

 Imidazole-based epidermal growth factor receptor (EGFR) inhibitors were computationally designed and synthesized.
Imidazole-based epidermal growth factor receptor (EGFR) inhibitors were computationally designed and synthesized.
Abstract
Imidazole-based epidermal growth factor receptor (EGFR) inhibitors were computationally designed and synthesized. All the compounds were assessed for their anti-proliferative activity against five cancer cell lines, viz., MDA-MB-231 (breast), T47D (breast) and MCF-7 (breast), A549 (lung) and HT-29 (colorectal). Compounds 2c and 2d emerged as better anticancer molecules with no toxicity towards normal cells. 2c and 2d inhibited EGFR enzymatic activity in vitro with IC50 values of 617.33 ± 0.04 nM and 710 ± 0.05 nM, respectively. In order to further improve the potency, we explored an unoccupied area of the ATP binding domain of EGFR and analysed an in silico interaction model of 2c and 2d–EGFR complexes that guided and allowed substitution of the 4-fluorophenyl ring (2c and 2d) with 4-(4-methylpiperazinyl)-3-nitrophenyl at the N-9 position, resulting in compound 3c with a better binding score and potent EGFR inhibitory activity (IC50: 236.38 ± 0.04 nM), which was comparable to the positive control erlotinib (239.91 ± 0.05 nM). 3c exhibited a great improvement in anticancer potency with inhibition of cell growth of all cancer cell lines at very low micromolar concentrations (IC50 = 1.98 to 4.07 μM). Further investigation revealed that 3c also induced an increase in ROS levels in cancer cells in a mitochondrial-independent manner and halted the cell cycle at the sub-G1 phase.
Introduction
Cancer accounts for a number of deaths worldwide and is the second leading cause of death after cardiovascular disorders.1 Cancer cells possess inherent hallmarks that include tissue invasion, metastasis, evasion of apoptosis, angiogenesis, inflammation and immortality2 that make them insensitive to conventional anticancer agents. Considering this scenario, the development of anticancer drugs targeting the major hallmarks was put forth.3 Amongst the various anticancer drug targets known, protein kinases are the most studied as druggable targets.4 The epidermal growth factor receptor (EGFR) belongs to the ErbB family of receptor tyrosine kinases5 and is one of the effective drug targets owing to its overexpression and elevation in multiple cancer subtypes including non-small cell lung cancer (NSCLC), breast cancer, and ovarian cancer.6 The EGFR is considered to be the driver of cell proliferation, migration, adhesion and cell survival which consequently lead to tumorigenesis via auto-phosphorylation by activating an intracellular signalling cascade.
Considering the centralized role of kinases, there had been a revolution in the development of EGFR inhibitors7 (Fig. 1). First-generation EGFR inhibitors such as erlotinib and gefitinib (quinazoline scaffold) were approved by the USFDA in 2002 and 2004, respectively, for the treatment of NSCLC.8 Eventually, the first-generation EGFR inhibitors were ineffective due to acquired point mutations, T790M and L858R, that hinder their binding at the ATP kinase domain of EGFR. This further gave way to the development of second-generation EGFR inhibitors (canertinib, dacomitinib and afatinib) which were able to interact with CYS797 via covalent binding and hinder the ATP binding at the active site.9 The clinical trials were promising initially but toxicity (skin and gastrointestinal) halted their further development.10 Thereafter, pyrimidine-based irreversible, third-generation EGFR inhibitors (WZ4002 and osimertinib) were developed; they possessed a much better safety profile than the second generation due to their lower affinity and binding for wild-type EGFR.11 Irreversible inhibition of tyrosine kinase activity results in continuous suppression of EGFR signalling as compared to reversible inhibition.12 The phase trials suggested that the prolonged use of third-generation inhibitors was associated with off-target side effects followed by C797S, BRAF and MEK mutations.13 Very recently, fourth-generation EGFR inhibitors (e.g. EAI045) have been disclosed to overcome the concomitant EGFR T790M and C797S mutations, but their safety profile in the clinic is yet to be established.14
Fig. 1. Chemical structures of some EGFR inhibitors.

A trisubstituted imidazole (11g) based on a p38α MAPK inhibitor template, recently reported by the Juchum group against mutated EGFR L858R/T790M/C797S, had a reasonable orientation in the active site of mutant EGFR kinase domain.15
We exercised target hopping of 11g along with molecular docking to design new compounds (Fig. 2), 1 and 2, based on the considerations that (a) the p-fluorophenyl residue at the N-1 position of imidazole (1) or the N-9 position of fused imidazole (2) is favoured and compatible not only with the gatekeeper residue (Thr766) but also with the mutated one (MET) in the binding pocket, thus making it suitable for clinically challenging EGFR in addition to its metabolic stability; (b) hydrogen bond interactions of designed compounds with MET769 are conserved; (c) another phenyl ring of 1 (substituted at N-5; amino group) or 2 (substituted at the C-2 position) orients towards the DFG motif (ASP831, PHE832 and GLY833) which also interacts with the p-hydroxyphenyl residue of 11g; (d) –CN (1) or –CONH2 (2) interacts with PRO770; and (e) they are easy to synthesize. Further, the designed compounds do not contain pyridinyl imidazoles in their framework which can potentially inhibit cytochrome p450 and lead to toxic side effects.16 In the present work, the rational design of 1 and 2 followed by their synthesis along with detailed in vitro biological evaluation including cancer cell based antiproliferative effects, enzymatic inhibition, alteration of ROS levels and cell cycle analysis are reported. Molecular docking at the EGFR ATP kinase domain further helped in achieving high EGFR potency in the nM range.
Fig. 2. Design of target compounds 1 and 2 based on target hopping of 11g. 1c, 2c1 (as a representative compound) and 11g were docked at the ATP binding domain of EGFR (PDB ID: ; 1M17).

Results and discussion
Synthesis procedures
For the synthesis of target compounds 1 (1a–1r) and 2 (2a–2d), a synthesis plan was designed (Scheme 1) that utilized the starting materials 6, 8 and 9 which in turn can be synthesized from a common precursor, ethyl N-((Z)-2-amino-1,2-dicyanovinyl)formimidate (4). The condensation of commercially available diaminomaleonitrile (3) (1.1 equiv.) with triethyl orthoformate (1.1 equiv.) yielded a formimidate ester, 4.174, when treated with p-fluororaniline and p-fluorobenzylamine, underwent a nucleophilic substitution reaction under the catalytic influence of aniline hydrochloride, yielding 5 and 7, respectively, which under strong basic conditions afforded ring cyclised products, 6 and 8, respectively (Scheme 1), while 5 when kept under mild basic conditions underwent ring cyclisation to form 9.
Scheme 1. Reagents and conditions: (i) CH(OEt)3, 1,4-dioxane, 80 °C, 6 h. (ii) Aniline hydrochloride, EtOH, rt, 3 h. (iii) KOH (1 M), rt, 6 h. (iv) p-TsOH, MeOH, rt, 2 h. (v) DBU, EtOH, rt, 2 h. (vi) H2SO4, MeOH, rt, 1 h.

Synthesis of target compounds 1a–1r
The synthesis of target compounds 1a–1j (n = 0) and 1k–1r (n = 1) was successfully achieved by reacting the corresponding aryl aldehydes (1 equiv.) with 6 and 8, respectively, in the presence of a catalytic amount of p-TsOH (10 mol%).18
Synthesis of target compounds 2a–2d
9 (1 equiv.) underwent cyclocondensation with aryl aldehydes (1 equiv.) under acidic conditions (1 drop of H2SO4) to afford the target compounds 2a–2d in good yields (>60%). Two types of products were observed while using the same conditions. In the case of the reaction with 3,4-dimethoxy- and 3,4,5-trimethoxybenzaldehydes, 2a and 2c, respectively, were obtained as partially oxidized products, whereas in the case of 4-methoxy- and 4-isopropyl benzaldehydes fully aromatized products, 2b and 2d, respectively, were formed. All the final compounds were fully characterized by mp, IR, NMR and HRMS techniques.
Biological evaluation
Cytotoxic potential of target compounds
To evaluate the cytotoxic potential of the target compounds (1a–1r and 2a–2d), the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)-based antiproliferative assay was performed against five cancer cell lines, viz., MDA-MB-231 (breast), T47D (breast) and MCF-7 (breast),19 A549 (lung) and HT-29 (colorectal). The respective cancer cells were treated with the target compounds at varying concentrations of 1, 5 and 25 μM and incubated for 48 h. Erlotinib was taken as a positive control. The results suggested that nine compounds 1i, 1k, 1m, 1o, 1p, and 2a–2d exhibited good antiproliferative activity in at least two cancer cell lines employed (Table 1).
Table 1. The antiproliferative potential of the target compounds (1 and 2) against various cancer cell lines.
| Inhibitory potential (IC50 (μM) ± SD)
a
of target compounds of growth of cancer cells | |||||
| Code | MDA-MB-231 | T47D | MCF-7 | A549 | HT-29 |
| 1a | 18.74 ± 0.23 | 13.74 ± 0.17 | 8.32 ± 0.32 | 9.88 ± 0.52 | 13.74 ± 0.17 |
| 1b | 12.34 ± 0.55 | 16.32 ± 0.44 | 7.09 ± 0.26 | 9.28 ± 0.31 | 16.32 ± 0.31 |
| 1c | 13.36 ± 0.14 | 15.86 ± 0.62 | 9.1 ± 0.44 | 18.12 ± 0.16 | 15.86 ± 0.16 |
| 1d | 13.6 ± 0.38 | 20.10 ± 0.21 | 9.99 ± 0.52 | 13.6 ± 0.23 | 20.10 ± 0.55 |
| 1e | 16.28 ± 0.52 | 20.39 ± 0.26 | 11.45 ± 0.29 | 16.28 ± 0.55 | 20.39 ± 0.18 |
| 1f | 17.09 ± 0.43 | 19.43 ± 0.47 | 9.33 ± 0.31 | 17.09 ± 0.22 | 19.43 ± 0.49 |
| 1g | 17.17 ± 0.2 | 15.26 ± 0.54 | 9.99 ± 0.16 | 17.17 ± 0.41 | 15.26 ± 0.32 |
| 1h | 21.45 ± 0.16 | 19.95 ± 0.63 | 7.85 ± 0.28 | >25 | 19.95 ± 0.23 |
| 1i | 4.27 ± 0.32 | 7.23 ± 0.22 | 14.65 ± 0.42 | 2.96 ± 0.17 | 7.23 ± 0.18 |
| 1j | 20.45 ± 0.25 | >25 | 9.87 ± 0.19 | 14.66 ± 0.32 | >25 |
| 1k | 2.87 ± 0.28 | 17.5 ± 0.76 | 9.04 ± 0.26 | 9.71 ± 0.23 | 16.5 ± 0.37 |
| 1l | 17.58 ± 0.68 | 22.42 ± 0.22 | 10.89 ± 0.24 | 13.13 ± 0.17 | 22.42 ± 0.41 |
| 1m | 3.44 ± 0.31 | 5.72 ± 0.62 | 10.64 ± 0.28 | 2.02 ± 0.26 | 5.72 ± 0.16 |
| 1n | 16.05 ± 0.34 | 21.48 ± 0.56 | 11.35 ± 0.36 | 10.87 ± 0.42 | 21.48 ± 0.27 |
| 1o | 2.87 ± 0.43 | 9.19 ± 0.78 | 3.87 ± 0.35 | 4.62 ± 0.30 | 9.19 ± 0.19 |
| 1p | 2.67 ± 0.14 | 6.14 ± 0.24 | 4.53 ± 0.27 | 1.32 ± 0.17 | 6.14 ± 0.42 |
| 1q | 10.54 ± 0.74 | 8.2 ± 0.21 | 11.09 ± 0.22 | 9.13 ± 0.14 | 13.78 ± 0.37 |
| 1r | >25 | >25 | 12.57 ± 0.18 | 24.34 ± 0.47 | >25 |
| 2a | 8.67 ± 0.15 | 10.68 ± 0.72 | 7.46 ± 0.49 | 2.29 ± 0.22 | 2.37 ± 0.18 |
| 2b | 1.22 ± 0.29 | 5.87 ± 0.36 | 3.99 ± 0.32 | 10.94 ± 0.27 | 11.03 ± 0.39 |
| 2c | 6.76 ± 0.32 | 9.96 ± 0.31 | 5.71 ± 0.21 | 2.29 ± 0.23 | 3.29 ± 0.19 |
| 2d | 2.29 ± 0.35 | 5.35 ± 0.22 | 3.46 ± 0.16 | 10.6 ± 0.41 | 11.02 ± 0.38 |
| Erlotinib | 5.46 ± 0.19 | 9.80 (ref. 24) | >30.00 (ref. 25) | 1.04 ± 0.15 | 4.63 ± 0.32 |
aData are expressed as mean values ± SD of three independent experiments.
The lead compounds 1i, 1k, 1m, 1o, 1p, and 2a–2d were further investigated for their toxicity toward human peripheral blood mononuclear cells (HPBMCs) using the MTT assay at 10 μM concentration. The results showed that compounds 1m and 1i possessed extreme toxicity, whereas compounds 2a and 1k were found to possess mild toxicity (Fig. 3A). However, compounds 1o, 1p, 2b, 2c and 2d did not induce any toxicity in HPBMCs and were further assessed for their cytotoxicity (if any) on normal breast cells (HBL-100; as control in the experiment). The results suggested that 2c and 2d were non-cytotoxic to HBL-100 (Fig. 3B and C) at 10 μM concentration.
Fig. 3. (A) MTT assay on HPBMCs using selected compounds 1i, 1k, 1m, 1o, 1p and 2a–2d; compounds 1i, 1k, 1m, and 2a showed toxicity towards the HPBMCs. The graph depicts the percentage survival of the cells after treatment. (B) MTT assay on normal breast cells (HBL-100) using compounds 1o, 1p, 2b, 2c, and 2d. The assay was conducted at a concentration of 10 μM and incubated for 48 h. The graph depicts the percentage survival of cells after treatment. (C) Disruption of HBL-100 cell morphology by 1o, 1p and 2b suggesting toxicity, whereas compounds 2c and 2d did not produce any morphological alteration towards HBL-100 cells, suggesting their non-cytotoxic potential.

2c and 2d, being the most promising anticancer molecules of the series, were chosen further to establish the mechanism of action as proposed by evaluating their ability to inhibit ATP-dependent phosphorylation of EGFR. The results suggested that 2c and 2d inhibited in vitro enzymatic activity of EGFR with IC50 of 617.33 ± 0.04 nM and 710 ± 0.05 nM, respectively, in comparison to the positive control, erlotinib (IC50 = 239.91 ± 0.05 nM) (Fig. 4).20
Fig. 4. (A) Dose–response curves showing percentage inhibition of phosphorylation in EGFR as induced by investigational compounds 2c and 2d in comparison with the positive control erlotinib. (B) The enzymatic inhibitory concentration (IC50) obtained as a consequence of inhibition in phosphorylation.

In order to achieve a low nM anti-EGFR IC50 value comparable to that of erlotinib, we utilised a molecular modelling tool to explore the unoccupied area of the ATP binding domain of EGFR and analysed an in silico interaction model of 2c and 2d–EGFR complexes that guided and allowed chain lengthening substitution of the 4-fluorophenyl ring (2c and 2d) with the 4-(4-methylpiperazinyl)-3-nitrophenyl) moiety at the N-9 position, which yielded compounds 3c and 3d, respectively (Fig. 5). As expected, the binding dock scores of 3c and 3d were higher than those of their corresponding parent compounds (2c; R isomer and 2d), and 3c was energetically more favoured in the EGFR binding site (dock score = –7.545) than 3d (dock score = –6.99). 3c maintained important and conserved interactions such as hydrogen bond formation (CONH2) with MET769 (1.8 Å), while a similar interaction in the case of 3d was weak and observed at a longer distance (2.4 Å). The N-methyl piperazinyl ring of 3c was found to be near the DFG motif (ASP831, PHE832 and GLY833), whereas the nitro group was observed to interact with ASP831. In addition to improvement in the binding affinity of the inhibitor, the N-methyl piperazinyl moiety could also potentiate the water solubility and bioavailability of 3c.21 Likewise, the –NO2 group upon bioreduction could selectively target hypoxic tumours by intercalating with DNA (in the case of hypoxic tumours);22 further it could be utilized upon conversion to NH2 and subsequent alkylation as a handle to design irreversible inhibitors of EGFR by making a covalent bond with remote cysteine residues.23
Fig. 5. Design of compounds based on chain lengthening of 2c and 2d. Designed compounds 3c and 3d were evaluated on the basis of the docking score and the important interactions; MET769 (H-bond) interaction was observed in 3c (1.8 Å) and 3d (2.4 Å).

Since 3c emerged to be the best fit, it was synthesized (Scheme 2) and assessed for anticancer evaluation in vitro.
Scheme 2. Synthesis of best fit compound 3c and reagents and conditions: (i) 4-fluoro-3-nitroaniline, aniline hydrochloride, EtOH, rt, 3 h, 82%, (ii) DBU, EtOH, rt, 2 h, 67%, (iii) 3,4-dimethoxybenzaldehyde, H2SO4, 1 drop MeOH, (iv) N-methyl piperazine, DMF, 50 °C, rt, 4 h, K2CO3.

3c was characterized by mp, IR, NMR and HRMS techniques. The 1H NMR spectrum showed characteristic amide NH2 protons as a singlet at δ 8.10. The protons of the N-methyl piperazine ring appeared as two singlets, each corresponding to 4 protons at δ 3.12 and δ 2.85, whereas N-methyl protons appeared at δ 2.21. The 13C NMR signal for carbonyl carbon of amide was observed at δ 165.04. The HRMS (TOF-ESI) spectrum confirmed the mass of the compound to be 519.2094 [M + H]+.
Compound 3c was anticancerous, induced cancer cell death by promoting oxidative stress in a mitochondrial independent manner, and was anti-EGFR
When tested against the same cancer cell lines, 3c inhibited their growth at a much lower concentration (IC50 = 1.98 to 4.07 μM; Table 2) as compared to its parent compound 2c (compare Table 1 with Table 2) and erlotinib, indicating its broad-spectrum anticancer activity. 3c was further assessed for its cytotoxicity against normal cells, i.e. HPBMCs and HBL-100 cells, and no noteworthy toxicity (Fig. 6A and B) was observed in 3c.263c was also able to elevate the ROS concentration inside MDA-MB-231 cancer cells when incubated for 48 h at a sub-IC50 concentration using the H2DCFDA assay (Fig. 6C and D). The alteration in ROS was measured and analysed using flow cytometer on FL2-H channel. Next, the JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide)-based mitochondrial membrane permeability assay indicated (Fig. 6E and F) that 3c caused an increase in the red/green ratio (membrane depolarization), suggesting the progression of cell death in a mitochondrial-independent pathway.27 Next, the effect of compound 3c on cell cycle progression using propidium iodide-based cell cycle analysis suggested that 3c induced cell cycle arrest at the sub-G1 phase (Fig. 6G and H). Furthermore, 3c inhibited in vitro the enzymatic activity of EGFR at a much lower concentration (Fig. 7A; compare with 2c) with an IC50 of 236.38 ± 0.04 nM which was comparable to that of the positive control, erlotinib (IC50 = 239.91 ± 0.05 nM) (Fig. 7B).
Table 2. Anti-proliferative effect of 3c on cancer cells.
| Inhibitory potential (IC50 (μM) ± SD)
a
of 3c | ||||||
| Entry | Breast cancer cells |
Lung cancer cells |
||||
| Code | MDAMB-231 | T47D | MCF-7 | A549 | H1299 | |
| 1 | 3c | 3.26 ± 0.11 | 4.07 ± 0.29 | 4.05 ± 0.22 | 1.98 ± 0.19 | 2.09 ± 0.24 |
| 2 | Erlotinib | 5.46 ± 0.19 | 9.8 (ref. 24) | >30.00 (ref. 25) | 1.04 ± 0.15 | 3.05 ± 0.23 |
aData are expressed as mean values ± SD of three independent experiments.
Fig. 6. MTT assay of 3c against HPBMC (A) and HBL100 cells (B). (C) and (D) depict the relative fluorescence altered by 3c under the influence of ROS generated in MDA-MB-231 cells as confirmed by the H2DCFDA-based assay and interpreted using a flow cytometer. (E) Quantification ratio of J-aggregates vs. J-monomers depicting mitochondrial membrane permeability as detected by JC-1 dye and analyzed via a flow cytometer. (F) The relative ratio of red/green (J-aggregates vs. monomers) altered by treatment with 3c incubated for 48 h (G) Graph depicting the effect of 3c on cell cycle progression in MDA-MB-231 cells after 48 h of treatment at sub-IC50 concentration. The result signifies sub-G1 arrest as identified using PI dye and analyzed using a flow cytometer. (H) Histogram shift defines the overlapping and similar pattern of arrest by 3c at the sub-G1 phase.

Fig. 7. (A) Dose–response curves showing the percentage inhibition of phosphorylation in EGFR induced by 3c and erlotinib. (B) The enzymatic inhibitory concentration (IC50) obtained as a consequence of inhibition in phosphorylation.

In order to explore the alternative anticancer mechanism(s) of 3c in addition to EGFR inhibition, we performed in silico binding studies of 3c with proteins which are mostly over-expressed in cancer such as Her-2, ERα, PR, etc. (Table S1 and Fig. S1: see the ESI†). However, the results were not promising and did not encourage us to carry out further in vitro binding studies. Cell cycle arrest at sub-G1 by 3c suggested that it could also be provoked by the inhibition of other mitogens such as CDK2, 4 or 6.28
However, experimental results suggested that 3c was not a strong inhibitor of CDKs (IC50 >500 nM with all CDKs, i.e. 2, 4, and 6), further endorsing EGFR inhibition as one of the major anticancer mechanisms.
Experimental section
General
Reagents of AR/GR quality were purchased from Sigma-Aldrich, S.D. Fine chemicals, Loba-Chemie Pvt. Ltd., and Sisco Research Laboratory and were used without further purification. A Mettler Toledo (for quantity measurements <10 mg) and a Sartorius analytical balance (BSA224S-CW) (for quantity measurements >10 mg) were used for weighing purposes. An SGW heating mantle, Tarson Spinot Digital, ILMVAC rotary digital evaporator, digital hot plate and NSW oven/vacuum oven were used during the course of the reaction. The progress of the reaction was monitored by TLC, using petroleum ether/ethyl acetate and chloroform/methanol as the mobile phase on pre-coated Merck TLC (TLC silica gel 60, F254) plates in a JSGW UV/fluorescence analysis cabinet and/or iodine chamber. Melting points were recorded on Stuart melting point apparatus (SMP-30) with open glass capillary tubes and were uncorrected. Infrared (IR) spectra of compounds were recorded with KBr on a Bruker FT-IR spectrophotometer. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded at Panjab University, Chandigarh, Indian Institute of Technology, Ropar, and the Central University of Rajasthan in CDCl3/DMSO-d6 on a Bruker Advance II (400 MHz)/or JEOL NMR spectrometer using TMS (δ = 0) as an internal standard. Mass spectra were recorded on a Shimadzu GCMS-QP2010 and DST-FIST funded GCMS instruments with EI mode at the Central Instrumentation Laboratory (CIL) and DPSN, the Central University of Punjab, and HRMS analysis was performed at IIT, Ropar (Punjab, India). The CDK assay was outsourced at Life Technologies SelectScreen™ Biochemical Profiling Lab.
Chemistry
N-(2-Amino-1,2-dicyano-vinyl)-formimidic acid ethyl ester (4)17
 To a suspension of 2,3-diaminomaleonitrile (3, 3 g, 27.75 mmol) in 1,4-dioxane (25 ml) was added triethyl orthoformate (4.5 mL). The reaction mixture was refluxed at 80 °C for 6 h (TLC). The dark brown mass formed after the evaporation of dioxane was extracted with diethyl ether (3 × 50 mL) and filtered. The yellow needle-shaped crystals 4 formed were used for the next step without any further purification. Yield: 82%; color: yellow; mp: 135 °C (reported 134–136 °C). IR (KBr, cm–1): 3418, 3390 (NH2 stretch), 2244, 2258 (CN stretch). 1H NMR (400 MHz, CDCl3, TMS = 0) δ: 1.37 (3H, t, J = 7.2 Hz), 4.29 (2H, q, J = 7.3 Hz), 7.99 (2H, s, D2O exchangeable NH2), 7.97 (1H, s).
To a suspension of 2,3-diaminomaleonitrile (3, 3 g, 27.75 mmol) in 1,4-dioxane (25 ml) was added triethyl orthoformate (4.5 mL). The reaction mixture was refluxed at 80 °C for 6 h (TLC). The dark brown mass formed after the evaporation of dioxane was extracted with diethyl ether (3 × 50 mL) and filtered. The yellow needle-shaped crystals 4 formed were used for the next step without any further purification. Yield: 82%; color: yellow; mp: 135 °C (reported 134–136 °C). IR (KBr, cm–1): 3418, 3390 (NH2 stretch), 2244, 2258 (CN stretch). 1H NMR (400 MHz, CDCl3, TMS = 0) δ: 1.37 (3H, t, J = 7.2 Hz), 4.29 (2H, q, J = 7.3 Hz), 7.99 (2H, s, D2O exchangeable NH2), 7.97 (1H, s).
5-Amino-1-(4-fluorophenyl)-1H-imidazole-4-carbonitrile (6)
 To a suspension of 4 (3 g, 21.42 mmol) in methanol (1.5 mL) were added p-fluoroaniline (1.85 ml, 17.14 mmol) and aniline hydrochloride (2.7 mg, 0.021 mmol). The reaction mixture was stirred for 3 h at rt (TLC). The precipitate so obtained (5) was filtered, washed with diethyl ether, dried and used for the next step without further purification. A suspension of 5 (1 g, 4.42 mmol) was stirred in 1 M KOH (15 mL) at rt for 6 h (TLC) to obtain 5-amino-1-(4-fluorophenyl)-1H-imidazole-4-carbonitrile (6). The crude product was extracted with EtOAc (3 × 20 mL) and further recrystallized from EtOAc to obtain the pure compound (6). Yield: 89%; color: brown solid; mp: 192–196 °C. IR (KBr, cm–1): 3334 and 3295 (NH2 stretch), 2211 (CN stretch), 1577 (NH2 bend), 1248 (C–F). 1H NMR (400 MHz, DMSO-d6) δ: 7.53–7.50 (2H, m), 7.42–7.38 (3H, m), 6.18 (2H, s, D2O exchangeable NH2). HRMS (TOF-ESI) calcd for C10H7FN4: 202.0655 [M]+; observed: 203.0719 [M + H]+.
To a suspension of 4 (3 g, 21.42 mmol) in methanol (1.5 mL) were added p-fluoroaniline (1.85 ml, 17.14 mmol) and aniline hydrochloride (2.7 mg, 0.021 mmol). The reaction mixture was stirred for 3 h at rt (TLC). The precipitate so obtained (5) was filtered, washed with diethyl ether, dried and used for the next step without further purification. A suspension of 5 (1 g, 4.42 mmol) was stirred in 1 M KOH (15 mL) at rt for 6 h (TLC) to obtain 5-amino-1-(4-fluorophenyl)-1H-imidazole-4-carbonitrile (6). The crude product was extracted with EtOAc (3 × 20 mL) and further recrystallized from EtOAc to obtain the pure compound (6). Yield: 89%; color: brown solid; mp: 192–196 °C. IR (KBr, cm–1): 3334 and 3295 (NH2 stretch), 2211 (CN stretch), 1577 (NH2 bend), 1248 (C–F). 1H NMR (400 MHz, DMSO-d6) δ: 7.53–7.50 (2H, m), 7.42–7.38 (3H, m), 6.18 (2H, s, D2O exchangeable NH2). HRMS (TOF-ESI) calcd for C10H7FN4: 202.0655 [M]+; observed: 203.0719 [M + H]+.
Representative procedure for the synthesis of compounds 1a–1j
A mixture of 6 (0.5 g, 2.48 mmol), 3,4,5-trimethoxybenzaldehyde (0.49 g, 2.48 mmol) and p-TsOH (0.042 g, 10 mol%) in methanol (10 mL) was stirred at rt for 2 h. The progress of the reaction was monitored by TLC, using a petroleum ether and ethyl acetate solvent system (4 : 1). The precipitate so obtained was filtered and washed with an excess of methanol. The compound was dried and further recrystallized from methanol.
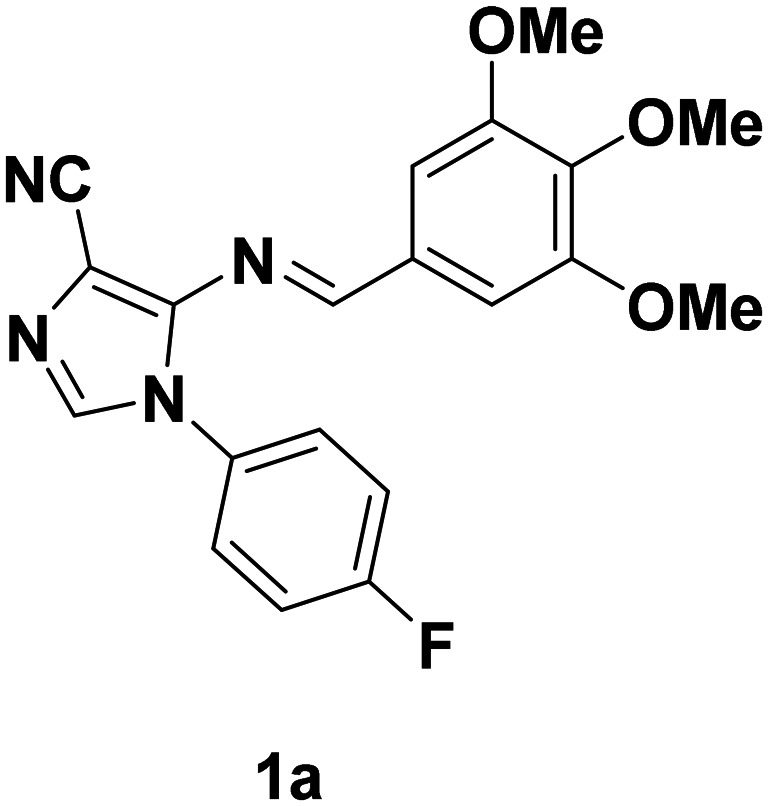
(E)-1-(4-Fluorophenyl)-5-((3,4,5-trimethoxybenzylidene)amino)-1H-imidazole-4 carbonitrile (1a)
 Yield: 82%; color: light yellow; mp: 120–122 °C. IR (KBr, cm–1): 2222 (CN), 1654 (C
N), 1236 (C–O), 1164 (C–F). 1H NMR (CDCl3, 400 MHz): 9.01 (1H, s), 7.67 (1H, s), 7.45–7.42 (2H, m), 7.27–7.21 (2H, m), 7.08 (2H, s), 3.93 (3H, s), 3.89 (6H, s). 13C NMR (100 MHz, CDCl3): 163.79, 161.35, 153.61, 146.94, 142.71, 136.41, 130.48, 130.34, 127.43, 127.34, 116.57, 116.34, 115.59, 106.57, 100.49, 61.17, 56.30. HRMS (TOF-ESI) calcd for C20H17FN4O3: 380.1285 [M]+; observed: 381.1366 [M + H]+.
Yield: 82%; color: light yellow; mp: 120–122 °C. IR (KBr, cm–1): 2222 (CN), 1654 (C
N), 1236 (C–O), 1164 (C–F). 1H NMR (CDCl3, 400 MHz): 9.01 (1H, s), 7.67 (1H, s), 7.45–7.42 (2H, m), 7.27–7.21 (2H, m), 7.08 (2H, s), 3.93 (3H, s), 3.89 (6H, s). 13C NMR (100 MHz, CDCl3): 163.79, 161.35, 153.61, 146.94, 142.71, 136.41, 130.48, 130.34, 127.43, 127.34, 116.57, 116.34, 115.59, 106.57, 100.49, 61.17, 56.30. HRMS (TOF-ESI) calcd for C20H17FN4O3: 380.1285 [M]+; observed: 381.1366 [M + H]+.
The mentioned procedure was further utilized for the rest of the compounds in the series. The physical data of the compounds are as follows:
(E)-5-((2,5-Dimethoxybenzylidene)amino)-1-(4-fluorophenyl)-1H-imidazole-4-carbonitrile (1b)
 Yield: 86%; color: bright yellow; mp: 125–127 °C. IR (KBr, cm–1): 2224 (CN), 1659 (C
N), 1221 (C–O), 1172 (C–F). 1H NMR (CDCl3, 400 MHz): 9.48 (1H, s), 7.66 (1H, s), 7.42–7.38 (3H, m), 7.22–7.16 (2H, m), 7.06–7.03 (1H, m), 6.89 (1H, m), 3.49 (3H, s), 3.12 (3H, s). 13C NMR (100 MHz, DMSO-d6): 189.59, 156.72, 153.57, 148.04, 133.19, 128.69, 128.58, 125.99, 124.76, 123.70, 117.52, 117.34, 117.11, 114.96, 110.82, 56.84. HRMS (TOF-ESI) calcd for C19H15FN4O2: 350.1179 [M]+; observed: 351.1250 [M + H]+.
Yield: 86%; color: bright yellow; mp: 125–127 °C. IR (KBr, cm–1): 2224 (CN), 1659 (C
N), 1221 (C–O), 1172 (C–F). 1H NMR (CDCl3, 400 MHz): 9.48 (1H, s), 7.66 (1H, s), 7.42–7.38 (3H, m), 7.22–7.16 (2H, m), 7.06–7.03 (1H, m), 6.89 (1H, m), 3.49 (3H, s), 3.12 (3H, s). 13C NMR (100 MHz, DMSO-d6): 189.59, 156.72, 153.57, 148.04, 133.19, 128.69, 128.58, 125.99, 124.76, 123.70, 117.52, 117.34, 117.11, 114.96, 110.82, 56.84. HRMS (TOF-ESI) calcd for C19H15FN4O2: 350.1179 [M]+; observed: 351.1250 [M + H]+.
(E)-1-(4-Fluorophenyl)-5-((4-nitrobenzylidene)amino)-1H-imidazole-4-carbonitrile (1c)
 Yield: 82%; color: yellow solid; mp: 112–114 °C. IR (KBr, cm–1): 2227 (CN), 1603 (C
N), 1519 (N
O), 1348 (N–O), 1116 (C–F).1H NMR (CDCl3, 400 MHz): 9.22 (1H, s), 8.29 (2H, d, J = 8 Hz), 7.96 (2H, d, J = 8 Hz), 7.71 (1H, s), 7.41–7.38 (2H, m), 7.29–7.24 (2H, m). HRMS (TOF-ESI) calcd for C17H10FN5O2: 335.0819 [M]+; observed: 336.0881 [M + H]+.
Yield: 82%; color: yellow solid; mp: 112–114 °C. IR (KBr, cm–1): 2227 (CN), 1603 (C
N), 1519 (N
O), 1348 (N–O), 1116 (C–F).1H NMR (CDCl3, 400 MHz): 9.22 (1H, s), 8.29 (2H, d, J = 8 Hz), 7.96 (2H, d, J = 8 Hz), 7.71 (1H, s), 7.41–7.38 (2H, m), 7.29–7.24 (2H, m). HRMS (TOF-ESI) calcd for C17H10FN5O2: 335.0819 [M]+; observed: 336.0881 [M + H]+.
(E)-1-(4-Fluorophenyl)-5-((4-hydroxy-3-methoxybenzylidene)amino)-1H-imidazole-4-carbonitrile (1d)
 Yield: 87% colour: dark yellow, mp: 105–107 °C. IR (KBr, cm–1): 3453 (OH), 2219 (CN), 1654 (C
N), 1215 (C–O), 1173 (C–F). 1H NMR (CDCl3, 400 MHz): 8.96 (1H, s), 7.61 (1H, s), 7.42–7.38 (3H, m), 7.22–7.18 (3H, m), 6.97 (1H, m), 6.08 (1H, s, D2O exchangeable OH), 3.89 (3H, s). 13C NMR (100 MHz, CDCl3): 164.04, 163.38, 160.88, 152.20, 148.14, 147.73, 136.05, 130.38, 127.13, 127.07, 126.82, 125.81, 116.26, 116.03, 115.97, 115.48, 110.29, 55.71. HRMS (TOF-ESI) calcd for C18H13FN4O2: 336.1023 [M]+; observed: 337.1081 [M + H]+.
Yield: 87% colour: dark yellow, mp: 105–107 °C. IR (KBr, cm–1): 3453 (OH), 2219 (CN), 1654 (C
N), 1215 (C–O), 1173 (C–F). 1H NMR (CDCl3, 400 MHz): 8.96 (1H, s), 7.61 (1H, s), 7.42–7.38 (3H, m), 7.22–7.18 (3H, m), 6.97 (1H, m), 6.08 (1H, s, D2O exchangeable OH), 3.89 (3H, s). 13C NMR (100 MHz, CDCl3): 164.04, 163.38, 160.88, 152.20, 148.14, 147.73, 136.05, 130.38, 127.13, 127.07, 126.82, 125.81, 116.26, 116.03, 115.97, 115.48, 110.29, 55.71. HRMS (TOF-ESI) calcd for C18H13FN4O2: 336.1023 [M]+; observed: 337.1081 [M + H]+.
(E)-1-(4-Fluorophenyl)-5-((4-hydroxybenzylidene)amino)-1H-imidazole-4-carbonitrile (1e)
 Yield: 78%; color: pale yellow; mp: 130–132 °C. IR (KBr, cm–1): 3449 (OH), 2224 (CN), 1666 (C
N), 1235 (C–O), 1166 (C–F). 1H NMR (CDCl3, 400 MHz): 9.66 (1H, s), 8.60 (1H, s), 7.38–7.35 (3H, m), 7.26–7.23 (2H, m), 7.41–7.38 (2H, m), 6.58–6.53 (2H, m). HRMS (TOF-ESI) calcd for C17H11FN4O: 306.0917 [M]+; observed: 307.0989 [M + H]+.
Yield: 78%; color: pale yellow; mp: 130–132 °C. IR (KBr, cm–1): 3449 (OH), 2224 (CN), 1666 (C
N), 1235 (C–O), 1166 (C–F). 1H NMR (CDCl3, 400 MHz): 9.66 (1H, s), 8.60 (1H, s), 7.38–7.35 (3H, m), 7.26–7.23 (2H, m), 7.41–7.38 (2H, m), 6.58–6.53 (2H, m). HRMS (TOF-ESI) calcd for C17H11FN4O: 306.0917 [M]+; observed: 307.0989 [M + H]+.
(E)-1-(4-Fluorophenyl)-5-((4-methoxybenzylidene)amino)-1H-imidazole-4-carbonitrile (1f)
 Yield: 85%, colour: yellow, mp: 134–136 °C. IR (KBr, cm–1): 1654 (C
N), 1247 (C–O). 1H NMR (CDCl3, 400 MHz): 9.00 (1H, s), 7.76 (2H, d, J = 8 Hz), 7.61 (1H, s), 7.42–7.38 (2H, m), 7.22–7.18 (2H, m), 6.94 (2H, d, J = 8 Hz), 3.86 (3H, s). 13C NMR (100 MHz, CDCl3): 164.41, 162.45, 161.25, 158.19, 153.70, 146.78, 145.72, 129.68, 129.17, 125.09, 125.00, 116.36, 116.13, 113.43, 54.93. MS (EI): m/z 319 [M – 1]+.
Yield: 85%, colour: yellow, mp: 134–136 °C. IR (KBr, cm–1): 1654 (C
N), 1247 (C–O). 1H NMR (CDCl3, 400 MHz): 9.00 (1H, s), 7.76 (2H, d, J = 8 Hz), 7.61 (1H, s), 7.42–7.38 (2H, m), 7.22–7.18 (2H, m), 6.94 (2H, d, J = 8 Hz), 3.86 (3H, s). 13C NMR (100 MHz, CDCl3): 164.41, 162.45, 161.25, 158.19, 153.70, 146.78, 145.72, 129.68, 129.17, 125.09, 125.00, 116.36, 116.13, 113.43, 54.93. MS (EI): m/z 319 [M – 1]+.
(E)-1-(4-Fluorophenyl)-5-((2-hydroxy-5-nitrobenzylidene)amino)-1H-imidazole-4-carbonitrile (1g)
 Yield: 84%, colour: creamish, mp: 128–130 °C. IR (KBr, cm–1): 2224 (CN), 1590 (C
N), 1451 (N
O), 1272 (N–O), 1070 (C–F). 1H NMR ((CDCl3 + DMSO-d6), 400 MHz): 10.77 (1H, s), 9.49 (1H, s), 7.97 (1H, d, J = 8 Hz), 7.83 (1H, s), 7.44–7.40 (2H, m), 7.26 (1H, d, J = 3 Hz), 7.21–7.15 (2H, m), 6.95–6.92 (1H, dd, J12 = 2.4 Hz, J13 = 8 Hz). HRMS (TOF-ESI) calcd for C17H10FN5O3: 351.0768 [M]+; observed: 352.0833 [M + H]+.
Yield: 84%, colour: creamish, mp: 128–130 °C. IR (KBr, cm–1): 2224 (CN), 1590 (C
N), 1451 (N
O), 1272 (N–O), 1070 (C–F). 1H NMR ((CDCl3 + DMSO-d6), 400 MHz): 10.77 (1H, s), 9.49 (1H, s), 7.97 (1H, d, J = 8 Hz), 7.83 (1H, s), 7.44–7.40 (2H, m), 7.26 (1H, d, J = 3 Hz), 7.21–7.15 (2H, m), 6.95–6.92 (1H, dd, J12 = 2.4 Hz, J13 = 8 Hz). HRMS (TOF-ESI) calcd for C17H10FN5O3: 351.0768 [M]+; observed: 352.0833 [M + H]+.
(E)-1-(4-Fluorophenyl)-5-((3-hydroxy-4-methoxybenzylidene)amino)-1H-imidazole-4-carbonitrile (1h)
 Yield: 82%, colour: off-white, mp: 110–112 °C. IR (KBr cm–1): 3452 (OH), 2222 (CN), 1654 (C
N), 1224 (C–O), 1021 (C–F). 1H NMR (CDCl3, 400 MHz): 8.98 (1H, s), 7.62 (1H, s), 7.44–7.39 (3H, m), 7.33–7.30 (1H, dd, J12 = 1.6 Hz, J13 = 8.5 Hz), 7.22–7.20 (2H, m), 6.91 (1H, d, J = 8 Hz), 5.72 (1H, s, D2O exchangeable OH), 3.99 (3H, s). 13C NMR (100 MHz, CDCl3): 164.09, 163.8, 160.74, 152.48, 147.58, 146.94, 136.17, 130.33, 128.04, 127.14, 127.05, 124.17, 116.34, 116.11, 113.88, 110.88, 99.32, 55.83. HRMS (TOF-ESI) calcd for C18H13FN4O2: 336.1023 [M]+; observed: 337.1094 [M + H]+.
Yield: 82%, colour: off-white, mp: 110–112 °C. IR (KBr cm–1): 3452 (OH), 2222 (CN), 1654 (C
N), 1224 (C–O), 1021 (C–F). 1H NMR (CDCl3, 400 MHz): 8.98 (1H, s), 7.62 (1H, s), 7.44–7.39 (3H, m), 7.33–7.30 (1H, dd, J12 = 1.6 Hz, J13 = 8.5 Hz), 7.22–7.20 (2H, m), 6.91 (1H, d, J = 8 Hz), 5.72 (1H, s, D2O exchangeable OH), 3.99 (3H, s). 13C NMR (100 MHz, CDCl3): 164.09, 163.8, 160.74, 152.48, 147.58, 146.94, 136.17, 130.33, 128.04, 127.14, 127.05, 124.17, 116.34, 116.11, 113.88, 110.88, 99.32, 55.83. HRMS (TOF-ESI) calcd for C18H13FN4O2: 336.1023 [M]+; observed: 337.1094 [M + H]+.
(E)-5-((3,4-Dimethoxybenzylidene)amino)-1-(4-fluorophenyl)-1H-imidazole-4-carbonitrile (1i)
 Yield: 82%, colour: cream, mp: 120–122 °C; IR (KBr, cm–1): 2222 (CN), 1659 (C
N), 1237 (C–O). 1H NMR (CDCl3, 400 MHz): 9.00 (1H, s), 7.63 (1H, s), 7.44–7.39 (3H, m), 7.38–7.36 (1H, m), 7.22–7.18 (2H, m), 6.94 (1H, d, J = 8 Hz), 3.99 (3H, s), 3.87 (3H, s). 13C NMR (100 MHz, DMSO-d6): 56.03, 56.39, 91.37, 110.04, 111.83, 117.07, 117.29, 117.56, 126.61, 128.12, 128.53, 128.63, 130.18, 133.14, 148.04, 149.72, 154.74, 163.63, 165.52. HRMS (TOF-ESI) calcd for C19H15FN4O: 350.1179 [M]+; observed: 351.1260 [M + H]+.
Yield: 82%, colour: cream, mp: 120–122 °C; IR (KBr, cm–1): 2222 (CN), 1659 (C
N), 1237 (C–O). 1H NMR (CDCl3, 400 MHz): 9.00 (1H, s), 7.63 (1H, s), 7.44–7.39 (3H, m), 7.38–7.36 (1H, m), 7.22–7.18 (2H, m), 6.94 (1H, d, J = 8 Hz), 3.99 (3H, s), 3.87 (3H, s). 13C NMR (100 MHz, DMSO-d6): 56.03, 56.39, 91.37, 110.04, 111.83, 117.07, 117.29, 117.56, 126.61, 128.12, 128.53, 128.63, 130.18, 133.14, 148.04, 149.72, 154.74, 163.63, 165.52. HRMS (TOF-ESI) calcd for C19H15FN4O: 350.1179 [M]+; observed: 351.1260 [M + H]+.
(E)-1-(4-Fluorophenyl)-5-((4-isopropylbenzylidene)amino)-1H-imidazole-4-carbonitrile (1j)
 Yield: 82%, colour: pale yellow, mp: 140–142 °C. IR (KBr, cm–1): 2223 (CN), 1653 (C
N), 1166 (C–F). 1H NMR (CDCl3, 400 MHz): 9.06 (1H, s), 7.75 (2H, d, J = 8 Hz), 7.64 (1H, s), 7.43–7.39 (2H, m), 7.32 (2H, d, J = 8 Hz), 7.24–7.19 (2H, m), 2.99–2.93 (1H, sep), 1.27 (3H, s), 1.25 (3H, s). MS (EI): m/z 332 [M]+.
Yield: 82%, colour: pale yellow, mp: 140–142 °C. IR (KBr, cm–1): 2223 (CN), 1653 (C
N), 1166 (C–F). 1H NMR (CDCl3, 400 MHz): 9.06 (1H, s), 7.75 (2H, d, J = 8 Hz), 7.64 (1H, s), 7.43–7.39 (2H, m), 7.32 (2H, d, J = 8 Hz), 7.24–7.19 (2H, m), 2.99–2.93 (1H, sep), 1.27 (3H, s), 1.25 (3H, s). MS (EI): m/z 332 [M]+.
Synthesis of 5-amino-1-(4-fluorobenzyl)-1H-imidazole-4-carbonitrile (8)
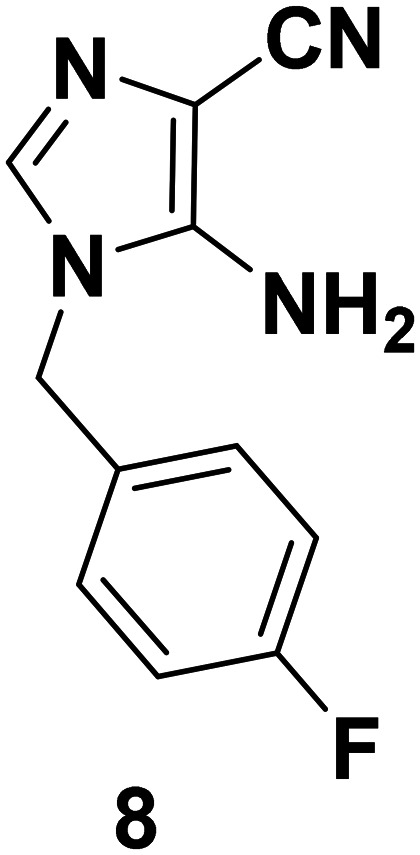 To a suspension of 4 (3 g, 21.42 mmol) in methanol (1.5 mL) were added p-fluorobenzylamine (1.85 ml, 17.14 mmol) and aniline hydrochloride (2.7 mg, 0.021 mmol). The reaction mixture was stirred at rt for 3 h. The precipitate so obtained was filtered, washed with diethyl ether and dried to afford 7 and used without any further purification. Yield: 78%. A suspension of 7 (1 g, 3.88 mmol) was stirred in 1 M KOH solution (15 mL) to obtain 5-amino-1-(4-fluorobenzyl)-1H-imidazole-4-carbonitrile (8). The reaction was monitored using TLC. The crude product was extracted with ethyl acetate (3 × 20 mL) and further recrystallized from ethyl acetate to obtain the pure compound (8). Yield: 68%; color: coffee brown solid; mp: 201–203 °C. IR (KBr, cm–1): 3395, 3321 (NH2 stretch), 2243 (CN stretch), 1561 (NH2 bend), 1124 (C–F). 1H NMR (400 MHz, CDCl3 + DMSO-d6) δ: 7.1–7.10 (2H, m), 6.97–6.93 (3H, m), 5.43 (2H, s, D2O exchangeable NH2), 4.93 (2H, s). 13C NMR (CDCl3 + DMSO-d6, 100 MHz): 163.27, 161.30, 147.23, 132.2, 132.22, 131.07, 129.27, 129.20, 116.54, 115.89, 115.72, 46.51. MS (EI): m/z 216 [M]+.
To a suspension of 4 (3 g, 21.42 mmol) in methanol (1.5 mL) were added p-fluorobenzylamine (1.85 ml, 17.14 mmol) and aniline hydrochloride (2.7 mg, 0.021 mmol). The reaction mixture was stirred at rt for 3 h. The precipitate so obtained was filtered, washed with diethyl ether and dried to afford 7 and used without any further purification. Yield: 78%. A suspension of 7 (1 g, 3.88 mmol) was stirred in 1 M KOH solution (15 mL) to obtain 5-amino-1-(4-fluorobenzyl)-1H-imidazole-4-carbonitrile (8). The reaction was monitored using TLC. The crude product was extracted with ethyl acetate (3 × 20 mL) and further recrystallized from ethyl acetate to obtain the pure compound (8). Yield: 68%; color: coffee brown solid; mp: 201–203 °C. IR (KBr, cm–1): 3395, 3321 (NH2 stretch), 2243 (CN stretch), 1561 (NH2 bend), 1124 (C–F). 1H NMR (400 MHz, CDCl3 + DMSO-d6) δ: 7.1–7.10 (2H, m), 6.97–6.93 (3H, m), 5.43 (2H, s, D2O exchangeable NH2), 4.93 (2H, s). 13C NMR (CDCl3 + DMSO-d6, 100 MHz): 163.27, 161.30, 147.23, 132.2, 132.22, 131.07, 129.27, 129.20, 116.54, 115.89, 115.72, 46.51. MS (EI): m/z 216 [M]+.
Representative procedure for the synthesis of compounds 1i–1r
To a suspension of 8 (0.1 g, 0.38 mmol, 1 equiv.) in methanol (10 ml), 3,4,5-trimethoxybenzaldehyde (0.074 g, 0.38 mmol, 1 equiv.) was added followed by addition of p-TsOH (0.006 g, 10 mol%). The reaction mixture was stirred for 2 h at rt. The completion of the reaction was confirmed by TLC, using a petroleum ether and ethyl acetate elution system (4 : 1). The precipitate so obtained was filtered and washed with an excess of methanol. The compound was dried and further recrystallized from methanol (1k).
(E)-1-(4-Fluorobenzyl)-5-((3,4,5-trimethoxybenzylidene)amino)-1H-imidazole-4-carbonitrile (1k)
 Yield: 84%; color: yellow color solid; mp: 159–161 °C. IR (KBr, cm–1): 2216 (CN), 1650 (C
N), 1238 (C–O), 1136 (C–F). 1H NMR (CDCl3, 400 MHz): 8.99 (1H, s), 7.46 (1H, s), 7.26–7.24 (2H, m), 7.16 (1H, s), 7.07–7.03 (3H, m), 5.22 (2H, s), 3.95 (3H, s), 3.94 (6H, s). HRMS (TOF-ESI) calcd for C21H19FN4O3: 394.1441 [M]+; observed: 395.1426 [M + H]+.
Yield: 84%; color: yellow color solid; mp: 159–161 °C. IR (KBr, cm–1): 2216 (CN), 1650 (C
N), 1238 (C–O), 1136 (C–F). 1H NMR (CDCl3, 400 MHz): 8.99 (1H, s), 7.46 (1H, s), 7.26–7.24 (2H, m), 7.16 (1H, s), 7.07–7.03 (3H, m), 5.22 (2H, s), 3.95 (3H, s), 3.94 (6H, s). HRMS (TOF-ESI) calcd for C21H19FN4O3: 394.1441 [M]+; observed: 395.1426 [M + H]+.
Using the above-mentioned procedure, the following compounds were synthesized. The physical data of the compounds are as follows:
(E)-5-((2,5-Dimethoxybenzylidene)amino)-1-(4-fluorobenzyl)-1H-imidazole-4-carbonitrile (1l)
 Yield: 81%; color: bright golden solid; mp: 178–180 °C. IR (KBr, cm–1): 2221 (CN), 1654 (C
N), 1224 (C–O), 1168 (C–F). 1H NMR (CDCl3, 400 MHz): 9.47 (1H, s), 7.57 (1H, s), 7.42 (1H, s), 7.24–7.21 (2H, m), 7.04–7.02 (3H, m), 6.92–6.89 (1H, m), 5.18 (2H, s) 3.88 (3H, s), 3.80 (3H, s). HRMS (TOF-ESI) calcd for C20H17FN4O2: 364.1336 [M]+; observed: 365.1409 [M + H]+.
Yield: 81%; color: bright golden solid; mp: 178–180 °C. IR (KBr, cm–1): 2221 (CN), 1654 (C
N), 1224 (C–O), 1168 (C–F). 1H NMR (CDCl3, 400 MHz): 9.47 (1H, s), 7.57 (1H, s), 7.42 (1H, s), 7.24–7.21 (2H, m), 7.04–7.02 (3H, m), 6.92–6.89 (1H, m), 5.18 (2H, s) 3.88 (3H, s), 3.80 (3H, s). HRMS (TOF-ESI) calcd for C20H17FN4O2: 364.1336 [M]+; observed: 365.1409 [M + H]+.
(E)-1-(4-Fluorobenzyl)-5-((4-nitrobenzylidene)amino)-1H-imidazole-4-carbonitrile (1m)
 Yield: 88%; color: orange color solid; mp: 110–112 °C; IR (KBr, cm–1): 2224 (CN), 1660 (C
N), 1517 (N
O), 1344 (N–O), 1228 (C–F). 1H NMR (CDCl3, 400 MHz): 9.03 (1H, s), 8.19 (2H, d, J = 8 Hz), 7.95–7.94 (2H, m), 7.49–7.46 (1H, m), 7.12–7.10 (2H, m), 6.90–6.87 (2H, m), 5.13 (2H, s). 13C NMR (CDCl3, 100 MHz): 160.26, 149.90, 145.08, 140.19, 137.58, 129.84, 129.55, 124.08, 116.11, 115.96, 115.56, 100.75, 89.73, 47.45. HRMS (TOF-ESI) calcd for C18H12FN5O2: 349.0975 [M]+; observed: 350.0986 [M + H]+.
Yield: 88%; color: orange color solid; mp: 110–112 °C; IR (KBr, cm–1): 2224 (CN), 1660 (C
N), 1517 (N
O), 1344 (N–O), 1228 (C–F). 1H NMR (CDCl3, 400 MHz): 9.03 (1H, s), 8.19 (2H, d, J = 8 Hz), 7.95–7.94 (2H, m), 7.49–7.46 (1H, m), 7.12–7.10 (2H, m), 6.90–6.87 (2H, m), 5.13 (2H, s). 13C NMR (CDCl3, 100 MHz): 160.26, 149.90, 145.08, 140.19, 137.58, 129.84, 129.55, 124.08, 116.11, 115.96, 115.56, 100.75, 89.73, 47.45. HRMS (TOF-ESI) calcd for C18H12FN5O2: 349.0975 [M]+; observed: 350.0986 [M + H]+.
(E)-1-(4-Fluorobenzyl)-5-((4-hydroxy-3-methoxybenzylidene)amino)-1H-imidazole-4-carbonitrile (1n)
 Yield: 79%; color: light grey; mp: 161–163 °C. IR (KBr, cm–1): 3451 (OH), 2218 (CN), 1654 (C
N), 1214 (C–O), 1158 (CF). 1H NMR (CDCl3, 400 MHz): 8.97 (1H, s), 7.44–7.43 (2H, m), 7.26–7.23 (4H, m), 7.06–7.02 (2H, m), 6.15 (1H, s), 5.21 (2H, s), 3.98 (3H, s). 13C NMR (100 MHz, DMSO-d6): 163.34, 152.21, 147.00, 146.91, 136.13, 11.46, 129.78, 129.71, 128.37, 124.19, 116.19, 115.96, 115.79, 113.88, 110.83, 55.98, 47.10. MS (EI): m/z 350 [M]+.
Yield: 79%; color: light grey; mp: 161–163 °C. IR (KBr, cm–1): 3451 (OH), 2218 (CN), 1654 (C
N), 1214 (C–O), 1158 (CF). 1H NMR (CDCl3, 400 MHz): 8.97 (1H, s), 7.44–7.43 (2H, m), 7.26–7.23 (4H, m), 7.06–7.02 (2H, m), 6.15 (1H, s), 5.21 (2H, s), 3.98 (3H, s). 13C NMR (100 MHz, DMSO-d6): 163.34, 152.21, 147.00, 146.91, 136.13, 11.46, 129.78, 129.71, 128.37, 124.19, 116.19, 115.96, 115.79, 113.88, 110.83, 55.98, 47.10. MS (EI): m/z 350 [M]+.
(E)-1-(4-Fluorobenzyl)-5-((4-methoxybenzylidene)amino)-1H-imidazole-4-carbonitrile (1o)
 Yield – 83%; color: dark yellow solid; mp: 125–127 °C. IR (KBr, cm–1): 2217 (CN), 1654 (C
N), 1221 (C–O), 1166 (C–F). 1H NMR (400 MHz, DMSO-d6) δ: 9.02 (1H, s), 7.90–7.88 (2H, d, J = 8 Hz), 7.43 (1H, s), 7.26–7.23 (2H, m), 7.06–7.00 (4H, m), 5.21 (2H, s), 3.90 (3H, s). 13C NMR (100 MHz, DMSO-d6) δ: 163.78, 163.55, 163.09, 161.68, 147, 136.04, 131.32, 129.69, 129.69, 128.05, 116.20, 116.03, 114.57, 99.36, 55.61, 47.28. HRMS (TOF-ESI) calcd for C19H15FN4O: 334.1229 [M]+; observed: 335.1236 [M + H]+.
Yield – 83%; color: dark yellow solid; mp: 125–127 °C. IR (KBr, cm–1): 2217 (CN), 1654 (C
N), 1221 (C–O), 1166 (C–F). 1H NMR (400 MHz, DMSO-d6) δ: 9.02 (1H, s), 7.90–7.88 (2H, d, J = 8 Hz), 7.43 (1H, s), 7.26–7.23 (2H, m), 7.06–7.00 (4H, m), 5.21 (2H, s), 3.90 (3H, s). 13C NMR (100 MHz, DMSO-d6) δ: 163.78, 163.55, 163.09, 161.68, 147, 136.04, 131.32, 129.69, 129.69, 128.05, 116.20, 116.03, 114.57, 99.36, 55.61, 47.28. HRMS (TOF-ESI) calcd for C19H15FN4O: 334.1229 [M]+; observed: 335.1236 [M + H]+.
(E)-1-(4-Fluorobenzyl)-5-((3-hydroxy-4-methoxybenzylidene)amino)-1H-imidazole-4-carbonitrile (1p)
 Yield: 83%; color: yellow solid; mp: 167–169 °C; IR (KBr, cm–1): 3450 (OH), 2220 (CN), 1654 (C
N), 1275 (C–O). 1H NMR (CDCl3, 400 MHz): 8.76 (1H, s), 8.23 (1H, brs), 7.42 (1H, s), 7.35 (1H, s), 7.28 (1H, s), 7.21–7.19 (2H, m), 7.15–7.12 (3H, m), 5.07 (2H, s), 3.83 (3H, s). 13C NMR (100 MHz, CDCl3): 163.34, 152.21, 147.00, 146.91, 136.13, 11.46, 129.78, 129.71, 128.37, 124.19, 116.19, 115.96, 115.79, 113.88, 110.83, 55.98, 47.10. HRMS (TOF-ESI) calcd for C19H15FN4O2: 350.1179 [M]+; observed: 351.1173 [M + H]+.
Yield: 83%; color: yellow solid; mp: 167–169 °C; IR (KBr, cm–1): 3450 (OH), 2220 (CN), 1654 (C
N), 1275 (C–O). 1H NMR (CDCl3, 400 MHz): 8.76 (1H, s), 8.23 (1H, brs), 7.42 (1H, s), 7.35 (1H, s), 7.28 (1H, s), 7.21–7.19 (2H, m), 7.15–7.12 (3H, m), 5.07 (2H, s), 3.83 (3H, s). 13C NMR (100 MHz, CDCl3): 163.34, 152.21, 147.00, 146.91, 136.13, 11.46, 129.78, 129.71, 128.37, 124.19, 116.19, 115.96, 115.79, 113.88, 110.83, 55.98, 47.10. HRMS (TOF-ESI) calcd for C19H15FN4O2: 350.1179 [M]+; observed: 351.1173 [M + H]+.
(E)-5-((3,4-Dimethoxybenzylidene)amino)-1-(4-fluorobenzyl)-1H-imidazole-4-carbonitrile (1q)
 Yield: 84%; color: light yellow solid; mp: 172–174 °C; IR (KBr, cm–1): 2217 (CN), 1654 (C
N), 1223 (C–O), 1167 (C–F). 1H NMR (CDCl3, 400 MHz): 8.97 (1H, s), 7.47–7.42 (3H, m), 7.24–7.23 (2H, m), 7.04 (2H, m), 6.95 (1H, d, J = 8 Hz), 5.19 (2H, s), 3.96 (3H, s), 3.94 (3H, s). 13C NMR (100 MHz, CDCl3): 163.78, 161.80, 149.15, 148.81, 145.53, 133.19, 129.81, 129.52, 129.49, 128.73, 128.66, 120.04, 117.00, 116.64, 116.47, 111.19, 111.13, 95.52, 55.94, 55.91, 49.30. HRMS (TOF-ESI) calcd for C20H17FN4O2: 364.1336 [M]+; observed: 365.1419 [M + H]+.
Yield: 84%; color: light yellow solid; mp: 172–174 °C; IR (KBr, cm–1): 2217 (CN), 1654 (C
N), 1223 (C–O), 1167 (C–F). 1H NMR (CDCl3, 400 MHz): 8.97 (1H, s), 7.47–7.42 (3H, m), 7.24–7.23 (2H, m), 7.04 (2H, m), 6.95 (1H, d, J = 8 Hz), 5.19 (2H, s), 3.96 (3H, s), 3.94 (3H, s). 13C NMR (100 MHz, CDCl3): 163.78, 161.80, 149.15, 148.81, 145.53, 133.19, 129.81, 129.52, 129.49, 128.73, 128.66, 120.04, 117.00, 116.64, 116.47, 111.19, 111.13, 95.52, 55.94, 55.91, 49.30. HRMS (TOF-ESI) calcd for C20H17FN4O2: 364.1336 [M]+; observed: 365.1419 [M + H]+.
(E)-1-(4-Fluorobenzyl)-5-((2,3,4-trimethoxybenzylidene)amino)-1H-imidazole-4-carbonitrile (1r)
 Yield: 85%; yellow color solid; mp: 158–160 °C; IR (KBr, cm–1): 2213 (CN), 1653 (C
N), 1217 (C–O), 1168 (C–F). 1H NMR (400 MHz, DMSO-d6): 9.35 (1H, s), 7.86 (1H, d, J = 7.2 Hz), 7.42 (1H, s), 7.26–7.22 (2H, m), 7.04 (2H, m), 6.80–6.87 (1H, m), 5.19 (2H, s), 4.03 (3H, s), 3.96 (3H, s), 3.90 (3H, s). 13C NMR (DMSO-d6, 100 MHz, δ with TMS = 0): 160.03, 158.26, 155.87, 147.92, 135.96, 129.67, 129.60, 122.88, 121.93, 116.19, 116.02, 107.93, 99.50, 62.50, 61.04, 56.26, 47.27. MS (EI): m/z 394 [M]+.
Yield: 85%; yellow color solid; mp: 158–160 °C; IR (KBr, cm–1): 2213 (CN), 1653 (C
N), 1217 (C–O), 1168 (C–F). 1H NMR (400 MHz, DMSO-d6): 9.35 (1H, s), 7.86 (1H, d, J = 7.2 Hz), 7.42 (1H, s), 7.26–7.22 (2H, m), 7.04 (2H, m), 6.80–6.87 (1H, m), 5.19 (2H, s), 4.03 (3H, s), 3.96 (3H, s), 3.90 (3H, s). 13C NMR (DMSO-d6, 100 MHz, δ with TMS = 0): 160.03, 158.26, 155.87, 147.92, 135.96, 129.67, 129.60, 122.88, 121.93, 116.19, 116.02, 107.93, 99.50, 62.50, 61.04, 56.26, 47.27. MS (EI): m/z 394 [M]+.
Synthesis of 5-amino-1-(4-fluorophenyl)-1H-imidazole-4-carbimidoyl cyanide (9)
 To a solution of 5 (1 g, 4.78 mmol) were added 1–2 drops of DBU to obtain 5-amino-1-(4-fluorophenyl)-1H-imidazole-4-carbimidoyl cyanide (9). The reaction mixture was stirred at rt for 2 h (TLC). The precipitate so obtained was filtered, washed with diethyl ether and dried to afford 9. Yield: 83%; color: apple green; mp: 120–122 °C. IR (KBr, cm–1): 2221 (CN stretch), 3421, 3321 (N–H), 1632 (C
N), 1220 (C–F). 1H NMR (400 MHz, DMSO-d6) δ: 11.08 (1H, s, NH), 7.52–7.50 (2H, m), 7.41–7.39 (3H, m), 6.67 (2H, s, NH2). 13C NMR (100 MHz, DMSO-d6) δ: 165.81, 163.53, 161.09, 148, 144.61, 143.76, 143.58, 132.69, 130.64, 128.16, 128.07, 117.38, 117.28. HRMS (TOF-ESI) calcd for C11H8FN5: 229.0764 [M]+; observed: 230.0837 [M + H]+.
To a solution of 5 (1 g, 4.78 mmol) were added 1–2 drops of DBU to obtain 5-amino-1-(4-fluorophenyl)-1H-imidazole-4-carbimidoyl cyanide (9). The reaction mixture was stirred at rt for 2 h (TLC). The precipitate so obtained was filtered, washed with diethyl ether and dried to afford 9. Yield: 83%; color: apple green; mp: 120–122 °C. IR (KBr, cm–1): 2221 (CN stretch), 3421, 3321 (N–H), 1632 (C
N), 1220 (C–F). 1H NMR (400 MHz, DMSO-d6) δ: 11.08 (1H, s, NH), 7.52–7.50 (2H, m), 7.41–7.39 (3H, m), 6.67 (2H, s, NH2). 13C NMR (100 MHz, DMSO-d6) δ: 165.81, 163.53, 161.09, 148, 144.61, 143.76, 143.58, 132.69, 130.64, 128.16, 128.07, 117.38, 117.28. HRMS (TOF-ESI) calcd for C11H8FN5: 229.0764 [M]+; observed: 230.0837 [M + H]+.
Representative procedure for the synthesis of compounds 2a–2d
To a mixture of 9 (0.1 g, 0.44 mmol, 1 equiv.) in methanol (5 ml) was added 3,4,5-trimethoxybenzaldehyde (1 equiv.) followed by addition of 1 drop of diluted sulfuric acid. The reaction mixture was stirred for 1 h at rt. The progress of the reaction was monitored by TLC, using a petroleum ether and ethyl acetate solvent system (2 : 1) as eluent. The precipitate so obtained was filtered and washed with excess methanol. The compound was dried and further recrystallized from methanol to afford pure 2a.
9-(4-Fluorophenyl)-2-(3,4,5-trimethoxyphenyl)-3,9-dihydro-2H-purine-6-carboxamide (2a)
 Yield: 76%; color: dark yellow; mp: 145–147 °C. IR (KBr, cm–1): 3355 and 3299 (NH2), 1643 (C
O), 1588 (C
N), 1228 (C–O), 1165 (C–F). 1H NMR (400 MHz, DMSO-d6) δ: 8.18 (1H, s), 8.15 (1H, s), 8.08 (1H, s), 7.91–7.88 (2H, m), 7.40–7.37 (1H, m), 7.32 (2H, m), 6.71 (2H, s), 6.1 (1H, s), 3.66 (6H, s), 3.58 (3H, s). 13C NMR (100 MHz, DMSO-d6) δ: 162.4, 161.45, 159.04, 157.91, 153.56, 153.04, 145.22, 139.74, 137.61, 133.80, 123.39, 123.31, 117.20, 116.98, 116.92, 116.64, 116.41, 103.86, 60.47, 56.56, 56.28. HRMS (TOF-ESI) calcd for C21H20FN5O4: 425.1499 [M]+; observed: 426.1589 [M + H]+; observed: 448.1589 [M + Na]+.
Yield: 76%; color: dark yellow; mp: 145–147 °C. IR (KBr, cm–1): 3355 and 3299 (NH2), 1643 (C
O), 1588 (C
N), 1228 (C–O), 1165 (C–F). 1H NMR (400 MHz, DMSO-d6) δ: 8.18 (1H, s), 8.15 (1H, s), 8.08 (1H, s), 7.91–7.88 (2H, m), 7.40–7.37 (1H, m), 7.32 (2H, m), 6.71 (2H, s), 6.1 (1H, s), 3.66 (6H, s), 3.58 (3H, s). 13C NMR (100 MHz, DMSO-d6) δ: 162.4, 161.45, 159.04, 157.91, 153.56, 153.04, 145.22, 139.74, 137.61, 133.80, 123.39, 123.31, 117.20, 116.98, 116.92, 116.64, 116.41, 103.86, 60.47, 56.56, 56.28. HRMS (TOF-ESI) calcd for C21H20FN5O4: 425.1499 [M]+; observed: 426.1589 [M + H]+; observed: 448.1589 [M + Na]+.
Using the above-mentioned protocol, the remaining compounds were synthesized. The physical data of the compounds are as follows:
9-(4-Fluorophenyl)-2-(4-methoxyphenyl)-9H-purine-6-carboxamide (2b)
 Yield: 65%; color: off white solid; mp: 115–117 °C. IR (KBr, cm–1): 3442, 3392 (NH2), 1658 (C
O), 1578 (C
N), 1245 (C–O), 1171 (C–F). 1H NMR (400 MHz, DMSO-d6) δ: 9.02 (1H, s), 8.48 (1H, s, CONH), 8.45–8.42 (2H, d, J = 8 Hz), 8.05 (1H, s, CONH), 8.03–7.98 (2H, m), 7.53–7.49 (2H, m), 7.05–7.03 (2H, m), 5.21 (2H, s), 3.80 (3H, s). 13C NMR (100 MHz, DMSO-d6) δ: 165.13, 163.05, 161.93, 160.6, 158.29, 154.38, 148.78, 147.30, 130.34, 129.69, 129.96, 126.42, 126.34, 117.19, 116.96, 114.52, 55.61. HRMS (TOF-ESI) calcd for C19H14FN5O2: 363.1131 [M]+; observed: 364.1280 [M + H]+; observed: 386.1087 [M + Na]+.
Yield: 65%; color: off white solid; mp: 115–117 °C. IR (KBr, cm–1): 3442, 3392 (NH2), 1658 (C
O), 1578 (C
N), 1245 (C–O), 1171 (C–F). 1H NMR (400 MHz, DMSO-d6) δ: 9.02 (1H, s), 8.48 (1H, s, CONH), 8.45–8.42 (2H, d, J = 8 Hz), 8.05 (1H, s, CONH), 8.03–7.98 (2H, m), 7.53–7.49 (2H, m), 7.05–7.03 (2H, m), 5.21 (2H, s), 3.80 (3H, s). 13C NMR (100 MHz, DMSO-d6) δ: 165.13, 163.05, 161.93, 160.6, 158.29, 154.38, 148.78, 147.30, 130.34, 129.69, 129.96, 126.42, 126.34, 117.19, 116.96, 114.52, 55.61. HRMS (TOF-ESI) calcd for C19H14FN5O2: 363.1131 [M]+; observed: 364.1280 [M + H]+; observed: 386.1087 [M + Na]+.
2-(3,4-Dimethoxyphenyl)-9-(4-fluorophenyl)-3,9-dihydro-2H-purine-6-carboxamide (2c)
 Yield: 62%; color: dark orange solid; mp: 133–135 °C. IR (KBr, cm–1): 3346, 3280 (NH2), 1651 (C
O), 1232 (C–O), 1171 (C–F). 1H NMR (400 MHz, DMSO-d6) δ: 8.14 (2H, s), 8.04 (1H, s), 7.93 (2H, s), 7.32 (2H, m), 7.03 (1H, s), 6.85 (2H, s), 6.12 (1H, s), 3.72 (3H, s), 3.68 (3H, s). 13C NMR (100 MHz, DMSO-d6) δ: 162.55, 148.96, 148.93, 145.02, 119.94, 118.77, 116.66, 116.54, 116.43, 111.77, 110.31, 110.26, 56.05, 55.92. HRMS (TOF-ESI) calcd for C20H18FN5O3: 395.1393 [M]+; observed: 396.1531 [M + H]+; observed: 418.1341 [M + Na]+.
Yield: 62%; color: dark orange solid; mp: 133–135 °C. IR (KBr, cm–1): 3346, 3280 (NH2), 1651 (C
O), 1232 (C–O), 1171 (C–F). 1H NMR (400 MHz, DMSO-d6) δ: 8.14 (2H, s), 8.04 (1H, s), 7.93 (2H, s), 7.32 (2H, m), 7.03 (1H, s), 6.85 (2H, s), 6.12 (1H, s), 3.72 (3H, s), 3.68 (3H, s). 13C NMR (100 MHz, DMSO-d6) δ: 162.55, 148.96, 148.93, 145.02, 119.94, 118.77, 116.66, 116.54, 116.43, 111.77, 110.31, 110.26, 56.05, 55.92. HRMS (TOF-ESI) calcd for C20H18FN5O3: 395.1393 [M]+; observed: 396.1531 [M + H]+; observed: 418.1341 [M + Na]+.
9-(4-Fluorophenyl)-2-(4-isopropylphenyl)-9H-purine-6-carboxamide (2d)
 Yield: 68%; color: off white solid; mp: 156–158 °C. IR (KBr, cm–1): 3313, 3299 (NH2), 1650 (C
O), 1548 (C
N), 1132 (C–F). 1H NMR (400 MHz, DMSO-d6) δ: 9.05 (1H, s), 8.47 (1H, s, CONH), 8.40 (2H, d, J = 8 Hz), 8.02 (1H, s, CONH), 8.01 (2H, m), 7.54–7.51 (2H, m), 7.36 (2H, d, J = 8 Hz), 2.95–2.91 (1H, sep), 1.20 (6H, d). 13C NMR (100 MHz, DMSO-d6) δ: 165.09, 163.08, 160.63, 158.44, 154.36, 151.74, 148.83, 147.56, 135.22, 130.49, 128.79, 127.12, 126.43, 126.35, 117.20, 116.97, 32.21, 24.22. HRMS (TOF-ESI) calcd for C21H18FN5O: 375.1495 [M]+; observed: 376.1566 [M + H]+.
Yield: 68%; color: off white solid; mp: 156–158 °C. IR (KBr, cm–1): 3313, 3299 (NH2), 1650 (C
O), 1548 (C
N), 1132 (C–F). 1H NMR (400 MHz, DMSO-d6) δ: 9.05 (1H, s), 8.47 (1H, s, CONH), 8.40 (2H, d, J = 8 Hz), 8.02 (1H, s, CONH), 8.01 (2H, m), 7.54–7.51 (2H, m), 7.36 (2H, d, J = 8 Hz), 2.95–2.91 (1H, sep), 1.20 (6H, d). 13C NMR (100 MHz, DMSO-d6) δ: 165.09, 163.08, 160.63, 158.44, 154.36, 151.74, 148.83, 147.56, 135.22, 130.49, 128.79, 127.12, 126.43, 126.35, 117.20, 116.97, 32.21, 24.22. HRMS (TOF-ESI) calcd for C21H18FN5O: 375.1495 [M]+; observed: 376.1566 [M + H]+.
Synthesis of 5-amino-1-(4-fluoro-3-nitrophenyl)-1H-imidazole-4-carbimidoylcyanide (11)
 To a suspension of 4 (3 g, 21.42 mmol) in methanol (10 mL) were added p-fluoro-3 nitroaniline (2.7 g, 17.30 mmol) and aniline hydrochloride (2.7 mg, 0.021 mmol). The reaction mixture was stirred for 3 h at rt. The precipitate so obtained was filtered, washed with diethyl ether and dried to afford 10 and was used without any further purification. Yield: 82%. To a solution of 10 (1 g, 3.64 mmol) were added 1–2 drops of DBU to obtain 5-amino-1-(4-fluorophenyl)-1H-imidazole-4-carbimidoyl cyanide (11). The reaction mixture was stirred for 2 h at rt. The precipitate so obtained was filtered, washed with diethyl ether and dried to afford 11, which was used for the next step without further purification. Yield: 67%; color: green color; mp: 120–122 °C. IR (KBr, cm–1): 3399 (N–H), 2280 (CN stretch), 1654 (C
N), 1538 (N
O), 1342 (N–O), 1261 (C–F). MS (EI): m/z 274 [M]+.
To a suspension of 4 (3 g, 21.42 mmol) in methanol (10 mL) were added p-fluoro-3 nitroaniline (2.7 g, 17.30 mmol) and aniline hydrochloride (2.7 mg, 0.021 mmol). The reaction mixture was stirred for 3 h at rt. The precipitate so obtained was filtered, washed with diethyl ether and dried to afford 10 and was used without any further purification. Yield: 82%. To a solution of 10 (1 g, 3.64 mmol) were added 1–2 drops of DBU to obtain 5-amino-1-(4-fluorophenyl)-1H-imidazole-4-carbimidoyl cyanide (11). The reaction mixture was stirred for 2 h at rt. The precipitate so obtained was filtered, washed with diethyl ether and dried to afford 11, which was used for the next step without further purification. Yield: 67%; color: green color; mp: 120–122 °C. IR (KBr, cm–1): 3399 (N–H), 2280 (CN stretch), 1654 (C
N), 1538 (N
O), 1342 (N–O), 1261 (C–F). MS (EI): m/z 274 [M]+.
Synthesis of 2-(3,4-dimethoxyphenyl)-9-(4-fluoro-3-nitrophenyl)-9H-purine-6-carboxamide (12)
 To a suspension of 11 (0.4 g, 1.45 mmol, 1 equiv.) in methanol (5 ml), 3,4-dimethoxybenzaldehyde (0.3 g, 1 equiv.) was added and the mixture was stirred at rt after the addition of 1 drop of H2SO4 for 1 h. The completion of the reaction was confirmed by TLC, and the product (12) was filtered and washed with methanol and was used for the next step without further purification. Yield: 85%; dark yellow color solid; mp: 172–174 °C; IR (KBr, cm–1): 2218 (CN), 1655 (C
O), 1512 (N
O), 1323 (N–O), 1216 (C–F). MS (EI): m/z 438 [M]+.
To a suspension of 11 (0.4 g, 1.45 mmol, 1 equiv.) in methanol (5 ml), 3,4-dimethoxybenzaldehyde (0.3 g, 1 equiv.) was added and the mixture was stirred at rt after the addition of 1 drop of H2SO4 for 1 h. The completion of the reaction was confirmed by TLC, and the product (12) was filtered and washed with methanol and was used for the next step without further purification. Yield: 85%; dark yellow color solid; mp: 172–174 °C; IR (KBr, cm–1): 2218 (CN), 1655 (C
O), 1512 (N
O), 1323 (N–O), 1216 (C–F). MS (EI): m/z 438 [M]+.
Synthesis of 2-(3,4-dimethoxyphenyl)-9-(4-(4-methylpiperazin-1-yl)-3-nitrophenyl)-9H-purine-6-carboxamide (3c)
 1-Methyl piperazine (0.42 mL, 4.378 mmol) and K2CO3 (2 equiv., 0.803 g) were added to 12 (200 mg, 2.91 mmol) dissolved in DMF (0.5 mL). The reaction mixture was stirred for 4 h (TLC) at 50 °C. After the completion of the reaction, the crude mixture was extracted with ethyl acetate (10 mL × 3) and water. The organic layer was evaporated under vacuum using a rotary evaporator to afford an orange colored compound which was recrystallized from methanol to afford pure 3c. Yield: 65%; color: orange solid; mp: 181–183 °C. IR (KBr, cm–1): 3320, 3291 (NH2), 1680 (C
O), 1620 (C
N), 1548 (N
O), 1372 (N–O). 1H NMR (400 MHz, DMSO-d6) δ: 9.15 (1H, s), 8.76 (1H, s), 8.53 (1H, s), 8.22–8.16 (2H, m), 8.10 (2H, s), 7.58 (1H, d, J = 8 Hz), 7.11 (1H, d, J = 8 Hz), 3.91 (3H, s), 3.85 (3H, s), 3.12 (4H, s), 2.85 (4H, s), 2.25 (3H, s). 13C NMR (100 MHz, DMSO-d6) δ: 165.04, 158.14, 154.05, 151.69, 149.20, 148.83, 146.69, 144.83, 141.94, 130.25, 129.83, 128.29, 127.80, 122.74, 122.04, 120.71, 111.92, 111.38, 56.06, 55.92, 54.90, 51.29, 46.17. HRMS (TOF-ESI) calcd for C25H26N8O5: 518.2026 [M]+; observed: 519.2094 [M + H]+.
1-Methyl piperazine (0.42 mL, 4.378 mmol) and K2CO3 (2 equiv., 0.803 g) were added to 12 (200 mg, 2.91 mmol) dissolved in DMF (0.5 mL). The reaction mixture was stirred for 4 h (TLC) at 50 °C. After the completion of the reaction, the crude mixture was extracted with ethyl acetate (10 mL × 3) and water. The organic layer was evaporated under vacuum using a rotary evaporator to afford an orange colored compound which was recrystallized from methanol to afford pure 3c. Yield: 65%; color: orange solid; mp: 181–183 °C. IR (KBr, cm–1): 3320, 3291 (NH2), 1680 (C
O), 1620 (C
N), 1548 (N
O), 1372 (N–O). 1H NMR (400 MHz, DMSO-d6) δ: 9.15 (1H, s), 8.76 (1H, s), 8.53 (1H, s), 8.22–8.16 (2H, m), 8.10 (2H, s), 7.58 (1H, d, J = 8 Hz), 7.11 (1H, d, J = 8 Hz), 3.91 (3H, s), 3.85 (3H, s), 3.12 (4H, s), 2.85 (4H, s), 2.25 (3H, s). 13C NMR (100 MHz, DMSO-d6) δ: 165.04, 158.14, 154.05, 151.69, 149.20, 148.83, 146.69, 144.83, 141.94, 130.25, 129.83, 128.29, 127.80, 122.74, 122.04, 120.71, 111.92, 111.38, 56.06, 55.92, 54.90, 51.29, 46.17. HRMS (TOF-ESI) calcd for C25H26N8O5: 518.2026 [M]+; observed: 519.2094 [M + H]+.
Biological evaluation
Cell culture
Cancer and normal cells were purchased from the National Cell Repository situated at NCCS, Pune. The cells were grown in DMEM containing 10% fetal bovine serum (FBS) and antibiotic solution (1× Penstrip, Invitrogen). The maintenance of cell growth was attained under standard conditions inside a CO2 incubator with 5% CO2 and 95% humidity at 37 °C. Cells were seeded at a density of between 1 and 3 × 104 cells per cm2 and sub-cultured using trypsin (trypsinisation) when 70–80% confluent.
2,3-(4,5-Dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay
The MTT assay was performed under normoxic conditions. The cells were counted using an automated cell counter and approximately 8 × 103 cells in 100 μl of DMEM were seeded in each well of 96 well plates.29 For human peripheral blood mononuclear cells (HPBMCs), RPMI medium was used instead of DMEM. Cells were further treated with the investigational compounds that were subjected to serial dilution (prior) in biological grade DMSO. The treatment was made at varying concentrations and for 48 h under standard conditions. After a specific time period the medium was discarded and MTT was added (10 μl MTT dye per well (5 mg ml–1 in PBS stock)). The cells were further incubated to allow for the formation of formazan; after the stipulated time of 4 h, the crystal was dissolved in DMSO and absorbance was read using a microplate reader at 570 nm. The results are presented as mean ± SD. The IC50 values of these interesting compounds were calculated. HPBMCs were cultured and used as per the protocol no. CUPB/cc/14/IEC/4483 approved by the Institutional Ethics Committee of the Central University of Punjab, Bathinda, according to guidelines issued by the Indian Council of Medical Research (ICMR), Govt. of India.
Reactive oxygen species (ROS) and mitochondrial membrane integrative assay
To perform ROS and mitochondrial permeability assay, H2DCFDA and JC-1 dyes were employed. Cells were cultured and treatment was given using the above-mentioned procedures. After the stipulated incubation time of 48 h, each dye was added separately and the fluorescence intensity was measured using a BD Accuri C6 flow cytometer.
Cell cycle analysis
Cells were cultured in 30 mm dishes and treated with the selected investigational compounds using the above-mentioned protocols. After the desired time period of 48 h, the media was removed and the cells were washed and trypsinized. Further, the cells were fixed using 70% ethanol to allow the passage of propidium iodide (PI) inside the cells. After fixing for 4 h at –20 °C, the cells were centrifuged (2200 rpm) and washed subsequently with 1X PBS. After this, RNAse was added and the cells were kept aside for 15 min; then PI was added to each vial and the cells were incubated for 30 min in the dark before analysing them in a flow cytometer.
Epidermal growth factor receptor (EGFR) inhibitory activity
The EGFR inhibitory activity of the investigational compounds was determined using a commercially available assay kit (z-Lyte kinase assay kit-tyr4 peptide (catalogue no. PV3193)). The assay was performed in accordance with the manufacturer's protocol and as per our previously published reports.20a,c The percentage inhibition of kinase was measured at Ex/Em 400, 445 and 520 nm using a microplate reader. The calculation to deduce emission ratio was done by dividing the coumarin emission (445 nm) by the fluorescein emission (520 nm).
The extent of phosphorylation was calculated using the given formula: where F100% = average fluorescein emission signal of the 100% Phos. Control; C100% = average coumarin emission signal of the 0% Phos. Control; C0% = average coumarin emission signal of the 100% Phos. Control; F0% = average fluorescein emission signal of the 0% Phos. Control.
where F100% = average fluorescein emission signal of the 100% Phos. Control; C100% = average coumarin emission signal of the 0% Phos. Control; C0% = average coumarin emission signal of the 100% Phos. Control; F0% = average fluorescein emission signal of the 0% Phos. Control.
Cyclin-dependent kinase assays
The CDK assay was outsourced at the Life Technologies SelectScreen™ Biochemical Profiling Lab, USA. The brief protocol is as follows:
For CDK2/cyclin A inhibitory assessment, a 2X CDK2/cyclin A/Ser/Thr 12 mixture was prepared in 50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MgCl2, and 1 mM EGTA. The final 10 μL kinase reaction mixture consisted of 1.22–10.3 ng CDK2/cyclin A and 2 μM Ser/Thr 12 in 50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MgCl2, and 1 mM EGTA. After the 1-hour kinase reaction incubation, 5 μL of a 1 : 4096 dilution of Development Reagent A was added.
For CDK4/cyclin D1 inhibitory assessment, a 2X CDK4/cyclin D1/Rb substrate mixture was prepared in 50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MnCl2, 1 mM EGTA, 2 mM DTT, and 0.02% NaN3. The final 10 μL kinase reaction mixture consisted of 7.5–30 ng CDK4/cyclin D1 and 1 μM Rb substrate in 32.5 mM HEPES pH 7.5, 0.005% BRIJ-35, 5 mM MnCl2, 0.5 mM EGTA, 1 mM DTT, and 0.01% NaN3. After the 1 hour kinase reaction incubation, 5 μL of detection mix was added. A similar protocol was followed for the CDK4/cyclin D3.
For CDK6/cyclin D1, a 2X CDK6/cyclin D1/Rb substrate mixture was prepared in 50 mM HEPES pH 7.5, 0.01% BRIJ-35, 10 mM MnCl2, 1 mM EGTA, 2 mM DTT, and 0.02% NaN3. The final 10 μL kinase reaction mixture consisted of 1.75–7 ng CDK6/cyclin D1 and 1 μM Rb substrate in 32.5 mM HEPES pH 7.5, 0.005% BRIJ-35, 5 mM MnCl2, 0.5 mM EGTA, 1 mM DTT, and 0.01% NaN3. After the 1 hour kinase reaction incubation, 5 μL of detection mix was added.
Molecular modelling studies
Molecular docking studies were carried out using the Maestro 11.4 molecular docking suite (Glide). All the designed ligands were imported in the Maestro-scratch project in a 2D format which were drawn using ChemDraw Professional. Ligands were prepared using Ligprep to convert in 3D format. The force field used was OPLS3 and all ionization states were generated at pH 7.0 ± 2.0. Tautomers were generated for each neutralised or ionized molecule. In the computation, specified chiralities were retained and generated, at the most 10 per ligand. The 3D structure of the EGFR was imported from the RCSB Protein Data Bank, PDB ID 1M17, using Protein Preparation Wizard in which the workspace structure was pre-processed where bond orders were assigned, the CCD database was used, hydrogens were added, and zero-order bonds to metal and disulphide bonds were created. Water beyond 5.00 Å was deleted and pre-processed. After pre-processing, the workspace was analysed and an active chain was selected in which prepared ligands were to be docked. Later on, the protein was refined which included optimization, removal of waters and finally restrained minimization. After the completion of ligand and protein preparation, receptor grid generation was done in which a grid is formed around an active site and a receptor-grid file is generated. Molecular docking was carried out by docking the prepared ligand molecule into the active ATP binding site of the minimized protein using the receptor grid file. The flexible docking was done using the extra precision mode and RMSD was computed for the input ligand(s).
Conclusion
In the current work, we emphasized target hopping of 11g along with molecular docking to design new fused imidazole derivatives. The rational design led to the synthesis of target molecules that were assessed for in vitro biological evaluation. Among all the imidazole-bearing compounds, two compounds 2c and 2d were found to possess selective anticancer potential against five cancer cell lines; they were devoid of toxicity towards normal cells and inhibited EGFR at the nM level. We further tuned the EGFR binding ability of the compounds by exploring a 3D interaction model of 2c and 2d–EGFR complexes and extending the chain length by substitution of the 4-fluorophenyl ring (2c and 2d) with 4-(4-methylpiperazinyl)-3-nitrophenyl) at the N-9 position, resulting in a best fit compound 3c with maximum binding interactions with residual amino acids. 3c was synthesized and found to possess more potent anti-EGFR and anticancer activities than the parent compound 2c and the positive control in addition to induction of oxidative stress in cancer cells and cell cycle arrest at the sub-G1 phase. Further, our efforts (in silico and in vitro) to explore the possibility of inhibition of other overexpressed proteins by 3c also suggested EGFR inhibition as one of the major anticancer mechanisms. We will be interested to determine the effect of 3c on mutated EGFR (T790M and L858R) and its efficacy in hypoxic lung cancer models (in vitro and in vivo) in the future and the results will be published in due course.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We are thankful to the Vice-Chancellor and Dean of Academic Affairs for providing the funds to support the present work. The authors thank CIL, CUPB, for data analysis. GJ thanks CSIR, New Delhi (Grant no. 05/1051(0011)/2018-EMR-I), MK thanks DST-SERB, New Delhi (Grant no. EMR/2017/002702/HS) and SA thanks Bristol Myers Squibb, USA (Grant No.53803645), for providing research fellowships. GC-MS analysis service using GC-MS equipment under DST-FIST support to the Department of Pharmaceutical Sciences and Natural Products is also duly acknowledged.
Footnotes
†Electronic supplementary information (ESI) available: Copies of spectra and docking results. See DOI: 10.1039/d0md00146e
References
- Siegel R. L., Miller K. D., Jemal A. Ca-Cancer J. Clin. 2019;69(1):7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- (a) Hanahan D., Weinberg R. A. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]; (b) Lazebnik Y. Nat. Rev. Cancer. 2010;10(4):232. doi: 10.1038/nrc2827. [DOI] [PubMed] [Google Scholar]
- (a) Rana A., Alex J. M., Chauhan M., Joshi G., Kumar R. Med. Chem. Res. 2015;24(3):903–920. [Google Scholar]; (b) Rowinsky E. K. Drugs. 2000;60(1):1–14. doi: 10.2165/00003495-200060001-00001. [DOI] [PubMed] [Google Scholar]
- Cohen P. Nat. Rev. Drug Discovery. 2002;1(4):309. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- Normanno N., De Luca A., Bianco C., Strizzi L., Mancino M., Maiello M. R., Carotenuto A., De Feo G., Caponigro F., Salomon D. S. Gene. 2006;366(1):2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- (a) Yewale C., Baradia D., Vhora I., Patil S., Misra A. Biomaterials. 2013;34(34):8690–8707. doi: 10.1016/j.biomaterials.2013.07.100. [DOI] [PubMed] [Google Scholar]; (b) Roskoski Jr R. Pharmacol. Res. 2014;79:34–74. doi: 10.1016/j.phrs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- (a) Mendelsohn J., Baselga J. J. Clin. Oncol. 2003;21(14):2787–2799. doi: 10.1200/JCO.2003.01.504. [DOI] [PubMed] [Google Scholar]; (b) Roeper J., Griesinger F. Curr. Opin. Oncol. 2019;31(1):1–7. doi: 10.1097/CCO.0000000000000495. [DOI] [PubMed] [Google Scholar]
- Liao B.-C., Lin C.-C., Yang J. C.-H. Curr. Opin. Oncol. 2015;27(2):94–101. doi: 10.1097/CCO.0000000000000164. [DOI] [PubMed] [Google Scholar]
- (a) Hossam M., Lasheen D. S., Abouzid K. A. Arch. Pharm. 2016;349(8):573–593. doi: 10.1002/ardp.201600063. [DOI] [PubMed] [Google Scholar]; (b) Lu X., Yu L., Zhang Z., Ren X., Smaill J. B., Ding K. Med. Res. Rev. 2018;38(5):1550–1581. doi: 10.1002/med.21488. [DOI] [PubMed] [Google Scholar]
- Ou S.-H. I. Crit. Rev. Oncol. Hematol. 2012;83(3):407–421. doi: 10.1016/j.critrevonc.2011.11.010. [DOI] [PubMed] [Google Scholar]
- Le T., Gerber D. E. Cancers. 2019;11(3):366. doi: 10.3390/cancers11030366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda M., Nakagawa K. Int. J. Mol. Sci. 2019;20(1):146. doi: 10.3390/ijms20010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C.-S., Kumarakulasinghe N. B., Huang Y.-Q., Ang Y. L. E., Choo J. R.-E., Goh B.-C., Soo R. A. Mol. Cancer. 2018;17(1):29. doi: 10.1186/s12943-018-0778-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Serrano A., Gella P., Jiménez E., Zugazagoitia J., Rodríguez L. P.-A. Drugs. 2018;78(9):893–911. doi: 10.1007/s40265-018-0916-4. [DOI] [PubMed] [Google Scholar]
- Juchum M., Günther M., Döring E., Sievers-Engler A., Lämmerhofer M., Laufer S. J. Med. Chem. 2017;60(11):4636–4656. doi: 10.1021/acs.jmedchem.7b00178. [DOI] [PubMed] [Google Scholar]
- Günther M., Juchum M., Kelter G., Fiebig H., Laufer S. Angew. Chem., Int. Ed. 2016;55(36):10890–10894. doi: 10.1002/anie.201603736. [DOI] [PubMed] [Google Scholar]
- Kumar R., Ujjinamatada R. K., Hosmane R. S. Org. Lett. 2008;10(20):4681–4684. doi: 10.1021/ol8020176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Kundu B., Sawant D., Chhabra R. J. Comb. Chem. 2005;7(2):317–321. doi: 10.1021/cc049851j. [DOI] [PubMed] [Google Scholar]; (b) Agarwal P. K., Sharma S. K., Sawant D., Kundu B. Tetrahedron. 2009;65(6):1153–1161. [Google Scholar]; (c) Mandadapu A. K., Saifuddin M., Agarwal P. K., Kundu B. Org. Biomol. Chem. 2009;7(13):2796–2803. doi: 10.1039/b905696c. [DOI] [PubMed] [Google Scholar]
- (a) Dai X., Cheng H., Bai Z., Li J. J. Cancer. 2017;8(16):3131. doi: 10.7150/jca.18457. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Radde B. N., Ivanova M. M., Mai H. X., Salabei J. K., Hill B. G., Klinge C. M. Biochem. J. 2015;465(1):49–61. doi: 10.1042/BJ20131608. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mooney L., Al-Sakkaf K., Brown B., Dobson P. Br. J. Cancer. 2002;87(8):909. doi: 10.1038/sj.bjc.6600541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Sawant D. M., Sharma S., Pathare R. S., Joshi G., Kalra S., Sukanya S., Maurya A. K., Metre R. K., Agnihotri V. K., Khan S. Chem. Commun. 2018;54(82):11530–11533. doi: 10.1039/c8cc05845h. [DOI] [PubMed] [Google Scholar]; (b) Chauhan M., Joshi G., Kler H., Kashyap A., Amrutkar S. M., Sharma P., Bhilare K. D., Banerjee U. C., Singh S., Kumar R. RSC Adv. 2016;6(81):77717–77734. [Google Scholar]; (c) Joshi G., Nayyar H., Kalra S., Sharma P., Munshi A., Singh S., Kumar R. Chem. Biol. Drug Des. 2017;90(5):995–1006. doi: 10.1111/cbdd.13027. [DOI] [PubMed] [Google Scholar]
- Wang P., Huang J., Wang K., Gu Y. Eur. J. Med. Chem. 2016;122:546–556. doi: 10.1016/j.ejmech.2016.07.020. [DOI] [PubMed] [Google Scholar]
- (a) Yang D., Tian H. Y., Zang T. N., Li M., Zhou Y., Zhang J. F. Sci. Rep. 2017;7(1):9174. doi: 10.1038/s41598-017-09525-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wilson W. R., Hay M. P. Nat. Rev. Cancer. 2011;11(6):393. doi: 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- (a) Hu C., Dong X. Curr. Med. Chem. 2019;26(31):5811–5824. doi: 10.2174/0929867325666180713124223. [DOI] [PubMed] [Google Scholar]; (b) Zhao Z., Bourne P. E. Drug Discovery Today. 2018;23(3):727–735. doi: 10.1016/j.drudis.2018.01.035. [DOI] [PubMed] [Google Scholar]
- Yamasaki F., Zhang D., Bartholomeusz C., Sudo T., Hortobagyi G. N., Kurisu K., Ueno N. T. Mol. Cancer Ther. 2007;6(8):2168–2177. doi: 10.1158/1535-7163.MCT-06-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindikar A., Deshpande G., Chaudhari U., Bhatia D., Joshi K. Journal of Pharmacology & Clinical Research. 2016;1(2):555557. [Google Scholar]
- Wu D., Yotnda P. J. Visualized Exp. 2011;(57):e3357. doi: 10.3791/3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Zorov D. B., Juhaszova M., Sollott S. J. Physiol. Rev. 2014;94(3):909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bonora M., Pinton P. Front. Oncol. 2014;4:302. doi: 10.3389/fonc.2014.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Ekholm S. V., Reed S. I. Curr. Opin. Cell Biol. 2000;12(6):676–684. doi: 10.1016/s0955-0674(00)00151-4. [DOI] [PubMed] [Google Scholar]; (b) Duronio R. J., Xiong Y. Cold Spring Harbor Perspect. Biol. 2013;5(3):a008904. doi: 10.1101/cshperspect.a008904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Mosmann T. J. Immunol. Methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]; (b) Sumantran V. N., Cellular chemosensitivity assays: An overview, in Cancer Cell Culture, Springer, 2011, pp. 219–236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.