

Abstract
Objectives
Evaluating potential relationships between progression‐free survival (PFS) and tumor gene expression patterns and mutational status was an exploratory objective of the phase 3 TOURMALINE‐MM1 study (NCT01564537) of ixazomib‐lenalidomide‐dexamethasone (IRd) vs placebo‐Rd in 722 patients with relapsed/refractory multiple myeloma (MM).
Methods
We utilized gene expression and mutation data from screening bone marrow aspirates to identify tumors with non‐canonical nuclear factor‐κB (NF‐κB) signaling pathway activation.
Results
DNA/RNA sequencing data were available for 339 (47.0%)/399 (55.2%) patients; 49/339 (14.5%) patients had non‐canonical NF‐κB pathway gene mutations (tumor‐necrosis‐factor receptor‐associated factor 2, 3 [TRAF2, TRAF3], baculoviral‐inhibitor‐of‐apoptosis repeat‐containing 2/3 [BIRC2/3]), and PFS was significantly longer with IRd vs placebo‐Rd in these patients (hazard ratio [HR] 0.23). In patients with lower TRAF3 expression (median not reached vs 11 months, HR 0.47) and higher NF‐κB‐inducing kinase (NIK) expression (median not reached vs 14 months, HR 0.45), both associated with non‐canonical NF‐κB pathway activation, PFS was significantly longer with IRd vs placebo‐Rd. TRAF3 expression was decreased in patients harboring t(4;14) and 1q21 amplification, suggesting increased non‐canonical NF‐κB pathway activation.
Conclusions
Adding ixazomib to Rd provides clinical benefit in MM tumors with increased non‐canonical NF‐κB pathway activity. This is a potential mechanism for activity in 1q21 amplified high‐risk tumors.
Keywords: DNA sequencing, gene expression profiling, multiple myeloma, mutation, nuclear factor kappa B, progression‐free survival, RNA sequencing, TRAF3
Novelty statement.
1. What is the new aspect of your work? Our analyses of the gene expression and mutation data generated in a phase 3 clinical study in relapsed/refractory multiple myeloma patients are the first to demonstrate increased non‐canonical NF‐κB pathway activation in a subset of 1q21 amplified tumors and suggest a potential correlation to increased clinical benefit of ixazomib plus lenalidomide‐dexamethasone in these patients.
2. What is the central finding of your work? The addition of ixazomib to lenalidomide‐dexamethasone provides greater progression‐free survival benefit in patients with relapsed/refractory multiple myeloma who have tumors with increased non‐canonical NF‐κB pathway activity, including patients with high‐risk abnormalities such as 1q21 amplification and t(4;14).
3. What is (or could be) the specific clinical relevance of your work? Our results may offer an understanding of the mechanisms underlying the observed clinical benefit of ixazomib in combination with lenalidomide‐dexamethasone in subgroups of relapsed/refractory multiple myeloma patients, including those with 1q21 amplification.
1. INTRODUCTION
The nuclear factor‐κB (NF‐κB) pathway has been implicated in the development of several malignant disorders, including multiple myeloma (MM). 1 Dysregulation of NF‐κB transcription factors due to mutations in NF‐κB‐regulating genes is associated with MM disease pathogenesis, 1 with frequent mutations in positive and negative regulators of NF‐κB seen in MM, 2 as highlighted in Figure 1. NF‐κB signaling is mediated by multiple different homo‐ or hetero‐dimeric transcription factors based on 5 key monomeric subunits, the most prevalent being RelA:p50 and RelB:p52, through the canonical (classical) and non‐canonical (alternative) pathways. In the former, signaling is stimulated by various cytokine receptors and results in the release of pre‐existing NF‐κB dimers, notably RelA:p50, that have been sequestered in the cytoplasm by inhibitory IκB proteins. In the latter, cell‐differentiating and organogenic stimuli result in signaling that triggers RelB NF‐κB activation during immune cell maturation. 1 Proteasomal cleavage of key regulatory intermediates is a critical event in the activation of both pathways. In both pathways, the regulatory intermediates—the inhibitory IκB proteins and the p100 regulator of RelB—are degraded by the proteasome, enabling translocation of NF‐κB dimers to the nucleus 1 ; thus, proteasome inhibition can result in inhibition of NF‐κB signaling. 3 , 4
Figure 1.
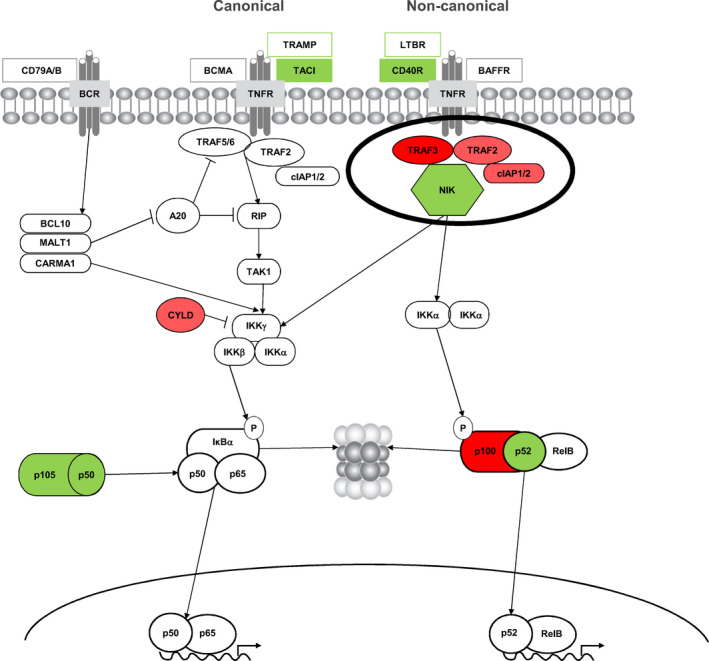
Frequent mutations in positive (green) and negative (red) regulators of the NF‐κB pathway in MM. TRAF3 is an important negative regulator and NIK an important positive regulator of the non‐canonical NF‐κB signaling pathway. Figure reproduced and adapted under the Creative Commons Attribution License (CCBY 3.0; https://creativecommons.org/licenses/by/3.0/) from Demchenko & Kuehl, Oncotarget 2010;1(1):59‐68. 2 MM, multiple myeloma; NF‐κB, nuclear factor kappa B
Through these canonical and non‐canonical pathways, NF‐κB signaling provides proliferation and survival signals in MM 1 , 5 , 6 , 7 , 8 by mediating the expression of hundreds of downstream target genes. NF‐κB signaling plays an important role in signaling pathways that control both pro‐inflammatory and pro‐survival pathways and in critical supportive interactions between the MM cells and the bone marrow microenvironment. 1 NF‐κB pathway activation in MM can result from cell extrinsic signaling, as well as from constitutive activation through mutations in key regulatory intermediates or via epigenetic modifications that result in changes in expression of key regulatory genes. 5 , 7 Overall, approximately 43.2% of MM tumors are thought to harbor an abnormality in this pathway. 8
While activation of the non‐canonical NF‐κB pathway is associated with proliferative and pro‐survival stimuli from the bone marrow microenvironment, constitutive activation would be expected to decrease dependence of tumor cells on the bone marrow niche and thereby contribute to the pathogenesis of MM. 6 Mutations in key components of the non‐canonical NF‐κB pathway have been identified in approximately 17% of MM, 1 , 6 with alterations in the expression of key regulatory intermediates occurring through chromosomal deletions/amplifications as well as epigenetic modifications. 5 For example, decreased expression of tumor‐necrosis‐factor receptor‐associated factor protein 3 (TRAF3) via chromosomal deletion and increased expression of NF‐κB‐inducing kinase (NIK) via chromosomal amplification have been shown to be associated with increased activation of the non‐canonical NF‐κB pathway. 1 , 5 , 6
Ixazomib, the first oral proteasome inhibitor, 9 , 10 is approved by healthcare authorities, in combination with lenalidomide‐dexamethasone (Rd), for the treatment of MM patients who have received at least 1 prior therapy. 11 , 12 Approval was based on the international, multicenter, double‐blind, placebo‐controlled, phase 3 TOURMALINE‐MM1 study (NCT01564537), 13 in which the triplet regimen of ixazomib plus Rd (IRd) demonstrated a clinically meaningful and statistically significant progression‐free survival (PFS) benefit vs placebo‐Rd, with a median PFS of 20.6 vs 14.7 months (hazard ratio [HR] 0.74, P = .012), while resulting in limited additional toxicity. Consistent PFS benefit with IRd vs placebo‐Rd was demonstrated in prespecified subgroup analyses of TOURMALINE‐MM1, 13 with notable efficacy found in patients with high‐risk cytogenetics 14 and pronounced effects seen in more heavily pretreated patients. 15
The evaluation of potential relationships between PFS and tumor gene expression patterns and mutational status in the NF‐κB pathway was a prespecified exploratory objective of TOURMALINE‐MM1. In the analyses reported herein, we utilized gene expression profile and mutation data to identify tumors with non‐canonical NF‐κB pathway activation and analyzed its impact on PFS in patients with relapsed/refractory MM who were treated with IRd or placebo‐Rd in the TOURMALINE‐MM1 trial.
2. METHODS
2.1. Study design and patients
The design of the randomized, double‐blind, placebo‐controlled TOURMALINE‐MM1 study has been reported previously. 13 Briefly, 722 patients with RRMM who had received 1‐3 prior lines of therapy were randomized (1:1) to receive IRd (n = 360) or placebo‐Rd (n = 362) until disease progression or unacceptable toxicity. All patients provided written informed consent. Patients received 28‐day treatment cycles comprising ixazomib 4.0 mg or matching placebo on days 1, 8, and 15, lenalidomide 25 mg on days 1‐21 (dose adjusted to 10 mg for patients with moderate renal impairment), and dexamethasone 40 mg on days 1, 8, 15, and 22. Randomization was stratified by number of prior therapies, prior proteasome inhibitor exposure, and International Staging System (ISS) disease stage. Per protocol, cutoff values for defining the presence of high‐risk cytogenetic abnormalities were based on the false‐positive rates (technical cutoffs) of the Kreatech fluorescence in situ hybridization (FISH) probes used; these were 5%, 3%, and 3% positive cells for del(17p), t(4;14), and 1q21 amplification (amp 1q21), respectively. 14 The primary endpoint was PFS by independent review committee assessment; key secondary endpoints were overall survival (OS) and OS in patients with del(17p). An exploratory study objective was to evaluate potential relationships between treatment outcomes and somatic mutations, as well as pathway activations, identified in patient tumor samples; here, we report these exploratory analyses utilizing PFS data from the first prespecified statistical analysis, as reported in the primary manuscript, after a median follow‐up of ~15 months. 13 OS data were not mature at this time, and follow‐up is ongoing; hence, analyses utilizing OS data are not reported herein.
2.2. Sample collection
Bone marrow aspirate collection and processing was carried out as described in another paper from TOURMALINE‐MM1 published recently by Di Bacco et al. 16 Briefly, samples were collected at screening from patients enrolled in the trial for targeted sequencing and for gene expression profiling of CD138‐positive cells. All analyses were done centrally at the Broad Institute, Cambridge, MA, as described below. Following isolation of CD138‐positive cells, frozen pellets were shipped to LabCorp Research (Triangle Park, NC) for DNA and RNA co‐extraction using the Qiagen AllPrep DNA/RNA kit. Extracted DNA and RNA were measured for quantity, quality, and purity. Each sample was aliquoted in two vials, frozen at −80°C, and shipped to the Clinical Research Sequencing Platform (CRSP) at the Broad Institute (Cambridge, MA) for sequencing. All RNA samples were quantified again using a Nanodrop spectrophotometer (Thermo Scientific), and their integrity was assessed using the Agilent Bioanalyzer and normalized before starting the sequencing. The minimum Nanodrop and BioAnalyzer input requirement was ≥250 ng RNA and an RNA integrity number (RIN) ≥6.
2.3. RNA sequencing and analysis
The methodology for RNA sequencing (RNAseq) and analysis is reported in the paper from TOURMALINE‐MM1 published recently by Di Bacco et al. 16 Briefly, cDNA library preparation using 500 base‐pair (bp) cDNA was done using an automated variant of the Illumina Tru‐seq (Cat # RS‐930‐2001 and RS‐122‐2002) protocol; after enrichment, libraries were quantified with quantitative polymerase chain reaction (qPCR) and then pooled in equimolar ratios. Pooled libraries were normalized and denatured prior to sequencing. After quality control, a minimum of 50 million pair‐end reads (~100 bp in length) per sample were generated for 399 patients (55.2% of the intent‐to‐treat population of 722 patients). Data were analyzed using the Broad Picard Pipeline, which includes de‐multiplexing and data aggregation (TopHat 1.4.1). The reads were aligned to the Human B37.3 reference sequence using the OSA2 aligner 17 developed by the OmicSoft Corporation (now Qiagen). RPKM (reads per kilobase of transcript per million mapped reads) values at both gene and transcript level were calculated using the RSEM 18 algorithm, and the gene‐level RPKM values were used for the expression analysis.
2.4. DNA sequencing and analysis
Somatic mutations were identified by sequencing both tumor and matched‐normal whole‐blood samples from an individual patient using a hybridization‐based targeted MM‐specific mutation panel with >400× target mean coverage per sample on ~1200 genes. The Broad Institute's MuTect pipeline (https://software.broadinstitute.org/cancer/cga/mutect) 19 was used to identify both single‐nucleotide polymorphisms (SNPs) and insertion‐deletions (indels), which were then subject to additional filtering. Specifically, mutations were annotated by the SNPEff program (http://snpeff.sourceforge.net/), and only those annotated as “Coding” in the “Effect” field were retained. Samples with high contamination of non‐myeloma cells and samples with more than 0.5% tumor material in normal tissue samples were removed. For quality filtering of the results, only SNPs with read depth (DP) at the mutation position in the sample of ≥100 and a Phred‐scaled quality score (QUAL) for the assertion made in alternative allele of ≥100 were retained; indels and those SNPs belonging to a list of manually curated important genes in MM were not subject to this filtering process.
2.5. Statistical analysis
Based on DNA sequencing (DNAseq) data, patients were stratified for analysis according to the presence or absence of genetic mutations in the four genes (TRAF2, TRAF3, baculoviral‐inhibitor‐of‐apoptosis repeat‐containing 2/3 [BIRC2/3]) known to be negative regulators of the non‐canonical NF‐κB pathway via regulation of NIK protein stability. 1 , 5 , 6 Based on RNAseq data, TRAF3 and NIK mRNA expression were evaluated, and patients were dichotomized for analysis into “high” or “low” expression groups, based on the global median across treatment arms. A NF‐κB10 index was developed based on RNA expression of 10 NF‐κB target genes (CD40, CIAP, CYLD, LTBR, NFKB1, NFKB2, NIK, TACI, TRAF2, TRAF3), utilizing the sum of the 10 expression levels in the log2 scale, 2 , 5 , 20 and patients were dichotomized for analysis into “high” or “low” NF‐κB10 index groups, based on the global median, as well as grouped into quartiles based on NF‐κB10 index.
PFS was evaluated with IRd and placebo‐Rd within patient groups defined by NF‐κB pathway mutation status, TRAF3 expression, NIK expression, and NF‐κB10 index. PFS was assessed using Kaplan‐Meier methodology, and comparisons were reported as hazard ratios (HR) with 95% confidence intervals (CI). The log‐rank test was used to determine statistical significance between pairs of survival curves. For comparisons of more than 2 PFS curves, the score test was used to calculate the P value to test for significance in overall difference among the curves, under the proportional hazard assumption.
3. RESULTS
3.1. Patients
RNAseq data were available for 399 (55.2%) patients (189 IRd, 210 placebo‐Rd), and DNAseq data were available for 339 (47.0%) patients (166 IRd, 173 placebo‐Rd) of the 722 patients in the ITT population. Baseline characteristics of patients with and without genomic sequence data were comparable (Tables S1 and S2).
3.2. Analysis of PFS by mutation status
Overall, 49 of 339 patients (14.5%) with DNAseq data had mutations in TRAF2, TRAF3, and BIRC2/3, including 28 of 166 IRd patients (16.9%) and 21 of 173 placebo‐Rd patients (12.1%). These genes are known to play a role in the negative regulation of the non‐canonical NF‐κB pathway by regulating NIK protein stability. 1 , 5 , 6 NIK is targeted for proteasomal degradation by the activity of a protein complex comprising TRAF3, TRAF2, and BIRC2/3 proteins. In turn, TRAF3 and TRAF2 are degraded and NIK rescued from constitutive destruction through non‐canonical stimuli; p100 is then marked by NIK for proteasome processing, leading to activation of the non‐canonical pathway. Loss‐of‐function mutations in these genes are known to result in increased NF‐κB activity. Kaplan‐Meier analyses of PFS in the DNAseq population according to mutation status, overall and with IRd and placebo‐Rd, are shown in Figure 2. In a pooled analysis across treatment groups, there was no significant difference in PFS between patients with vs without NF‐κB pathway mutations (HR 1.2, median 17.5 months vs not reached; Figure 2A).
Figure 2.
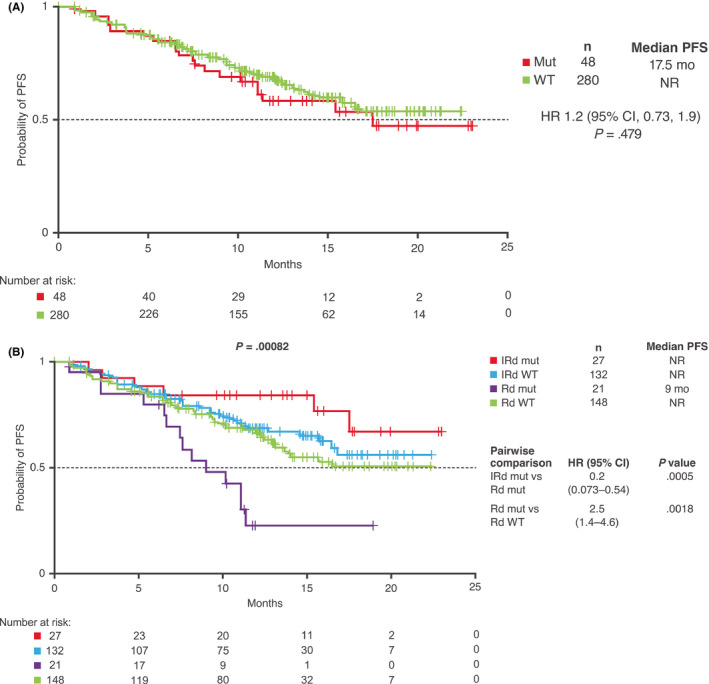
PFS according to NF‐κB mutation status. Kaplan‐Meier analysis of PFS in the DNAseq‐evaluable population (A) according to presence (mut) or absence (WT) of mutations in the non‐canonical NF‐κB pathway, and (B) with IRd and placebo‐Rd according to mutation status. CI, confidence interval; HR, hazard ratio; IRd, ixazomib‐lenalidomide‐dexamethasone; mut, NF‐κB pathway mutation; NF‐κB, nuclear factor kappa B; NR, not reached; PFS, progression‐free survival; Rd, placebo‐lenalidomide‐dexamethasone; WT, wild‐type NF‐κB pathway
Further analysis of PFS according to mutation status and treatment received demonstrated a significant impact on PFS (P = .00082; Figure 2B). Median PFS was not reached in both IRd subgroups and in the placebo‐Rd wild‐type subgroup but was 9 months in the subgroup of placebo‐Rd patients with mutations. PFS was significantly longer with IRd vs placebo‐Rd in patients with non‐canonical NF‐κB pathway mutations (HR 0.20), and the presence of non‐canonical NF‐κB pathway mutations conferred significantly shorter PFS with placebo‐Rd compared to the wild‐type subgroup (HR 2.5; Figure 2B). There were no significant differences in PFS with IRd by mutation status (HR 0.59; 95% CI, 0.25, 1.4; mutation vs wild‐type, P = .22) or between the IRd and placebo‐Rd wild‐type subgroups (HR 0.83; 95% CI, 0.53, 1.3; IRd wild‐type vs placebo‐Rd wild‐type, P = .39).
3.3. Analysis of PFS by mRNA expression
Kaplan‐Meier analyses of PFS in the RNAseq population according to expression of TRAF3, a negative regulator of the non‐canonical NF‐κB pathway, are shown in Figure 3. In a pooled analysis across treatment groups, with patients dichotomized around the global median TRAF3 expression level, PFS was significantly longer among patients with higher vs lower TRAF3 expression (HR 0.65, median 21 vs 14 months; Figure 3A).
Figure 3.
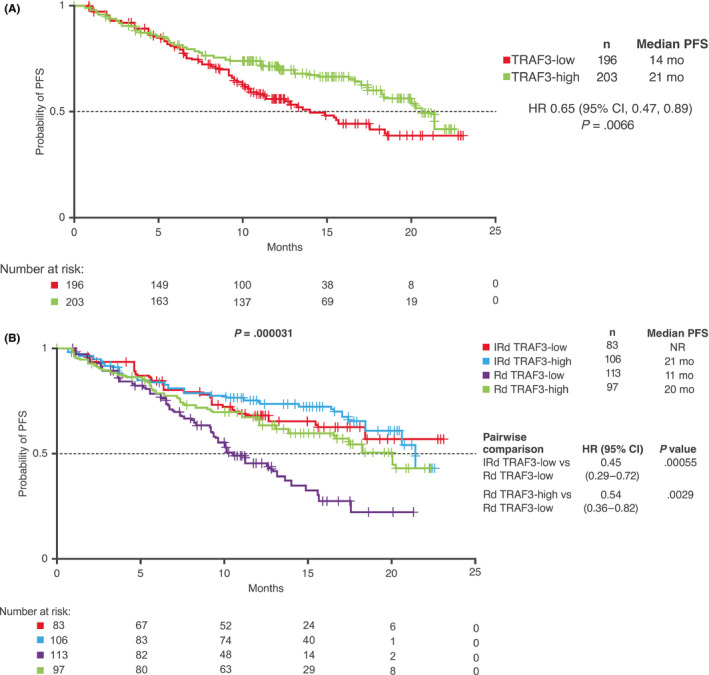
PFS according to TRAF3 expression. Kaplan‐Meier analysis of PFS in the RNAseq‐evaluable population (A) based on TRAF3 expression, dichotomized as TRAF3‐high and TRAF3‐low based on the global median and (B) with IRd and placebo‐Rd according to TRAF3 expression. CI, confidence interval; HR, hazard ratio; IRd, ixazomib‐lenalidomide‐dexamethasone; NR, not reached; PFS, progression‐free survival; Rd, placebo‐lenalidomide‐dexamethasone; RNAseq, RNA sequencing
Further analysis of PFS according to TRAF3 expression and treatment received demonstrated significant differences between groups (P = .000031; Figure 3B). Median PFS with IRd was not reached and 21 months in patients with lower and higher levels of TRAF3 expression, respectively, and with placebo‐Rd the respective medians were 11 and 20 months. PFS was significantly longer with IRd vs placebo‐Rd in patients with lower TRAF3 expression levels (HR 0.45), and PFS was significantly longer with placebo‐Rd among patients with higher vs lower TRAF3 expression (HR 0.54; Figure 3B). There were no significant differences in PFS with IRd by TRAF3 expression (HR 0.87; 95% CI, 0.52, 1.5; low vs high TRAF3 expression, P = .61) or between the IRd and placebo‐Rd subgroups with higher TRAF3 expression levels (HR 0.71; 95% CI, 0.44, 1.1; IRd vs placebo‐Rd, P = .15).
Kaplan‐Meier analyses of PFS in the RNAseq population according to expression of NIK, a positive regulator of the non‐canonical NF‐κB pathway, dichotomized around the global median, are shown in Figure 4A. NIK expression and treatment received demonstrated an overall significant impact on PFS (P = .00081). Median PFS with IRd was not reached and 18 months in patients with higher and lower levels of NIK expression, respectively (HR 0.56), and with placebo‐Rd the median PFS was 16 and 13 months in these subgroups, respectively. PFS was significantly longer with IRd vs placebo‐Rd in patients with higher NIK expression levels (HR 0.41; Figure 4A). There were no significant differences in PFS with placebo‐Rd according to NIK expression (HR 1.0; 95% CI, 0.70, 1.6; NIK‐high vs NIK‐low, P = .82), or between IRd and placebo‐Rd in patients with lower levels of NIK expression (HR 0.76; 95% CI, 0.50, 1.2; IRd vs placebo‐Rd, P = .22).
Figure 4.

PFS according to NIK expression. Kaplan‐Meier analysis of PFS with IRd and placebo‐Rd in the RNAseq‐evaluable population based on (A) NIK expression, dichotomized as NIK‐high and NIK‐low based on the global median, and (B) NIK expression by quartile, showing only the lowest (q1) and highest (q4) NIK expression quartiles in each treatment group. CI, confidence interval; HR, hazard ratio; IRd, ixazomib‐lenalidomide‐dexamethasone; NR, not reached; PFS, progression‐free survival; q, quartile; Rd, placebo‐lenalidomide‐dexamethasone; RNAseq, RNA sequencing
Further analysis of PFS among patients in the highest and lowest quartiles of NIK expression, according to treatment received, also demonstrated an overall significant impact on PFS (P = .00481; Figure 4B). Median PFS with IRd was not reached and 18 months in patients in the highest and lowest NIK expression quartiles, respectively, and with placebo‐Rd the respective medians were 16 and 13 months. PFS was significantly longer with IRd vs placebo‐Rd in patients in the highest NIK expression quartile (HR 0.36), as well as among patients in the highest vs lowest NIK expression quartiles in the IRd group (HR 0.42; Figure 4B). There were no significant differences in PFS between IRd and placebo‐Rd in patients in the lowest NIK expression quartile (HR 0.85; 95% CI, 0.47, 1.5; IRd vs placebo‐Rd; P = .577) or between the highest and lowest NIK expression quartiles in the placebo‐Rd group (HR 1.0; 95% CI, 0.58, 1.8; P = .90).
3.4. Analysis of PFS by NF‐κB target gene expression
Values for the NF‐κB10 index, based on RNA expression levels of 10 NF‐κB target genes, 2 , 5 , 20 were determined for all patients in the RNAseq‐evaluable population, with higher index values indicating greater non‐canonical NF‐κB pathway activation. Kaplan‐Meier analyses of PFS according to NF‐κB10 index values are shown in Figure 5.
Figure 5.
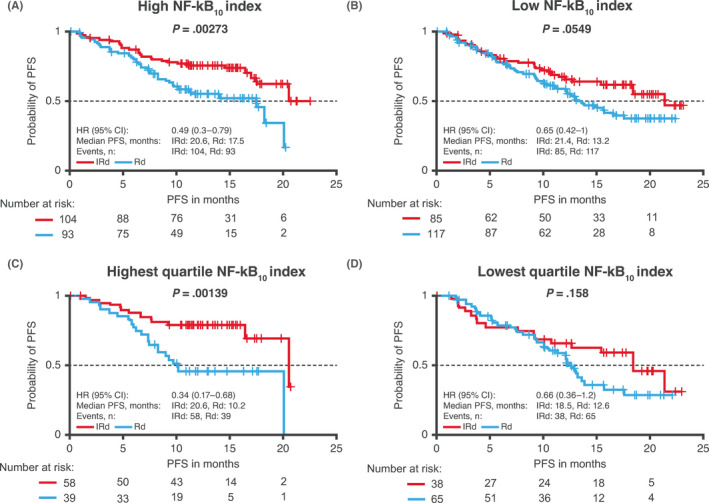
PFS according to NF‐κB10 index. Kaplan‐Meier analysis of PFS with IRd and placebo‐Rd in the RNAseq‐evaluable population based on NF‐κB10 index, 5 dichotomized as (A) high and (B) low NF‐κB10 index based on the global median and analyzed in patients in the (C) highest and (D) lowest quartiles of NF‐κB10 index values. CI, confidence interval; HR, hazard ratio; IRd, ixazomib‐lenalidomide‐dexamethasone; NF‐κB, nuclear factor kappa B; PFS, progression‐free survival; Rd, placebo‐lenalidomide‐dexamethasone; RNAseq, RNA sequencing
With patients dichotomized around the global median NF‐κB10 index value, the PFS benefit with IRd vs placebo‐Rd was greater among patients with higher NF‐κB10 index values (HR 0.49; median 20.6 vs 17.5 months) than among those with lower NF‐κB10 index values (HR 0.65; median 21.4 vs 13.2 months), with the difference not reaching statistical significance in the latter group. Similarly, the PFS benefit with IRd vs placebo‐Rd was greater among patients in the highest quartile of NF‐κB10 index values (HR 0.34; median 20.6 vs 10.2 months) than among those in the lowest quartile (HR 0.66; median 18.5 vs 12.6 months).
3.5. mRNA expression and PFS in patients with high‐risk cytogenetic abnormalities
In the TOURMALINE‐MM1 study, IRd demonstrated a significant PFS benefit vs placebo‐Rd in an expanded high‐risk group of patients defined as those with del(17p), t(4;14), t(14;16), and/or amp 1q21. 14 The association between level of TRAF3 expression and the presence of the high‐risk cytogenetic abnormalities del(17p), t(4;14), and amp 1q21 is shown in Figure 6. (Due to the small number of patients—1%—with t(14,16) in the IRd arm of TOURMALINE‐MM1, 14 this translocation was not included in the present analysis). Compared with patients with standard‐risk cytogenetics, expression of TRAF3 was significantly higher in patients with del(17p) (P = .0013) and significantly lower in patients with amp 1q21 (P = .0000012). Expression of TRAF3 was also lower in patients with t(4;14) compared to patients with standard‐risk cytogenetics, but the difference was not statistically significant (P = .099).
Figure 6.
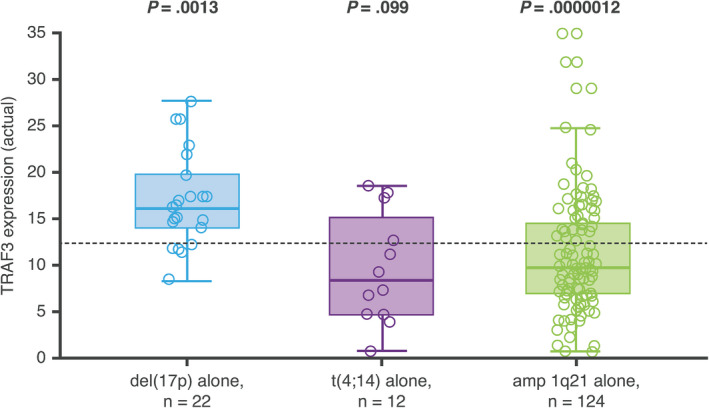
TRAF3 expression in patients with del(17p), t(4;14), or amp 1q21 alone, compared to median TRAF3 expression in patients with standard‐risk cytogenetics (dashed line)
4. DISCUSSION
The findings of these analyses of PFS among patients with RRMM enrolled in the phase 3 TOURMALINE‐MM1 study show that the addition of ixazomib to Rd provides clinical benefit in MM patients with tumors with increased non‐canonical NF‐κB pathway activity. Specifically, increased IRd activity was seen in patients with low TRAF3 expression and with high NIK expression, both of which have been previously reported to be associated with activation of the non‐canonical NF‐κB pathway 5 , 6 , 20 ; of note, our findings showed an overall association between low TRAF3 expression and poor prognosis. Increased IRd activity was also seen in patients with loss‐of‐function mutations in genes that negatively regulate the non‐canonical NF‐κB pathway. Furthermore, with regards to the findings showing increased IRd activity in patients with higher NF‐κB10 index values indicating greater non‐canonical pathway activity, it should be noted that the genes in this index may also be upregulated by the canonical NF‐κB pathway. Given the regulation of the non‐canonical pathway by protein degradation, being able to correlate gene and protein expression would have contributed greatly to the current analysis. However, we were not able to evaluate protein expression in this study due to the lack of appropriate samples. Inhibition of non‐canonical NF‐κB signaling has long been acknowledged as a key component of the mechanism of action of proteasome inhibition in MM. 3 , 4 The first‐in‐class agent bortezomib has been shown to inhibit inducible NF‐κB activity but paradoxically also to enhance constitutive NF‐κB activation via the canonical pathway. In fact, other proteasome inhibitors have also shown similar activation of the canonical NF‐κB pathway though the exact mechanism underlying this is unclear. 21 , 22
There is evidence of significant cross‐talk between the canonical and non‐canonical pathways, especially through NIK activation, 20 though our data suggest that ixazomib might be able to blunt this NIK‐mediated enhancement of the canonical pathway. It has been proposed that dual inhibition of the canonical and non‐canonical pathways is required for full inhibition of NF‐κB activity, and this has been suggested as a treatment strategy. 23 , 24 Indeed, studies combining bortezomib and IKKβ‐targeted inhibitors have shown synergistic increases in cytotoxicity in breast cancer cells. 25 Similarly, combining a specific NIK inhibitor 20 , 26 , 27 with canonical pathway inhibitors has also been suggested as a potential treatment strategy. 28
Our findings are consistent with previous reports of enhanced activity of bortezomib in MM patients with low TRAF3 expression, 6 that is, with constitutive activation of the non‐canonical NF‐κB pathway. However, to our knowledge this is the first analysis that demonstrates the specific differential activity imparted by addition of a proteasome inhibitor to treatment in the setting of non‐canonical NF‐κB pathway activation in a large, randomized, placebo‐controlled, phase 3 study in RRMM patients.
This is also the first analysis to demonstrate specific activation of the non‐canonical NF‐κB pathway in the context of two different high‐risk cytogenetic abnormalities. Our findings from the TOURMALINE‐MM1 study demonstrated that tumors bearing t(4;14) had decreased TRAF3 expression, which was an expected observation based on previously reported genome‐wide screening analyses in 91 MM patients with cytogenetic abnormalities. 29 In these studies, TRAF3 was identified as one of the genes affected by homozygous deletions in the 14q32.32 chromosome location, which were seen in 7.7% of patients. 29 However, our data also provide the novel observation that MM tumors bearing the amp 1q21 abnormality are associated with decreased TRAF3 expression, potentially resulting in increased non‐canonical NF‐κB pathway activity. The mechanism underlying the decreased TRAF3 expression in amp 1q21 patient tumors needs to be further explored.
Previous subgroup analyses of the TOURMALINE‐MM1 study have demonstrated that IRd has substantial differential activity vs placebo‐Rd in patients with high‐risk cytogenetic abnormalities, including those with del(17p), t(4;14), and t(14;16), resulting in a median PFS of 21.4 vs 9.7 months (HR 0.543, P = .021). 14 A similar benefit was demonstrated in an expanded high‐risk subgroup that incorporated patients with amp 1q21. 14 More broadly, prior studies have suggested that proteasome inhibitors have specific activity in patients with high‐risk cytogenetics, 30 , 31 including t(4;14), 32 , 33 and indeed the International Myeloma Working Group consensus on the treatment of patients with high‐risk cytogenetics recommends the use of a triplet regimen comprising a proteasome inhibitor, lenalidomide or pomalidomide, and dexamethasone. 31 Our data suggest that the ability of ixazomib to inhibit the non‐canonical NF‐κB pathway is one of the mechanisms contributing to the activity of IRd in patients bearing t(4;14) and amp1q21. In particular, decreased TRAF3 expression in the context of amp1q21 might define a subset of tumors that are exquisitely dependent on the non‐canonical NF‐κB pathway for survival and are thus responsive to IRd. By contrast, placebo‐Rd appeared to be unable to overcome the adverse prognostic impact of amp 1q21 in the context of low TRAF3 expression levels, possibly associated with its lack of inhibitory activity in the context of constitutive non‐canonical NF‐κB pathway signaling.
Our analysis has a number of limitations. DNAseq and RNAseq data were obtained from only 47.0% and 55.2% of patients, respectively, in the TOURMALINE‐MM1 ITT population, suggesting a potential for patient selection bias. However, baseline patient and disease characteristics—including age, disease stage, performance status, and relative proportions of patients with high‐risk versus standard‐risk cytogenetics—were comparable between patients with or without DNAseq data and RNAseq data, as shown in the supplementary tables. A limitation of the targeted sequencing approach that we employed for these analyses is that we were unable to evaluate copy number changes and chromosomal deletions, alterations frequently observed in TRAF3 and NIK genes; additionally, as previously noted, we were not able to evaluate protein expression due to a lack of appropriate samples. Other future directions for research in this area include conducting similar analyses in patients receiving ixazomib in combination with agents other than lenalidomide, as well as determining whether similar effects are seen in patients with newly diagnosed MM.
In conclusion, our analysis of DNAseq data from RRMM patients enrolled in the TOURMALINE‐MM1 trial demonstrated that 14.5% harbored mutations in the non‐canonical NF‐κB pathway. The presence of these mutations was associated with specific significant PFS benefit with IRd vs placebo‐Rd. Similarly, analyses of RNAseq data showed specific PFS benefit with IRd in patients with low TRAF3 expression or high NIK expression, both markers of elevated non‐canonical NF‐κB pathway activity. Decreased TRAF3 expression was seen in tumors with t(4;14) and amp 1q21 abnormalities, providing a potential rationale for the activity of IRd in these high‐risk patients. Together with the findings reported in the article from TOURMALINE‐MM1 published recently by Di Bacco et al, 16 our results from these exploratory endpoints of TOURMALINE‐MM1 provide hypothesis‐generating data that offer insights into potential markers associated with more substantial benefit from the addition of ixazomib to Rd in patients with RRMM.
CONFLICT OF INTEREST
ABD: Employment, Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. JZ: Employment, Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. LS: Employment, Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. BL: Employment, Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. DB: Employment, Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. JL: Employment, Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. HA‐L: Research funding: Amgen, Celgene, Sanofi, Takeda; Membership of advisory committees: Abbvie, Amgen, Celgene, Janssen, Sanofi, Takeda. NJB: No conflicts to disclose. PM: Advisory board member and honoraria: Takeda, Celgene. PGR: Advisory Committee Member: Karyopharm, Oncopeptides, Celgene, Takeda, Janssen, Sanofi. Research support: Oncopeptides, Celgene, Takeda, BMS. ADB: Employment, Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
AUTHOR CONTRIBUTIONS
ABD, LS, DB, HA‐L, NJB, PM, PGR, and ADB designed the research. ABD, JZ, LS, DB, HA‐L, NJB, PM, PGR, and ADB conducted the research. HA‐L, PM, and PGR contributed patients/materials. ABD, JZ, LS, BL, JL, HA‐L, NJB, PM, PGR, and ADB analyzed and interpreted the data. ABD, JZ, LS, BL, and JL processed the data. ABD and LS wrote the manuscript. All authors reviewed and edited the manuscript. All authors approved the final draft for submission.
Supporting information
Table S1‐S2
ACKNOWLEDGMENTS
We thank all patients and their families, and investigators at all clinical sites for their participation in the study. We would also like to acknowledge Steve Hill, PhD, of FireKite, an Ashfield company, part of UDG Healthcare plc, for writing support during the development of this manuscript, which was funded by Millennium Pharmaceuticals, Inc, and complied with Good Publication Practice 3 ethical guidelines (Battisti et al, Ann Intern Med 2015;163:461–4), and Renda Ferrari, PhD, of Millennium Pharmaceuticals, Inc, Cambridge, MA, USA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, for editorial support. This study was sponsored by Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Dash AB, Zhang J, Shen L, et al. Clinical benefit of ixazomib plus lenalidomide‐dexamethasone in myeloma patients with non‐canonical NF‐κB pathway activation. Eur J Haematol. 2020;105:274–285. 10.1111/ejh.13435
This trial was registered at www.clinicaltrials.gov as: NCT01564537.
DATA AVAILABILITY STATEMENT
Takeda makes patient‐level, de‐identified data sets and associated documents available after applicable marketing approvals and commercial availability have been received, an opportunity for the primary publication of the research has been allowed, and other criteria have been met as set forth in Takeda's Data Sharing Policy (see https://www.takedaclinicaltrials.com for details). To obtain access, researchers must submit a legitimate academic research proposal for adjudication by an independent review panel, who will review the scientific merit of the research and the requestor's qualifications and conflict of interest that can result in potential bias. Once approved, qualified researchers who sign a data sharing agreement are provided access to these data in a secure research environment.
REFERENCES
- 1. Roy P, Sarkar UA, Basak S. The NF‐kappaB activating pathways in multiple myeloma. Biomedicines. 2018;6(2).59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Demchenko YN, Kuehl WM. A critical role for the NFkB pathway in multiple myeloma. Oncotarget. 2010;1(1):59‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4(5):349‐360. [DOI] [PubMed] [Google Scholar]
- 4. Chauhan D, Tian Z, Zhou B, et al. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res. 2011;17(16):5311‐5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Annunziata CM, Davis RE, Demchenko Y, et al. Frequent engagement of the classical and alternative NF‐kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12(2):115‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Keats JJ, Fonseca R, Chesi M, et al. Promiscuous mutations activate the noncanonical NF‐kappaB pathway in multiple myeloma. Cancer Cell. 2007;12(2):131‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roy P, Mukherjee T, Chatterjee B, Vijayaragavan B, Banoth B, Basak S. Non‐canonical NFkappaB mutations reinforce pro‐survival TNF response in multiple myeloma through an autoregulatory RelB:p50 NFkappaB pathway. Oncogene. 2017;36(10):1417‐1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shah V, Sherborne AL, Walker BA, et al. Prediction of outcome in newly diagnosed myeloma: a meta‐analysis of the molecular profiles of 1905 trial patients. Leukemia. 2018;32(1):102‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gupta N, Hanley MJ, Xia C, Labotka R, Harvey RD, Venkatakrishnan K. Clinical pharmacology of ixazomib: the first oral proteasome inhibitor. Clin Pharmacokinet. 2019;58(4):431‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Richardson PG, Zweegman S, O'Donnell EK, et al. Ixazomib for the treatment of multiple myeloma. Expert Opin Pharmacother. 2018;19(17):1949‐1968. [DOI] [PubMed] [Google Scholar]
- 11. Millennium Pharmaceuticals Inc . NINLARO® (ixazomib) capsules, for oral use. 2016. (November 2016). https://www.ninlaro.com/prescribing‐information.pdf. Accessed 05/04/2019.
- 12. Takeda Pharma A/S . NINLARO® (ixazomib) capsules, summary of product characteristics. 2016. (November 2016). https://www.ema.europa.eu/en/documents/product‐information/ninlaro‐epar‐product‐information_en.pdf. Accessed 05/04/2019.
- 13. Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;374(17):1621‐1634. [DOI] [PubMed] [Google Scholar]
- 14. Avet‐Loiseau H, Bahlis NJ, Chng WJ, et al. Ixazomib significantly prolongs progression‐free survival in high‐risk relapsed/refractory myeloma patients. Blood. 2017;130(24):2610‐2618. [DOI] [PubMed] [Google Scholar]
- 15. Mateos MV, Masszi T, Grzasko N, et al. Impact of prior therapy on the efficacy and safety of oral ixazomib‐lenalidomide‐dexamethasone vs. placebo‐lenalidomide‐dexamethasone in patients with relapsed/refractory multiple myeloma in TOURMALINE‐MM1. Haematologica. 2017;102(10):1767‐1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Di Bacco A, Bahlis NJ, Munshi NC, et al. c‐MYC expression and maturity phenotypes are associated with outcome benefit from addition of ixazomib to lenalidomide‐dexamethasone in myeloma. Eur J Haematol. 2020. 10.1111/ejh.13405. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu J, Ge H, Newman M, Liu K. OSA: a fast and accurate alignment tool for RNA‐Seq. Bioinformatics. 2012;28(14):1933‐1934. [DOI] [PubMed] [Google Scholar]
- 18. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinform. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31(3):213‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Demchenko YN, Glebov OK, Zingone A, Keats JJ, Bergsagel PL, Kuehl WM. Classical and/or alternative NF‐kappaB pathway activation in multiple myeloma. Blood. 2010;115(17):3541‐3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hideshima T, Ikeda H, Chauhan D, et al. Bortezomib induces canonical nuclear factor‐kappaB activation in multiple myeloma cells. Blood. 2009;114(5):1046‐1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fristedt Duvefelt C, Lub S, Agarwal P, et al. Increased resistance to proteasome inhibitors in multiple myeloma mediated by cIAP2–implications for a combinatorial treatment. Oncotarget. 2015;6(24):20621‐20635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fabre C, Mimura N, Bobb K, et al. Dual inhibition of canonical and noncanonical NF‐kappaB pathways demonstrates significant antitumor activities in multiple myeloma. Clin Cancer Res. 2012;18(17):4669‐4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Muraoka H, Yoshimura C, Kawabata R, et al. Activity of TAS4464, a novel NEDD8 activating enzyme E1 inhibitor, against multiple myeloma via inactivation of nuclear factor kappaB pathways. Cancer Sci. 2019;110(12):3802‐3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hideshima H, Yoshida Y, Ikeda H, et al. IKKbeta inhibitor in combination with bortezomib induces cytotoxicity in breast cancer cells. Int J Oncol. 2014;44(4):1171‐1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sun SC. The noncanonical NF‐kappaB pathway. Immunol Rev. 2012;246(1):125‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ramakrishnan P, Wang W, Wallach D. Receptor‐specific signaling for both the alternative and the canonical NF‐kappaB activation pathways by NF‐kappaB‐inducing kinase. Immunity. 2004;21(4):477‐489. [DOI] [PubMed] [Google Scholar]
- 28. Demchenko YN, Brents LA, Li Z, Bergsagel LP, McGee LR, Kuehl MW. Novel inhibitors are cytotoxic for myeloma cells with NFkB inducing kinase‐dependent activation of NFkB. Oncotarget. 2014;5(12):4554‐4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smetana J, Frohlich J, Zaoralova R, et al. Genome‐wide screening of cytogenetic abnormalities in multiple myeloma patients using array‐CGH technique: a Czech multicenter experience. Biomed Res Int. 2014;2014:209670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sonneveld P, Goldschmidt H, Rosinol L, et al. Bortezomib‐based versus nonbortezomib‐based induction treatment before autologous stem‐cell transplantation in patients with previously untreated multiple myeloma: a meta‐analysis of phase III randomized, controlled trials. J Clin Oncol. 2013;31(26):3279‐3287. [DOI] [PubMed] [Google Scholar]
- 31. Sonneveld P, Avet‐Loiseau H, Lonial S, et al. Treatment of multiple myeloma with high‐risk cytogenetics: a consensus of the International Myeloma Working Group. Blood. 2016;127(24):2955‐2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. El‐Ghammaz AM, Abdelwahed E. Bortezomib‐based induction improves progression‐free survival of myeloma patients harboring 17p deletion and/or t(4;14) and overcomes their adverse prognosis. Ann Hematol. 2016;95(8):1315‐1321. [DOI] [PubMed] [Google Scholar]
- 33. Avet‐Loiseau H, Leleu X, Roussel M, et al. Bortezomib plus dexamethasone induction improves outcome of patients with t(4;14) myeloma but not outcome of patients with del(17p). J Clin Oncol. 2010;28(30):4630‐4634. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S2
Data Availability Statement
Takeda makes patient‐level, de‐identified data sets and associated documents available after applicable marketing approvals and commercial availability have been received, an opportunity for the primary publication of the research has been allowed, and other criteria have been met as set forth in Takeda's Data Sharing Policy (see https://www.takedaclinicaltrials.com for details). To obtain access, researchers must submit a legitimate academic research proposal for adjudication by an independent review panel, who will review the scientific merit of the research and the requestor's qualifications and conflict of interest that can result in potential bias. Once approved, qualified researchers who sign a data sharing agreement are provided access to these data in a secure research environment.