 29.2 ± 1.2 and 27.7 ± 2.4 g, respectively. The sterility of GF mice was confirmed by microscopic and culture examination.
All experiments using animals were performed under the supervision of the Institutional Animal Care and Use Committee of Yakult Central Institute and approved by the head of the Yakult Central Institute (approval number: 19‐0047). All animals were cared for and used under a program approved by AAALAC International.
2.3. Microsomal preparation
Livers were soaked with a 250 mM sucrose solution. After homogenization, the homogenate was centrifuged at 9000 × g for 10 min at 4°C. Subsequently, the supernatant was ultracentrifuged at 100000 × g for 45 min at 4°C. The microsomal pellets were resuspended in 0.1 M Tris buffer (pH 7.4) containing 10 mM EDTA‐2K and 20% glycerol. The microsomes were stored in a −80°C freezer until use. The total protein concentration was determined by a Pierce™ BCA protein assay kit (Thermo Fisher Scientific) according to the manufacturer's protocol.
2.4. Cyp and Ugt activities
The Cyp enzyme activity in liver microsomes was determined using the P450‐Glo™ assay (Promega) according to the manufacturer's protocol with slight modifications. Briefly, an appropriate concentration of luminogenic substrate (Luciferin IPA for Cyp3a and Luciferin H for Cyp2c) was preincubated for 10 min at 37°C with liver microsomes in 0.2 M potassium phosphate buffer (pH 7.4). The reactions were initiated by adding NADPH regeneration systems (Promega). The incubation times for Cyp3a and Cyp2c were 10 and 30 min, respectively. At the end of the incubation, an equal volume of luciferin detection reagent was added and incubated at room temperature for 20 min. The signals were measured by a LUMIstar OPTIMA microplate reader (BMG LABTECH). The value range was within the range of the standard curve generated using various concentrations of microsomes. Positive controls were Supersomes‐recombinant enzymes of Human CYP3A4 and 2C9 (Corning) for Luciferin IPA and Luciferin H, respectively. Likewise, the negative control was Supersomes‐CYP minus (Corning).
The Ugt enzyme activity in liver microsomes was determined using the Ugt‐Glo™ assay kit (Promega) according to the manufacturer's instructions and a previously published method (Porro, Bockor, De Caneva, Bortolussi, & Muro, 2014). Briefly, a luminogenic substrate (UGT multi‐substrate) was incubated with microsomes and uridine‐diphosphate‐glucuronic acid trisodium salt (UDPGA) in a supplied buffer at 37°C for 150 min. At the end of the incubation, an equal volume of luciferin detection reagent was added at room temperature. Signals were then allowed to stabilize for 20 min at room temperature before measuring the luminescence. Data are shown as the percentage of the substrate consumed. The value range was within the range of the standard curve generated using various concentrations of microsomes. Positive controls were pooled microsomes prepared from ICR male mice (Corning). Negative controls were pure water.
2.5. In vitro metabolite activities
Midazolam hydroxylase activity was assessed using a previously described method (Perloff et al., 2000) with slight modifications. Initially, the microsomes were preincubated with midazolam at a final concentration of 0.5–200 μM and NADPH regeneration system solution B in potassium phosphate buffer at 37°C for 5 min. The reaction was initiated by adding NADPH regeneration system solution A. After incubation at 37°C for 5 min, the reaction was stopped by adding acetonitrile containing phenacetin as an internal standard. Reaction samples were then placed on ice for 5 min before centrifugation at 14800 × g for 10 min. The resultant supernatant was injected into the HPLC system. The analysis was performed on an HPLC system GL7400 (GL Sciences, Inc.) equipped with an Inertsil ODS‐2 column (4.6 mm × 150 mm; particle size 5 μm; GL Sciences) using a UV detector at a wavelength of 220 nm. The coefficient of determination of the calibration curve was ≥ 0.999. For 1′‐OH and 4‐OH midazolam, the data were fit to the substrate inhibition model (Gandhi et al., 2012) and Michaelis–Menten model, respectively, using GraphPad Prism version 7.0 for Windows (GraphPad Software). Kinetic parameters, maximum velocity (Vmax), the Michaelis–Menten constant (Km), and intrinsic clearance (CLint) were determined.
2.6. PK analysis
The SPF and GF mice were administered 12 mL/kg of midazolam injection (60 mg/kg of midazolam). At 5, 30, 60, 120, and 180 min after administration, the saphenous vein was punctured, and about 65 μL of blood was collected using a heparin‐treated hematocrit tube. To recover the circulating blood volume, physiological saline was injected subcutaneously after 120 min. After the collection of blood at 180 min, the mice were exsanguinated under isoflurane anesthesia. Brain, liver, and kidney samples were collected in polypropylene tubes and stored at −80°C until use. Blood was centrifuged at 15000 rpm for 1 min at 4°C, and plasma was stored at −80°C.
Sample extraction was performed according to previous reports (Juřica et al., 2007; Miyamoto et al., 2015), with minor modifications. Specifically, 200 μL of 0.1 M sodium hydroxide, 50 μL of 4 μg/mL diazepam as an internal standard, and 4 mL of diethyl ether were added to 450 μL of 20‐fold diluted plasma or 15‐ to 60‐fold homogenized tissues. After 10 min of vortexing, the mixture was centrifuged at 3000 rpm for 15 min for plasma extraction or 45 min for homogenized tissue extraction at room temperature. After freezing at −30°C for 35 min, the organic layer was decanted into a conical glass. After blowing nitrogen in a heating block at 40°C, the residue was dissolved in 150 μL of mobile phase and filtered through a 0.45 μm centrifugal filter (Ultrafree‐MC HV 0.45UM; Merck) to obtain samples.
For HPLC, the same device described above was used. The column used was a LUNA C18 (2) (4.6 mm × 150 mm; particle size 5 μm; Phenomenex), and the guard column was a security guard cartridge C1 (Phenomenex). Acetonitrile and 10 mM sodium acetate buffer (pH 4.7) were used at a ratio of 37:63 as the mobile phase, and the flow rate was 1.5 mL/min. The detection wavelength was 220 nm, and the injection volume of the sample was 50 μL. The coefficient of determination of the calibration curve was ≥ 0.99. The parameters were the maximum plasma concentration (Cmax), the area under the plasma concentration–time curve from time zero to infinity (AUC0–inf), and the elimination half‐life (T1/2). AUC0–inf was calculated by the linear trapezoidal method, and from the final observation time to the infinite time was calculated based on the final observation concentration. T1/2 was calculated using three consecutive data from the last measurement point.
2.7. Statistical analysis
Data are presented as the mean ± standard deviation. Significant differences between SPF and GF mice were determined by Student's t‐test or Welch's t‐test in response to homoscedasticity or heteroscedasticity, respectively. A p‐value less than 0.05 was considered statistically significant. All statistical analyses were performed using Bell Curve for Excel (Social Survey Research Information Co., Ltd).
3. RESULTS
3.1. Hepatic Cyp and Ugt activities in SPF and GF mice
To confirm the difference in Cyp and Ugt activity between SPF and GF mice, hepatic Cyp and Ugt activity were determined. The Cyp3a activity was clearly about eight‐fold lower in the GF mice than in the SPF mice (Figure 1a). In contrast, the Cyp2c activity in the GF mice was 1.5‐fold higher than in the SPF mice (Figure 1b). Additionally, significantly higher Ugt activity was observed in the GF mice compared with the SPF mice (Figure 1c).
FIGURE 1.
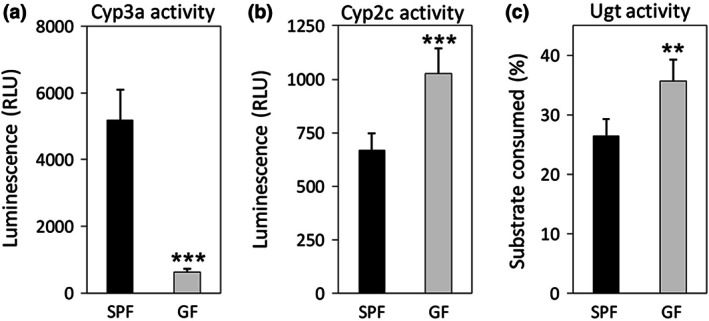
Activities of (a) Cyp3a, (b) Cyp2c, and (c) Ugt in liver microsomes prepared from specific pathogen‐free (SPF) and germ‐free (GF) mice (n = 5 per group) using a chemiluminescence assay. Data are expressed as the mean ± standard deviation. Asterisk (*) represents a statistically significant difference between SPF and GF mice. **p < 0.01, ***p < 0.001
3.2. Kinetics of hepatic midazolam hydroxylase activities in SPF and GF mice
The midazolam hydroxylase activities were determined in liver microsomes prepared from SPF and GF mice. The Michaelis–Menten plots of 1′‐OH and 4‐OH midazolam in these microsomes are shown in Figure 2. For both metabolites, GF mice exhibited lower metabolic activity compared with SPF mice (Figure 2). The Vmax, Km, and CLint values are listed in Table 1. The GF mice showed significantly lower values for Vmax and CLint in the 1′‐hydroxylation pathway compared with the SPF mice, whereas the Km was significantly higher in the GF mice than in the SPF mice. In the 4‐hydroxylation pathway, the GF mice showed a significantly lower Vmax, Km, and CLint compared with the SPF mice.
FIGURE 2.
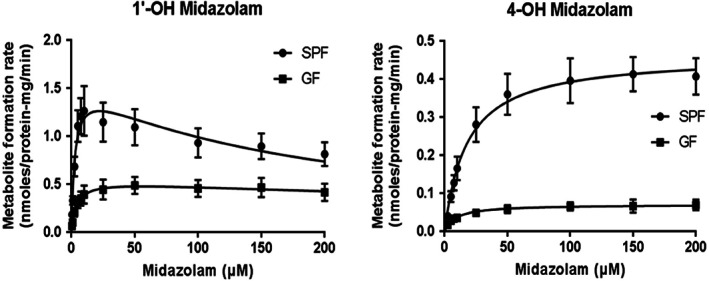
Midazolam hydroxylase activities in live microsomes prepared from specific pathogen‐free (SPF) and germ‐free (GF) mice (n = 5 per group). Data are expressed as the mean ± standard deviation
TABLE 1.
Kinetic parameters of midazolam hydroxylase activities in liver microsomes prepared from SPF and GF mice
| SPF | GF | |
|---|---|---|
| 1′‐OH Midazolam | ||
| Vmax (nmol/min/mg) | 1.57 ± 0.31 | 0.55 ± 0.11*** |
| Km (μM) | 2.71 ± 0.28 | 4.05 ± 0.41*** |
| CLint (μL/min/mg) | 577 ± 58 | 135 ± 28*** |
| 4‐OH Midazolam | ||
| Vmax (nmol/min/mg) | 0.46 ± 0.05 | 0.07 ± 0.01*** |
| Km (μM) | 18.12 ± 1.55 | 9.27 ± 3.43*** |
| CLint (μL/min/mg) | 25.86 ± 4.91 | 8.09 ± 2.15*** |
Note: Data are expressed as the mean ± standard deviation.
Abbreviations: CLint, intrinsic clearance; GF, germ‐free; Km, Michaelis–Menten constant; SPF, specific pathogen‐free; Vmax, maximum velocity.
Asterisk (*) represents a statistically significant difference between SPF and GF mice. ***p < 0.001.
3.3. Midazolam PK test
Since there were significant differences in midazolam hydroxylase activities in the SPF and GF liver microsomes, a midazolam PK study was conducted. The midazolam plasma concentration versus time profiles in SPF and GF mice are shown in Figure 3. The plasma concentration of midazolam in SPF mice was 9.83 ± 2.56 μg/mL at 5 min after administration. At 30 min, it began to decrease, and by 180 min, it had reached 0.73 ± 0.35 μg/mL (Figure 3). In contrast, the plasma concentration of midazolam in GF mice was 13.88 ± 3.97 μg/mL at 5 min after administration and 5.94 ± 1.46 μg/mL at 180 min after administration, and all measurement points were higher than those in the SPF mice (Figure 3).
FIGURE 3.
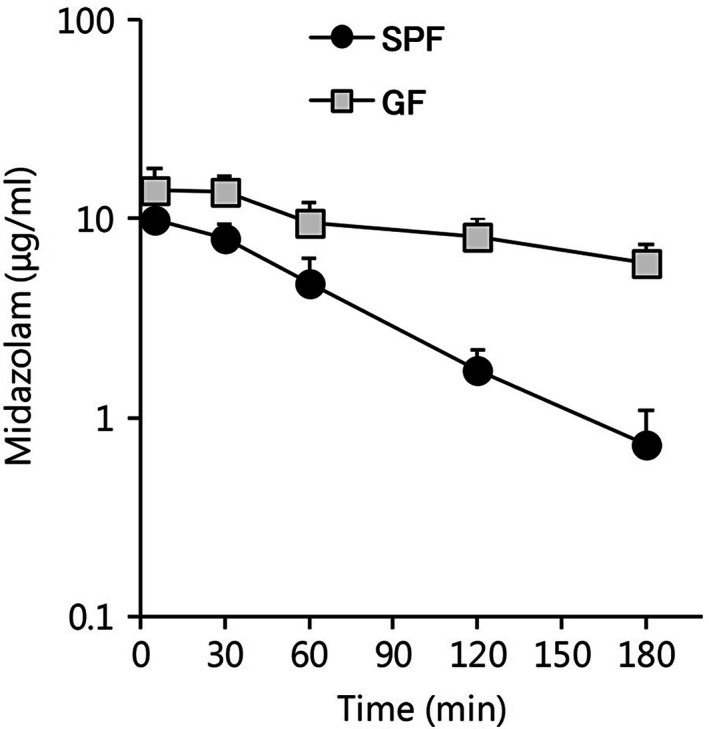
Midazolam pharmacokinetics in specific pathogen‐free (SPF) and germ‐free (GF) mice (n = 6 per group). Midazolam plasma concentration versus time profiles from 5 to 180 min following an intraperitoneal administration of midazolam (60 mg/kg). Data are expressed as the mean ± standard deviation
All PK parameters in the SPF and GF mice are shown in Table 2. Cmax was significantly higher in the GF mice than in the SPF mice. In addition, AUC0–inf was significantly higher in the GF mice (53.20 ± 11.67 µg·h/mL) than in the SPF mice (12.55 ± 2.05 µg·h/mL). Furthermore, T1/2 was significantly longer in the GF mice (2.96 ± 0.51 h) than in the SPF mice (0.73 ± 0.08 h).
TABLE 2.
Midazolam pharmacokinetic parameters in SPF and GF mice (n = 6 per group)
| Parameters | SPF | GF |
|---|---|---|
| Cmax (µg/mL) | 10.05 ± 2.43 | 14.44 ± 2.99* |
| AUCinf (µg·h/mL) | 12.55 ± 2.05 | 53.20 ± 11.67*** |
| T1/2 (h) | 0.73 ± 0.08 | 2.96 ± 0.51*** |
Note: Data are expressed as mean ± standard deviation.
Abbreviations: AUC, area under the plasma concentration; Cmax, maximum plasma concentration; GF, germ‐free; SPF, specific pathogen‐free; T1/2, elimination half‐life.
Asterisk (*) represents a statistically significant difference between SPF and GF mice. *p < 0.05, ***p < 0.001.
3.4. Tissue accumulation of midazolam
Midazolam concentrations in the brain, liver, and kidney at 180 min after administration are shown in Figure 4. The concentration of midazolam in the brain was significantly higher in GF mice (8.52 ± 4.26 µg/g) compared with SPF mice (0.63 ± 0.28 µg/g). Similarly, the concentrations of midazolam in the liver and kidney (22.16 ± 10.99 and 15.11 ± 5.68 µg/g) harvested from the GF mice were significantly higher than those in the SPF mice (2.27 ± 0.91 and 2.12 ± 0.72 µg/g).
FIGURE 4.
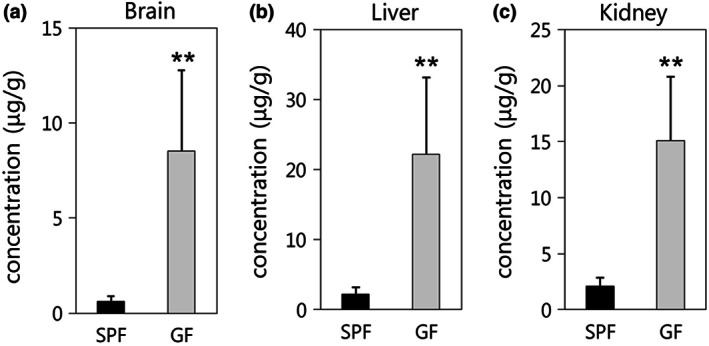
Midazolam concentrations in the (a) brain, (b) liver, and (c) kidney of specific pathogen‐free (SPF) and germ‐free (GF) mice 180 min after the administration of 60 mg/kg midazolam (n = 6 per group). Data are expressed as the mean ± standard deviation. Asterisk (*) represents a statistically significant differences between SPF and GF mice. **p < 0.01
4. DISCUSSION
Several studies have shown that gut microbiota regulate the expression and activity of hepatic Cyp3a. In this study using SPF and GF mice, we report that the gut microbiota affects hepatic Cyp3a enzyme activity and midazolam metabolism in vitro and in vivo, altering the levels of midazolam in target tissues.
First, we investigated hepatic Cyp3a, Cyp2c, and Ugt activity in SPF and GF mice. Hepatic Cyp3a activity in GF mice was about eight‐fold lower than in SPF mice (Figure 1a). This result was similar to previous studies (Selwyn et al., 2016; Toda et al., 2009). Therefore, the microbiota likely regulates hepatic Cyp3a activity. Additionally, Cyp2c activity in GF mice was higher than in SPF mice (Figure 1b). Previous studies have reported that the gene expression of some Cyp2c isoforms in the liver was higher in GF mice than in SPF mice (Li et al., 2017; Selwyn et al., 2015). Therefore, we suggest that gut microbiota may modulate hepatic Cyp2c activity via regulation of its gene expression. A previous study demonstrated that some Cyp2c isoforms were up‐regulated in Cyp3a knockout mice (Van Waterschoot et al., 2008). Thus, in the absence of gut microbiota, Cyp2c activity may partially compensate for the decreased Cyp3a activity. Furthermore, Ugt activity in GF mice was higher than in SPF mice (Figure 1c). There is limited information available with regard to the impact of gut microbiota on Ugt activity (Collins & Patterson, 2020). However, 15% of marketed drugs are metabolized by UGT (Williams et al., 2004), and thus understanding the effects of gut microbiota on Ugt activity is of great significance. Since the present study used a multi‐UGT enzyme and did not discriminate between isoforms, further studies are needed to identify the Ugt isoforms that are influenced by gut microbiota. Together, these results suggest that the gut microbiota affected the activity of hepatic Cyp3a, Cyp2c, and Ugt.
Given that hepatic Cyp3a activity was evidently different between the SPF and GF mice, in vitro midazolam 1′‐hydroxylation and 4‐hydroxylation activity were evaluated using liver microsomes. We observed that CLint and Vmax were lower in the GF mice than in the SPF mice in both pathways (Table 1), likely due to the lower hepatic Cyp3a activity in GF mice. Specifically, the difference in Vmax between SPF and GF mice was greater in the 4‐hydroxylation activity than in the 1′‐hydroxylation activity. This result was attributed to the fact that Cyp2c plays an important role in the 1′‐hydroxylation of midazolam in mice (Perloff et al., 2000; Van Waterschoot et al., 2008), and in the present study, GF mice exhibited higher Cyp2c activity than SPF mice (Figure 1b). Thus, it is possible that Cyp2c partially compensates for the midazolam 1′‐hydroxylation in GF mice. These results indicated that midazolam hydroxylase activity in the liver was modified by the gut microbiota.
In the PK study, the AUC0–inf and T1/2 of midazolam were about four‐fold higher in GF mice than in SPF mice (Table 2). These results were in agreement with the results of the in vitro experiment, suggesting that low levels of hepatic Cyp3a activity in GF mice decrease drug metabolism in vivo. A previous study reported that purified diets affected Cyp3a activity and the PK of triazolam (Tajima et al., 2013), suggesting that changes in the intestinal environment caused by the purified diet may have reduced the hepatic Cyp3a activity. However, several studies have demonstrated that the expression level and activity of hepatic Cyp3a are influenced by the type of diet (Kang, Song, Lee, Yoo, & Shin, 2008; Peters & Teel, 2003). The SPF and GF mice used in the present study were fed the same diet, and thus we could exclude any potential diet effects. As demonstrated from the above results, the in vivo ability of Cyp3a to metabolize midazolam was affected by the gut microbiota.
The concentration of midazolam in the brain, which is the target tissue of midazolam, was significantly higher in GF mice than in SPF mice (Figure 4a). Therefore, the gut microbiota affects both the plasma concentration and tissue accumulation of midazolam. In general, the drug concentration in a target tissue strongly correlates with drug efficacy. Hence, the results suggest that the gut microbiota may also alter the efficacy of midazolam via the host's drug metabolism. As mentioned above, more than half of all marketed drugs are metabolized by CYP3A (Zuber et al., 2002). Therefore, the results of this study suggest that the metabolism and tissue accumulation of many drugs are likely affected by the gut microbiota. To improve the efficacy and safety of these drugs, further studies are needed to better understand the consequences of the gut microbiome‐mediated regulation of metabolic enzymes.
5. CONCLUSION
The present study demonstrated that the gut microbiota influenced the metabolic ability of Cyp3a both in vitro and in vivo and altered the tissue accumulation of midazolam.
CONFLICT OF INTEREST
There are no conflicts of interest.
ACKNOWLEDGEMENTS
The authors would like to thank Takashi Sasaki for technical assistance with the experiments and Enago (www.enago.jp) for the English language review. This research did not receive any specific grant from funding agencies in the public, commercial, or not‐for‐profit sectors. All the authors are employees of Yakult Honsha Co, Ltd.
References
REFERENCES
- Clemente, J. C. , Ursell, L. K. , Parfrey, L. W. , & Knight, R. (2012). The impact of the gut microbiota on human health: An integrative view. Cell, 148(6), 1258–1270. 10.1016/j.cell.2012.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, S. L. , & Patterson, A. D. (2020). The gut microbiome: An orchestrator of xenobiotic metabolism. Acta Pharmaceutica Sinica B, 10(1), 19–32. 10.1016/j.apsb.2019.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi, A. S. , Guo, T. , Shah, P. , Moorthy, B. , Chow, D. L. , Hu, M. , & Ghose, R. (2012). CYP3A‐dependent drug metabolism is reduced in bacterial inflammation in mice. British Journal of Pharmacology, 166(7), 2176–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt, D. J. , von Moltke, L. L. , Harmatz, J. S. , Chen, G. , Weemhoff, J. L. , Jen, C. , … Zinny, M. A. (2003). Time course of recovery of cytochrome P450 3A function after single doses of grapefruit juice. Clinical Pharmacology & Therapeutics, 74(2), 121–129. 10.1016/S0009-9236(03)00118-8 [DOI] [PubMed] [Google Scholar]
- Juřica, J. , Dostálek, M. , Konečný, J. , Glatz, Z. , Hadašová, E. , & Tomandl, J. (2007). HPLC determination of midazolam and its three hydroxy metabolites in perfusion medium and plasma from rats. Journal of Chromatography. B, Analytical Technologies in the Biomedical and Life Sciences, 852(1–2), 571–577. 10.1016/j.jchromb.2007.02.034 [DOI] [PubMed] [Google Scholar]
- Kang, H. J. , Song, I. S. , Lee, S. S. , Yoo, M. A. , & Shin, J. G. (2008). Effects of dietary salt on the expression of drug transporters, cytochrome P4503a, and nuclear receptors in rats. Xenobiotica; the Fate of Foreign Compounds in Biological Systems, 38(2), 147–155. 10.1080/00498250701744674 [DOI] [PubMed] [Google Scholar]
- Kirn, R. B. , Wandel, C. , Leake, B. , Cvetkovic, M. , Fromm, M. F. , Dempsey, P. J. , … Wood, A. J. (1999). Interrelationship between substrates and inhibitors of human CYP3A and P‐glycoprotein. Pharmaceutical Research, 16(3), 408–414. [DOI] [PubMed] [Google Scholar]
- Klieber, S. , Ngo, R. , Arabeyre‐Fabre, C. , Meunier, V. , Sadoun, F. , Fedeli, O. , … Maurel, P. (2008). Contribution of the N‐glucuronidation pathway to the overall in vitro metabolic clearance of midazolam in humans. Drug Metabolism and Disposition, 36(5), 851–862. 10.1124/dmd.107.019539.MDZ [DOI] [PubMed] [Google Scholar]
- Kuno, T. , Hirayama‐Kurogi, M. , Ito, S. , & Ohtsuki, S. (2016). Effect of intestinal flora on protein expression of drug‐metabolizing enzymes and transporters in the liver and kidney of germ‐free and antibiotics‐treated mice. Molecular Pharmaceutics, 13(8), 2691–2701. 10.1021/acs.molpharmaceut.6b00259 [DOI] [PubMed] [Google Scholar]
- Kuno, T. , Hirayama‐Kurogi, M. , Ito, S. , & Ohtsuki, S. (2018). Reduction in hepatic secondary bile acids caused by short‐term antibiotic‐induced dysbiosis decreases mouse serum glucose and triglyceride levels. Scientific Reports, 8(1), 1253 10.1038/s41598-018-19545-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C. Y. , Lee, S. , Cade, S. , Kuo, L. J. , Schultz, I. R. , Bhatt, D. K. , … Cui, J. Y. (2017). Novel interactions between gut microbiome and host drug‐processing genes modify the hepatic metabolism of the environmental chemicals polybrominated diphenyl ethers. Drug Metabolism and Disposition: the Biological Fate of Chemicals, 45(11), 1197–1214. 10.1124/dmd.117.077024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, S. V. , & Pedersen, O. (2016). The human intestinal microbiome in health and disease. New England Journal of Medicine, 375(24), 2369–2379. 10.1056/NEJMra1600266 [DOI] [PubMed] [Google Scholar]
- Miyamoto, H. , Matsueda, S. , Moritsuka, A. , Shimokawa, K. , Hirata, H. , Nakashima, M. , … Nishida, K. (2015). Evaluation of hypothermia on the in vitro metabolism and binding and in vivo disposition of midazolam in rats. Biopharmaceutics and Drug Disposition, 36(7), 481–489. 10.1002/bdd.1960 [DOI] [PubMed] [Google Scholar]
- Perloff, M. D. , Von Moltke, L. L. , Court, M. H. , Kotegawa, T. , Shader, R. I. , & Greenblatt, D. J. (2000). Midazolam and triazolam biotransformation in mouse and human liver microsomes: Relative contribution of CYP3A and CYP2C isoforms. Journal of Pharmacology and Experimental Therapeutics, 292(2), 618–628. [PubMed] [Google Scholar]
- Peters, L. P. , & Teel, R. W. (2003). Effects of high sucrose diet on body and liver weight and hepatic enzyme content and activity in the rat. In Vivo, 17(1), 61–65. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/126557922 [PubMed] [Google Scholar]
- Porro, F. , Bockor, L. , De Caneva, A. , Bortolussi, G. , & Muro, A. F. (2014). Generation of Ugt1‐deficient murine liver cell lines using TALEN technology. PLoS One, 9(8), e104816 10.1371/journal.pone.0104816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selwyn, F. P. , Cheng, S. L. , Bammler, T. K. , Prasad, B. , Vrana, M. , Klaassen, C. , & Cui, J. Y. (2015). Developmental regulation of drug‐processing genes in livers of germ‐free mice. Toxicological Sciences, 147(1), 84–103. 10.1093/toxsci/kfv110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selwyn, F. P. , Cheng, S. L. , Klaassen, C. D. , & Cui, J. Y. (2016). Regulation of hepatic drug‐metabolizing enzymes in germ‐free mice by conventionalization and probiotics. Drug Metabolism and Disposition: the Biological Fate of Chemicals, 44(2), 262–274. 10.1124/dmd.115.067504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, M. , Ikarashi, N. , Igeta, S. , Toda, T. , Ishii, M. , Tanaka, Y. , … Sugiyama, K. (2013). Different diets cause alterations in the enteric environment and trigger changes in the expression of hepatic cytochrome P450 3A, a drug‐metabolizing enzyme. Biological and Pharmaceutical Bulletin, 36(4), 624–634. 10.1248/bpb.b12-01005 [DOI] [PubMed] [Google Scholar]
- Toda, T. , Saito, N. , Ikarashi, N. , Ito, K. , Yamamoto, M. , Ishige, A. , … Sugiyama, K. (2009). Intestinal flora induces the expression of Cyp3a in the mouse liver. Xenobiotica; the Fate of Foreign Compounds in Biological Systems, 39(4), 323–334. 10.1080/00498250802651984 [DOI] [PubMed] [Google Scholar]
- Valdes, A. M. , Walter, J. , Segal, E. , & Spector, T. D. (2018). Role of the gut microbiota in nutrition and health. BMJ, 361, k2179 10.1136/bmj.k2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Waterschoot, R. A. B. , Van Herwaarden, A. E. , Lagas, J. S. , Sparidans, R. W. , Wagenaar, E. , Van Der Kruijssen, C. M. M. , … Schinkel, A. H. (2008). Midazolam metabolism in cytochrome P450 3A knockout mice can be attributed to up‐regulated CYP2C enzymes. Molecular Pharmacology, 73(3), 1029–1036. 10.1124/mol.107.043869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, J. A. , Hyland, R. , Jones, B. C. , Smith, D. A. , Hurst, S. , Goosen, T. C. , … Ball, S. E. (2004). Drug–drug interactions for UDP‐glucuronosyltransferase substrates: A pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metabolism and Disposition: the Biological Fate of Chemicals, 32(11), 1201–1208. 10.1124/dmd.104.000794 [DOI] [PubMed] [Google Scholar]
- Zimmermann, M. , Zimmermann‐Kogadeeva, M. , Wegmann, R. , & Goodman, A. L. (2019). Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature, 570(7762), 462–467. 10.1038/s41586-019-1291-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber, R. , Anzenbacherová, E. , & Anzenbacher, P. (2002). Cytochromes P450 and experimental models of drug metabolism. Journal of Cellular and Molecular Medicine, 6(2), 189–198. 10.1111/j.1582-4934.2002.tb00186.x [DOI] [PMC free article] [PubMed] [Google Scholar]